

Abstract
The human body requires about 1-2 mg of iron per day for its normal functioning, and dietary iron is the only source for this essential metal. Since humans do not possess a mechanism for the active excretion of iron, the amount of iron in the body is determined by the amount absorbed across the proximal small intestine and, consequently, intestinal iron absorption is a highly regulated process. In recent years, the liver has emerged as a central regulator of both iron absorption and iron release from other tissues. It achieves this by secreting a peptide hormone called hepcidin that acts on the small intestinal epithelium and other cells to limit iron delivery to the plasma. Hepcidin itself is regulated in response to various systemic stimuli including variations in body iron stores, the rate of erythropoiesis, inflammation and hypoxia, the same stimuli that have been known for many years to modulate iron absorption. This review will summarize recent findings on the role played by the liver and hepcidin in the regulation of body iron absorption.
Keywords: Iron, Homeostasis, Intestinal iron absorption, Liver, Hepcidin
INTRODUCTION
Intestinal epithelial cells or enterocytes take up the predominating ferric iron from the diet through the combined action of an iron reductase (duodenal cytochrome B or DcytB is a strong candidate) and a ferrous iron transporter known as divalent metal-ion transporter (DMT1) on the brush border membrane[1,2]. Heme iron, on the other hand, appears to be absorbed through a separate system, and a recently identified apical membrane protein, HCP1, has emerged as a candidate heme transporter[3]. Irrespective of the form in which iron crosses the brush border, enterocytes export iron into the circulation by the combined action of an iron reductase, hephaestin, and a basolateral membrane iron transporter, ferroportin (FPN). The newly absorbed iron is then bound to circulating transferrin which distributes it around the body to sites of utilization and storage.
The amount of iron transported across the enterocytes is ultimately influenced by body iron requirements. Thus, for example, when body iron levels are low or when erythropoietic demand is increased, iron absorption is elevated. The factors that alter iron absorption exert their effects by influencing the duodenal expression of the major iron transport molecules, particularly DMT1, Dcytb1 and ferroportin[1,2,4]. Early kinetic studies suggested that it was the efflux of iron out of the enterocytes and into the circulation that was rate limiting for absorption[5], and more recent molecular studies have provided support for this concept[6,7]. In particular, this work suggested that basolateral iron transfer by ferroportin was most likely the primary regulatory step. But, how signals from distant sites modulate iron release from enterocytes has until recently remained poorly understood. The missing link in this regulatory pathway has now emerged as the liver-derived peptide hormone hepcidin, and thus we know that the liver plays a central role in the regulation of body iron homeostasis. Hepcidin regulates plasma iron levels by controlling the cell surface expression of ferroportin, and this in turn limits the efflux of iron from enterocytes, macrophages and a number of other cell types.
HEPCIDIN
Hepcidin was first discovered as an antimicrobial peptide in human blood ultrafiltrate[8] and urine samples[9]. The gene encoding hepcidin (HAMP) is very strongly expressed in the liver. But, weak expression has also been detected in heart, spinal cord, stomach, intestine and lungs[8-10]. The first evidence linking hepcidin to iron metabolism came almost simultaneously from two groups. One group, using suppressive subtractive hybridization, demonstrated that the levels of the hepcidin transcript were greatly increased in the livers of iron loaded mice[10]. The other group inadvertently engineered a mouse strain with negligible hepcidin expression (considered in more detail below), and found that these animals accumulated high levels of body iron[11]. HAMP was mapped to human chromosome 19, and it encodes an 84 amino acid pre-pro-peptide[9,10]. Pre-pro-hepcidin is ultimately processed into several smaller peptides that consist of the 20, 22 or 25 C-terminal amino acids[9]. The 25 amino acid peptide has eight cysteine residues forming four intramolecular disulfide bonds, and is the biologically active form of hepcidin[9,12]. The eight cysteine residues are highly conserved among species from zebra fish to humans (Figure 1). Two-dimensional nuclear magnetic resonance (NMR) spectroscopy showed that hepcidin forms a distorted hairpin-like structure, and NMR diffusion studies demonstrated that the 25 residue peptide forms aggregates involving the first five N-terminal residues[13]. The 20 and 22 amino acid forms, that lack the N-terminal residues, do not form aggregates and have much reduced iron regulatory capacity[12,13]. Removal of individual disulfide bonds did not reduce the hepcidin function significantly in vitro. However, serial deletions of the N-terminal residues progressively reduced hepcidin activity[12].
Figure 1.

Sequence alignment of hepcidin from various species.
The first animal model describing the relationship between hepcidin and iron homeostasis came serendipitously from the knockout of an adjacent gene encoding upstream stimulatory factor 2 (USF2)[11]. The USF2 knockout mice of this strain (Paris USF2) had very low levels of hepcidin transcript in the liver and developed multi-organ iron overload (but with relative sparing the spleen) and high transferrin saturation, a phenotype consistent with the human iron loading disease hereditary hemochromatosis[11]. These results provided the first indication that hepcidin was a negative regulator of iron uptake from the intestine and of iron release from macrophages. Another USF2 knockout mouse strain (Houston USF2)[14] had normal hepcidin levels, and showed no aberrations in iron metabolism[15], indicating that the Paris USF2 knockout phenotype was due to hepcidin deficiency. Since these original studies, a specific hepcidin knockout mouse has been generated, and it also shows an iron loading phenotype[16]. In contrast, mice over expressing hepcidin display decreased body iron levels and a microcytic hypochromic anaemia typical of severe iron deficiency[15]. The majority of these mice die within a few hours after birth suggesting that hepcidin also inhibits placental iron transport[15]. A similar situation has been described in humans, and patients with hepatic adenomas producing abnormally high levels of hepcidin suffer from a severe iron refractory anaemia that only resolved after resection of the tumour[17]. As might be expected from these results, injection of synthetic hepcidin peptide into mice leads to inhibition of intestinal iron absorption and consequently hypoferremia[18]. Taken together, these data provide strong evidence that hepcidin is the central regulator of body iron levels.
In humans, mutations in the HAMP gene result in a severe form of iron loading disease that presents at early age, and is aptly named juvenile hemochromatosis (JH). HAMP-associated JH is inherited in an autosomal recessive manner, and two mutations have been described (93delG and C166T) that are associated with iron loading when present in the homozygous state[13,19]. While heterozygosity for these mutations alone does not lead to iron loading, compound heterozygosity between two other HAMP mutations (Met50del IVS2+1(-G) and G71D) and C282Y, the most common mutation in patients with HFE-associated iron loading, has been reported to result in hemochromatosis[20]. Thus HAMP mutations may act as modifiers of the HFE-associated hemochromatosis phenotype.
