

Abstract
This review explores the chief genetic and epigenetic events that promote pathological progression in colorectal carcinogenesis. This article discusses the molecular and pathological basis for classifying colorectal neoplasia into suppressor, mutator and methylator pathways. These differing mechanisms of genomic instability are associated with specific cancer characteristics, and may provide the opportunity for more effective prevention and surveillance strategies in the future. This is the first review in a series of five topics outlining important and developing aspects of colorectal cancer.
Keywords: Colorectal cancer; Neoplastic processes; Adenoma-carcinoma sequence; Serrated adenoma; Adenomas; Hyperplastic polyps, Microsatellite instability; CpG Island methylator phenotype
INTRODUCTION
Rapid integration of new endoscopic and molecular techniques into clinical practice, has established colorectal medicine in the vanguard of translational research. Close collaboration between surgeons, gastroenterologists, oncologists, pathologists, geneticists and molecular scientists has advanced our understanding of the histopathological and molecular stages in colorectal cancer (CRC). CRCs develop from apparently normal mucosa into a benign precursor stage, the premalignant polyp, and can progress to invasive disease[1]. The molecular mechanisms driving this process and the important pathological landmarks that characterize CRC, however, are being continually refined.
In 1988, Vogelstein et al[2] published their molecular analysis of 172 colorectal neoplasias, including adenomas from patients with familial adenomatous polyposis (FAP), sporadic adenomas, and CRCs. They characterized several of the sentinel molecular events occurring in progressive pathological stages, from adenoma to cancer. This research generated the ‘traditional pathway’ model that integrated these events into a coherent adenoma-carcinoma sequence[3]. Conceptually, this traditional model of the adenoma-carcinoma sequence is extremely attractive. It is relatively uncomplicated, it is likely to explain the growth of many cancers and, within the context of CRC screening, supports the clinical practice of removing adenomas to reduce, albeit very slightly, the incidence of invasive neoplasia[4]. The natural history of adenomas, hyperplastic polyps, and carcinomas, however, is extremely variable. The discovery of one such subset of phenotypically distinct CRC, the microsatellite instability-high (MSI-H) group, represents another key milestone in the scientific history of CRC[5]. MSI-H CRCs show particular histopathological characteristics, a proclivity for the proximal colon, relatively rapid progression from adenoma to invasive cancer, and generally have a better prognosis for a given stage of disease[6]. MSI-H cancers result from failure of the mismatch repair (MMR) system, which is critical in maintaining genetic fidelity following DNA replication[7]. MMR dysfunction may be caused by germline mutation of MMR genes as in Lynch syndrome, also known as hereditary non-polyposis CRC (HNPCC) or somatic (epigenetic) inactivation of an MMR gene, most commonly MLH1[8]. There are other colorectal lesions, however, that do not strictly conform to either of these pathways. Some cancers exhibit unique pathological features and distinct molecular signatures that translate into specific natural histories. The discovery of epigenetic mechanisms in colorectal carcinogenesis has necessitated the development of new frameworks and pathways[9]. More precise characterization of colorectal carcinogenesis and a better appreciation of the interplay between genetic predisposition, environmental exposure and luminal events, will generate new opportunities to improve screening, surveillance, chemoprevention and therapeutic strategies.
This article reviews recent developments in defining colorectal cancer pathways. In particular, we identify the chief genetic events that advance neoplasia, outline the histopathological correlates of these molecular perturbations and, where possible, contrast these differing mechanisms to emphasize the clinical value of dissecting out these road maps to cancer.
CANCER: A DISTURBANCE OF MUTATION, REPAIR AND EXPRESSION
Colorectal carcinogenesis requires a previously normal cell to accumulate multiple genetic alterations and establish successive clones, each characterized by relative growth advantage. This growth advantage may be conferred through an increased rate of proliferation, impaired apoptosis, or both. Most cancers probably require between 6 to 10 clonal events to reach their final malignant phenotype, which also requires the acquisition of metastatic traits[10]. In addition to simple growth advantage, the “successful” pre-cancerous clone must develop a cellular environment permissive of future mutation[7]. This process, referred to as genomic instability, ensures that subsequent strategic mutations occur at increasingly greater likelihood. Genomic instability is critical in carcinogenesis. It accelerates the neoplastic evolutionary process, by increasing the mutation rate induced by the background mutagenic challenge. Without genomic instability, the acquisition of new mutations would occur far too slowly for a cancer to develop during a person’s lifetime[11]. Genomic integrity in a normal cell is under careful scrutiny. The cell cycle and mitotic spindle checkpoints are critical in this process to ensure that cell proliferation only follows correct replication and organization of genetic material, respectively. If the genetic damage is too great for repair, the cell avoids propagating the damaged DNA by undergoing apoptosis. When one particular type of genomic instability predominates and neoplasias progress through characteristic histopathological stages with similar genetic alterations, then it is appropriate to consider such a process as a discrete pathway.
There are two chief categories of genomic instability in colorectal cancer. The most common is chromosomal instability (CIN), in which the requisite genetic events occur through the accumulation of numerical or structural chromosomal abnormalities (aneuploidy)[10]. The other main type of genomic instability is microsatellite instability (MSI), which is a consequence of impaired recognition and repair of mismatched bases in the daughter strand of DNA during DNA replication. Microsatellites are nucleotide repeat sequences scattered throughout the genome and MSI refers to discrepancies in the number of nucleotide repeats found within these microsatellite regions in tumour versus germline DNA (see below). MSI testing reveals the mismatch repair dysfunction, which is responsible for generating the genetic events involved in the MSI pathway of CRC. The mutual exclusivity of the pathways associated with CIN or MSI suggests that genomic instability is necessary and that either pathway is sufficient to drive colorectal carcinogenesis. More recently, epigenetic factors have been implicated in the development of certain subsets of cancers and polyps[9]. Careful characterization of the epigenetics, particularly promoter sequence methylation, has led to the definition of the CpG Island Methylator Phenotype (CIMP+) cancer and a proposed novel pathway, the serrated neoplasia pathway[12].
