

Abstract
DNA damage response (DDR) and the centrosome cycle are 2 of the most critical cellular processes affecting the genome stability in animal cells. Yet the cross-talks between DDR and the centrosome are poorly understood. Here we showed that deficiency of the breast cancer 1, early onset gene (BRCA1) induces centrosome amplification in non-stressed cells as previously reported while attenuating DNA damage-induced centrosome amplification (DDICA) in cells experiencing prolonged genotoxic stress. Mechanistically, the function of BRCA1 in promoting DDICA is through binding and recruiting polo-like kinase 1 (PLK1) to the centrosome. In a recent study, we showed that FancJ also suppresses centrosome amplification in non-stressed cells while promoting DDICA in both hydroxyurea and mitomycin C treated cells. FancJ is a key component of the BRCA1 B-complex. Here, we further demonstrated that, in coordination with BRCA1, FancJ promotes DDICA by recruiting both BRCA1 and PLK1 to the centrosome in the DNA damaged cells. Thus, we have uncovered a novel role of BRCA1 and FancJ in the regulation of DDICA. Dysregulation of DDR or centrosome cycle leads to aneuploidy, which is frequently seen in both solid and hematological cancers. BRCA1 and FancJ are known tumor suppressors and have well-recognized functions in DNA damage checkpoint and DNA repair. Together with our recent findings, we demonstrated here that BRCA1 and FancJ also play an important role in centrosome cycle especially in DDICA. DDICA is thought to be an alternative fail-safe mechanism to prevent cells experiencing severe DNA damage from becoming carcinogenic. Therefore, BRCA1 and FancJ are potential liaisons linking early DDR with the DDICA. We propose that together with their functions in DDR, the role of BRCA1 and FancJ in the activation of DDICA is also crucial for their tumor suppression functions in vivo.
Keywords: BRCA1, centrosome amplification, DNA damage response, FancJ, interstrand cross-link, PLK1
Abbreviations
- ATM
ataxia telangiectasia mutated
- ATR
ataxia telangiectasia Rad3-related
- BRCA1
breast cancer gene 1
- CIN
chromosome instability
- DDICA
DNA damage induced centrosome amplification
- DDR
DNA damage response
- GFP
green fluorescent protein
- HR
homologous recombination
- HU
hydroxyurea
- ICL
interstrand cross-linkers
- MIN
microsatellite instability
- MMC
mitomycin C
- MT
microtubule
- PCM
pericentriolar materials
- PLK1
Polo-like kinase 1
- UTR
untranslated region
- WCL
whole-cell lysate
Introduction
Genomic instability is a critical enabling characteristic of cancer1 and can be classified broadly into 2 major groups: chromosomal instability (CIN) and microsatellite instability (MIN).2 Animal cells are constantly exposed to genotoxic insults, from either endogenous or exogenous sources, which then activate a variety of DNA damage checkpoint and DNA repair processes that are collectively called DNA damage response (DDR).3 DDR is essential to maintain a stable genome of animal cells, and deficiency of genes in DDR pathways often causes cancer.4 The numerical and structural integrity of the centrosome in animal cells are also critical for their genome stability. As demonstrated recently by 2 labs, cells with more than 2 centrosomes (centrosome amplification) have increased frequency of merotelic kinetochore attachment during mitosis, which in turn induces lagging chromosome and aneuploidy.5,6 Centrosome amplification is frequently observed in a variety of solid and hematological cancers.7 It is now generally accepted that defects of either DDR or the centrosome can induce aneuploidy and therefore are likely the major causes of tumorigenesis.8,9
The centrosome is an important organelle and functions primarily as the microtubule (MT) organizing center in animal cells.10,11 Most animal cells have either one or 2 centrosomes, depending on the cell cycle stage, and the number and structure of the centrosome are highly regulated during each cell cycle. Similar to the replication of DNA, the centrosome duplicates once and only once per cell cycle. Most studies have focused on the centrosome cycle in non-stressed cells and the molecular mechanism behind it has been well elucidated.10
Sporadic evidence suggests a connection between DDR and the centrosome. For example, deficiency of centrosomal proteins, such as pericentrin (PCNT), CEP131, CEP152, and CEP164, causes DDR defects.12-15 On the other hand, deficiency of certain DDR proteins, including BRCA1, induces centrosome amplification.16,17 Further supporting this connection, it has long been observed that a variety of genotoxic stress can induce pronounced centrosome amplification (DNA damage induced centrosome amplification, or DDICA).18-20 DDICA has been reported in a variety of organisms, including fly, chicken, rodent, and human. In Drosophila, DDICA is dependent on Chk2 but not Chk1.20 In chicken and mammalian cells, known DDR proteins involved in regulating DDICA include ATM, BRIT1/MCPH1, Chk1, MDC1, and PLK1.17,21-25 However, the detailed mechanism behind the regulation of DDICA is still a mystery.
BRCA1 is one of the most important DDR factors and is involved in a variety of DDR processes.26,27 BRCA1 also has potential roles in the centrosome cycle. BRCA1 localizes to the centrosome28-31 and regulates centrosome related processes; for example, recruiting TPX2 to the spindle poles32 and mono-ubiquitinating γ-Tubulin.33 The former fine-tunes the mitotic spindle pole assembly, while the latter inhibits the MT nucleation function of the centrosome during S phase. Most importantly, centrosome amplification and aneuploidy are observed in both BRCA1-deficient mouse models as well as human breast cancers, suggesting that the centrosomal function of BRCA1 may also be critical for its tumor suppression functions.34-37 Nonetheless, whether and how BRCA1 plays a role in DDICA is unknown.
We showed recently that another key DDR factor, FancJ (also called BACH1, or BRIP1), is also involved in regulating centrosome biogenesis and DDICA.38 FancJ belongs to the Fanconi Anemia (FA) family of genes, whose major function is to repair interstrand cross-linkers (ICL) induced genotoxic lesions.39 Our studies showed that FancJ inhibits centrosome amplification in non-stressed cells. In addition, by acting upstream of PLK1, FancJ also promotes both hydroxyurea (HU) and mitomycin C (MMC)-induced centrosome amplification. Consistent with our findings, Nalepa and colleagues also observed a higher percentage of fibroblast cells taken from FA patients including FancJ patients showing centrosome amplification compared to that from healthy individuals.40
Though the exact physiological implication is still unknown, DDICA is thought to be another fail-safe mechanism to commit cells experiencing severe DNA damages to cell death and thereby plays a potential role in suppressing tumorigenesis.10 Here we showed that deficiency of BRCA1 induces centrosome amplification in non-stressed cells as previously reported while attenuating both HU- and MMC-induced centrosome amplification. Intriguingly, MMC stimulates the centrosome relocalization of BRCA1 in late G2 cells. The function of BRCA1 in promoting DDICA is through binding and recruiting PLK1 to the centrosome in response to DNA damage. Finally, we demonstrated that in coordination with BRCA1, FancJ promotes DDICA by recruiting both BRCA1 and PLK1 to the centrosome in response to DNA damage. We propose that the role of BRCA1 and FancJ in promoting DDICA may contribute to their tumor suppression functions in vivo.
