

Abstract
AIM: The genomes of Helicobacter pylori (H. pylori) from different individuals are different. This project was to identify the strain specific DNA sequences between two clinical H. pylori isolates by suppression subtractive hybridization (SSH).
METHODS: Two clinical H. pylori isolates, one from gastric ulcer (GU, tester) and the other from non-ulcer dyspepsia (NUD, driver), were cultured and the genomic DNA was prepared and submitted to Alu I digestion. Then two different adaptors were ligated respectively to the 5’-end of two aliquots of the tester DNA fragments and SSH was made between the tester and driver DNA. The un-hybridized tester DNA sequences were amplified by two sequential PCR and cloned into pGEM-T-Easy Vector. The tester strain specific inserts were screened and disease related DNA sequences were identified by dot blotting.
RESULTS: Among the 240 colonies randomly chosen, 50 contained the tester strain specific DNA sequences. Twenty three inserts were sequenced and the sizes ranged from 261 bp to 1036 bp. Fifteen inserts belonged to the H.pylori plasmid pHPO100 that is about 3.5 kb and codes a replication protein A. Other inserts had patches of homologous to the genes of H.pylori in GenBank. Various patterns of dot blots were given and no GU strain unique DNA sequences were found when 4 inserts were used as probes to screen the genomic DNA from 27 clinical isolates, 8 from GU, 12 from duodenum ulcer (DU), 4 from GU-DU, 2 from NUD and 1 from gastric cancer (GC). But a 670 bp DNA fragment (GU198) that was a bit homologous to the 3’-end of the gene of thymidylate kinase was positive in 7 GU strains (7/8), 3 GU-DU strains (3/4) and 3 DU strains (3/12). A 384 bp fragment (GU79) of the replication gene A (repA) was positive only in 4 H.pylori isolates, 2 from GU and 2 from GU-DU.
CONCLUSION: Differences exist in the genes of different H.pylori isolates. SSH is very effective to screen H.pylori strain specific DNA sequences between two clinical isolates, and some of these sequences may have clinical significance.
INTRODUCTION
Helicobacter pylori (H. pylori) is a microaerophilic Gram-negative bacterium that colonizes the stomach in more than half of the world population. It is the causative agent of chronic gastritis and contributes to peptic ulcer[1,2] and also plays an important role in the pathogenesis of gastric cancer[3-16]. When one is infected by this bacterium, the clinical outcome depends on the interaction of virulent effects of the bacterium, the host response, and the environment[18]. The genomes of H.pylori from different individuals are quite different and each one contains about 1600 genes, among which almost 320 genes are dispensable and 100 genes are strain unique[17-19]. Genes that are present in one strain and absent or substantially different in the others can be of great biological interest[20].
H. pylori strains with the cag pathogenicity island (PAI) can induce more severe inflammation, proliferation and apoptosis in the gerbil mucosa than do the strains partial or complete lack of the cag PAI[21]. In contrast to the CagA- H. pylori infection, CagA+ H. pylori infection is associated with a higher prevalence of p53 mutation in gastric adenocarcinoma[22]. In addition to the cag island, other polymorphic loci that appear clinically relevant have to be identified.
We screened the strain specific DNA sequences between two clinical isolates and tried further to identify the disease related sequences. Different methods, such as microarray[19,21], and subtractive hybridization[18,23,24] could be used for bacterial strain specific DNA screening. Suppression subtractive hybridization (SSH), in which the genomic DNA sample containing the sequences of interest is called “tester”, and the reference sample is called “driver”, is a powerful approach for assessing the DNA sequence differences among the closely related bacterial strains.