CHROMOSOMAL INSTABILITY PATHWAY (CIN) OR SUPPRESSOR PATHWAY
Approximately 70%-85% of CRCs generally develop via the ‘traditional’ pathway, which has also been referred to as the CIN, or ‘suppressor’, pathway[13]. The earliest identifiable lesion in this pathway is probably the dysplastic aberrant crypt focus (ACF), a microscopic mucosal lesion, that precedes the development of a macroscopic polyp[14]. The ‘traditional’ pathway is associated with mutation in APC or loss of 5q (APC gene), mutation of K-ras, loss of 18q, and finally, deletion of 17p, which contains the important tumour suppressor gene p53 ( (Figure 1)[13]. However, only a very small minority of CRCs characterized by CIN possess a full complement of these molecular abnormalities[15]. It is possible that several of these “steps” can be bypassed by other genetic events to deliver the necessary biological consequences[16]. Sequencing of the human genome has provided the opportunity to analyze tumour-specific mutations in far greater detail and a recent study discovered many potentially important CRC mutations outside of the genes referred to above[17]. Some of these mutations may serve as additional or alternative steps in the adenoma-carcinoma sequence. Nevertheless, it is still worthwhile to consider the traditional CIN pathway genes, as well as the Wnt-signaling pathway affected by APC dysfunction, in more detail.
Figure 1.
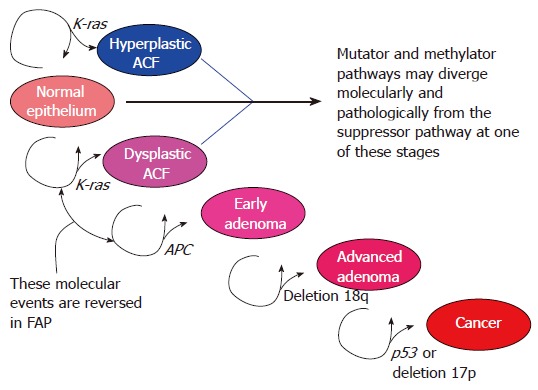
Sporadic traditional/CIN/suppressor pathway: sequential pathological stages and molecular events.
APC (5q21)
The adenomatous polyposis coli (APC) gene is large, containing 15 exons with several functional domains, and is thus a prime target for mutagenesis. Pathogenic mutations in APC frequently truncate the APC protein, which in turn interferes with its binding to β-catenin. APC binding to β-catenin is an important step in dampening down the Wnt-signaling pathway[18]. Wnt signaling normally helps to regulate growth, apoptosis and differentiation and thus is particularly relevant in embryogenesis, as well as maintaining tissue specific stem cell compartments, including the stem cell compartment found at the base of colonic crypts[19]. Corruption of this pathway can contribute to carcinogenesis in adulthood, with FAP providing a dramatic example. Normal activation of the Wnt-pathway requires binding of Wnt proteins, possibly secreted by neighbouring subepithelial cells[20], to the cell surface receptor complex of the Frizzled receptor and the LDL-receptor-related protein (LRP) co-receptor. Binding to the receptor complex induces LRP-axin interaction as well as phosphorylation of Disheveled, another cytoplasmic protein. Activated Disheveled interferes with the usual APC-axin-GSK3β-induced degradation of cytoplasmic β-catenin, which is then available to associate with Tcf, translocate to the nucleus as a transcription factor, and promote the expression of several important cell cycle regulating genes, including cyclin D1 and c-Myc. Thus APC mutations that interfere with APC-β-catenin binding impair the normal degradation of β-catenin and result in a constitutively active Wnt-signaling pathway. Occasionally β-catenin mutation, which renders the protein resistant to APC-degradation, can act as an alternative to APC mutation, emphasizing the importance of Wnt-signaling in CRC[20].
Loss of functional APC might also interfere with the normal regulation of mitosis, contributing to CIN[21]. During metaphase there is careful organization of the sister chromatids to ensure their successful distribution to the daughter cells. Several cellular components, including the kinetochore, carefully coordinate this organization of the chromatin, to ensure correct delivery during cell division. APC is a kinetochore-bound microtubule-associated protein, and along with EB1, is important in promoting correct chromosomal alignment and subsequent chromosomal segregation[22]. APC deficient cells do not adequately detect chromosomal abnormalities during metaphase, yet can still proceed into anaphase (separation stage of mitosis) potentially generating CIN[23]. There is some suggestion that the contribution of APC to CIN may be related to the site of somatic APC mutation[24]. Through this mechanism, impairment of APC might be important in the loss of other strategic loci[23].
In patients with FAP, all of their cells harbour a germline mutation in one APC allele. Initiation of the adenoma-carcinoma sequence in these patients follows subsequent inactivation of the remaining normal copy of APC, often by deletion but occasionally through somatic mutation. Thus, the function of the APC protein is altered in all FAP-associated ACF. In addition, K-ras mutation is seldom found in this familial form of dysplastic ACF, whereas 82% of nondysplastic and 63% of dysplastic sporadic ACF have K-ras mutation and APC mutation is notably absent[14,25]. β-catenin immunofluorescence studies suggest that Wnt-activation occurs in FAP-related, but not in sporadic ACF. These findings support the concept that APC and the Wnt-pathway is less important in the earliest stages of sporadic CRC development via the traditional pathway[14]. The frequency of APC or β-catenin mutation in early adenomas has been reported as high as 80%[14], although the rate of APC mutation is significantly lower in some other series and often depends on the histopathological type of adenoma[9]. Mutation of APC is found in approximately 60% of cancers arising from the colon and 82% of rectal cancers[9]. Abnormalities in APC and the Wnt-pathway may represent a convenient mechanism to advance sporadic ACF into early adenomas or as a means of advancing very early lesions into more advanced stages. The fact that abnormalities in APC are not universal in adenomas or even in cancers, however, indicates that other genetic alterations may serve as molecular surrogates, either within the traditional pathway, or through other mechanisms of colorectal carcinogenesis[26,27].
K-ras (12p12)
The K-ras proto-oncogene encodes a GTP-binding protein and when mutated can cause a loss of inherent GTPase activity. Thus, hydrolysis of active GTP into inactive GDP is impaired, causing constitutive signaling through the downstream pathway[28]. Activating K-ras mutations are found in 35%-42% of colorectal cancers, and in a similar number of advanced adenomas, but is less common in small adenomas[28]. This is despite the fact that K-ras mutations are frequent in sporadic dysplastic ACF (63%), suggesting that although K-ras mutation confers a growth advantage to its daughter cells, it is neither sufficient nor necessary to drive carcinogenesis[25]. The role of K-ras is not unique to the traditional pathway and K-ras mutations are present in both adenomas and hyperplastic polyps, whilst APC mutation occurs specifically in adenomas[29]. As discussed below, K-ras has a developing role in the serrated neoplasia pathway.