RESULTS
BRCA1 promotes DNA damage-induced centrosome amplification
Among its many biological functions, BRCA1 has well-established roles in regulating the centrosome cycle as well as centrosome related processes.41 siRNA depletion or dominant negative of BRCA1 in human cells and genetic knockout of BRCA1 in mice induce pronounced centrosome amplification and aneuploidy.33-36,42 Using different siRNAs (BRCA1-pool siRNA, which consists of 4 different siRNA, BRCA1-siRNA-A, -B, -C, and –D; and BRCA1-UTR siRNA, which targets the 3‘-UTR region of the human BRCA1 gene), we confirmed that in 2 different human cell lines, U2-OS and Hs587T, depletion of BRCA1 indeed induces centrosome amplification in non-stressed cells (Figs. S1B–D and S2). γ-Tubulin, a key component of the pericentriolar material (PCM) of the centrosome, and Centrin 2, a key component of the centriole, were used as the centrosome markers in these experiments and throughout our studies. A variety of genotoxic stresses are known to induce pronounced centrosome amplification.18–20 We recently showed that prolonged treatment of 2 ICLs, MMC and cis-platin, also induces pronounced centrosome amplification,38 suggesting that the induction of centrosome amplification is likely an integral part of DDR. Because BRCA1 is such an important DDR protein and is modified and stabilized during sustained DNA damage,26,27,43 we then investigated whether BRCA1 is also involved in DDICA. Interestingly, depletion of BRCA1 reduced the MMC- and HU-induced centrosome amplification by about 30–50% (Fig. 1A–C; Figs. S1A–C, 1E and F, and S2). Because the BRCA1-UTR siRNA specifically targets the 3’UTR region of human BRCA1 mRNA,44 expression of the BRCA1 cDNA efficiently rescued the BRCA1 protein (Fig. 1D). Overexpression of the BRCA1 cDNA fully rescued the reduced centrosome amplification in BRCA1-UTR siRNA transfected cells (Fig. 1E). Moreover, the percentage of cells with amplified centrosome in BRCA1 UTR-siRNA/BRCA1 cells is even 30% higher than that in Control siRNA/GFP cells, suggesting that overexpression of BRCA1 may further stimulate the DDICA. Indeed, overexpression of BRCA1 in Control siRNA transfected cells increased the centrosome amplification approximately 30–40% compared to the overexpression of GFP alone (Fig. 1E, the first 2 columns). This stimulating effect of BRCA1 on the centrosome amplification is DNA damage-dependent because overexpression of BRCA1 in non-damaged cells does not affect the centrosome amplification (Fig. 4D, the 0 hr samples). Together with previous studies, these data suggest that BRCA1 suppresses centrosome amplification in non-stressed cells while stimulates DDICA in cells experiencing prolonged DNA damages.
Figure 1.

BRCA1 promotes mitomycin C-induced centrosome amplification. (A) Representative images of MMC induced centrosome amplification. U2-OS cells were treated with 0.5 μM MMC for 72 hours. Cells were fixed in methanol and then stained with antibodies against γ-Tubulin (green) and Centrin-2 (red). Nuclei were stained with DAPI (blue). (B and C) Depletion of BRCA1 attenuates MMC induced centrosome amplification. U2-OS cells were transfected with either Control siRNA or siRNA against BRCA1 (BRCA1-pool or BRCA1-UTR) and then split into 2 sets. One set of cells was collected for western blot analysis (B). The second set of cells was treated with 0.5 μM MMC for 72 hours and then fixed in methanol and stained with antibodies against γ-Tubulin. More than 300 cells were counted and the percentage of cells with more than 2 centrosomes was quantified (C). (D and E) Expression of siRNA-resistant BRCA1 cDNA rescues the centrosome amplification defects in BRCA1 depleted cells. U2-OS cells were transfected with either Control siRNA (C) or siRNA against BRCA1 (BRCA1-UTR, UTR) and then transfected with plasmid expressing either Green Fluorescent Protein (GFP) or GFP-BRCA1 (BRCA1). These cells were then split into 2 sets. One set of cells was used for protein gel blot analysis (D). The second set of cells was treated with 0.5 μM MMC for 72 hours and then fixed in methanol and stained with antibodies against γ-Tubulin. More than 300 cells were counted and the percentage of cells with more than 2 centrosomes was quantified (E). Immunoblotting antibodies are indicated on the right. All error bars are standard deviation obtained from 3 different experiments. Standard 2-sided t test: **P < 0.01, ***P < 0.001. NS, not significant.
Figure 4 (See previous page).

BRCA1 and FancJ cooperatively promote mitomycin C-induced centrosome amplification. (A and B) Co-depletion of BRCA1 and FancJ further attenuates MMC-induced centrosome amplification. U2-OS cells were transfected with either Control siRNA (C), siRNA against BRCA1 (B), FancJ (J), or both BRCA1 and FancJ (B + J) and then split into 2 sets. One set was collected for protein gel blot analysis (A). The second set of cells was treated with 0.5 μM MMC for 72 hours and then fixed in methanol and stained with antibodies against γ-Tubulin. More than 300 cells were counted and the percentage of cells with more than 2 centrosomes was quantified (B). (C and D) Overexpression of BRCA1 or FancJ or both stimulates the MMC-induced centrosome amplification. U2-OS cells were transfected with plasmid expressing either GFP, GFP-BRCA1 (B), FLAG-FancJ (J), or both GFP-BRCA1 and FLAG-FancJ (B + J). 48 hours after transfection, the cells were then pooled and split into 5 sets (Set #1 to #5). Cells of Set #1 were collected for western blot analysis (C). For cells of Sets #2 to #5, they were either left untreated (Set #2, 0 h) or treated with 0.5 μM MMC for the indicated time (Set #3 to #5), and then were fixed in methanol and stained with antibodies against γ-Tubulin. More than 300 cells were counted and the percentage of cells with more than 2 centrosomes was quantitated (D). (E) Inhibition of PLK1 in BRCA1 and FancJ co-depleted cells further attenuates the centrosome amplification defects. U2-OS cells were transfected with either Control siRNA (C) or siRNA against both BRCA1 and FancJ (B + J) and then split into 2 sets. One set of cells was treated with 0.5 μM MMC for 72 hours (-BI). In the second set, cells were first treated with 0.5 μM MMC for 12 hours, followed by the addition of 100 nM BI-2536 (+BI). Sixty hours later, cells were fixed and stained with antibodies against γ-Tubulin. More than 300 cells were counted and the percentage of cells with more than 2 centrosomes was quantified. (F and G) Expression of the constitutive active PLK1 rescues the centrosome amplification defects in BRCA1 and FancJ co-depleted cells. The 3 different U2-OS cell lines expressing different PLK1 variants as in Figure 3 were first transfected with either Control siRNA (C) or siRNA against both BRCA1 and FancJ (B + J). Dox was added to the cells to induce the expression of PLK1 variants. The knockdown efficiency of BRCA1 and FancJ were monitored by protein gel blot (F). In addition, cells were treated with 0.5 μM MMC for 72 hours, then fixed in methanol and stained with antibodies against γ-Tubulin. More than 300 cells were counted and the percentage of cells with more than 2 centrosomes was quantified (G). Immunoblotting antibodies are indicated on the right. All error bars are standard deviation obtained from 3 different experiments. Standard 2-sided t test: *P < 0.05, **P < 0.01, ***P < 0.001. NS, not significant.
Mitomycin C induces centrosome relocalization of BRCA1
Our recent work demonstrated that FancJ localizes to the centrosome during G1 and S phase (cells with either one γ-Tubulin dot or 2 closely positioned γ-Tubulin dots), while the centrosomal staining of FancJ is significantly weaker during G2 and M phase (cells with 2 further separated γ-Tubulin dots).38 However, when treated with MMC, the centrosomal staining of FancJ increased in almost 100% of G2 cells (MMC reduces the percentage of M phase cells to almost zero). More intriguingly, FancJ predominantly localizes to the mother centrosome. FancJ is a key component of BRCA1 B-complex.45,46 Next, we examined whether MMC also regulates the centrosome localization of BRCA1. Because of the conflicting reports related to the centrosome localization of BRCA1,28-30,47 Fukasawa and colleagues revisited this issue recently.31 Using 2 different antibodies against BRCA1, siRNA depletion as well as tagged protein, they have convincingly demonstrated that BRCA1 indeed localizes to the centrosome in MCF7 cells. Using one of the antibodies in their studies, BRCA1-Ab2, we also confirmed the specificity of the BRCA1-Ab2 antibody and centrosome localization of BRCA1 in U2-OS cells (Fig. S3). In U2-OS cells, we observed strong centrosome staining of BRCA1 in G1 and S cells (Fig. S3 and data not shown). However, the centrosomal BRCA1 signal is dramatically reduced in more than half of the untreated G2 cells (Fig. 2). Intriguingly, when treated with MMC, 90% of G2 cells showed strong centrosomal BRCA1 staining, suggesting that, consistent with data shown in Fig. 1, BRCA1 most likely play a direct role in MMC-induced centrosome amplification.
Figure 2.

Mitomycin C stimulates BRCA1 centrosome localization in G2 cells. U2-OS cells were either left untreated (-MMC) or treated with 0.5 μM MMC overnight (+MMC). Cells were then fixed in methanol and stained with antibodies against γ-Tubulin (green) and BRCA1 (red). Nuclei were stained with DAPI (blue). Representative images of G2 cells are shown in (A). More than 100 cells with the G2 centrosome staining pattern (2 further separated centrosomes) were counted and quantified (B). All error bars are standard deviation obtained from 4 different experiments. Standard 2-sided t test: **P < 0.01.