MATERIALS AND METHODS
H.pylori growth and genomic DNA extraction
H. pylori clinical isolates preserved in the brain heart infusion broth (BHI, Gibco) supplemented with 100 mL/L horse serum (Gibco) and 200 mL/L glycerol (BDH) were inoculated on the H.pylori selective chocolate blood agar containing 40 g/L blood agar base No.2 (Oxoid) and 50 mL/L horse blood (Gibco). Antibiotics were also used at the following concentrations: vancomycin (Sigma) 3 mg/L, trimethoprim (Sigma) 5 mg/L, nalidixic acid (Sigma) 10 mg/L and amphotericin B (Sigma) 2 mg/L. The plates were incubated in microaerobic atmosphere (50 mL/L CO2) in a CO2 incubator (Forma Scientific) at 37 °C for up to 5 days. H.pylori genomic DNA was extracted following Bow’s method[25].
Driver and tester DNA preparations
The subtractive DNA library was made by using the PCR-select bacterial genome subtraction kit (Clontech) with Akopyants NS’s method (Proc Natl Acad Sci USA. 1998;95: 13108) as reference, but with some change at certain procedures. Genomic DNA of an H.pylori strain from gastric ulcer (GU) was used as the tester, and DNA of a strain from non-ulcer dyspepsia (NUD) was used as the driver. The sequences of the adaptors and primers were listed as followings: adaptor 1, 5’-CTAATACGACTCACTATAGGGCTCGA GCGGCCGCCCGGGCAGGT-3’,5’-ACCTGCCCGG-3’,adaptor 2R, 5’-CTAATACGACTCACTATAGGGCAGCGTGGTCGCGGCCGAGGT-3’, 5’-ACCTCGGCCG-3’, P1, 5’-CTAATACGACTCACTATAGGGC-3’, NP1, 5’-TCGAGCGGCCGCCCGGGCAGGT-3’, NP2, 5’-AGCGTGGTCGCGGCCGAGGT-3’. In brief, 4 µg of the tester or driver genomic DNA was digested with 40 units of AluI (New England Biolabs) in 400 µL reaction volume at 37 °C for 16 hours. The DNA fragments were then extracted with phenol and precipitated with ethanol, and resuspended in sterile distilled water at a final concentration of 300 mg/L. Two aliquots of tester DNA (120 ng each) were ligated separately to the two adaptors (2 µmol/L final concentration) at 16 °C for 16 hours, each in a total volume of 10 µL, with 1 µL (New Engl and Biolabs, 400 units) of T4 DNA ligase in the buffer supplied by the manufacturer. After ligation, 1 µL of 0.2 mol/L EDTA was added, and the sample was heated at 70 °C for 5 minutes to inactivate the ligase and then stored at -20 °C.
Suppression subtractive hybridization
Two microliters of the driver DNA fragments (600 ng) were added to 1 µL (12 ng) of each adaptor-ligated tester DNA (50:1 ratio). One microliter of 4×hybridization buffer (2 mol/L NaCl, 200 mmol/L Hepes, pH 8.0, 0.8 mmol/L EDTA) was added to each tube, and the solution was overlaid with a drop of mineral oil. The DNA fragments were then denatured at 98 °C for 2 minutes, and allowed to anneal at 65 °C for 1.5 hours. After the first hybridization, the two samples (one with adaptor 1, the other with adaptor 2R) were combined and 300 ng more heat-denatured driver DNA dissolved in 2 µL of 1× hybridization buffer was added. The 10 µL mixture was allowed to hybridize at 65 °C for an additional 14 hours. After being diluted to 200 µL with dilution buffer (50 mmol/L, NaCl, 20 mmol/L Hepes, pH8.3, 0.2 mmol/L EDTA), the sample was heated at 65 °C for 10 minutes, and stored at -20 °C.