Clearly hepcidin plays a major role in the regulation of intestinal iron absorption. But, how does it exert its effects? Soon after the link between hepcidin and iron was recognised, a close inverse correlation between HAMP expression and iron absorption and the expression of duodenal iron transporter transcripts was described[21]. It was suspected that hepcidin interacted with a receptor on the basolateral surface of the enterocytes, thereby activating one or more signal transduction pathways that ultimately led to changes in the expression of the iron transport genes. The truth turned out to be elegantly simple. Hepcidin acts by directly binding to the sole basolateral iron export molecule, ferroportin, and causing its internalisation and subsequent degradation[22]. Thus ferroportin is the hepcidin “receptor”. This loss of ferroportin on the cell surface reduces iron export from the cells leading to intracellular iron accumulation. As ferroportin is responsible for iron export from both enterocytes and macrophages, loss of this protein will result in reduced supply of iron to the plasma and, hence, will cause hypoferremia and, ultimately, anaemia. Consistent with this mechanism is the observation that mice lacking hepcidin show decreased iron in the spleen, an organ rich in macrophages, in the face of increased hepatic iron[15]. Similarly, J774 mouse macrophages treated with hepcidin peptide showed decreased levels of ferroportin and reduced the efflux of iron[23]. Hepcidin likely acts on iron export from other cell types, such as hepatocytes, in a similar fashion, and this can explain its key role in regulating iron traffic into and around the body.
Since hepcidin interacts with FPN, it might be expected that mutations in FPN that alter this interaction could essentially mimic hepcidin deficiency. This has been found to be the case. Two classes of FPN mutations have been identified in human subjects and both lead to iron loading. However, there are subtle differences in the phenotypes, with one being consistent with reduced iron transport by the protein and, the other consistent with impaired interaction with hepcidin[24].
SYSTEMIC FACTORS THAT REGULATE HEPCIDIN
Consistent with its role as a central regulator of body iron metabolism, hepcidin levels are modulated by the same factors that alter iron homeostasis. Changes in body iron stores, the rate of erythropoiesis, inflammation and hypoxia all influence iron absorption and iron release from macrophages and these are the major systemic factors that regulate HAMP mRNA levels in the liver.
Hepcidin levels are increased in response to oral and parenteral iron loading and decreased under iron deficient conditions[10]. This inverse relationship is seen with chronic changes in body iron status. But, it can also occur quite quickly, and HAMP mRNA levels in the liver can decrease within days of transferring rats from a control to iron deficient diet[21]. The regulation of hepcidin by body iron levels acts as a feedback mechanism to allow sufficient iron to enter the plasma when demand is high, but to limit iron intake/release in times of iron sufficiency. How hepcidin responds to changes in body iron levels is incompletely understood. Since hepcidin expression is largely restricted to the liver, it is highly likely that the hepatocyte is the site of action of the regulatory stimulus. But, whether hepatocyte iron levels per se play a primary role or whether an external signal is involved in unclear. This will be considered in more detail in the following section.
Interestingly, in vitro loading of hepatocyte cell lines and primary hepatocytes with iron decreases HAMP mRNA expression, the opposite effect to that seen in vivo[25]. Why this is the case has proved difficult to resolve. One possibility is that the iron supplied to the cultured cells is of a different form to that presented to the liver in vivo. This may be the case, but the in vitro reduction in hepcidin expression is seen when both iron salts and transferrin-bound iron is presented to the cells. A second possibility is that during the isolation and culture procedure liver-derived cells lose some factor that is critical for their normal physiological response to iron. Since the same effect is seen on freshly isolated primary hepatocytes this appears unlikely. But, it remains possible. Potentially the most satisfying explanation for the observations is that the response of the liver to iron in vivo requires the interaction of two or more types of cells, and that this interaction is lost after the cells are isolated. The liver macrophages or Kupffer cells are strong candidates for cells that might interact with hepatocytes to regulate iron homeostasis.But, several studies have now shown that when animals are depleted of macrophages (including Kupffer cells) their livers respond normally by increasing hepcidin expression in response to iron[26,27]. Thus it appears that macrophages are not required for hepcidin regulation in the liver in response to iron in vivo. A final possibility is that reduced expression of hepcidin is the normal physiological response of hepcidin to iron, and that the in vivo situation is complicated by a range of interacting stimuli that influence expression of the HAMP gene. Further work is required to resolve this issue.
The largest single sink for iron in the body is haemoglobin in the red blood cells, and consequently iron demand is closely linked to the rate of erythropoiesis. Thus when erythropoiesis is stimulated, following phenylhydrazine-induced hemolysis for example, hepcidin expression is suppressed to allow increased iron flow into the plasma and consequently to the developing red cells[15,28]. This hepcidin response is observed only in the presence of erythropoiesis as suppression of erythropoiesis by irradiation or by post-transfusion polycythemia leads to increased hepcidin levels[29,30]. The regulation of hepcidin mRNA levels by erythropoiesis is independent of direct erythropoietin effects[30], and is likely to reflect several stimuli. The iron requirement of the erythroid marrow is certainly a major factor. But, hypoxia too is also likely to be important as reduced haemoglobin may reduce oxygen delivery to the tissues. The response of hepcidin to hypoxia is considered in more detail below. In a recent study by Ganz and colleagues, it was concluded that in addition to iron requirements and hypoxia, there is an erythropoiesis-specific factor that affects hepcidin expression[30]. However, this factor has yet to be characterized.
Another situation where body iron homeostasis is perturbed is during inflammation or infection. Under these circumstances, iron absorption declines and iron is sequestered in macrophages, with the consequence that the plasma iron level is decreased (hypoferremia). With chronic inflammation or infection, anemia may result, and this condition is often called the anemia of chronic disease[31]. Consistent with the reduction in plasma iron is the demonstration that inflammatory stimuli positively regulate hepcidin levels[32-35]. Increased hepcidin means decreased iron entry into the plasma. That hepcidin is responsible for the hypoferremia accompanying inflammation has been shown by studies with Hamp null mice. These animals mount a standard inflammatory response to a stimulus such as bacterial lipopolysaccharide (LPS), but the expected hypoferremia does not occur[32].
One of the major mediators of the inflammatory response is the cytokine IL-6. IL-6 infusion in humans or administration to experimental animals leads to an increase in hepcidin production and decrease in serum iron levels within a few hours[36]. A time course analysis in human subjects injected with LPS revealed a strong temporal correlation between increases in serum IL-6 and urinary hepcidin, and the decrease in serum iron[34]. Similarly, IL-6, other pro-inflammatory cytokines like IL-1α and IL-1β and LPS stimulate hepcidin in primary hepatocytes and hepatoma cell lines[36,37].
Many inflammatory processes have a systemic component, and these are able to influence hepcidin expression in hepatocytes. However, there is increasing evidence that hepcidin may also be relevant in the local, extra-hepatic setting. For example, in an in vivo murine granulomatous pouch model of infection the host animals responded to bacterial infection by upregulating hepcidin at the local level, presumably to limit availability of iron to the pathogens in the immediate vicinity[35]. The cells responsible for the local production of hepcidin are unknown, but may be infiltrating macrophages and neutrophils. Indeed hepcidin expression has been detected in myeloid cells in response to systemic infection[38]. Hepcidin production also has been demonstrated in adipose tissue. But, again the responsible cell type is not known[39].
At the whole body level, hypoxia is usually associated with a reduced amount of circulating hemoglobin, and the body’s response to this deficiency is to stimulate erythropoiesis. This in turn requires an increased iron supply. It thus comes as no surprise that hepcidin levels drop 2-4 d after animals are placed in a hypoxic chamber[32], and that luminal iron uptake is increased in the small intestine under the same conditions[40]. Some of the in vivo effects of hypoxia cannot be attributed to the direct repression of hepcidin expression by low oxygen as increased erythropoiesis may reduce hepcidin levels by other mechanisms[30]. But, the demonstration that hypoxia down regulates hepcidin mRNA levels in human hepatoma cell lines[32] suggests that the HAMP gene itself may be regulated by hypoxia. In addition, hypoxia could also trigger a stress response in cells and animals, and HAMP is a known stress response gene[33].