BRCA1 promotes mitomycin C-induced centrosome amplification through binding and regulating PLK1
We and others have shown that the activation of HU- and MMC-induced DDICA is largely dependent on PLK1.21,38,48,49 Our recent work further demonstrated that FancJ acts upstream of PLK1 during MMC-induced DDICA.38 FancJ is also part of the BRCA1 B-complex.45,46 We therefore hypothesized that BRCA1 may also regulate PLK1 during DDICA. We first examined whether BRCA1 associates with PLK1. Similar to our recent findings that HU induces robust interaction between BRCA1 and PLK1,50 as seen in Fig. 3A, MMC also induces robust interaction between BRCA1 and PLK1. Interestingly, the kinetics of this interaction track very well with the MMC-induced centrosome amplification, which also occurs around 48 hours after the addition of MMC and peaks around 72 hours (Fig. 4D).38 Next, we tested whether BRCA1 also acts upstream of PLK1 during MMC-induced DDICA. For this, we took advantage of the 3 doxycycline (Dox)-inducible PLK1 variants in U2-OS cells generated by Medema and colleagues51: a wild-type PLK1 (WT), a kinase-dead PLK1 (K82R), and a constitutively active PLK1 (T210D) (Fig. 3B). These 3 cell lines were first transfected with either Control siRNA or siRNA against BRCA1, and subsequently the expression of PLK1 was induced with Dox. The cells were then treated with MMC for 72 hours and then stained with anti-γ-tubulin antibody to mark the centrosome. BRCA1 was efficiently depleted in all 3 cell lines (Fig. 3C). Interestingly, the expression of PLK1-T210D fully rescued the reduced centrosome amplification in BRCA1 depleted cells, while PLK1-WT partially rescued the defects (Fig. 3D). In contrast, the kinase-dead PLK1 failed to rescue this defect. These data strongly suggest that similar to FancJ, BRCA1 also binds and acts upstream of PLK1 during the MMC-induced centrosome amplification.
Figure 3.
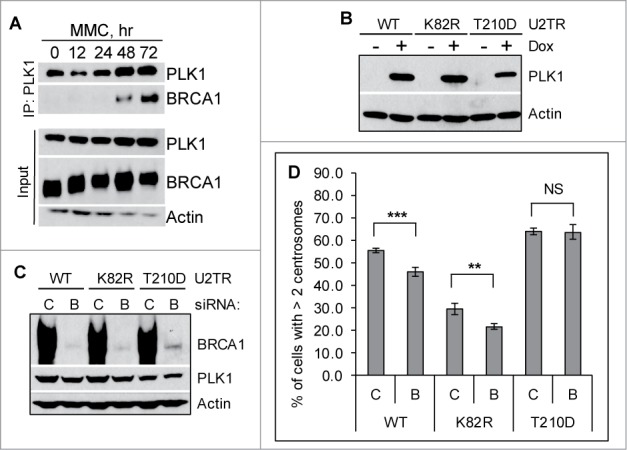
BRCA1 functions upstream of PLK1 in promoting mitomycin C-induced centrosome amplification. (A) MMC induces interaction between BRCA1 and PLK1. U2-OS cells were either left untreated (0 hr) or treated with 0.5 μM MMC for the indicated time. Equal amounts of lysate were used for IP with a monoclonal antibody against PLK1. (B–D) Expression of the constitutive active PLK1 rescues the centrosome amplification defects in BRCA1 depleted cells. Three different U2-OS cell lines expressing different PLK1 variants (WT: wild-type PLK1; K82R: kinase dead PLK1; T210D: constitutively active PLK1) under the control of a doxycycline (Dox)-inducible promoter were first transfected with either Control siRNA (C) or siRNA against BRCA1 (BRCA1-pool, B). Dox was added to one set of the cells to induce the expression of PLK1 variants (+). The induction of PLK1 (B) and the knockdown efficiency of BRCA1 (C) were monitored by western blot. In addition, cells were treated with 0.5 μM MMC for 72 hours, then fixed in methanol and stained with antibodies against γ-Tubulin. More than 300 cells were counted and the percentage of cells with more than 2 centrosomes was quantified (D). All error bars are standard deviation obtained from 4 different experiments. Standard 2-sided t test: **P < 0.01, ***P < 0.001. NS, not significant.
BRCA1 and FancJ cooperatively promote mitomycin C-induced centrosome amplification
FancJ is part of the BRCA1 B-complex.45,46 MMC induces centrosome re-localization of both BRCA1 (Fig. 2) and FancJ.38 Both of them also bind and act upstream of PLK1 during DDICA (Fig. 3).38 These data suggest an intimate relationship between BRCA1 and FancJ during the DDICA. To investigate the potential cooperative function of BRCA1 and FancJ in DDICA, we depleted FancJ, BRCA1, or both with siRNA and then treated cells with MMC and quantified the percentage of cells with amplified centrosome. As shown in Fig. 4A and B, depletion of BRCA1 alone reduced the percentage of cells with centrosome amplification more than depletion of FancJ alone while depletion of both BRCA1 and FancJ reduced the most, suggesting that BRCA1 and FancJ have partial additive functions in this process. In addition, we also examined the effects of overexpressing BRCA1 alone, FancJ alone, or both on MMC-induced centrosome amplification. As seen in Fig. 4C and D, overexpression of BRCA1 alone, FancJ alone, or both has no impact on centrosome amplification in untreated cells (0 hour samples). However, overexpression of either BRCA1 or FancJ could further stimulate MMC-induced centrosome amplification. Overexpression of both has no further stimulating effects. Another intriguing observation in these experiments is that overexpression of BRCA1, FancJ, or both is not sufficient to maximize MMC-induced centrosome amplification in the earlier timepoints. For example, the level of centrosome amplification seen in BRCA1, FancJ, or both overexpressed cells at 48 hours is still lower than that of 72 hours. This data suggests that other unknown events or factors are still needed to induce maximum centrosome amplification when treated with MMC. None of these observations are due to pronounced cell cycle changes (Figs. S4 and S5).
To test whether BRCA1 and FancJ contribute to 100% of the activation of PLK1, we treated BRCA1 and FancJ co-depleted cells with a commonly used PLK1 inhibitor, BI-2536.52 As seen in Fig. 4E, treatment of the BRCA1 and FancJ co-depleted cells with BI-2536 further reduced the MMC-induced centrosome amplification, suggesting that, in addition to BRCA1 and FancJ, one or more other factors may be also involved in the regulation of PLK1 during DDICA. Consistent with both BRCA1 and FancJ function upstream of PLK1, once again the reduced centrosome amplification in the BRCA1 and FancJ co-depleted cells was fully rescued by the constitutively active PLK1 (T210D) but not with the kinase-dead PLK1 (K82R) (Fig. 4F and G). In summary, the data shown in Fig. 4 indicates that BRCA1 and FancJ act cooperatively to promote the activation of PLK1 during MMC-induced centrosome amplification.
BRCA1 and FancJ promote centrosomal recruitment of PLK1 in response to mitomycin C treatment
Having established that BRCA1 and FancJ act upstream of PLK1 during DDICA, we next investigated the molecular mechanism behind this process. We first examined whether BRCA1 and FancJ regulate the centrosome localization of PLK1. For this, the centrosome in either BRCA1 depleted cells, or FancJ depleted cells, or both depleted cells was stained with anti-PLK1 antibody. The centrosome intensity of PLK1 was measured and further normalized against the centrosome intensity of γ-Tubulin, which is not affected by the status of BRCA1 or FancJ (Fig. S6). Amazingly, depletion of either BRCA1 alone, FancJ alone, or both attenuated the centrosome localization of PLK1 only in MMC treated cells (Fig. 5A and B) but not in non-treated cells (Fig. S7A). Conversely, overexpression of either BRCA1 alone, FancJ alone, or both enhanced the centrosome staining of PLK1, once again only in MMC treated cells (Figs. 5C and D) but not in untreated cells (Fig. S7B). Taken together, these data strongly suggest that BRCA1 and FancJ promote MMC-induced centrosome amplification by recruiting or retaining PLK1 at the centrosome.
Figure 5.