The first and second PCRs
The first PCR mixture (25 µL) containing 1 µL of the above diluted DNA, 1 µL of 10 µmol/L PCR primer P1, 2 µL of 2.5 mmol/L dNTPs, 2.5 µL of 10×PCR reaction buffer, and 5 µL of the Advantage 2 Polymerase Mix (Clontech) was incubated in a thermal cycler (Perkin-Elmer 2400) at 72 °C for 2 minutes and subjected to 25 cycles at 94 °C for 30 s, at 66 °C for 30 s, and at 72 °C for 1.5 minutes. Seven microliters of the PCR products were analyzed by 15 g/L agarose gel electrophoresis. The products were then diluted 40-fold in 10 mmol/L Tris·HCl (pH 7.5). The second PCR mixture (25 µL) contained 1 µL of the diluted first PCR products, 1 µL of the nest PCR primers (NP1 and NP2, 10 µmol/L each), 2 µL of 2.5 mmol/L dNTPs, 2.5 µL of 10×PCR reaction buffer, and 5 µL of the Advantage 2 Polymerase Mix. The cycling program was 12 cycles at 94 °C for 30 s, at 68 °C for 30 s, and at 72 °C for 1.5 minutes, followed by further extension at 72 °C for 5 minutes. The PCR products were analyzed by 15 g/L agarose gel electrophoresis, purified by using the Quaquick Spin PCR Purification Kit (Qiagen) and cloned into the pGEM- T Easy Vector (Promega) following the protocols. The recombinant plasmids were transformed into E.coli Top10, which was then cultured overnight on the selective agar plates containing 20 µL of 50 g/L ampicillin, 35 µL of 100 mmol/L IPTG and 40 µL of 20 g/L X-gal. White colonies were randomly picked and suspended in 100 µL of Luria-Bertani medium containing ampicillin in an eppendorf tube and cultured at 37 °C for 2 hours. One microliter of the cell suspension, after being frozen and thawed three times, was used as templates and the inserts were amplified under condition as in the second PCR except for 25 cycles. The sizes of the inserts were identified by agarose gel electrophoresis.
Strain specific insert screening and disease related DNA sequence identification
One microliter of each PCR product (10 ng) was dotted on the Hybond N+ membrane (Amersham) in duplicating forms and DNA fixation was carried out by irradiation under a UV transilluminator (Vilber Lourmat) for 5 minutes. The AluI-digested DNA fragments of the tester and driver H.pylori were used as probes and dot blotting was preformed using the ECL direct nucleic acid labeling and detection system (Amersham Phamacia Biotech). The pre-hybridization and hybridization were carried out in the hybridization oven (Amersham) at 42 °C for 1 hour and 12 hours respectively. After stringently washing the Hybond N+ membrane (twice for 20 minutes in 6 mol/L Urea, 4 g/L SDS and 0.1×SSC, and twice for 5 minutes in 2× SSC), the chemiluminenscence signals were detected by exposure of the Hyperfilm to the membrane for 5 to 30 minutes. The inserts that gave positive results to the tester probes and negative to the driver probes were sequenced using a Big Dye Terminator DNA sequencing kit (Perkin-Elmer) and ABI automated sequencer. The sequences were then submitted to gene and protein homologous analysis. In the same way, by dotting 100 ng of each genomic DNA from different H.pylori isolates on the membrane and using the inserts interested as probes, the disease related DNA sequences were identified. In the above cases, 1 ng of each probe DNA was dotted on the membrane to be used as positive control.
RESULTS
Tester strain specific DNA sequencing and homologous analysis
After SSH between the tester and driver DNA fragments, about 900 colonies grew on the ampicillin plates and two thirds of them were white in color. Two hundred and forty white colonies were randomly chosen and the inserts were amplified using the primers as in the nest PCR. By dotting and fixing the equal amount of each PCR product in replica form on the membrane, dot blotting was carried out using the AluI digested DNA fragments as probes (Figure 1). Fifty tester strain specific DNA sequences were screened and 23 of them were sequenced. The sizes of the inserts ranged from 261 to 1036 bp. Blast analysis showed that, of the 23 inserts sequenced, 15 belonged to parts of the H.pylori plasmid pHPO100 (Table 1 only shows GU79) and 2 of them contained the same fragment. The size of this plasmid is 3520 bp, which codes a replication protein A (RepA, 1842-2765) and an unknown protein (2762-3520). The other 8 inserts were homologous to genes of H.pylori in GenBank to different extent (Table 1).