SMAD2, SMAD4 and DCC
DCC, SMAD2 and SMAD4 are all located at 18q21.1, and allelic loss at this site is found in up to 60% of CRCs[2]. SMAD2 and SMAD4 are involved in the TGF-β signaling pathway, important in regulating growth as well as apoptosis. DCC encodes a transmembrane receptor that promotes apoptosis, in the absence of its ligand, netrin-1. Historically, DCC has been implicated as the key driver of carcinogenesis from this cluster. In contrast to SMAD4, however, mutations in DCC and SMAD2 are rare in CRC, thus SMAD4 may be more influential in colorectal carcinogenesis[30]. Furthermore, germline mutation of SMAD4 can cause juvenile polyposis syndrome, which is associated with CRC[31].
p53 (17p13)
Loss of p53, usually through allelic loss of 17p, is often a late event in the traditional pathway, and heralds the transition from pre-invasive to invasive disease. Several studies have shown that the frequency of p53 abnormalities, either mutation or loss of heterozygosity (LOH), increases relative to the histological stage of the lesion within the pathway. Thus, abnormalities are found in 4%-26% of adenomas, 50% of adenomas with invasive foci, and in 50%-75% of CRCs[28]. The functional p53 protein, which is stabilized by DNA damage, is an important transcription factor. It acts to increase the expression of cell-cycle retarding genes, to slow the cell cycle and provide sufficient time for DNA repair during occasions of genetic insult. When genetic damage is too great for the cell to repair, p53 induces pro-apoptotic genes[32].
To summarize the events above, a widely accepted interpretation of the sporadic CIN pathway would follow that the initial lesion is the dysplastic ACF, which often contains K-ras mutation. Acquisition of an APC mutation, or loss of 5q, then contributes to the growth of the ACF into an adenoma. It is unclear why the frequency of K-ras mutation in non-advanced (< 1 cm, tubular) adenomas is relatively low (12%-30%), despite being reported in 63% of dysplastic ACF[14]. It could be that APC mutations occur more readily in cells that are wild type for K-ras, so most APC wild type/K-ras mutant ACF are destined not to progress. It is possible that the APC wild type/K-ras mutant ACF lesions are diverted from the traditional pathway into other pathological streams. Or that the APC mutant/K-ras mutant lesions, when they develop, endow their respective clone with significant advantage, so that they pass more quickly into advanced pathologies, such as tubulovillous adenomas. This would cause an artefactual under-representation of K-ras mutation in earlier adenoma stages. From this last hypothesis, it would follow that although APC is the gateway or shortcut to adenoma development, K-ras is a molecular shortcut to developing advanced adenomas, a prelude to cancer. The frequency of K-ras and APC abnormality then remains relatively stable for the rest of the pathway. LOH at 18q and loss of function of DCC, SMAD2 and perhaps most importantly SMAD4, may further advance the pathway by interfering with apoptosis, allowing accumulation of mutations that had hitherto induced cell-cycle arrest or programmed cell death. Finally, acquisition of p53 mutation or functionally equivalent LOH at 17p accompanies the transition of the benign lesion into invasive disease (Figure 1).
There is increasing evidence that the mitotic spindle checkpoint is an important factor in CIN[10]. This checkpoint serves to ensure that all the chromosomes are correctly aligned in metaphase, before advancing into anaphase. A partially compromised checkpoint provides the cell with a relative growth advantage. The mitotic-spindle-checkpoint genes hBUB1 and hBUBR1 are occasionally mutated in colorectal cancer[33]. Loss of entire chromosomes may accompany overexpression of particular genes that affect the number of centrosomes within the dividing cell[34]. For instance, overexpression of either STK15 kinase or POLO-like kinase (PLK1) can cause aneuploidy in cell lines[35,36]. The G1/S checkpoint genes p53, pRb, p16INK4A[37] and p21Waf1/Cip1[38], are also important in preventing aneuploidy, and abnormalities of these genes have all been implicated in CRC. However, a thorough explanation of the mechanisms behind CIN awaits further study.
The traditional model of the adenoma-carcinoma sequence has provided a foundation for molecular classification of colorectal carcinogenesis and has established a reference against which one may contrast other CRC molecular profiles. The shortcomings of the traditional model include a failure to explain why the majority of benign adenomas never transform into invasive cancers. One study suggested that in large polyps (> 1 cm) the cumulative risk of cancer was only 2.5%, 8% and 24% at 5, 10 and 20 years, respectively[39]. From an autopsy series, the prevalence of asymptomatic polyps in those greater than 75 years was 40%-60%, whilst the prevalence of CRC was approximately 3%[40]. Thus in approximately 19 of every 20 cases[41], the sequence would be better described as an adenoma-adenoma pathway. This probably reflects bottlenecks in carcinogenesis that chance and genomic instability are only occasionally able to overcome. In contrast the cancer risk is much higher in polyps originating in patients with HNPCC[42], and some histopathological groups from other pathways are similarly associated with a greater risk for cancer development[43]. It is uncertain whether advanced adenomas, characterized by high-grade dysplasia, villous architecture and size (≥ 1 cm) simply represent more advanced stages within the sequential advancement of colorectal neoplasia, or whether these advanced changes are predetermined. Are there multiple tracks to take within this pathway, even though CIN is the key mechanism driving genomic instability? There are several possibilities for the heterogeneous natural history of these adenomas, including luminal factors and the resultant environmental mutational load that is fuelling the genetic aberration. The variability might also relate to other modifying genetic, microbiological or immunological factors that temper progression through the sequence. Finally, the traditional pathway does not explain the contribution of non-dysplastic precursor lesions, such as hyperplastic polyps, to CRC.