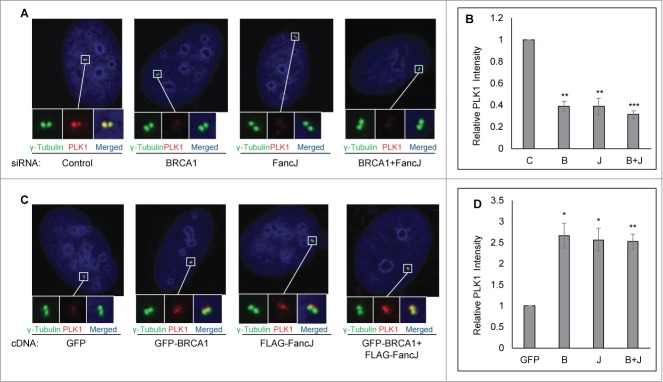
BRCA1 and FancJ cooperatively promote centrosome localization of PLK1 in mitomycin C treated cells. (A and B) Depletion of BRCA1, or FancJ, or both impedes centrosome localization of PLK1. U2-OS cells were first transfected with either Control siRNA (C), siRNA against BRCA1 (B), FancJ (J), or both BRCA1 and FancJ (B + J). Cells were treated with 0.5 μM MMC for 48 hours and then fixed in methanol and stained with antibodies against γ-Tubulin (green) and PLK1 (red). Nuclei were stained with DAPI (blue). (C and D) Overexpression of BRCA1, or FancJ, or both stimulates centrosome localization of PLK1. U2-OS cells were transfected with plasmid expressing either GFP, GFP-BRCA1 (B), FLAG-FancJ (J), or both GFP-BRCA1 and FLAG-FancJ (B + J). Cells were treated with 0.5 μM MMC for 48 hours and then fixed in methanol and stained with antibodies against γ-Tubulin (green) and PLK1 (red). Nuclei were stained with DAPI (blue). The intensity of centrosomal PLK1 in both (A) and (C) were measured and normalized against the intensity of γ-Tubulin in more than 30 cells. All error bars are standard deviation obtained from 3 different experiments. Standard 2-sided t test: *P < 0.05, **P < 0.01, ***P < 0.001.
FancJ promotes centrosome localization of BRCA1 in response to mitomycin C treatment
Next, we examined whether BRCA1 and FancJ affect each other's centrosome localization during DDICA. The centrosome intensity of FancJ was quantified in BRCA1 depleted cells. Conversely, the centrosome intensity of BRCA1 was quantified in FancJ depleted cells. The centrosomal intensity of BRCA1 and FancJ is normalized against the intensity of γ-Tubulin. Depletion of BRCA1 has no obvious impact on the centrosome intensity of FancJ irrespective of MMC treatment (Fig. 6A and B; Fig. S8A). Intriguingly, depletion of FancJ attenuated the centrosome staining of BRCA1 in MMC-treated cells but not untreated cells (Fig. 6C and D; Fig. S8B), suggesting that FancJ is partially responsible for the recruitment or retention of BRCA1 at the centrosome in response to MMC treatment.
Figure 6.

FancJ enhances centrosome localization of BRCA1 in mitomycin C treated cells. (A and B) Depletion of BRCA1 does not affect the centrosome localization of FancJ. U2-OS cells were first transfected with either Control siRNA or siRNA against BRCA1. Cells were treated with 0.5 μM MMC for 48 hours and then were fixed in methanol and stained with antibodies against γ-Tubulin (green) and FancJ (red). Nuclei were stained with DAPI (blue). The intensity of centrosomal FancJ were measured and normalized against the intensity of γ-Tubulin in more than 30 cells. (C and D) Depletion of FancJ attenuates the centrosome localization of BRCA1. U2-OS cells were first transfected with either Control siRNA or siRNA against FancJ. Cells were treated with 0.5 μM MMC for 48 hours and then were fixed in methanol and stained with antibodies against γ-Tubulin (green) and BRCA1 (red). Nuclei were stained with DAPI (blue). The intensity of centrosomal BRCA1 were measured and normalized against the intensity of γ-Tubulin in more than 30 cells. All error bars are standard deviation obtained from 3 different experiments. Standard 2-sided t test: **P < 0.01. NS, not significant.
Both the RING domain and the BRCT domain are important for BRCA1 to promote MMC-induced centrosome amplification and to recruit PLK1 to the centrosome
Finally we investigated which domain of BRCA1 is important for it to promote MMC-induced centrosome amplification and to recruit PLK1 to the centrosome. BRCA1 contains 3 important domains (Fig. 7A): a RING domain at its N-terminus (1–302 aa), a coiled-coil domain (CC, 1393–1476 aa), and 2 BRCT domains at its C-terminus (1528–1863 aa). The RING domain is commonly found in enzymes that catalyze the conjugation of ubiquitin. The BRCT domain is primarily involved in protein-protein interactions and binds phosphorylated peptides, such as phosphorylated Abraxas, FancJ/BACH1, and CtIP.26 Endogenous BRCA1 was first depleted using the BRCA1-UTR siRNA. Cells were then transfected with either GFP tagged Full-length BRCA1 or different GFP-tagged truncated BRCA1. The endogenous BRCA1 is detected with BRCA1-Ab1 antibody, the epitope of which resides in the RING domain. Therefore BRCA1-Ab1 will not detect the BRCA1 RING mutants, Δ1–302 and 303–1526 (Fig. S9B, lane 7 and 9 in panel 2 and 3 from the top). Exogenously expressed proteins were detected by anti-GFP antibody (Fig. S9B, panel 4 and 5 from the top). It was previously shown that the RING domain of BRCA1 dimerizes with the RING domain of BARD1 and is critical for their stability.53–55 Indeed when different BRCA1 variants were transfected into U2-OS cells, the 2 RING domain mutants, Δ1–302 and 303–1526, expressed at very low level (Fig. S9, Lane 7 and 9 in panels 4 and 5). Conversely, deletion of residues 305–770 or 775–1292 seems to have stabilizing effects (Lanes 5 and 6 in panels 2 to 5). As seen in Fig. 7B, expression of the full-length BRCA1 or BRCA1 missing only the RING domain alone or the BRCT domain alone fully rescued the MMC-induced centrosome amplification defects, while Δ305–770 or Δ775–1292 rescued partially. However deletion of both RING domain and BRCT domain failed to rescue the MMC-induced centrosome amplification defects, suggesting that both the RING domain and the BRCT domain are required for BRCA1 to activate the DDICA. Consistent with these observations, only the deletion of both RING domain and BRCT domain failed to stimulate the centrosome localization of PLK1 (Fig. S10).
Figure 7.
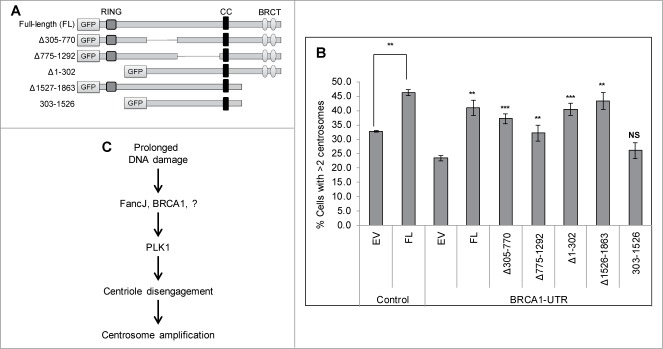
Both the RING domain and BRCT domain of BRCA1 are important for it to stimulate the MMC induced centrosome amplification. (A) Diagram of the domain structure of BRCA1. Full-length as well as different truncation variants of BRCA1 are tagged at the N-terminus with the Green Fluorescent Protein (GFP). (B) U2-OS cells were first transfected with either Control siRNA (C), or siRNA against BRCA1 (BRCA1-UTR) and then transfected with plasmid expressing either empty vector (EV), or GFP tagged full-length BRCA1 (FL), or different GFP-tagged truncation variants. Cells were treated with 0.5 μM MMC for 72 hours and then fixed in methanol and stained with antibodies against γ-Tubulin. More than 300 cells were counted and the percentage of cells with more than 2 centrosomes was quantified. All error bars are standard deviation obtained from 3 different experiments. Standard 2-sided t test: **P < 0.01. NS, not significant. The t test for BRCA1-UTR transfected cells were done compared to EV/BRCA1-UTR cells. (C) A proposed model of how DDICA is regulated by BRCA1 and FancJ.
The interaction between BRCA1 and FancJ is important for their functions in promoting the centrosome amplification in MMC-treated cells.