Figure 1.
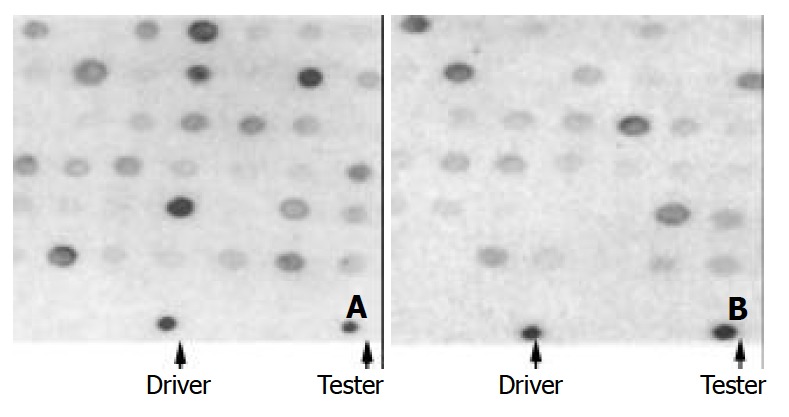
Dot blotting to screen the tester strain specific inserts by using the Alu I digested tester DNA (A) and driver DNA (B) as probes. The arrowheads indicate the spots of positive controls.
Table 1.
Homologous analysis of some strain specific DNA sequences
| Inserts | GenBank accesion No. | Homolog/genes or proteins | DNA matches |
| GU79 | AF056496 | H.pylori plasmid pHPO100/ RepA, 2e-53 | 1882-2265 |
| GU150 | AE000629 | Carbamoyl-phosphate synthetase, 3e-29 | 1029-1229 |
| GU185 | AE000595 | Putative outer membrane p1, 7e-24 | 2993-3177 |
| GU198 | AE001560 | Thymidylate kinase, 2e-10 | 640-717 |
| GU210 | AE000590 | Hypothetical protein , 3e-33 | 789-396 |
| GU212 | AE000531 | Type II restriction enzyme R protein, 4e-26 | 1-597 |
| GU223 | AE000647 | Hypothetical protein, 4e-16 | 7724-8193 |
| GU234 | AE000650 | Site specific DNA-methyltransferase, 3e-54 | 9070-8752 |
| GU235 | AE000547 | Toxin-like outer membrane protein, 2e-64 | 13943-14494 |
Disease related DNA sequence screening
We next chose 4 tester strain specific DNA sequences as probes to screen the genomic DNA of H.pylori with different clinical background, 8 from GU, 12 from duodenum ulcer (DU), 4 from GU-DU, 2 from NUD and 1 from gastric cancer (GC), to know if some of them were specific for the GU isolates. We found that the 4 different inserts gave 4 different dot blotting profiles (Figure 2) and there were no GU strain unique sequences. But a DNA fragment GU198 (670 bp) that was a bit (78 bp) homologous to the 3’-end of the gene of hymidylate kinase was positive in 7 of the 8 GU strains, 3 of the 4 GU-DU strains, and 3 of the 12 DU strains. It was also positive in one of the 2 NUD strains and the GC strain. The replication gene A fragment GU79 (384 bp) was positive in 4 H.pylori isolates, 2 from GU and 2 from GU-DU.
Figure 2.

Dot blotting for identification the disease related DNA sequences in 27 different clinical isolates by using GU210 (A), GU79 (B), GU198 (C) and GU235 (D) as probes. The arrow-heads indicate the spots of positive controls.