MICROSATELLITE INSTABILITY (MSI) OR MUTATOR PATHWAY
The MSI or mutator pathway is the other chief mechanism for genomic instability in CRC. Approximately 20% of CRCs display this “mutator” phenotype, in which failure of the mismatch repair system interrupts normal review and repair of DNA following replication. The MMR system is composed of at least 7 proteins, hMLH1, hMLH3, hMSH2, hMSH3, hMSH6, hPMS1 and hPMS2, which associate with specific partners to form functional heterodimers[7]. hMLH1 and hMSH2 are essential components of the human mismatch repair machinery and form five functional heterodimeric proteins (hMSH2-hMSH3; hMSH2-hMSH6; hMLH1-hPMS1; hMLH1-hPMS2; hMLH1-hMLH3). DNA polymerase is susceptible to making errors in short repeat sequences, thus MMR dysfunction, which fails to repair these, results in detectable differences between tumour and germline DNA in the number of copies found in these repeat sequences (microsatellites). Many colon cancers, however, have frameshift mutations at a small number of microsatellites, thus a standardized panel of microsatellites was devised to provide uniformity of definition for research and clinicopathological practice[44]. The panel includes two mononucleotide (BAT25 and BAT26) and three dinucleotide microsatellites (D5S346, D2S123, and D17S250). MSI-H is defined as MSI at ≥ 2 (40%) of the five specified sites, MSI-L (low) MSI at 1 site, and microsatellite stable (MSS) when no instability is demonstrated. MSI leads to a dramatic increase in genetic errors and several microsatellites are present in genes implicated in colorectal carcinogenesis, such as TGF-βRII, Bax, Caspase 5, MSH3, MSH6, β-catenin, APC, IGF-II, and E2F4[1]. MSI-H cancers are invariably diploid, as MMR dysfunction provides the mechanism of genomic instability without the need for CIN, which characterizes the traditional pathway.
MSI-H tumours occur in HNPCC (Lynch syndrome), in which germline mutation in a particular MMR gene provides each cell with only one functional copy of the protein. Following a second somatic hit, the cell is rendered deficient in the specific MMR protein product, and thus incapable of accurate mismatch repair. MSI is one possible pathological screening test, to help direct genetic testing for HNPCC. The majority of MSI-H CRCs, however, are sporadic, the consequence of epigenetic silencing of MLH1[45]. Most, but not all, MSI-H tumours, whether sporadic or inherited, share similar biology, as characterized in Table 1. hMSH2 and hMLH1 are the two most frequently affected genes in HNPCC, whilst hMLH1 is targeted for epigenetic silencing in sporadic MSI-H CRCs.
Table 1.
Characteristics of different CRC pathways
| Predominant pathway | MSI/CIMP status | Site of cancer | Prognosis | CIN | Germline MMR mutation | Somatic BRAF mutation | MLH1 methylation | MGMT methylation | Immediate precursor |
| Mutator | MSI-H/CIMP- | Proximal | Better | No | Yes | No | No | No | Advanced adenoma |
| Methylator/Mutator | MSI-H/CIMP+ | Proximal | Better | No | No | Yes | Yes | No | Serrated polyp |
| Methylator | Non-MSI-H/CIMP+ | Proximal | Worse | No | No | Yes | No | No | Serrated polyp |
| “Alternate” methylator | MSI-L/CIMP+ | Distal | Unclear | No | No | No (K-ras) | No | Yes | Serrated polyp |
| Suppressor | Non-MSI-H/CIMP- | Distal | Standard | Yes | No | No | No | No | Advanced adenoma |
MSI: Microsatellite instability; CIMP: CpG island methylator phenotype.
What of the MSI-L cancers that display only a mild mutator phenotype? In HNPCC, germline mutations of MLH1 or MSH2 cause MSI-H cancers, whilst MSI-L lesions may be associated with MSH6 mutations[46]. Clinicopathologically, MSI-L cancers are similar to MSS tumours, but have a higher prevalence of K-ras mutation (54% vs 27%) and a lower frequency of 5q LOH (23% vs 48%), whilst sharing MSS cancers’ reliance on APC[47]. Interestingly, the rate of K-ras mutation in MSI-L cancers is higher than in either MSS or MSI-H (7%), rather than at an intermediate level, expected if MSI-L were merely an interface between these other two pathways. The DNA repair gene MGMT removes mutagenic adducts from guanine, and inactivation of MGMT is associated with a particular G to A transversion in K-ras. Given the association with K-ras, methylation of MGMT was evaluated in sporadic CRCs stratified for MSI status. Methylation of the MGMT promoter was significantly associated with MSI-L (64%), compared to either MSS (26%) or MSI-H (13%) tumours (P = 0.0001)[48]. It is possible that epigenetic silencing of MGMT could overwhelm the reparative capacity of the MMR apparatus, causing MSI-L cancers. Thus, epigenetic silencing of MGMT expression provides a potentially unifying mechanism for both the K-ras mutations and the low level of MSI found in this group. These findings support the hypothesis that MSI-L tumours may constitute a unique molecular pathway.
To investigate whether the adenoma is the precursor lesion in the mutator, as it is in the traditional pathway, MSI testing was performed on several different sporadic polyps, including tubular adenomas, hyperplastic polyps, serrated adenomas and mixed polyps (adjacent, yet discrete hyperplastic and adenomatous components). The MSI-L phenotype was demonstrated in a high proportion of hyperplastic polyps, serrated adenomas and mixed polyps, 29%, 53% and 83%, respectively, but only in 13% of tubular adenomas[49]. It is postulated that hyperplastic polyps and other serrated lesions may act as histologically distinct precursors to the sporadic mutator pathway cancers[9]. In HNPCC, however, the precursor lesion is still likely to be the adenoma. MSI is often, albeit not universally, found in adenomas identified in patients with HNPCC[50]. In one study the rate of MSI-H was 89% in adenomas found within 5 cm of a cancer, and 77% of those located > 5 cm from cancer[50]. MSI, of at least one marker, can occasionally be found in ACF[51], and MSI-L ACF are associated with methylation of MGMT[51]. This lends support to the concept introduced above, that sporadic MSI-L cancers associated with MGMT methylation may represent a unique entity characterized by a common cluster of molecular and pathological events, some of which can be identified even at the earliest stages of carcinogenesis.