The factors that regulate hepcidin levels described above vary in their relative strength, and in certain situations, where more than one stimulus is present, one may predominate. A good example of this is in β-thalassemia. In both mice and humans with this disorder, hepcidin levels are initially low despite increased levels of storage iron, and thus the erythropoietic stimulus is predominating[41-43]. However, as the disease advances the effect of the increasing iron stores become relatively stronger and hepcidin levels increase. A similar situation is found in hypotransferrinaemic mice[17] and in iron loaded animals treated with PHZ to induce anaemia and erythropoiesis[32] where the erythropoietic stimulus predominates.
THE MOLECULAR BASIS OF HEPCIDIN REGULATION
While the major physiological factors that alter hepcidin expression have been identified, how these stimuli signal the liver to alter hepcidin expression, and how the changes in expression are brought about is complex and only partially understood. Important clues in dissecting these regulatory pathways have come from the analysis of human iron loading disorders and their equivalent murine models. Patients with mutations in the HFE, transferrin receptor 2 (TfR2), hemojuvelin (HJV) or HAMP genes all show a histological pattern of iron deposition that is similar and consistent with elevated iron absorption. Furthermore, HFE, TfR2 and HAMP all show their highest expression in the liver and HJV shows strong expression in this organ. These similarities suggested that the proteins these genes encode may form part of the same regulatory pathway. This has now proved to be the case.
Classical, adult onset hereditary hemochromatosis (HH) results from mutations in the HFE gene, and both human patients and mouse strains with a disrupted HFE gene show periportal iron deposition with relative sparing of the Kupffer cells[44,45]. HFE is expressed on the cell surface as a complex with β2-microglobulin (β2m) and mouse models of β2m deficiency show a similar iron loading phenotype[46-48]. Since hepcidin expression is increased with iron loading, it was expected that disruption of the HFE gene would lead to enhanced hepcidin levels. When this was investigated, however, the opposite was seen. Human patients with HFE-associated hemochromatosis showed significantly lower levels of liver hepcidin expression than control subjects[49]. In Hfe knockout mice, hepcidin levels were similar to those of wild-type animals. But, the level remained low even when the knockout animals were fed a high iron diet[50]. Taken together, these observations show that HFE is an upstream regulator of hepcidin, and not a downstream target as was previously believed[49]. Proof of this was provided by Nicolas and colleagues who showed that mice over expressing hepcidin, but null at the Hfe locus, did not accumulate excess iron[51]. Despite low hepcidin levels in patients with HH, hepcidin levels still increase as the body iron load increases indicating that hepcidin regulation is not completely disrupted when HFE is mutated[49,52]. This is consistent with the milder phenotype of patients with HFE-associated HH compared to those with mutations in the HAMP gene[24].
If HFE acts as an upstream regulator of hepcidin, how does it respond to changes in body iron demand? The answer to this question is not clear. But, the level of circulating diferric transferrin has emerged as a potential regulator[53,54]. HFE is able to bind to TfR1, the major cell-surface diferric transferrin binding protein[55,56], and HFE and transferrin are able to compete for TfR1 binding. Such competition could modulate the amount of HFE that is not bound to TfR1 that in turn could transduce a signal to alter hepcidin expression. It is also possible that HFE interferes with the cellular uptake of transferrin-bound iron, and that this in turn affects HAMP expression[57]. Support for diferric transferrin as a signal to alter HAMP expression has come from the demonstration that another transferrin binding protein, TfR2, also is involved in hepcidin regulation.
Transferrin receptor 2 encodes a protein that shares 45% identity with TfR1[58]. However, in contrast to its widely expressed homolog, TfR2 expression is restricted to the liver, spleen, brain and heart, with highest expression in the liver[58]. Mutations in TfR2 lead to iron overload in human patients[59] with a clinical picture similar to classical HFE-associated hereditary hemochromatosis[60]. The same phenotype is seen in TfR2 knockout and mutant mice[61,62]. As in HFE-associated iron loading, hepcidin expression is decreased in the liver of TfR2 mutant mice[62,63], and patients with TfR2-related hemochromatosis also show decreased urinary hepcidin levels[64]. The severity of TfR2-related hemochromatosis is much less than that associated with mutations in HAMP or HJV, but is similar to HFE-associated hemochromatosis.
Like HFE, TfR2 appears to be an upstream regulator of hepcidin. But, how it exerts its effects is unknown. It has been reported that, unlike TfR1, TfR2 does not bind to HFE, and shows 25 times less affinity for transferrin compared to TfR1[65] making it unlikely that TfR2 makes a major contribution to cellular iron uptake. Indeed, in Tfr2 knockout mice, the liver accumulates iron very efficiently indicating that TfR2 is not essential for hepatic iron uptake. In contrast, a recent study has shown that when TfR2 and HFE are over expressed together in the same cells they can be co-immunoprecipitated[66]. Whether this interaction is physiologically relevant or represents an overexpression artefact remains to be determined. Interestingly, two independent studies have demonstrated that TfR2 protein levels are upregulated by diferric transferrin, but do not respond to apotransferrin or non-transferrin bound iron[67,68]. This effect of diferric transferrin is not observed at the transcript level, and appears to be post-translational[68]. Recently, it has been reported that the interaction between diferric transferrin and TfR2 activates ERK1/ERK2 and p38 MAP kinase, but only when TfR2 is present on the lipid rafts of the exosomes[69]. However, the precise mechanisms and signalling pathway of TfR2-hepcidin axis are not yet understood and remain to be further explored.
The third important protein known to be involved in the regulation of hepcidin is hemojuvelin (HJV, RGMc). HJV, is a member of the repulsive guidance molecule (RGM) family, and shares some common features with RGMa and RGMb, including a C-terminal glycosylphosphatidylinositol-linked membrane anchor (GPI-anchor), N-terminal signal sequence, proteolytic cleavage site and partial von Willebrand factor type D domain[70,71]. Mutations in the HJV gene cause severe iron overload, and lead to juvenile hemochromatosis due to greatly decreased hepcidin levels in human patients[72] and mice[73,74]. Moreover, knocking down HJV with siRNA in Hep3B cells leads to a decrease in hepcidin levels[74]. Interestingly, HJV expression is strongest in heart and skeletal muscle, but also shows moderate expression in the liver[72], where HAMP is most strongly expressed. Treating primary hepatocytes with soluble HJV led to a decrease in hepcidin levels suggesting that a binding competition exists between soluble and cell-associated hemojuvelin[75]. In addition, increasing iron concentrations led to a decrease in soluble HJV (sHJV) in cells over expressing HJV[75], indicating that iron could regulate HJV at the post-transcriptional level.