DISCUSSION
A remarkable feature of H.pylori is the high diversity of its genomic DNA[26-29]. Clinical isolates of H.pylori from different individuals show enormous variations in their genomes or genes[30-33]. The genetic diversity is represented in many forms, including point mutations and inserted sequences[34]. Such variation has given rise to the notion that H. pylori has a very plastic genome, and that such plasticity confers a selective advantage on a bacterium that must co-evolve with its host over the course of decades[17]. Based on this, it is conceivable that there are different DNA sequences between two genomes of H.pylori with different clinical background.
SSH is a powerful technique that has been applied to research in many different fields. In studies of eukaryotic systems, application of subtraction techniques typically focuses on differential gene expression between two cDNA populations rather than differences between genomes (Diatchenko L. Proc Natl Acad Sci USA 1996; 93: 6025). This is because eukaryotic genomes are too complex for existing subtraction technologies. In contrast, bacterial genomes are considerably smaller, and are even less complex than many eukaryotic cDNA populations. Thus, subtraction methods can be used to identify sequences that are present in one bacterial genome, but absent in another. It requires only about several micrograms of genomic DNA, takes only several days, and does not involve the physical separation of single strain and double strain molecules. Furthermore, the suppression PCR prevents undesirable amplification while enrichment of target molecules proceeds. Even so, the conditions of the SSH were carefully optimized in this experiment. By comparing the genomic DNA digestion efficiency of Hae III, Rsa I and Alu I, Alu I was finally selected. The time of the adaptor ligation should be within 16 hours to avoid ligation between the DNA fragments. The ratio of tester and driver DNA was 1:50 in the first hybridization. In this way, we established a GU strain specific DNA sequence library.
When we used the tester and driver DNA fragments as probes to hybridize separately with different PCR products of the subtracted inserts, the tester specific DNA sequences were screened. Of the 23 sequenced DNA fragments, 15 were homologous to the H.pylori plasmid pHPO100. The size of this plasmid is about 3.5 kb and codes a replication protein A, which appears to be the predominant plasmid replication protein of H. pylori and has the highly conserved (76%-96%) amino acid sequence and may play an important role in the DNA sequence exchange between plasmid and the chromosome[35]. Thus the protein may increase the gene diversity and have the pathogenic significance. Other inserts had patches of homologous to the genes of different H.pylori proteins in GenBank. The results indicate that SSH can be used to screen the strain specific DNA sequences between clinical isolates even though the dominant subtracted fragments come from the plasmid of the tester strain.
When we tried to find out if some of the tester strain specific DNA sequences were unique to GU strains, no such DNA sequences were identified. Whereas a DNA fragment GU198 that is partly homologous to the gene of thymidylate kinase was positive in 7 of the 8 GU strains, 3 of the 4 GU-DU strains, and only 3 of the 12 DU strains. This is also of clinical significance. Study on identification of this DNA fragment is still going on.
Several aspects regarding the identification of disease related DNA sequences need to be discussed. First, the tester strain specific DNA sequences mean that, comparing with the driver strain, these sequences are unique to the tester strain, other strains from GU patients may share or may not share these sequences. Second, since the strain specific genes account for about 6% of the whole genes of a given H.pylori strain[19], more colonies should be screened and more DNA fragments be used to identify the disease specific DNA sequences. Third, dot blotting can be used as a primary screening method, and, if possible, Southern blotting be employed for further identification. Fourth, it is better to choose the plasmid free H. pylori for the strain specific DNA sequence screening. Since the copies of a plasmid in the bacterium are often more than that of a gene in its genome, amplification of the subtracted gene sequences coming from the genome would be suppressed in the PCR and, as a result, the dominant strain specific DNA sequences would belong to the plasmid. Although about 50% of H. pylori strains carry cryptic plasmids ranging in size from 2 to about 100 kb[35,36], the role of these plasmids is not well understood. In this investigation, the repA gene was positive in 4 H.pylori strains, 2 from GU (2/8), and 2 from GU-DU (2/ 4). This may be due to the homologous of the genomic DNA to the repA or the insertion of the repA fragment into the genome (De Ungria MC. Plasmid 1999; 41:97) or the plasmid DNA being not eliminated during genomic DNA preparation. Study on the relationship between repA gene and the disease state of the patients is also necessary.