METHYLATOR PATHWAY
DNA methylation is an epigenetic means of regulating gene transcription. Methylation of DNA can occur at cytosine bases when cytosine and guanosine occur in a dinucleotide pair, i.e. CpG. DNA methylation occurs throughout the genome, and many interspersed repetitive DNA elements, such as Alu, LINE and SINE families of sequences, are methylated during health. In contrast, CpG islands found within the promoter sequence of many genes are usually unmethylated[52]. Specific promoter sequence methylation can occur physiologically to silence particular genes, such as in X-chromosome inactivation, while disordered promoter methylation may also occur pathologically as an important event in carcinogenesis[53]. In CRC there is often both global hypomethylation of the genome, largely in the repeat DNA sequences and simultaneous hypermethylation of the promoter regions of strategic genes[54]. Promoter sequence methylation interrupts gene expression by directly inhibiting transcription factor binding, as well as influencing histone acetylation, and thus accessibility of the gene to the necessary transcriptional machinery[55]. In carcinogenesis, epigenetic silencing of gene transcription by CpG island methylation is biologically equivalent to acquiring an inactivating mutation. Thus, methylation can occur as the first, second or both hits, to silence important tumour suppressor genes.
Several genes are associated with CpG island methylation in CRC (Type C genes, MLH1, p16), whilst other genes are predominantly methylated in normal colorectal mucosa, and only occasionally methylated in cancer (Type A)[53,56]. In normal mucosa the type A genes (MINT6, MINT24, MINT32, ER) are associated with increasing age[53,56]. The mechanism driving these epigenetic events is uncertain. It is possible that environmental and luminal factors contribute to colorectal DNA methylation. The interface between diet, genetic predisposition, luminal environment and colorectal methylation will be an important area for future colorectal epigenetic research[53].
Currently there are two main methylation marker panels. These panels are used in an analogous fashion to the NIH MSI panel to classify cancers as CpG Island Methylator Phenotype positive or negative (CIMP + or -). Depending on the markers used, 24%-51% of all CRCs are CIMP+[56-59]. The first panel that was proposed, and the one that many studies have used, includes analysis of the promoter regions of the genes MLH1, p16, MINT 1, 2, and 3[56,60]. CIMP+ cancers often occur in older women, with a predominance of proximal colonic lesions. Sporadic MSI-H cancers share a similar phenotype, but CIMP+ characteristics remain even in the setting of non-MSI-H tumours[61], and although MSI-H cancers are associated with a good clinical outcome, CIMP+/non-MSI-H tumours generally have a poorer prognosis[62]. CIMP+ cancers also share common molecular events. Most CIMP+ cancers contain a BRAF mutation, but those that don’t have a BRAF mutation often have a mutation in K-ras[52,63]. BRAF and K-ras mutations occur in a mutually exclusive fashion, suggesting that a pathway common to both is critical to the development of these cancers. The RAS-RAF-MEK-ERK signaling pathway is important in apoptosis and in particular anoikis, the process of apoptosis following loss of the epithelial connection to the basement membrane[64]. Failure of anoikis is important in the development of hyperplastic polyps and serrated adenomas, which are the postulated precursors of CIMP+ CRCs[65]. BRAF and K-ras mutations interrupt the RAS-RAF-MEK-ERK signaling pathway at different levels, impairing normal anoikis. As alluded to above, the hypothesis of the serrated neoplasia pathway, in which serrated polyps (admixed polyps, hyperplastic polyps, and serrated adenomas) are precursors to CIMP+ CRCs, is further supported by the recent finding that 78% of sessile serrated adenomas exhibit BRAF mutation[66].
The main limitation of the originally proposed CIMP panel outlined above is its inability to reliably classify cancers into well defined subsets[67]. Therefore, an alternative panel of markers (CACNA1G, IGF2, NEUROG1, RUNX3, SOCS1) has been proposed[52]. In this new panel, methylation is defined quantitatively and cancers are defined according to the percentage methylation ratio (PMR), with CIMP+ CRCs having a PMR of > 10 at 3 or more of the 5 sites[52]. Using this new panel, CRCs distribute bimodally into CIMP+ and CIMP- cases, with an even closer correlation between CIMP+ and BRAF mutation.
CIMP+ CRCs are associated with methylation and frequent silencing of MLH1 with resultant high levels of MSI. Thus, there is a clear relationship between the sporadic mutator (MSI) and methylator (CIMP) pathways. However, CIMP is at least as common in non-MSI-H cancers[52], and although these lesions are also associated with BRAF mutation, the epigenetic silencing of MLH1 does not occur. These CRCs are associated with a worse prognosis[64,68]. The additional events regulating both prognosis and MLH1 methylation and thus MSI status are unclear. As discussed above, there is an association between MGMT methylation and K-ras mutation in a subset of non-MSI-H/CIMP+ cancers. It is possible that mutation of BRAF with or without methylation of MLH1 may define one methylator pathway, whilst the development of K-ras mutation and methylation of MGMT, could characterize an “alternate methylator” subtype, giving rise to MSI-L cancers[61].
CONCLUSION
The discussion above has outlined three pathways to CRC (Table 1). Firstly, there is the CIN pathway, or the suppressor pathway, characterized by stepwise mutation or deletion of K-ras, APC, SMAD2, SMAD4 and DCC, and p53. The Wnt-signaling pathway is likely to be important in many of these cancers, and there is a developing role of APC as a regulator of CIN. Fortunately, the vast majority of cancers in this pathway, never fully realize their neoplastic potential. This pathway begins with the dysplastic ACF, passes through simple and then advanced adenomatous stages, and, in a minority, produces an MSS cancer. The second pathway is the mutator pathway which, in its purest form (HNPCC), results from a germline mutation in a mismatch repair gene. The majority of MSI-H cancers, however, occur sporadically following the epigenetic silencing of MLH1. Cancers from this pathway, which is really a combination of the mutator and methylator pathways, may begin as hyperplastic ACF, becoming right sided hyperplastic polyps, serrated adenomas, and ultimately MSI-H, proximal colonic cancers. BRAF mutation and associated failure of anoikis may be important at least in the early stages of this pathway. The final pathway, the methylator pathway, is usually associated with BRAF mutation with or without epigenetic silencing of MLH1 to contribute to MSI-H (mutator/methylator pathway, above) or non-MSI-H CRC, respectively. There may be an alternate methylator pathway, without BRAF mutation, but rather with the acquisition or maintenance of K-ras mutation, and methylation of MGMT. The precursor lesions for these ultimately MSI-L cancers may also be serrated polyps, but this is an area requiring further research.