Since BRCA1 and FancJ interact with each other45 and both play an important role in promoting centrosome amplification in MMC-treated cells, we finally tested whether their interaction is important for their functions during DDICA. FancJ binds the BRCT domain of BRCA1 and phosphorylation of Serine-990 of FancJ stimulates its interaction with BRCA1.45,56 Mutating Serine-990 to alanine (FancJ-S990A) reduces its interaction with BRCA1.56 In addition, we also tested 2 other mutants of FancJ: the helicase mutant (FancJ-K52R) and the MLH1 binding mutant (FancJ-K141/142A) as comparison.57 As seen in Fig. S11, both FancJ-WT and FancJ-K52R stimulate the centrosome amplification in MMC-treated cells whereas FancJ-K141/142A and FancJ-S990A cannot. These results suggest that: (1) the DNA unwinding activity of FancJ is not required for its functions in DDICA; (2) The interaction between FancJ and BRCA1 is important for its functions in DDICA. How the interaction between FancJ and MLH1 contributes to its role in DDICA warrants further investigation.
A BRCT mutant of BRCA1, BRCA1-M1775R, impairs the interaction between BRCA1 and FancJ.58,59 Next we tested whether BRCA1-M1775R affects the function of BRCA1 in promoting centrosome amplification in MMC-treated cells. As comparison, we also examined 2 RING domain mutants of BRCA1: BRCA1-C61G and BRCA1-D67Y. Brodies and colleagues showed that though BRCA1-C61G does not affect the centrosome localization of BRCA1, it does affect its functions in suppressing IR-induced centrosome amplification in MCF7 cells.60 Additionally, Ransburg and colleagues showed that BRCA1-C61G affects the homologous recombination (HR) function of BRCA1 whereas BRCA1-D67Y does not.61 We first depleted BRCA1 with BRCA1-UTR siRNA in U2-OS cells and then transfected these cells with different BRCA1 variants. As seen in Fig. S12, BRCA1-WT, BRCA1-C61G fully and BRCA1-D67Y partially rescued the centrosome amplification defect of BRCA1-depleted and MMC-treated cells whereas BRCA1-M1775R did not. These results suggest that the E3 ligase activity of BRCA1 is not required for its functions in DDICA. On the other hand the interaction between BRCA1 and FancJ is critical for their functions in promoting the DDICA.
DISCUSSION
Both BRCA1 and FancJ have well-established roles in DDR, such as checkpoint response and DNA repair.26,27,62,63 We showed recently that in addition to UV, 6-thioguanine, aphidicolin, HU, camptothecin, etoposide, belomyin, and IR,18–20,64 interstrand crosslinking agents such as MMC and cis-platin also induce robust centrosome amplification,38 suggesting that, in addition to DNA damage checkpoint and DNA repair, centrosome amplification is likely an integral part of the DDR. DNA damage checkpoint and DNA repair are activated early, often within minutes or hours after the DNA damage,65 while DDICA takes place much later (within days).18,38 Though the exact physiological function of DDICA is still unresolved, observations made in fly embryos19,20 suggest that DDICA may be an alternative fail-safe mechanism to ensure the elimination of cells experiencing severe and irreparable DNA damages.10
We recent found that deficiency of FancJ induces centrosome amplification in non-stressed cells while attenuating both HU- and MMC-induced centrosome amplification.38 Because FancJ is also a key component of the BRCA1 B-complex,46 in this study we investigated the role of BRCA1 in centrosome biogenesis with a focus on DDICA. Similar to previous reports, we found that in U2-OS cells BRCA1 also localizes to the centrosome (Fig. S3)31 and deficiency of BRCA1 doubled the percentage of cells with amplified centrosomes (Figs. S1 and S2), suggesting that BRCA1 suppresses centrosome amplification in non-stressed cells. Counter-intuitively, however, depletion of BRCA1 in U2-OS as well as Hs587T attenuated both HU- and MMC-induced centrosome amplifications by 30% to 50% (Figs. 1C; FigS. S1 and S2). Consistent with the loss-of-function analysis, overexpression of either BRCA1 or FancJ further stimulates DDICA but has no effects on the centrosome biogenesis in non-stressed cells (Figs. 1D and 4E). Therefore, in both non-stressed cells and DNA damaged cells, the regulation of centrosome biogenesis by BRCA1 is similar to that of FancJ, albeit the effects of FancJ on the MMC induced centrosome amplification is somewhat milder than that of BRCA1 (Fig. 4B), suggesting that they likely act in the same pathway. As we discussed previously,38 assuming that DDICA functions as an alternative fail-safe mechanism in severely damaged cells,10 promoting centrosome amplification during DDICA would be consistent with the role of BRCA1 and FancJ as tumor suppressors.
Fukasawa and colleagues found that in MCF7 cells BRCA1 localizes to the centrosome throughout the cell cycle. Using the same anti-BRCA1 antibody (BRCA1-Ab2), we observed a somewhat different centrosome staining pattern of BRCA1 in U2-OS cells. We observed centrosome staining of BRCA1 in G1, S, and early G2 cells (Fig. S3 and data not shown). However, we found that in 60% of late G2 or mitotic U2-OS cells (with 2 further separated γ-Tubulin dots), centrosomal BRCA1 staining is dramatically reduced (Fig. 2). Intriguingly, when treated with MMC, close to 90% of late G2 centrosomes showed positive BRCA1 staining on both centrosomes (MMC reduces mitotic cells close to zero). We observed a similar but not identical centrosome staining pattern of FancJ when treated with MMC.38 FancJ localizes to most of G1, S, and early G2 cells, but is almost completely absent in late G2 and mitotic centrosomes. When treated with MMC, FancJ relocalizes to the late G2 centrosome but predominantly to the mother centrosome. We do not yet know the biological implication of the difference between BRCA1 and FancJ. We did notice that BRCA1 seems to have more profound effects on MMC-induced centrosome amplification than FancJ (Fig. 4B). Whether this is due to BRCA1 localizing to both centrosome while FancJ localizing predominantly to the mother centrosome in MMC treated cells needs further investigation. Nonetheless, when treated with MMC, relocalizing to the G2 centrosome is consistent with a direct role of BRCA1 and FancJ in promoting DDICA. Another study by Brodie and colleagues also confirmed the centrosome localization of BRCA1.60 Instead of promoting centrosome amplification as shown in this study in HU- or MMC-treated U2-OS and Hs587T cells, they found that BRCA1 suppresses the centrosome amplification in IR-treated MCF7 cells. The discrepancy between the 2 studies could be due to different genotoxic stress or cell lines used.
Previously we showed that FancJ interacts with and acts upstream of PLK1 during DDICA.38 In a separate study, we showed that HU induces pronounced interaction between BRCA1 and PLK1.50 Here we found that MMC also induces robust interaction between BRCA1 and PLK1 (Fig. 3A). Similar to FancJ, the constitutive active PLK1 (T210D) also fully rescued the centrosome amplification defects in BRCA1 depleted cells, indicating that BRCA1 also binds and acts upstream of PLK1 during DDICA (Figs. 3D; Fig. S1G). Furthermore, we propose that BRCA1 helps to recruit or retain PLK1 at the centrosome during DDICA because depletion of BRCA1 reduced the PLK1 centrosome staining while overexpression of BRCA1 increases the PLK1 centrosome staining (Fig. 5). Intriguingly, BRCA1 does not affect the centrosome staining of PLK1 in non-stressed cells (Fig. S6), suggesting that the effect of BRCA1 on the centrosome biogenesis in non-stressed cells is less likely through PLK1. The mechanism behind these differential regulations on PLK1 warrants further investigation. We speculate that the damage induced phosphorylation of BRCA1 may be involved.43
Both BRCA1 and FancJ have well-established roles in DNA damage checkpoint and DNA repair which take place early during DDR. DDICA occurs much later (often a few days later).18,38 Together with our recent findings, we propose a tentative model of the activation of DDICA (Fig. 7C): During the early phase of DNA damage response, BRCA1 and FancJ are phosphorylated and activated by the 3 major PI3K-like kinases, ATM, ATR, and DNA-PK. Activated BRCA1 and FancJ execute their DNA damage checkpoint and DNA repair functions. In cells that have experienced severe and sustained DNA damages, FancJ then recruits BRCA1 and PLK1 to the centrosome. Together with other unknown factors, BRCA1 and FancJ then activate PLK1 at the centrosome. Subsequently, PLK1 activates centriole disengagement, which then promotes centrosome amplification.21,66
Both BRCA1 and FancJ are well-established tumor suppressors.27,63 Throughout tumorigenesis, cancer cells experience sustained replicative and other forms of genotoxic stress.67,68 In light of the new findings presented here, we propose that the attenuated DDICA in BRCA1 or FancJ mutant cells likely increases their chance of survival after DNA damage. Combined with their compromised checkpoint and DNA repair capability, these cells would have a much greater chance of becoming tumorigenic.