Anyway, we have successfully screened some tester strain specific DNA sequences by SSH, and this study is one of the few attempts trying to identify the disease specific genes using H.pylori clinical isolates[23]. Though we failed to get the disease specific DNA sequences, we believe that, at least, some tester strain specific sequences may have significantly high positive rates in strains with similar background to the tester strain. This may aid in the effective diagnosis and treatment of H. pylori infection and have the potential value for pathogenic investigation of this bacterium.
ACKNOWLEDGEMENTS
Mr. Mun-Fai Loke, from Department of Microbiology, National University of Singapore, prepared some of the H. pylori clinical isolates.
Footnotes
Supported by the NMRC-Sponsored Project, Grant number: R-182-000-037-213
Edited by Zhang JZ
References
- 1.Israel DA, Peek RM. pathogenesis of Helicobacter pylori-induced gastric inflammation. Aliment Pharmacol Ther. 2001;15:1271–1290. doi: 10.1046/j.1365-2036.2001.01052.x. [DOI] [PubMed] [Google Scholar]
- 2.Sanders MK, Peura DA. Helicobacter pylori-Associated Diseases. Curr Gastroenterol Rep. 2002;4:448–454. doi: 10.1007/s11894-002-0019-x. [DOI] [PubMed] [Google Scholar]
- 3.Dawsey SM, Mark SD, Taylor PR, Limburg PJ. Gastric cancer and H pylori. Gut. 2002;51:457–458. doi: 10.1136/gut.51.3.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blaser MJ. Linking Helicobacter pylori to gastric cancer. Nat Med. 2000;6:376–377. doi: 10.1038/74627. [DOI] [PubMed] [Google Scholar]
- 5.Unger Z, Molnár B, Prónai L, Szaleczky E, Zágoni T, Tulassay Z. Mutant p53 expression and apoptotic activity of Helicobacter pylori positive and negative gastritis in correlation with the presence of intestinal metaplasia. Eur J Gastroenterol Hepatol. 2003;15:389–393. doi: 10.1097/00042737-200304000-00009. [DOI] [PubMed] [Google Scholar]
- 6.Yang Y, Deng CS, Peng JZ, Wong BC, Lam SK, Xia HH. Effect of Helicobacter pylori on apoptosis and apoptosis related genes in gastric cancer cells. Mol Pathol. 2003;56:19–24. doi: 10.1136/mp.56.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang RT, Wang T, Chen K, Wang JY, Zhang JP, Lin SR, Zhu YM, Zhang WM, Cao YX, Zhu CW, et al. Helicobacter pylori infection and gastric cancer: evidence from a retrospective cohort study and nested case-control study in China. World J Gastroenterol. 2002;8:1103–1107. doi: 10.3748/wjg.v8.i6.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meining A, Riedl B, Stolte M. Features of gastritis predisposing to gastric adenoma and early gastric cancer. J Clin Pathol. 2002;55:770–773. doi: 10.1136/jcp.55.10.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lan J, Xiong YY, Lin YX, Wang BC, Gong LL, Xu HS, Guo GS. Helicobacter pylori infection generated gastric cancer through p53-Rb tumor-suppressor system mutation and telomerase reactivation. World J Gastroenterol. 2003;9:54–58. doi: 10.3748/wjg.v9.i1.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cai L, Yu SZ, Zhang ZF. Helicobacter pylori infection and risk of gastric cancer in Changle County,Fujian Province,China. World J Gastroenterol. 2000;6:374–376. doi: 10.3748/wjg.v6.i3.374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu HF, Liu WW, Fang DC, Wang GA, Teng XC. Relationship between Helicobacter pylori infection and gastric precancerous lesions: a follow-up study. Shijie Huaren XiaohuaZazhi. 2002;10:912–915. [Google Scholar]