Many challenges remain in the understanding of colorectal carcinogenesis, including discovery of the mechanisms driving CIN and DNA methylation. In addition, further study of the molecular events found in apparently normal mucosa would be worthwhile. Normal appearing, non-resection mucosa escapes the surgeon’s knife but should not escape the attention of colorectal researchers as this tissue may provide insight into the key stimuli that initiate and then influence cancer pathways. A better understanding of the sequence of events from normal to early and advanced pre-invasive lesions will have clear benefit when recommending surveillance strategies for patients, as well as in identifying potential targets for screening and therapy. Ultimately, knowing the environmental and luminal factors that may initiate or exacerbate these pathways will be critical in the development of preventative strategies.
Footnotes
Supported by a Gastroenterological Society of Australia post-graduate scholarship
S- Editor Ma N L- Editor Roberts SE E- Editor Ma WH
References
- 1.Bresalier RS. Malignant neoplasms of the large intestine. In: Feldman M, Friedman LS, Sleisenger MH, editors. Sleisenger & Fordtran's gastrointestinal and liver disease: pathophysiology, diagnosis, management. 7th ed. Philadelphia: Saunders; 2002. pp. 2215–2261. [Google Scholar]
- 2.Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525–532. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 3.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 4.Mandel JS, Church TR, Bond JH, Ederer F, Geisser MS, Mongin SJ, Snover DC, Schuman LM. The effect of fecal occult-blood screening on the incidence of colorectal cancer. N Engl J Med. 2000;343:1603–1607. doi: 10.1056/NEJM200011303432203. [DOI] [PubMed] [Google Scholar]
- 5.Thibodeau SN, Bren G, Schaid D. Microsatellite instability in cancer of the proximal colon. Science. 1993;260:816–819. doi: 10.1126/science.8484122. [DOI] [PubMed] [Google Scholar]
- 6.Söreide K, Janssen EA, Söiland H, Körner H, Baak JP. Microsatellite instability in colorectal cancer. Br J Surg. 2006;93:395–406. doi: 10.1002/bjs.5328. [DOI] [PubMed] [Google Scholar]
- 7.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 8.Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, Nakagawa H, Sotamaa K, Prior TW, Westman J, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer) N Engl J Med. 2005;352:1851–1860. doi: 10.1056/NEJMoa043146. [DOI] [PubMed] [Google Scholar]
- 9.Jass JR, Whitehall VL, Young J, Leggett BA. Emerging concepts in colorectal neoplasia. Gastroenterology. 2002;123:862–876. doi: 10.1053/gast.2002.35392. [DOI] [PubMed] [Google Scholar]
- 10.Rajagopalan H, Lengauer C. Aneuploidy and cancer. Nature. 2004;432:338–341. doi: 10.1038/nature03099. [DOI] [PubMed] [Google Scholar]
- 11.Loeb LA. Mutator phenotype may be required for multistage carcinogenesis. Cancer Res. 1991;51:3075–3079. [PubMed] [Google Scholar]
- 12.Jass JR. Serrated adenoma of the colorectum and the DNA-methylator phenotype. Nat Clin Pract Oncol. 2005;2:398–405. doi: 10.1038/ncponc0248. [DOI] [PubMed] [Google Scholar]
- 13.Grady WM. Genomic instability and colon cancer. Cancer Metastasis Rev. 2004;23:11–27. doi: 10.1023/a:1025861527711. [DOI] [PubMed] [Google Scholar]
- 14.Takayama T, Ohi M, Hayashi T, Miyanishi K, Nobuoka A, Nakajima T, Satoh T, Takimoto R, Kato J, Sakamaki S, et al. Analysis of K-ras, APC, and beta-catenin in aberrant crypt foci in sporadic adenoma, cancer, and familial adenomatous polyposis. Gastroenterology. 2001;121:599–611. doi: 10.1053/gast.2001.27203. [DOI] [PubMed] [Google Scholar]
- 15.Smith G, Carey FA, Beattie J, Wilkie MJ, Lightfoot TJ, Coxhead J, Garner RC, Steele RJ, Wolf CR. Mutations in APC, Kirsten-ras, and p53--alternative genetic pathways to colorectal cancer. Proc Natl Acad Sci USA. 2002;99:9433–9438. doi: 10.1073/pnas.122612899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Young GP, Rozen P, Levin B. How does colorectal cancer develop. 1st ed 2002: pp. 23–37. [Google Scholar]
- 17.Sjöblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 18.Cadigan KM, Liu YI. Wnt signaling: complexity at the surface. J Cell Sci. 2006;119:395–402. doi: 10.1242/jcs.02826. [DOI] [PubMed] [Google Scholar]
- 19.Kuhnert F, Davis CR, Wang HT, Chu P, Lee M, Yuan J, Nusse R, Kuo CJ. Essential requirement for Wnt signaling in proliferation of adult small intestine and colon revealed by adenoviral expression of Dickkopf-1. Proc Natl Acad Sci USA. 2004;101:266–271. doi: 10.1073/pnas.2536800100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gregorieff A, Clevers H. Wnt signaling in the intestinal epithelium: from endoderm to cancer. Genes Dev. 2005;19:877–890. doi: 10.1101/gad.1295405. [DOI] [PubMed] [Google Scholar]
- 21.Fodde R, Kuipers J, Rosenberg C, Smits R, Kielman M, Gaspar C, van Es JH, Breukel C, Wiegant J, Giles RH, et al. Mutations in the APC tumour suppressor gene cause chromosomal instability. Nat Cell Biol. 2001;3:433–438. doi: 10.1038/35070129. [DOI] [PubMed] [Google Scholar]