Materials and Methods
Plasmids
Plasmids expressing C-terminal HA and FLAG-tagged FancJ-WT were generously provided by Dr. Sharon B. Cantor. Plasmids expressing GFP-tagged full-length BRCA1 and different truncation mutants of BRCA1 were generously provided by Dr. Natsuko Chiba.
siRNAs
siCONTROL: Non-targeting pool (Dharmacon, D-001810-10-20) was used as a negative control for all siRNA transfections. FancJ siRNA (Dharmacon, L-010587-00-0005). BRCA1 was depleted with ON-TARGETplus SMARTpool siRNA against BRCA1 (Dharmacon, L003461-00-0020), BRCA1-B (Dharmacon, CCAAAGCGAGCAAGAGAAU), BRCA1-C (Dharmacon, UGAUAAAGCUCCAGCAGGA), and BRCA1-UTR (Dharmacon, GCUCCUCUCACUCUUCAGU). Human cells were transfected with 50 nM siRNA twice using RNAiMAX (Invitrogen).
Chemicals
Hydroxyurea (Sigma, H8627), BI2536 (Selleck, S1109), mitomycin C (Research Products International, M92010-0.01), and doxycycline (Research Products International, D43020).
Antibodies
BRCA1: Ab-1 (Calbiochem, OP92) and Ab-2 (Calbiochem, OP93). Mouse GFP (Clontech, clone JL-8). Rabbit GFP (Invitrogen, A-11122). HA (Convance, MMS 101P). FLAG (Sigma, clone M2). Actin (Santa Cruz, sc-1616). PLK1m (Millipore, 05–844) and PLK1r (Bethyl, A300-251A). Centrin 2 (Santa Cruz, sc-27793-R). γ-Tubulin (Sigma, T5326 and T3195). GAPDH (Bethyl, A300–641A). FancJ (Bethyl, A300–561A).
Cell lines and cell culture
Hs587T and U2-OS cells were purchased from ATCC. All cells were grown in D-MEM supplemented with 10% fetal bovine serum (FBS), Penicillin and Streptomycin. All cells were cultivated at 37°C in a humidified incubator with 5% CO2.
Cell lysis and immunoprecipitation
For whole cell lysates (WCL), cells were lysed in NETN-150 buffer (20 mM Tris-HCl, pH 8.0; 150 mM NaCl; 1 mM EDTA; 0.5% NP-40) containing a cocktail of phosphatase and protease inhibitors (Sigma). For immunoprecipitation: Equal amounts of cell lysate were incubated with primary antibody and protein A Sepharose CL-4B beads (GE Healthcare, 17-078-01) with rotation at 4°C overnight.
Centrosome staining
Cells grown on a glass cover-slip (Fisher, 12-544-10) were washed twice with 1X PBS and then permeablized in ice cold 0.5% Triton X-100 in 1X PBS for 2 min. Cells were then washed twice with 1X PBS and fixed with 100% methanol at −20°C for 5 min. Cells were washed twice with 1X PBS. Cells were incubated in 1% gelatin at room temperature for 10 min. Cells were washed twice with 1X PBS. Cells were incubated in 0.02 M glycine at room temperature for 3 min. Cells were washed twice with 1X PBS. Cells were incubated in primary antibody at room temperature for one hour. Primary antibody was prepared in 1X PBS with 1% BSA. Cells were washed twice with 1X PBS. Cells were incubated in secondary antibody in the dark at room temperature for one hour. Secondary antibody was prepared in 1X PBS with 1% BSA. Cells were washed twice with 1X PBS. Cells finally were mounted using Prolong Gold antifade reagent with DAPI (Invitrogen, P36931). All the images were acquired using an Olympus Fluorescent Microscope, BX60, equipped with a Nikon DS-Qi1camera and analyzed with Nikon NIS-Element software. All the images taken with BX60 are from a single focal plane. For the quantification of centrosome staining, the staining intensities of γ-Tubulin, BRCA1, FancJ, and PLK1 at the centrosomes were measured in 30 to 40 cells of both Control siRNA transfected as well as siRNA against BRCA1 or FancJ transfected cells. The intensity of each centrosome staining of BRCA1, FancJ, and PLK1 was normalized against the intensity of the corresponding γ-Tubulin. The data was processed and plotted using Prism GraphPad 6.02. The error bars are the Tukey's confidence limits.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to Drs. Sharon Cantor, Natsuko Chiba and Erin Niggs for reagents and to Robin Miskimins for critical reading of the manuscript. This research was partially supported by the grant from Fanconi Anemia Research Fund (to DZ).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646-74; PMID: 21376230; http://dx.doi.org/ 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 2. Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature 1997; 386:623-7; PMID:9121588; http://dx.doi.org/ 10.1038/386623a0 [DOI] [PubMed] [Google Scholar]
- 3. Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell 2010; 40:179-204; PMID:20965415; http://dx.doi.org/ 10.1016/j.molcel.2010.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Curtin NJ. DNA repair dysregulation from cancer driver to therapeutic target. Nat Rev Cancer 2012; 12:801-17; PMID:23175119; http://dx.doi.org/ 10.1038/nrc3399 [DOI] [PubMed] [Google Scholar]
- 5. Ganem NJ, Godinho SA, Pellman D. A mechanism linking extra centrosomes to chromosomal instability. Nature 2009; 460:278-82; PMID:19506557; http://dx.doi.org/ 10.1038/nature08136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Silkworth WT, Nardi IK, Scholl LM, Cimini D. Multipolar spindle pole coalescence is a major source of kinetochore mis-attachment and chromosome mis-segregation in cancer cells. PLoS One 2009; 4:e6564; PMID:19668340; http://dx.doi.org/ 10.1371/journal.pone.0006564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chan JY. A clinical overview of centrosome amplification in human cancers. Int J Biol Sci 2011; 7:1122-44; PMID:22043171; http://dx.doi.org/ 10.7150/ijbs.7.1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rajagopalan H, Lengauer C. Aneuploidy and cancer. Nature 2004; 432:338-41; PMID:15549096; http://dx.doi.org/ 10.1038/nature03099 [DOI] [PubMed] [Google Scholar]
- 9. Anderhub SJ, Kramer A, Maier B. Centrosome amplification in tumorigenesis. Cancer Lett 2012; 322:8-17; PMID:22342684; http://dx.doi.org/ 10.1016/j.canlet.2012.02.006 [DOI] [PubMed] [Google Scholar]
- 10. Nigg EA, Stearns T. The centrosome cycle: Centriole biogenesis, duplication and inherent asymmetries. Nat Cell Biol 2011; 13:1154-60; PMID:21968988; http://dx.doi.org/ 10.1038/ncb2345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bornens M. The centrosome in cells and organisms. Science 2012; 335:422-6; PMID:22282802; http://dx.doi.org/ 10.1126/science.1209037 [DOI] [PubMed] [Google Scholar]
- 12. Chaki M, Airik R, Ghosh AK, Giles RH, Chen R, Slaats GG, Wang H, Hurd TW, Zhou W, Cluckey A, et al. Exome capture reveals ZNF423 and CEP164 mutations, linking renal ciliopathies to DNA damage response signaling. Cell 2012; 150:533-48; PMID: 22863007; http://dx.doi.org/ 10.1016/j.cell.2012.06.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kalay E, Yigit G, Aslan Y, Brown KE, Pohl E, Bicknell LS, Kayserili H, Li Y, Tuysuz B, Nurnberg G, et al. CEP152 is a genome maintenance protein disrupted in Seckel syndrome. Nat Genet 2011; 43:23-6; PMID:21131973; http://dx.doi.org/ 10.1038/ng.725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Griffith E, Walker S, Martin CA, Vagnarelli P, Stiff T, Vernay B, Al Sanna N, Saggar A, Hamel B, Earnshaw WC, et al. Mutations in pericentrin cause Seckel syndrome with defective ATR-dependent DNA damage signaling. Nat Genet 2008; 40:232-6; PMID: 18157127; http://dx.doi.org/ 10.1038/ng.2007.80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Staples CJ, Myers KN, Beveridge RD, Patil AA, Lee AJ, Swanton C, Howell M, Boulton SJ, Collis SJ. The centriolar satellite protein Cep131 is important for genome stability. J Cell Sci 2012; 125:4770-9; PMID: 22797915; http://dx.doi.org/ 10.1242/jcs.104059 [DOI] [PubMed] [Google Scholar]