- 12.Miehlke S, Kirsch C, Dragosics B, Gschwantler M, Oberhuber G, Antos D, Dite P, Läuter J, Labenz J, Leodolter A, et al. Helicobacter pylori and gastric cancer: current status of the Austrain Czech German gastric cancer prevention trial (PRISMA Study) World J Gastroenterol. 2001;7:243–247. doi: 10.3748/wjg.v7.i2.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yao YL, Xu B, Song YG, Zhang WD. Overexpression of cyclin E in Mongolian gerbil with Helicobacter pylori-induced gastric precancerosis. World J Gastroenterol. 2002;8:60–63. doi: 10.3748/wjg.v8.i1.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Z, Yuan Y, Gao H, Dong M, Wang L, Gong YH. Apoptosis, proliferation and p53 gene expression of H. pylori associated gastric epithelial lesions. World J Gastroenterol. 2001;7:779–782. doi: 10.3748/wjg.v7.i6.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xue FB, Xu YY, Wan Y, Pan BR, Ren J, Fan DM. Association of H. pylori infection with gastric carcinoma: a Meta analysis. World J Gastroenterol. 2001;7:801–804. doi: 10.3748/wjg.v7.i6.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu HF, Liu WW, Wang GA, Teng XC. Effect of Helicobacter pylori infection on Bax protein expression in patients with gastric precancerous lesions. World J Gastroenterol. 2005;11:5899–5901. doi: 10.3748/wjg.v11.i37.5899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Björkholm BM, Oh JD, Falk PG, Engstrand LG, Gordon JI. Genomics and proteomics converge on Helicobacter pylori. Curr Opin Microbiol. 2001;4:237–245. doi: 10.1016/s1369-5274(00)00197-1. [DOI] [PubMed] [Google Scholar]
- 18.Agron PG, Macht M, Radnedge L, Skowronski EW, Miller W, Andersen GL. Use of subtractive hybridization for comprehensive surveys of prokaryotic genome differences. FEMS Microbiol Lett. 2002;211:175–182. doi: 10.1111/j.1574-6968.2002.tb11221.x. [DOI] [PubMed] [Google Scholar]
- 19.Salama N, Guillemin K, McDaniel TK, Sherlock G, Tompkins L, Falkow S. A whole-genome microarray reveals genetic diversity among Helicobacter pylori strains. Proc Natl Acad Sci USA. 2000;97:14668–14673. doi: 10.1073/pnas.97.26.14668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blaser MJ, Berg DE. Helicobacter pylori genetic diversity and risk of human disease. J Clin Invest. 2001;107:767–773. doi: 10.1172/JCI12672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Israel DA, Salama N, Arnold CN, Moss SF, Ando T, Wirth HP, Tham KT, Camorlinga M, Blaser MJ, Falkow S, et al. Helicobacter pylori strain-specific differences in genetic content, identified by microarray, influence host inflammatory responses. J Clin Invest. 2001;107:611–620. doi: 10.1172/JCI11450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shibata A, Parsonnet J, Longacre TA, Garcia MI, Puligandla B, Davis RE, Vogelman JH, Orentreich N, Habel LA. CagA status of Helicobacter pylori infection and p53 gene mutations in gastric adenocarcinoma. Carcinogenesis. 2002;23:419–424. doi: 10.1093/carcin/23.3.419. [DOI] [PubMed] [Google Scholar]