HJV acts as a co-receptor for the bone morphogenetic proteins (BMPs), in a similar fashion to other molecules of the Rgm family, and HJV mutants have impaired BMP signalling[76]. BMPs represent a subfamily of transforming growth factor-beta (TGF-β) ligands that signal by binding to and bringing together typeIand type II BMP receptors on the cell surface, and then propagating the signal through phosphorylation of the Smad proteins[77] (Figure 2). BMPs phosphorylate receptor-regulated Smads 1, 5 and 8 that in turn form heteromers with the co-mediator Smad 4. The activated complex then translocates to the nucleus and, in combination with other factors, regulates target genes such as HAMP[77]. Cells transfected with the co-receptor HJV or treated with the ligand BMP-2 showed increased levels of hepcidin, and BMP-2 induction was enhanced in the presence of HJV[76]. Other BMPs, BMP-4 and BMP-9, have been shown to have a similar effect on hepcidin expression independent of HFE and TfR2 status[78]. This suggests that HJV acts via a HFE/TfR2 independent pathway to alter HAMP levels. Further insight into this aspect of hepcidin regulation comes from the studies of the liver-specific Smad 4 knockout mouse. These animals showed markedly decreased levels of hepcidin, increased duodenal transporters and iron overload[79]. Overexpression of Smad 4 led to transcriptional activation of HAMP due to epigenetic modification of histone H3 protein[79]. Smad 4-deficient hepatocytes showed no increase in hepcidin levels upon treatment with BMP, iron, TGF-β or IL-6[79]. This response to BMP is expected as Smad 4 acts downstream in the signalling pathway[76]. However, the result with IL-6 is interesting as it indicates that the IL-6 pathway and the BMP-Smad pathway converge at some point. Overall, these data indicate that the BMP/SMAD pathway plays an important role in the regulation of HAMP gene expression. Whether this is the major pathway operating or other pathways are dominant remains to be determined, as the relationship between HFE, TfR2 and HJV (Figure 2).
Figure 2.
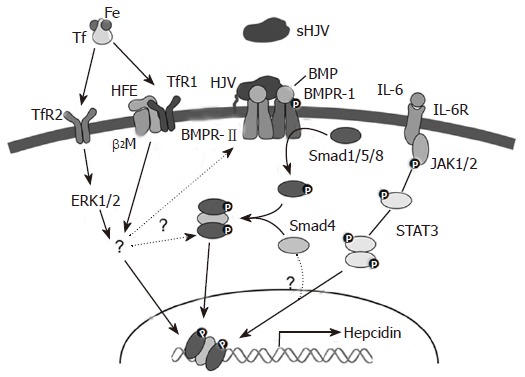
Signalling pathways for HAMP regulation.
Pro-inflammatory molecules like LPS and FCA positively regulate hepcidin expression by inducing the expression of cytokines such as IL-6. This induction of hepcidin by pro-inflammatory cytokines appears to be independent of HFE, β2m and TfR2[80,81], although one study provided evidence that these proteins may play some role[82]. The differences in these studies could be due to the difference in the timing and dosage of the treatments. IL-6 alters HAMP transcription through the classical JAK-STAT pathway[83,84]. This pathway ultimately leads to activation of STAT3 that binds to an element in the proximal 100 bases of the HAMP promoter. The interesting observation that the IL-6 dependent stimulation of the HAMP gene is abrogated in Smad 4 knockout mice indicates that Smad 4 is involved in the signalling process as well. This has yet to be investigated in detail, as have the mechanisms by which other pro-inflammatory cytokines stimulate HAMP expression.
Although it is widely considered that the HAMP gene is regulated predominantly at the level of transcription, relatively few promoter analyses have been carried out. As noted above, STAT3 is known to bind to the promoter, and several other transcription factors have been identified. C/EBPα, a liver-enriched transcription factor, has been shown to bind the HAMP promoter 230-250 bp upstream of the transcription start site[85], and may drive basal transcriptional activity of the gene. Supporting such a role, C/EBPα knockout mice showed decreased hepcidin levels and increased iron staining in their livers[85]. In another study, the role of the basic helix-loop-helix leucine zipper (bHLH-ZIP) family of transcriptional regulators, and notably USF2, in HAMP transcription was studied in order to investigate the reasons behind the lack of hepcidin expression in the USF2 knock out mouse. It was demonstrated using site-directed mutagenesis, chromatin immunoprecipitaton assays and mobility shift assays that USF1/USF2 and c-Myc/Max bind to E-boxes in the hepcidin promoter, and regulate its transcription[86]. Because some genes with E-boxes show rhythmicity, it has been proposed that hepcidin might also be under pulsatile or rhythmic transcriptional control. However, the signals and cellular pathways that lead to the activation of these proteins in the context of hepcidin regulation remain to be resolved.
A MODEL FOR THE LIVER-DEPENDENT REGULATION OF IRON HOMEOSTASIS
The discussion above has highlighted various factors that regulate hepcidin. and the current knowledge about the molecular mechanisms behind their effects. We previously proposed a model to explain the regulation of hepcidin in a physiological context[53], and present an updated version of the model here (Figure 3). In this model, signals that alter body iron homeostasis (iron stores, erythropoiesis, inflammation, hypoxia) act on the hepatocytes in the liver to modulate HAMP gene expression. Some of these stimuli may act directly on the liver cells (e.g. hypoxia), while others may act indirectly e.g. iron stores may act through changing the levels of diferric transferrin in the circulation. How the signals alter HAMP expression at the molecular level is incompletely understood. HFE, TfR2 and HJV are clearly involved in the process and, in the case of the latter; signalling through the BMP/SMAD pathway is a likely mode of action. Pro-inflammatory cytokines such as IL-6 act through the JAK/STAT pathway to regulate HAMP transcription. Hepcidin secreted by the liver acts on mature enterocytes in the proximal small intestine to reduce iron export into the circulation. Thus, high hepcidin means reduced iron absorption and vice versa. Hepcidin also acts on macrophages, hepatocytes, and likely other cells in the body to regulate their release of iron, so it plays a universal role in iron homeostasis.
Figure 3.
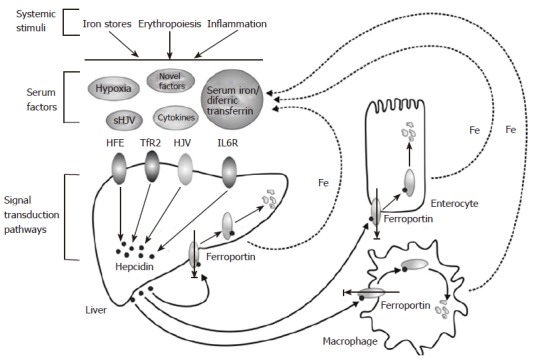
A model for the liver-dependent regulation of iron homeostasis.
Hepcidin levels show a positive correlation with transferrin saturation that is independent of the liver iron content[21] indicating that the levels of iron bound to transferrin may be a major signal that regulates the expression of hepcidin in the liver. Diferric transferrin is essential for iron delivery to the tissues making it suitable as an indicator of plasma iron status. Transferrin saturation thus decreases with iron deficiency and increases with iron loading. Diferric transferrin competes with HFE for binding to TfR1, and their binding sites overlap. But, it has a higher affinity for its principal receptor than HFE[87], and thus out-competes HFE for binding to TfR1. This leaves HFE “free” on the cell surface to initiate a signal to stimulate hepcidin expression. Similarly, diferric transferrin binds to TfR2, although with lower affinity, and sends a signal via the proposed ERK1/2 and MAPK pathways[69]. The signal transduction pathways driven by HFE and TfR2 that lead to the regulation of hepcidin expression are not yet clear. HFE and TfR2 may form a stable complex with each other as has been proposed[66] or may interact with HJV/BMPRs to propagate the signalling via the Smad1, 5, 8/Smad4 pathway. But, the precise details have yet to be elucidated.