- 22.Green RA, Wollman R, Kaplan KB. APC and EB1 function together in mitosis to regulate spindle dynamics and chromosome alignment. Mol Biol Cell. 2005;16:4609–4622. doi: 10.1091/mbc.E05-03-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Draviam VM, Shapiro I, Aldridge B, Sorger PK. Misorientation and reduced stretching of aligned sister kinetochores promote chromosome missegregation in EB1- or APC-depleted cells. EMBO J. 2006;25:2814–2827. doi: 10.1038/sj.emboj.7601168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giaretti W, Venesio T, Prevosto C, Lombardo F, Ceccarelli J, Molinu S, Risio M. Chromosomal instability and APC gene mutations in human sporadic colorectal adenomas. J Pathol. 2004;204:193–199. doi: 10.1002/path.1623. [DOI] [PubMed] [Google Scholar]
- 25.Cheng L, Lai MD. Aberrant crypt foci as microscopic precursors of colorectal cancer. World J Gastroenterol. 2003;9:2642–2649. doi: 10.3748/wjg.v9.i12.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Salahshor S, Kressner U, Pâhlman L, Glimelius B, Lindmark G, Lindblom A. Colorectal cancer with and without microsatellite instability involves different genes. Genes Chromosomes Cancer. 1999;26:247–252. [PubMed] [Google Scholar]
- 27.Huang J, Papadopoulos N, McKinley AJ, Farrington SM, Curtis LJ, Wyllie AH, Zheng S, Willson JK, Markowitz SD, Morin P, et al. APC mutations in colorectal tumors with mismatch repair deficiency. Proc Natl Acad Sci USA. 1996;93:9049–9054. doi: 10.1073/pnas.93.17.9049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leslie A, Carey FA, Pratt NR, Steele RJ. The colorectal adenoma-carcinoma sequence. Br J Surg. 2002;89:845–860. doi: 10.1046/j.1365-2168.2002.02120.x. [DOI] [PubMed] [Google Scholar]
- 29.Jen J, Powell SM, Papadopoulos N, Smith KJ, Hamilton SR, Vogelstein B, Kinzler KW. Molecular determinants of dysplasia in colorectal lesions. Cancer Res. 1994;54:5523–5526. [PubMed] [Google Scholar]
- 30.Woodford-Richens KL, Rowan AJ, Gorman P, Halford S, Bicknell DC, Wasan HS, Roylance RR, Bodmer WF, Tomlinson IP. SMAD4 mutations in colorectal cancer probably occur before chromosomal instability, but after divergence of the microsatellite instability pathway. Proc Natl Acad Sci USA. 2001;98:9719–9723. doi: 10.1073/pnas.171321498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bevan S, Woodford-Richens K, Rozen P, Eng C, Young J, Dunlop M, Neale K, Phillips R, Markie D, Rodriguez-Bigas M, et al. Screening SMAD1, SMAD2, SMAD3, and SMAD5 for germline mutations in juvenile polyposis syndrome. Gut. 1999;45:406–408. doi: 10.1136/gut.45.3.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mills AA. p53: link to the past, bridge to the future. Genes Dev. 2005;19:2091–2099. doi: 10.1101/gad.1362905. [DOI] [PubMed] [Google Scholar]
- 33.Cahill DP, Lengauer C, Yu J, Riggins GJ, Willson JK, Markowitz SD, Kinzler KW, Vogelstein B. Mutations of mitotic checkpoint genes in human cancers. Nature. 1998;392:300–303. doi: 10.1038/32688. [DOI] [PubMed] [Google Scholar]
- 34.Doxsey S. The centrosome--a tiny organelle with big potential. Nat Genet. 1998;20:104–106. doi: 10.1038/2392. [DOI] [PubMed] [Google Scholar]
- 35.Galaktionov K, Lee AK, Eckstein J, Draetta G, Meckler J, Loda M, Beach D. CDC25 phosphatases as potential human oncogenes. Science. 1995;269:1575–1577. doi: 10.1126/science.7667636. [DOI] [PubMed] [Google Scholar]
- 36.Zhou H, Kuang J, Zhong L, Kuo WL, Gray JW, Sahin A, Brinkley BR, Sen S. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat Genet. 1998;20:189–193. doi: 10.1038/2496. [DOI] [PubMed] [Google Scholar]
- 37.Khan SH, Wahl GM. p53 and pRb prevent rereplication in response to microtubule inhibitors by mediating a reversible G1 arrest. Cancer Res. 1998;58:396–401. [PubMed] [Google Scholar]
- 38.Stewart ZA, Leach SD, Pietenpol JA. p21(Waf1/Cip1) inhibition of cyclin E/Cdk2 activity prevents endoreduplication after mitotic spindle disruption. Mol Cell Biol. 1999;19:205–215. doi: 10.1128/mcb.19.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stryker SJ, Wolff BG, Culp CE, Libbe SD, Ilstrup DM, MacCarty RL. Natural history of untreated colonic polyps. Gastroenterology. 1987;93:1009–1013. doi: 10.1016/0016-5085(87)90563-4. [DOI] [PubMed] [Google Scholar]
- 40.Pollock AM, Quirke P. Adenoma screening and colorectal cancer. BMJ. 1991;303:1202. doi: 10.1136/bmj.303.6811.1202-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jass JR, Young J, Leggett BA. Evolution of colorectal cancer: change of pace and change of direction. J Gastroenterol Hepatol. 2002;17:17–26. doi: 10.1046/j.1440-1746.2002.02635.x. [DOI] [PubMed] [Google Scholar]
- 42.Järvinen HJ, Aarnio M, Mustonen H, Aktan-Collan K, Aaltonen LA, Peltomäki P, De La Chapelle A, Mecklin JP. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology. 2000;118:829–834. doi: 10.1016/s0016-5085(00)70168-5. [DOI] [PubMed] [Google Scholar]
- 43.Grossman S, Milos ML, Tekawa IS, Jewell NP. Colonoscopic screening of persons with suspected risk factors for colon cancer: II. Past history of colorectal neoplasms. Gastroenterology. 1989;96:299–306. doi: 10.1016/0016-5085(89)91551-5. [DOI] [PubMed] [Google Scholar]
- 44.Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58:5248–5257. [PubMed] [Google Scholar]
- 45.Deng G, Chen A, Hong J, Chae HS, Kim YS. Methylation of CpG in a small region of the hMLH1 promoter invariably correlates with the absence of gene expression. Cancer Res. 1999;59:2029–2033. [PubMed] [Google Scholar]
- 46.Parc YR, Halling KC, Wang L, Christensen ER, Cunningham JM, French AJ, Burgart LJ, Price-Troska TL, Roche PC, Thibodeau SN. HMSH6 alterations in patients with microsatellite instability-low colorectal cancer. Cancer Res. 2000;60:2225–2231. [PubMed] [Google Scholar]