- 16. Shimada M, Komatsu K. Emerging connection between centrosome and DNA repair machinery. J Radiat Res (Tokyo) 2009; 50:295-301; http://dx.doi.org/ 10.1269/jrr.09039 [DOI] [PubMed] [Google Scholar]
- 17. Loffler H, Lukas J, Bartek J, Kramer A. Structure meets function-centrosomes, genome maintenance and the DNA damage response. Exp Cell Res 2006; 312:2633-40; PMID:16854412; http://dx.doi.org/ 10.1016/j.yexcr.2006.06.008 [DOI] [PubMed] [Google Scholar]
- 18. Balczon R, Bao L, Zimmer WE, Brown K, Zinkowski RP, Brinkley BR. Dissociation of centrosome replication events from cycles of DNA synthesis and mitotic division in hydroxyurea-arrested Chinese hamster ovary cells. J Cell Biol 1995; 130:105-15; PMID:7790366; http://dx.doi.org/ 10.1083/jcb.130.1.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sibon OC, Kelkar A, Lemstra W, Theurkauf WE. DNA-replicationDNA-damage-dependent centrosome inactivation in Drosophila embryos. Nat Cell Biol 2000; 2:90-5; PMID:10655588; http://dx.doi.org/ 10.1038/35000041 [DOI] [PubMed] [Google Scholar]
- 20. Takada S, Kelkar A, Theurkauf WE. Drosophila checkpoint kinase 2 couples centrosome function and spindle assembly to genomic integrity. Cell 2003; 113:87-99; PMID:12679037; http://dx.doi.org/ 10.1016/S0092-8674(03)00202-2 [DOI] [PubMed] [Google Scholar]
- 21. Loncarek J, Hergert P, Khodjakov A. Centriole reduplication during prolonged interphase requires procentriole maturation governed by Plk1. Curr Biol 2010; 20:1277-82; PMID:20656208; http://dx.doi.org/ 10.1016/j.cub.2010.05.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rai R, Phadnis A, Haralkar S, Badwe RA, Dai H, Li K, Lin SY. Differential regulation of centrosome integrity by DNA damage response proteins. Cell Cycle 2008; 7:2225-33; PMID:18635967; http://dx.doi.org/ 10.4161/cc.7.14.6303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bourke E, Dodson H, Merdes A, Cuffe L, Zachos G, Walker M, Gillespie D, Morrison CG. DNA damage induces Chk1-dependent centrosome amplification. EMBO Rep 2007; 8:603-9; PMID:17468739; http://dx.doi.org/ 10.1038/sj.embor.7400962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dodson H, Bourke E, Jeffers LJ, Vagnarelli P, Sonoda E, Takeda S, Earnshaw WC, Merdes A, Morrison C. Centrosome amplification induced by DNA damage occurs during a prolonged G2 phase and involves ATM. EMBO J 2004; 23:3864-73; PMID:15359281; http://dx.doi.org/ 10.1038/sj.emboj.7600393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brown JA, Bourke E, Liptrot C, Dockery P, Morrison CG. MCPH1BRIT1 limits ionizing radiation-induced centrosome amplification. Oncogene 2010; 29:5537-44; PMID:20661222; http://dx.doi.org/ 10.1038/onc.2010.302 [DOI] [PubMed] [Google Scholar]
- 26. Huen MS, Sy SM, Chen J. BRCA1 and its toolbox for the maintenance of genome integrity. Nat Rev Mol Cell Biol 2010; 11:138-48; PMID:20029420; http://dx.doi.org/ 10.1038/nrm2831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. O'Donovan PJ, Livingston DM. BRCA1 and BRCA2: breastovarian cancer susceptibility gene products and participants in DNA double-strand break repair. Carcinogenesis 2010; 31:961-7; PMID:20400477; http://dx.doi.org/ 10.1093/carcin/bgq069 [DOI] [PubMed] [Google Scholar]
- 28. Hsu LC, White RL. BRCA1 is associated with the centrosome during mitosis. Proc Natl Acad Sci U S A 1998; 95:12983-8; PMID:9789027; http://dx.doi.org/ 10.1073/pnas.95.22.12983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lotti LV, Ottini L, D'Amico C, Gradini R, Cama A, Belleudi F, Frati L, Torrisi MR, Mariani-Costantini R. Subcellular localization of the BRCA1 gene product in mitotic cells. Genes Chromosomes Cancer 2002; 35:193-203; PMID:12353262; http://dx.doi.org/ 10.1002/gcc.10105 [DOI] [PubMed] [Google Scholar]
- 30. Maul GG, Jensen DE, Ishov AM, Herlyn M, Rauscher FJ, 3rd. Nuclear redistribution of BRCA1 during viral infection. Cell Growth Differ 1998; 9:743-55; PMID:9751118 [PubMed] [Google Scholar]
- 31. Tarapore P, Hanashiro K, Fukasawa K. Analysis of centrosome localization of BRCA1 and its activity in suppressing centrosomal aster formation. Cell Cycle 2012; 11:2931-46; PMID:22833046; http://dx.doi.org/ 10.4161/cc.21396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Joukov V, Groen AC, Prokhorova T, Gerson R, White E, Rodriguez A, Walter JC, Livingston DM. The BRCA1BARD1 heterodimer modulates ran-dependent mitotic spindle assembly. Cell 2006; 127:539-52; PMID: 17081976; http://dx.doi.org/ 10.1016/j.cell.2006.08.053 [DOI] [PubMed] [Google Scholar]
- 33. Starita LM, Machida Y, Sankaran S, Elias JE, Griffin K, Schlegel BP, Gygi SP, Parvin JD. BRCA1-dependent ubiquitination of gamma-tubulin regulates centrosome number. Mol Cell Biol 2004; 24:8457-66; PMID:15367667; http://dx.doi.org/ 10.1128/MCB.24.19.8457-8466.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xu X, Wagner KU, Larson D, Weaver Z, Li C, Ried T, Hennighausen L, Wynshaw-Boris A, Deng CX. Conditional mutation of Brca1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation. Nat Genet 1999; 22:37-43; PMID: 10319859; http://dx.doi.org/ 10.1038/8743 [DOI] [PubMed] [Google Scholar]
- 35. Xu X, Weaver Z, Linke SP, Li C, Gotay J, Wang XW, Harris CC, Ried T, Deng CX. Centrosome amplification and a defective G2-M cell cycle checkpoint induce genetic instability in BRCA1 exon 11 isoform-deficient cells. Mol Cell 1999; 3:389-95; PMID:10198641; http://dx.doi.org/ 10.1016/S1097-2765(00)80466-9 [DOI] [PubMed] [Google Scholar]
- 36. Weaver Z, Montagna C, Xu X, Howard T, Gadina M, Brodie SG, Deng CX, Ried T. Mammary tumors in mice conditionally mutant for Brca1 exhibit gross genomic instability and centrosome amplification yet display a recurring distribution of genomic imbalances that is similar to human breast cancer. Oncogene 2002; 21:5097-107; PMID:12140760; http://dx.doi.org/ 10.1038/sj.onc.1205636 [DOI] [PubMed] [Google Scholar]
- 37. Shimomura A, Miyoshi Y, Taguchi T, Tamaki Y, Noguchi S. Association of loss of BRCA1 expression with centrosome aberration in human breast cancer. J Cancer Res Clin Oncol 2009; 135:421-30; PMID: 18813953; http://dx.doi.org/ 10.1007/s00432-008-0472-5 [DOI] [PubMed] [Google Scholar]
- 38. Zou J, Tian F, Li J, Pickner W, Long M, Rezvani K, Wang H, Zhang D. FancJ regulates interstrand crosslinker induced centrosome amplification through the activation of polo-like kinase 1. Biology Open 2013; 2:1022-31; PMID:24167712; http://dx.doi.org/ 10.1242/bio.20135801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kottemann MC, Smogorzewska A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 2013; 493:356-63; PMID:23325218; http://dx.doi.org/ 10.1038/nature11863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nalepa G, Enzor R, Sun Z, Marchal C, Park SJ, Yang Y, Tedeschi L, Kelich S, Hanenberg H, Clapp DW. Fanconi anemia signaling network regulates the spindle assembly checkpoint. J Clin Invest 2013; 123:3839-47; PMID:23934222; http://dx.doi.org/ 10.1172/JCI67364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Parvin JD. The BRCA1-dependent ubiquitin ligase, gamma-tubulin, and centrosomes. Environ Mol Mutagen 2009; 50:649-53; PMID:19274767; http://dx.doi.org/ 10.1002/em.20475 [DOI] [PubMed] [Google Scholar]