In iron deficiency, TfR1 and transferrin levels increase, and transferrin saturation drops. The balance shifts towards monoferric transferrin that has lower binding affinity for TfR1 compared to diferric transferrin. This results in increased binding of HFE to TfR1, decreased signalling, and consequently lower hepcidin production. Similarly, as the plasma iron level falls, TfR1 binds residual diferric transferrin more efficiently than TfR2. This down regulates the TfR2-ERK signalling pathway, and thus hepcidin synthesis. When body iron levels are high the opposite pattern is seen, and hepcidin expression is increased. Stimulated erythropoiesis is another major stimulus for iron absorption, and at least one mechanism by which it might exert its effects on reducing hepcidin expression is via reduced transferrin saturation.
CONCLUSION
Recent developments in understanding the molecular mechanisms of iron homeostasis have greatly enhanced our knowledge of iron absorption in the gut. The most important advance in this area has been the recognition that the liver-derived peptide hepcidin responds to variations in body iron demand, and acts on the proximal small intestine to regulate iron efflux into the plasma. This shows that the liver plays a central role in the regulation of body iron homeostasis. Various systemic stimuli including iron stores, the rate of erythropoiesis, and oxygen levels regulate hepcidin expression, and consequently iron absorption. But, how systemic signals are received by the liver, and how these signals are transduced into changes in HAMP gene expression are incompletely understood. Strong evidence now suggests that signalling through the BMP/Smad pathway plays a major role in regulating hepcidin. But, how universal this pathway is has yet to be resolved.
Footnotes
S- Editor Liu Y L- Editor Alpini GD E- Editor Li JL
References
- 1.McKie AT, Barrow D, Latunde-Dada GO, Rolfs A, Sager G, Mudaly E, Mudaly M, Richardson C, Barlow D, Bomford A, et al. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science. 2001;291:1755–1759. doi: 10.1126/science.1057206. [DOI] [PubMed] [Google Scholar]
- 2.Gunshin H, Mackenzie B, Berger UV, Gunshin Y, Romero MF, Boron WF, Nussberger S, Gollan JL, Hediger MA. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature. 1997;388:482–488. doi: 10.1038/41343. [DOI] [PubMed] [Google Scholar]
- 3.Shayeghi M, Latunde-Dada GO, Oakhill JS, Laftah AH, Takeuchi K, Halliday N, Khan Y, Warley A, McCann FE, Hider RC, et al. Identification of an intestinal heme transporter. Cell. 2005;122:789–801. doi: 10.1016/j.cell.2005.06.025. [DOI] [PubMed] [Google Scholar]
- 4.McKie AT, Marciani P, Rolfs A, Brennan K, Wehr K, Barrow D, Miret S, Bomford A, Peters TJ, Farzaneh F, et al. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol Cell. 2000;5:299–309. doi: 10.1016/s1097-2765(00)80425-6. [DOI] [PubMed] [Google Scholar]
- 5.Anderson GJ, Vulpe CD. Regulation of intestinal iron transport. In: Templeton D, ed , editors. Molecular and Cellular Iron Transport. New York: Marcel Dekker; 2001. [Google Scholar]
- 6.Frazer DM, Wilkins SJ, Becker EM, Murphy TL, Vulpe CD, McKie AT, Anderson GJ. A rapid decrease in the expression of DMT1 and Dcytb but not Ireg1 or hephaestin explains the mucosal block phenomenon of iron absorption. Gut. 2003;52:340–346. doi: 10.1136/gut.52.3.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen H, Su T, Attieh ZK, Fox TC, McKie AT, Anderson GJ, Vulpe CD. Systemic regulation of Hephaestin and Ireg1 revealed in studies of genetic and nutritional iron deficiency. Blood. 2003;102:1893–1899. doi: 10.1182/blood-2003-02-0347. [DOI] [PubMed] [Google Scholar]
- 8.Krause A, Neitz S, Mägert HJ, Schulz A, Forssmann WG, Schulz-Knappe P, Adermann K. LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett. 2000;480:147–150. doi: 10.1016/s0014-5793(00)01920-7. [DOI] [PubMed] [Google Scholar]
- 9.Park CH, Valore EV, Waring AJ, Ganz T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem. 2001;276:7806–7810. doi: 10.1074/jbc.M008922200. [DOI] [PubMed] [Google Scholar]
- 10.Pigeon C, Ilyin G, Courselaud B, Leroyer P, Turlin B, Brissot P, Loréal O. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem. 2001;276:7811–7819. doi: 10.1074/jbc.M008923200. [DOI] [PubMed] [Google Scholar]
- 11.Nicolas G, Bennoun M, Devaux I, Beaumont C, Grandchamp B, Kahn A, Vaulont S. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc Natl Acad Sci USA. 2001;98:8780–8785. doi: 10.1073/pnas.151179498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nemeth E, Preza GC, Jung CL, Kaplan J, Waring AJ, Ganz T. The N-terminus of hepcidin is essential for its interaction with ferroportin: structure-function study. Blood. 2006;107:328–333. doi: 10.1182/blood-2005-05-2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hunter HN, Fulton DB, Ganz T, Vogel HJ. The solution structure of human hepcidin, a peptide hormone with antimicrobial activity that is involved in iron uptake and hereditary hemochromatosis. J Biol Chem. 2002;277:37597–37603. doi: 10.1074/jbc.M205305200. [DOI] [PubMed] [Google Scholar]
- 14.Sirito M, Lin Q, Deng JM, Behringer RR, Sawadogo M. Overlapping roles and asymmetrical cross-regulation of the USF proteins in mice. Proc Natl Acad Sci USA. 1998;95:3758–3763. doi: 10.1073/pnas.95.7.3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nicolas G, Bennoun M, Porteu A, Mativet S, Beaumont C, Grandchamp B, Sirito M, Sawadogo M, Kahn A, Vaulont S. Severe iron deficiency anemia in transgenic mice expressing liver hepcidin. Proc Natl Acad Sci USA. 2002;99:4596–4601. doi: 10.1073/pnas.072632499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lesbordes-Brion JC, Viatte L, Bennoun M, Lou DQ, Ramey G, Houbron C, Hamard G, Kahn A, Vaulont S. Targeted disruption of the hepcidin 1 gene results in severe hemochromatosis. Blood. 2006;108:1402–1405. doi: 10.1182/blood-2006-02-003376. [DOI] [PubMed] [Google Scholar]
- 17.Weinstein DA, Roy CN, Fleming MD, Loda MF, Wolfsdorf JI, Andrews NC. Inappropriate expression of hepcidin is associated with iron refractory anemia: implications for the anemia of chronic disease. Blood. 2002;100:3776–3781. doi: 10.1182/blood-2002-04-1260. [DOI] [PubMed] [Google Scholar]