- 47.Jass JR, Biden KG, Cummings MC, Simms LA, Walsh M, Schoch E, Meltzer SJ, Wright C, Searle J, Young J, et al. Characterisation of a subtype of colorectal cancer combining features of the suppressor and mild mutator pathways. J Clin Pathol. 1999;52:455–460. doi: 10.1136/jcp.52.6.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Whitehall VL, Walsh MD, Young J, Leggett BA, Jass JR. Methylation of O-6-methylguanine DNA methyltransferase characterizes a subset of colorectal cancer with low-level DNA microsatellite instability. Cancer Res. 2001;61:827–830. [PubMed] [Google Scholar]
- 49.Iino H, Jass JR, Simms LA, Young J, Leggett B, Ajioka Y, Watanabe H. DNA microsatellite instability in hyperplastic polyps, serrated adenomas, and mixed polyps: a mild mutator pathway for colorectal cancer? J Clin Pathol. 1999;52:5–9. doi: 10.1136/jcp.52.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Müller A, Beckmann C, Westphal G, Bocker Edmonston T, Friedrichs N, Dietmaier W, Brasch FE, Kloor M, Poremba C, Keller G, et al. Prevalence of the mismatch-repair-deficient phenotype in colonic adenomas arising in HNPCC patients: results of a 5-year follow-up study. Int J Colorectal Dis. 2006;21:632–641. doi: 10.1007/s00384-005-0073-6. [DOI] [PubMed] [Google Scholar]
- 51.Greenspan EJ, Cyr JL, Pleau DC, Levine J, Rajan TV, Rosenberg DW, Heinen CD. Microsatellite instability in aberrant crypt foci from patients without concurrent colon cancer. Carcinogenesis. 2007;28:769–776. doi: 10.1093/carcin/bgl209. [DOI] [PubMed] [Google Scholar]
- 52.Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, Kang GH, Widschwendter M, Weener D, Buchanan D, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38:787–793. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- 53.Rashid A, Issa JP. CpG island methylation in gastroenterologic neoplasia: a maturing field. Gastroenterology. 2004;127:1578–1588. doi: 10.1053/j.gastro.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 54.Bariol C, Suter C, Cheong K, Ku SL, Meagher A, Hawkins N, Ward R. The relationship between hypomethylation and CpG island methylation in colorectal neoplasia. Am J Pathol. 2003;162:1361–1371. doi: 10.1016/S0002-9440(10)63932-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 56.Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA. 1999;96:8681–8686. doi: 10.1073/pnas.96.15.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hawkins N, Norrie M, Cheong K, Mokany E, Ku SL, Meagher A, O'Connor T, Ward R. CpG island methylation in sporadic colorectal cancers and its relationship to microsatellite instability. Gastroenterology. 2002;122:1376–1387. doi: 10.1053/gast.2002.32997. [DOI] [PubMed] [Google Scholar]
- 58.van Rijnsoever M, Grieu F, Elsaleh H, Joseph D, Iacopetta B. Characterisation of colorectal cancers showing hypermethylation at multiple CpG islands. Gut. 2002;51:797–802. doi: 10.1136/gut.51.6.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Samowitz WS, Albertsen H, Herrick J, Levin TR, Sweeney C, Murtaugh MA, Wolff RK, Slattery ML. Evaluation of a large, population-based sample supports a CpG island methylator phenotype in colon cancer. Gastroenterology. 2005;129:837–845. doi: 10.1053/j.gastro.2005.06.020. [DOI] [PubMed] [Google Scholar]
- 60.Toyota M, Ho C, Ahuja N, Jair KW, Li Q, Ohe-Toyota M, Baylin SB, Issa JP. Identification of differentially methylated sequences in colorectal cancer by methylated CpG island amplification. Cancer Res. 1999;59:2307–2312. [PubMed] [Google Scholar]
- 61.Whitehall VL, Wynter CV, Walsh MD, Simms LA, Purdie D, Pandeya N, Young J, Meltzer SJ, Leggett BA, Jass JR. Morphological and molecular heterogeneity within nonmicrosatellite instability-high colorectal cancer. Cancer Res. 2002;62:6011–6014. [PubMed] [Google Scholar]
- 62.Issa JP. CpG island methylator phenotype in cancer. Nat Rev Cancer. 2004;4:988–993. doi: 10.1038/nrc1507. [DOI] [PubMed] [Google Scholar]
- 63.Samowitz WS, Sweeney C, Herrick J, Albertsen H, Levin TR, Murtaugh MA, Wolff RK, Slattery ML. Poor survival associated with the BRAF V600E mutation in microsatellite-stable colon cancers. Cancer Res. 2005;65:6063–6069. doi: 10.1158/0008-5472.CAN-05-0404. [DOI] [PubMed] [Google Scholar]
- 64.Erhardt P, Schremser EJ, Cooper GM. B-Raf inhibits programmed cell death downstream of cytochrome c release from mitochondria by activating the MEK/Erk pathway. Mol Cell Biol. 1999;19:5308–5315. doi: 10.1128/mcb.19.8.5308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Higuchi T, Jass JR. My approach to serrated polyps of the colorectum. J Clin Pathol. 2004;57:682–686. doi: 10.1136/jcp.2003.015230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Spring KJ, Zhao ZZ, Karamatic R, Walsh MD, Whitehall VL, Pike T, Simms LA, Young J, James M, Montgomery GW, et al. High prevalence of sessile serrated adenomas with BRAF mutations: a prospective study of patients undergoing colonoscopy. Gastroenterology. 2006;131:1400–1407. doi: 10.1053/j.gastro.2006.08.038. [DOI] [PubMed] [Google Scholar]
- 67.Yamashita K, Dai T, Dai Y, Yamamoto F, Perucho M. Genetics supersedes epigenetics in colon cancer phenotype. Cancer Cell. 2003;4:121–131. doi: 10.1016/s1535-6108(03)00190-9. [DOI] [PubMed] [Google Scholar]
- 68.Ward RL, Cheong K, Ku SL, Meagher A, O'Connor T, Hawkins NJ. Adverse prognostic effect of methylation in colorectal cancer is reversed by microsatellite instability. J Clin Oncol. 2003;21:3729–3736. doi: 10.1200/JCO.2003.03.123. [DOI] [PubMed] [Google Scholar]