- 23.Kersulyte D, Velapatiño B, Dailide G, Mukhopadhyay AK, Ito Y, Cahuayme L, Parkinson AJ, Gilman RH, Berg DE. Transposable element ISHp608 of Helicobacter pylori: nonrandom geographic distribution, functional organization, and insertion specificity. J Bacteriol. 2002;184:992–1002. doi: 10.1128/jb.184.4.992-1002.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kersulyte D, Mukhopadhyay AK, Shirai M, Nakazawa T, Berg DE. Functional organization and insertion specificity of IS607, a chimeric element of Helicobacter pylori. J Bacteriol. 2000;182:5300–5308. doi: 10.1128/jb.182.19.5300-5308.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hua JS, Zheng PY, Fong TK, Khin MM, Bow H. Helicobacter pylori acquistion of metronidazole resistance by natural transformation in vitro. World J Gastroenterol. 1998;4:385–387. doi: 10.3748/wjg.v4.i5.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suerbaum S. Genetic variability within Helicobacter pylori. Int J Med Microbiol. 2000;290:175–181. doi: 10.1016/S1438-4221(00)80087-9. [DOI] [PubMed] [Google Scholar]
- 27.Yakoob J, Hu GL, Fan XG, Yang HX, Liu SH, Tan DM, Li TG, Zhang Z. Diversity of Helicobacter pylori among Chinese persons with H. pylori infection. APMIS. 2000;108:482–486. doi: 10.1034/j.1600-0463.2000.d01-86.x. [DOI] [PubMed] [Google Scholar]
- 28.Israel DA, Salama N, Krishna U, Rieger UM, Atherton JC, Falkow S, Peek RM. Helicobacter pylori genetic diversity within the gastric niche of a single human host. Proc Natl Acad Sci USA. 2001;98:14625–14630. doi: 10.1073/pnas.251551698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nobusato A, Uchiyama I, Kobayashi I. Diversity of restriction-modification gene homologues in Helicobacter pylori. Gene. 2000;259:89–98. doi: 10.1016/s0378-1119(00)00455-8. [DOI] [PubMed] [Google Scholar]
- 30.Tomasini ML, Zanussi S, Sozzi M, Tedeschi R, Basaglia G, De Paoli P. Heterogeneity of cag genotypes in Helicobacter pylori isolates from human biopsy specimens. J Clin Microbiol. 2003;41:976–980. doi: 10.1128/JCM.41.3.976-980.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ji WS, Hu JL, Qiu JW, Peng DR, Shi BL, Zhou SJ, Wu KC, Fan DM. Polymorphism of flagellin A gene in Helicobacter pylori. World J Gastroenterol. 2001;7:783–787. doi: 10.3748/wjg.v7.i6.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pride DT, Meinersmann RJ, Blaser MJ. Allelic Variation within Helicobacter pylori babA and babB. Infect Immun. 2001;69:1160–1171. doi: 10.1128/IAI.69.2.1160-1171.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Azuma T, Yamakawa A, Yamazaki S, Fukuta K, Ohtani M, Ito Y, Dojo M, Yamazaki Y, Kuriyama M. Correlation between variation of the 3' region of the cagA gene in Helicobacter pylori and disease outcome in Japan. J Infect Dis. 2002;186:1621–1630. doi: 10.1086/345374. [DOI] [PubMed] [Google Scholar]
- 34.Nobusato A, Uchiyama I, Ohashi S, Kobayashi I. Insertion with long target duplication: a mechanism for gene mobility suggested from comparison of two related bacterial genomes. Gene. 2000;259:99–108. doi: 10.1016/s0378-1119(00)00456-x. [DOI] [PubMed] [Google Scholar]
- 35.Hofreuter D, Haas R. Characterization of two cryptic Helicobacter pylori plasmids: a putative source for horizontal gene transfer and gene shuffling. J Bacteriol. 2002;184:2755–2766. doi: 10.1128/JB.184.10.2755-2766.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hosaka Y, Okamoto R, Irinoda K, Kaieda S, Koizumi W, Saigenji K, Inoue M. Characterization of pKU701, a 2.5-kb plasmid, in a Japanese Helicobacter pylori isolate. Plasmid. 2002;47:193–200. doi: 10.1016/s0147-619x(02)00003-3. [DOI] [PubMed] [Google Scholar]