- 18.Laftah AH, Ramesh B, Simpson RJ, Solanky N, Bahram S, Schümann K, Debnam ES, Srai SK. Effect of hepcidin on intestinal iron absorption in mice. Blood. 2004;103:3940–3944. doi: 10.1182/blood-2003-03-0953. [DOI] [PubMed] [Google Scholar]
- 19.Roetto A, Papanikolaou G, Politou M, Alberti F, Girelli D, Christakis J, Loukopoulos D, Camaschella C. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat Genet. 2003;33:21–22. doi: 10.1038/ng1053. [DOI] [PubMed] [Google Scholar]
- 20.Merryweather-Clarke AT, Cadet E, Bomford A, Capron D, Viprakasit V, Miller A, McHugh PJ, Chapman RW, Pointon JJ, Wimhurst VL, et al. Digenic inheritance of mutations in HAMP and HFE results in different types of haemochromatosis. Hum Mol Genet. 2003;12:2241–2247. doi: 10.1093/hmg/ddg225. [DOI] [PubMed] [Google Scholar]
- 21.Frazer DM, Wilkins SJ, Becker EM, Vulpe CD, McKie AT, Trinder D, Anderson GJ. Hepcidin expression inversely correlates with the expression of duodenal iron transporters and iron absorption in rats. Gastroenterology. 2002;123:835–844. doi: 10.1053/gast.2002.35353. [DOI] [PubMed] [Google Scholar]
- 22.Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 23.Knutson MD, Oukka M, Koss LM, Aydemir F, Wessling-Resnick M. Iron release from macrophages after erythrophagocytosis is up-regulated by ferroportin 1 overexpression and down-regulated by hepcidin. Proc Natl Acad Sci USA. 2005;102:1324–1328. doi: 10.1073/pnas.0409409102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pietrangelo A. Hereditary hemochromatosis. Biochim Biophys Acta. 2006;1763:700–710. doi: 10.1016/j.bbamcr.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 25.Gehrke SG, Kulaksiz H, Herrmann T, Riedel HD, Bents K, Veltkamp C, Stremmel W. Expression of hepcidin in hereditary hemochromatosis: evidence for a regulation in response to the serum transferrin saturation and to non-transferrin-bound iron. Blood. 2003;102:371–376. doi: 10.1182/blood-2002-11-3610. [DOI] [PubMed] [Google Scholar]
- 26.Lou DQ, Lesbordes JC, Nicolas G, Viatte L, Bennoun M, Van Rooijen N, Kahn A, Renia L, Vaulont S. Iron- and inflammation-induced hepcidin gene expression in mice is not mediated by Kupffer cells in vivo. Hepatology. 2005;41:1056–1064. doi: 10.1002/hep.20663. [DOI] [PubMed] [Google Scholar]
- 27.Montosi G, Corradini E, Garuti C, Barelli S, Recalcati S, Cairo G, Valli L, Pignatti E, Vecchi C, Ferrara F, et al. Kupffer cells and macrophages are not required for hepatic hepcidin activation during iron overload. Hepatology. 2005;41:545–552. doi: 10.1002/hep.20620. [DOI] [PubMed] [Google Scholar]
- 28.Frazer DM, Inglis HR, Wilkins SJ, Millard KN, Steele TM, McLaren GD, McKie AT, Vulpe CD, Anderson GJ. Delayed hepcidin response explains the lag period in iron absorption following a stimulus to increase erythropoiesis. Gut. 2004;53:1509–1515. doi: 10.1136/gut.2003.037416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vokurka M, Krijt J, Sulc K, Necas E. Hepcidin mRNA levels in mouse liver respond to inhibition of erythropoiesis. Physiol Res. 2006;55:667–674. doi: 10.33549/physiolres.930841. [DOI] [PubMed] [Google Scholar]
- 30.Pak M, Lopez MA, Gabayan V, Ganz T, Rivera S. Suppression of hepcidin during anemia requires erythropoietic activity. Blood. 2006;108:3730–3735. doi: 10.1182/blood-2006-06-028787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Means RT. Hepcidin and cytokines in anaemia. Hematology. 2004;9:357–362. doi: 10.1080/10245330400018540. [DOI] [PubMed] [Google Scholar]
- 32.Nicolas G, Chauvet C, Viatte L, Danan JL, Bigard X, Devaux I, Beaumont C, Kahn A, Vaulont S. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest. 2002;110:1037–1044. doi: 10.1172/JCI15686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nemeth E, Valore EV, Territo M, Schiller G, Lichtenstein A, Ganz T. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood. 2003;101:2461–2463. doi: 10.1182/blood-2002-10-3235. [DOI] [PubMed] [Google Scholar]
- 34.Kemna E, Pickkers P, Nemeth E, van der Hoeven H, Swinkels D. Time-course analysis of hepcidin, serum iron, and plasma cytokine levels in humans injected with LPS. Blood. 2005;106:1864–1866. doi: 10.1182/blood-2005-03-1159. [DOI] [PubMed] [Google Scholar]
- 35.Motley ST, Morrow BJ, Liu X, Dodge IL, Vitiello A, Ward CK, Shaw KJ. Simultaneous analysis of host and pathogen interactions during an in vivo infection reveals local induction of host acute phase response proteins, a novel bacterial stress response, and evidence of a host-imposed metal ion limited environment. Cell Microbiol. 2004;6:849–865. doi: 10.1111/j.1462-5822.2004.00407.x. [DOI] [PubMed] [Google Scholar]
- 36.Nemeth E, Rivera S, Gabayan V, Keller C, Taudorf S, Pedersen BK, Ganz T. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest. 2004;113:1271–1276. doi: 10.1172/JCI20945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee P, Peng H, Gelbart T, Wang L, Beutler E. Regulation of hepcidin transcription by interleukin-1 and interleukin-6. Proc Natl Acad Sci USA. 2005;102:1906–1910. doi: 10.1073/pnas.0409808102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peyssonnaux C, Zinkernagel AS, Datta V, Lauth X, Johnson RS, Nizet V. TLR4-dependent hepcidin expression by myeloid cells in response to bacterial pathogens. Blood. 2006;107:3727–3732. doi: 10.1182/blood-2005-06-2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bekri S, Gual P, Anty R, Luciani N, Dahman M, Ramesh B, Iannelli A, Staccini-Myx A, Casanova D, Ben Amor I, et al. Increased adipose tissue expression of hepcidin in severe obesity is independent from diabetes and NASH. Gastroenterology. 2006;131:788–796. doi: 10.1053/j.gastro.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 40.Leung PS, Srai SK, Mascarenhas M, Churchill LJ, Debnam ES. Increased duodenal iron uptake and transfer in a rat model of chronic hypoxia is accompanied by reduced hepcidin expression. Gut. 2005;54:1391–1395. doi: 10.1136/gut.2004.062083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Adamsky K, Weizer O, Amariglio N, Breda L, Harmelin A, Rivella S, Rachmilewitz E, Rechavi G. Decreased hepcidin mRNA expression in thalassemic mice. Br J Haematol. 2004;124:123–124. doi: 10.1046/j.1365-2141.2003.04734.x. [DOI] [PubMed] [Google Scholar]