- 42. Schlegel BP, Starita LM, Parvin JD. Overexpression of a protein fragment of RNA helicase A causes inhibition of endogenous BRCA1 function and defects in ploidy and cytokinesis in mammary epithelial cells. Oncogene 2003; 22:983-91; PMID:12592385; http://dx.doi.org/ 10.1038/sj.onc.1206195 [DOI] [PubMed] [Google Scholar]
- 43. Tian F, Sharma S, Zou J, Lin SY, Wang B, Rezvani K, Wang H, Parvin JD, Ludwig T, Canman CE, et al. BRCA1 promotes the ubiquitination of PCNA and recruitment of translesion polymerases in response to replication blockade. Proc Natl Acad Sci U S A 2013; 110:13558-63; PMID:23901102; http://dx.doi.org/ 10.1073/pnas.1306534110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kais Z, Chiba N, Ishioka C, Parvin JD. Functional differences among BRCA1 missense mutations in the control of centrosome duplication. Oncogene 2012; 31:799-804; PMID:21725363; http://dx.doi.org/ 10.1038/onc.2011.271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cantor SB, Bell DW, Ganesan S, Kass EM, Drapkin R, Grossman S, Wahrer DC, Sgroi DC, Lane WS, Haber DA, et al. BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell 2001; 105:149-60; PMID: 11301010; http://dx.doi.org/ 10.1016/S0092-8674(01)00304-X [DOI] [PubMed] [Google Scholar]
- 46. Wang B, Matsuoka S, Ballif BA, Zhang D, Smogorzewska A, Gygi SP, Elledge SJ. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science 2007; 316:1194-8; PMID: 17525340; http://dx.doi.org/ 10.1126/science.1139476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hut HM, Rembacz KP, van Waarde MA, Lemstra W, van Cappellen WA, Kampinga HH, Sibon OC. Dysfunctional BRCA1 is only indirectly linked to multiple centrosomes. Oncogene 2005; 24:7619-23; PMID:16205648; http://dx.doi.org/ 10.1038/sj.onc.1208859 [DOI] [PubMed] [Google Scholar]
- 48. Inanc B, Dodson H, Morrison CG. A centrosome-autonomous signal that involves centriole disengagement permits centrosome duplication in G2 phase after DNA damage. Mol Biol Cell 2010; 21:3866-77; PMID:20861312; http://dx.doi.org/ 10.1091/mbc.E10-02-0124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liu X, Erikson RL. Activation of Cdc2cyclin B and inhibition of centrosome amplification in cells depleted of Plk1 by siRNA. Proc Natl Acad Sci U S A 2002; 99:8672-6; PMID:12077309; http://dx.doi.org/ 10.1073/pnas.132269599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zou J, Rezvani K, Wang H, Lee KS, Zhang D. BRCA1 downregulates the kinase activity of Polo-like kinase 1 in response to replication stress. Cell Cycle 2013; 12:2255-65; PMID:24067368; http://dx.doi.org/ 10.4161/cc.25349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Macurek L, Lindqvist A, Lim D, Lampson MA, Klompmaker R, Freire R, Clouin C, Taylor SS, Yaffe MB, Medema RH. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature 2008; 455:119-23; PMID:18615013; http://dx.doi.org/ 10.1038/nature07185 [DOI] [PubMed] [Google Scholar]
- 52. Steegmaier M, Hoffmann M, Baum A, Lenart P, Petronczki M, Krssak M, Gurtler U, Garin-Chesa P, Lieb S, Quant J, et al. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr Biol 2007; 17:316-22; PMID:17291758; http://dx.doi.org/ 10.1016/j.cub.2006.12.037 [DOI] [PubMed] [Google Scholar]
- 53. Hashizume R, Fukuda M, Maeda I, Nishikawa H, Oyake D, Yabuki Y, Ogata H, Ohta T. The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J Biol Chem 2001; 276:14537-40; PMID:11278247; http://dx.doi.org/ 10.1074/jbc.C000881200 [DOI] [PubMed] [Google Scholar]
- 54. Joukov V, Chen J, Fox EA, Green JB, Livingston DM. Functional communication between endogenous BRCA1 and its partner, BARD1, during Xenopus laevis development. Proc Natl Acad Sci U S A 2001; 98:12078-83; PMID:11593018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Brzovic PS, Rajagopal P, Hoyt DW, King MC, Klevit RE. Structure of a BRCA1-BARD1 heterodimeric RING-RING complex. Nat Struct Biol 2001; 8:833-7; PMID:11573085; http://dx.doi.org/ 10.1038/nsb1001-833 [DOI] [PubMed] [Google Scholar]
- 56. Yu X, Chini CC, He M, Mer G, Chen J. The BRCT domain is a phospho-protein binding domain. Science 2003; 302:639-42; PMID:14576433; http://dx.doi.org/ 10.1126/science.1088753 [DOI] [PubMed] [Google Scholar]
- 57. Peng M, Litman R, Xie J, Sharma S, Brosh RM, Jr., Cantor SB. The FANCJMutLalpha interaction is required for correction of the cross-link response in FA-J cells. EMBO J 2007; 26:3238-49; PMID:17581638; http://dx.doi.org/ 10.1038/sj.emboj.7601754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Clapperton JA, Manke IA, Lowery DM, Ho T, Haire LF, Yaffe MB, Smerdon SJ. Structure and mechanism of BRCA1 BRCT domain recognition of phosphorylated BACH1 with implications for cancer. Nat Struct Mol Biol 2004; 11:512-8; PMID:15133502; http://dx.doi.org/ 10.1038/nsmb775 [DOI] [PubMed] [Google Scholar]
- 59. Williams RS, Lee MS, Hau DD, Glover JN. Structural basis of phosphopeptide recognition by the BRCT domain of BRCA1. Nat Struct Mol Biol 2004; 11:519-25; PMID:15133503; http://dx.doi.org/ 10.1038/nsmb776 [DOI] [PubMed] [Google Scholar]
- 60. Brodie KM, Henderson BR. Characterization of BRCA1 protein targeting, dynamics, and function at the centrosome: a role for the nuclear export signal, CRM1, and Aurora A kinase. J Biol Chem 2012; 287:7701-16; PMID:22262852; http://dx.doi.org/ 10.1074/jbc.M111.327296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ransburgh DJ, Chiba N, Ishioka C, Toland AE, Parvin JD. Identification of breast tumor mutations in BRCA1 that abolish its function in homologous DNA recombination. Cancer Res 2010; 70:988-95; PMID:20103620; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-2850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hiom K. FANCJ: solving problems in DNA replication. DNA Repair (Amst) 2010; 9:250-6; PMID: 20122882; http://dx.doi.org/ 10.1016/j.dnarep.2010.01.005 [DOI] [PubMed] [Google Scholar]
- 63. Cantor SB, Guillemette S. Hereditary breast cancer and the BRCA1-associated FANCJBACH1BRIP1. Future Oncol 2011; 7:253-61; PMID:21345144; http://dx.doi.org/ 10.2217/fon.10.191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Robinson HM, Black EJ, Brown R, Gillespie DA. DNA mismatch repair and Chk1-dependent centrosome amplification in response to DNA alkylation damage. Cell Cycle 2007; 6:982-92; PMID:17404511; http://dx.doi.org/ 10.4161/cc.6.8.4111 [DOI] [PubMed] [Google Scholar]
- 65. Nelms BE, Maser RS, MacKay JF, Lagally MG, Petrini JH. In situ visualization of DNA double-strand break repair in human fibroblasts. Science 1998; 280:590-2; PMID:9554850; http://dx.doi.org/ 10.1126/science.280.5363.590 [DOI] [PubMed] [Google Scholar]
- 66. Tsou MF, Wang WJ, George KA, Uryu K, Stearns T, Jallepalli PV. Polo kinase and separase regulate the mitotic licensing of centriole duplication in human cells. Dev Cell 2009; 17:344-54; PMID:19758559; http://dx.doi.org/ 10.1016/j.devcel.2009.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005; 434:864-70; PMID:15829956; http://dx.doi.org/ 10.1038/nature03482 [DOI] [PubMed] [Google Scholar]
- 68. Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA, Jr., Kastrinakis NG, Levy B, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005; 434:907-13; PMID:15829965; http://dx.doi.org/ 10.1038/nature03485 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.