- 42.Weizer-Stern O, Adamsky K, Amariglio N, Rachmilewitz E, Breda L, Rivella S, Rechavi G. mRNA expression of iron regulatory genes in beta-thalassemia intermedia and beta-thalassemia major mouse models. Am J Hematol. 2006;81:479–483. doi: 10.1002/ajh.20549. [DOI] [PubMed] [Google Scholar]
- 43.Papanikolaou G, Tzilianos M, Christakis JI, Bogdanos D, Tsimirika K, MacFarlane J, Goldberg YP, Sakellaropoulos N, Ganz T, Nemeth E. Hepcidin in iron overload disorders. Blood. 2005;105:4103–4105. doi: 10.1182/blood-2004-12-4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dupic F, Fruchon S, Bensaid M, Borot N, Radosavljevic M, Loreal O, Brissot P, Gilfillan S, Bahram S, Coppin H, et al. Inactivation of the hemochromatosis gene differentially regulates duodenal expression of iron-related mRNAs between mouse strains. Gastroenterology. 2002;122:745–751. doi: 10.1053/gast.2002.31877. [DOI] [PubMed] [Google Scholar]
- 45.Herrmann T, Muckenthaler M, van der Hoeven F, Brennan K, Gehrke SG, Hubert N, Sergi C, Gröne HJ, Kaiser I, Gosch I, et al. Iron overload in adult Hfe-deficient mice independent of changes in the steady-state expression of the duodenal iron transporters DMT1 and Ireg1/ferroportin. J Mol Med (Berl) 2004;82:39–48. doi: 10.1007/s00109-003-0508-x. [DOI] [PubMed] [Google Scholar]
- 46.de Sousa M, Reimão R, Lacerda R, Hugo P, Kaufmann SH, Porto G. Iron overload in beta 2-microglobulin-deficient mice. Immunol Lett. 1994;39:105–111. doi: 10.1016/0165-2478(94)90094-9. [DOI] [PubMed] [Google Scholar]
- 47.Rothenberg BE, Voland JR. beta2 knockout mice develop parenchymal iron overload: A putative role for class I genes of the major histocompatibility complex in iron metabolism. Proc Natl Acad Sci USA. 1996;93:1529–1534. doi: 10.1073/pnas.93.4.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Muckenthaler MU, Rodrigues P, Macedo MG, Minana B, Brennan K, Cardoso EM, Hentze MW, de Sousa M. Molecular analysis of iron overload in beta2-microglobulin-deficient mice. Blood Cells Mol Dis. 2004;33:125–131. doi: 10.1016/j.bcmd.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 49.Bridle KR, Frazer DM, Wilkins SJ, Dixon JL, Purdie DM, Crawford DH, Subramaniam VN, Powell LW, Anderson GJ, Ramm GA. Disrupted hepcidin regulation in HFE-associated haemochromatosis and the liver as a regulator of body iron homoeostasis. Lancet. 2003;361:669–673. doi: 10.1016/S0140-6736(03)12602-5. [DOI] [PubMed] [Google Scholar]
- 50.Ahmad KA, Ahmann JR, Migas MC, Waheed A, Britton RS, Bacon BR, Sly WS, Fleming RE. Decreased liver hepcidin expression in the Hfe knockout mouse. Blood Cells Mol Dis. 2002;29:361–366. doi: 10.1006/bcmd.2002.0575. [DOI] [PubMed] [Google Scholar]
- 51.Nicolas G, Viatte L, Lou DQ, Bennoun M, Beaumont C, Kahn A, Andrews NC, Vaulont S. Constitutive hepcidin expression prevents iron overload in a mouse model of hemochromatosis. Nat Genet. 2003;34:97–101. doi: 10.1038/ng1150. [DOI] [PubMed] [Google Scholar]
- 52.Gehrke SG, Herrmann T, Kulaksiz H, Merle U, Bents K, Kaiser I, Riedel HD, Stremmel W. Iron stores modulate hepatic hepcidin expression by an HFE-independent pathway. Digestion. 2005;72:25–32. doi: 10.1159/000087400. [DOI] [PubMed] [Google Scholar]
- 53.Frazer DM, Anderson GJ. The orchestration of body iron intake: how and where do enterocytes receive their cues? Blood Cells Mol Dis. 2003;30:288–297. doi: 10.1016/s1079-9796(03)00039-1. [DOI] [PubMed] [Google Scholar]
- 54.Wilkins SJ, Frazer DM, Millard KN, McLaren GD, Anderson GJ. Iron metabolism in the hemoglobin-deficit mouse: correlation of diferric transferrin with hepcidin expression. Blood. 2006;107:1659–1664. doi: 10.1182/blood-2005-07-2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Feder JN, Penny DM, Irrinki A, Lee VK, Lebrón JA, Watson N, Tsuchihashi Z, Sigal E, Bjorkman PJ, Schatzman RC. The hemochromatosis gene product complexes with the transferrin receptor and lowers its affinity for ligand binding. Proc Natl Acad Sci USA. 1998;95:1472–1477. doi: 10.1073/pnas.95.4.1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lebrón JA, Bennett MJ, Vaughn DE, Chirino AJ, Snow PM, Mintier GA, Feder JN, Bjorkman PJ. Crystal structure of the hemochromatosis protein HFE and characterization of its interaction with transferrin receptor. Cell. 1998;93:111–123. doi: 10.1016/s0092-8674(00)81151-4. [DOI] [PubMed] [Google Scholar]
- 57.Lebrón JA, West AP, Bjorkman PJ. The hemochromatosis protein HFE competes with transferrin for binding to the transferrin receptor. J Mol Biol. 1999;294:239–245. doi: 10.1006/jmbi.1999.3252. [DOI] [PubMed] [Google Scholar]
- 58.Kawabata H, Yang R, Hirama T, Vuong PT, Kawano S, Gombart AF, Koeffler HP. Molecular cloning of transferrin receptor 2. A new member of the transferrin receptor-like family. J Biol Chem. 1999;274:20826–20832. doi: 10.1074/jbc.274.30.20826. [DOI] [PubMed] [Google Scholar]
- 59.Camaschella C, Roetto A, Calì A, De Gobbi M, Garozzo G, Carella M, Majorano N, Totaro A, Gasparini P. The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat Genet. 2000;25:14–15. doi: 10.1038/75534. [DOI] [PubMed] [Google Scholar]
- 60.Girelli D, Bozzini C, Roetto A, Alberti F, Daraio F, Colombari R, Olivieri O, Corrocher R, Camaschella C. Clinical and pathologic findings in hemochromatosis type 3 due to a novel mutation in transferrin receptor 2 gene. Gastroenterology. 2002;122:1295–1302. doi: 10.1053/gast.2002.32984. [DOI] [PubMed] [Google Scholar]
- 61.Fleming RE, Ahmann JR, Migas MC, Waheed A, Koeffler HP, Kawabata H, Britton RS, Bacon BR, Sly WS. Targeted mutagenesis of the murine transferrin receptor-2 gene produces hemochromatosis. Proc Natl Acad Sci USA. 2002;99:10653–10658. doi: 10.1073/pnas.162360699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wallace DF, Summerville L, Lusby PE, Subramaniam VN. First phenotypic description of transferrin receptor 2 knockout mouse, and the role of hepcidin. Gut. 2005;54:980–986. doi: 10.1136/gut.2004.062018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kawabata H, Fleming RE, Gui D, Moon SY, Saitoh T, O'Kelly J, Umehara Y, Wano Y, Said JW, Koeffler HP. Expression of hepcidin is down-regulated in TfR2 mutant mice manifesting a phenotype of hereditary hemochromatosis. Blood. 2005;105:376–381. doi: 10.1182/blood-2004-04-1416. [DOI] [PubMed] [Google Scholar]
- 64.Nemeth E, Roetto A, Garozzo G, Ganz T, Camaschella C. Hepcidin is decreased in TFR2 hemochromatosis. Blood. 2005;105:1803–1806. doi: 10.1182/blood-2004-08-3042. [DOI] [PubMed] [Google Scholar]
- 65.West AP, Bennett MJ, Sellers VM, Andrews NC, Enns CA, Bjorkman PJ. Comparison of the interactions of transferrin receptor and transferrin receptor 2 with transferrin and the hereditary hemochromatosis protein HFE. J Biol Chem. 2000;275:38135–38138. doi: 10.1074/jbc.C000664200. [DOI] [PubMed] [Google Scholar]
- 66.Goswami T, Andrews NC. Hereditary hemochromatosis protein, HFE, interaction with transferrin receptor 2 suggests a molecular mechanism for mammalian iron sensing. J Biol Chem. 2006;281:28494–28498. doi: 10.1074/jbc.C600197200. [DOI] [PubMed] [Google Scholar]
- 67.Robb A, Wessling-Resnick M. Regulation of transferrin receptor 2 protein levels by transferrin. Blood. 2004;104:4294–4299. doi: 10.1182/blood-2004-06-2481. [DOI] [PubMed] [Google Scholar]
- 68.Johnson MB, Enns CA. Diferric transferrin regulates transferrin receptor 2 protein stability. Blood. 2004;104:4287–4293. doi: 10.1182/blood-2004-06-2477. [DOI] [PubMed] [Google Scholar]
- 69.Calzolari A, Raggi C, Deaglio S, Sposi NM, Stafsnes M, Fecchi K, Parolini I, Malavasi F, Peschle C, Sargiacomo M, et al. TfR2 localizes in lipid raft domains and is released in exosomes to activate signal transduction along the MAPK pathway. J Cell Sci. 2006;119:4486–4498. doi: 10.1242/jcs.03228. [DOI] [PubMed] [Google Scholar]
- 70.Samad TA, Srinivasan A, Karchewski LA, Jeong SJ, Campagna JA, Ji RR, Fabrizio DA, Zhang Y, Lin HY, Bell E, et al. DRAGON: a member of the repulsive guidance molecule-related family of neuronal- and muscle-expressed membrane proteins is regulated by DRG11 and has neuronal adhesive properties. J Neurosci. 2004;24:2027–2036. doi: 10.1523/JNEUROSCI.4115-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Niederkofler V, Salie R, Sigrist M, Arber S. Repulsive guidance molecule (RGM) gene function is required for neural tube closure but not retinal topography in the mouse visual system. J Neurosci. 2004;24:808–818. doi: 10.1523/JNEUROSCI.4610-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Papanikolaou G, Samuels ME, Ludwig EH, MacDonald ML, Franchini PL, Dubé MP, Andres L, MacFarlane J, Sakellaropoulos N, Politou M, et al. Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nat Genet. 2004;36:77–82. doi: 10.1038/ng1274. [DOI] [PubMed] [Google Scholar]
- 73.Niederkofler V, Salie R, Arber S. Hemojuvelin is essential for dietary iron sensing, and its mutation leads to severe iron overload. J Clin Invest. 2005;115:2180–2186. doi: 10.1172/JCI25683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Huang FW, Pinkus JL, Pinkus GS, Fleming MD, Andrews NC. A mouse model of juvenile hemochromatosis. J Clin Invest. 2005;115:2187–2191. doi: 10.1172/JCI25049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lin L, Goldberg YP, Ganz T. Competitive regulation of hepcidin mRNA by soluble and cell-associated hemojuvelin. Blood. 2005;106:2884–2889. doi: 10.1182/blood-2005-05-1845. [DOI] [PubMed] [Google Scholar]
- 76.Babitt JL, Huang FW, Wrighting DM, Xia Y, Sidis Y, Samad TA, Campagna JA, Chung RT, Schneyer AL, Woolf CJ, et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat Genet. 2006;38:531–539. doi: 10.1038/ng1777. [DOI] [PubMed] [Google Scholar]
- 77.Shi Y, Massagué J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 78.Truksa J, Peng H, Lee P, Beutler E. Bone morphogenetic proteins 2, 4, and 9 stimulate murine hepcidin 1 expression independently of Hfe, transferrin receptor 2 (Tfr2), and IL-6. Proc Natl Acad Sci USA. 2006;103:10289–10293. doi: 10.1073/pnas.0603124103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang RH, Li C, Xu X, Zheng Y, Xiao C, Zerfas P, Cooperman S, Eckhaus M, Rouault T, Mishra L, et al. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab. 2005;2:399–409. doi: 10.1016/j.cmet.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 80.Lee P, Peng H, Gelbart T, Beutler E. The IL-6- and lipopolysaccharide-induced transcription of hepcidin in HFE-, transferrin receptor 2-, and beta 2-microglobulin-deficient hepatocytes. Proc Natl Acad Sci USA. 2004;101:9263–9265. doi: 10.1073/pnas.0403108101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Frazer DM, Wilkins SJ, Millard KN, McKie AT, Vulpe CD, Anderson GJ. Increased hepcidin expression and hypoferraemia associated with an acute phase response are not affected by inactivation of HFE. Br J Haematol. 2004;126:434–436. doi: 10.1111/j.1365-2141.2004.05044.x. [DOI] [PubMed] [Google Scholar]
- 82.Roy CN, Custodio AO, de Graaf J, Schneider S, Akpan I, Montross LK, Sanchez M, Gaudino A, Hentze MW, Andrews NC, et al. An Hfe-dependent pathway mediates hyposideremia in response to lipopolysaccharide-induced inflammation in mice. Nat Genet. 2004;36:481–485. doi: 10.1038/ng1350. [DOI] [PubMed] [Google Scholar]
- 83.Wrighting DM, Andrews NC. Interleukin-6 induces hepcidin expression through STAT3. Blood. 2006;108:3204–3209. doi: 10.1182/blood-2006-06-027631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Verga Falzacappa MV, Vujic Spasic M, Kessler R, Stolte J, Hentze MW, Muckenthaler MU. STAT3 mediates hepatic hepcidin expression and its inflammatory stimulation. Blood. 2007;109:353–358. doi: 10.1182/blood-2006-07-033969. [DOI] [PubMed] [Google Scholar]
- 85.Courselaud B, Pigeon C, Inoue Y, Inoue J, Gonzalez FJ, Leroyer P, Gilot D, Boudjema K, Guguen-Guillouzo C, Brissot P, et al. C/EBPalpha regulates hepatic transcription of hepcidin, an antimicrobial peptide and regulator of iron metabolism. Cross-talk between C/EBP pathway and iron metabolism. J Biol Chem. 2002;277:41163–41170. doi: 10.1074/jbc.M202653200. [DOI] [PubMed] [Google Scholar]
- 86.Bayele HK, McArdle H, Srai SK. Cis and trans regulation of hepcidin expression by upstream stimulatory factor. Blood. 2006;108:4237–4245. doi: 10.1182/blood-2005-07-027037. [DOI] [PubMed] [Google Scholar]
- 87.West AP, Giannetti AM, Herr AB, Bennett MJ, Nangiana JS, Pierce JR, Weiner LP, Snow PM, Bjorkman PJ. Mutational analysis of the transferrin receptor reveals overlapping HFE and transferrin binding sites. J Mol Biol. 2001;313:385–397. doi: 10.1006/jmbi.2001.5048. [DOI] [PubMed] [Google Scholar]