

Abstract
Background: Mitochondrial dysfunction has long been considered a major contributor to aging and age-related diseases. Harman’s Mitochondrial Free Radical Theory of Aging postulated that somatic mitochondrial DNA mutations that accumulate over the life span cause excessive production of reactive oxygen species that damage macromolecules and impair cell and tissue function. Indeed, studies have shown that maximal oxidative capacity declines with age while reactive oxygen species production increases. Harman’s hypothesis has been seriously challenged by recent studies showing that reactive oxygen species evoke metabolic health and longevity, perhaps through hormetic mechanisms that include autophagy. The purpose of this review is to scan the ever-growing literature on mitochondria from the perspective of aging research and try to identify priority questions that should be addressed in future research.Methods: A systematic search of peer-reviewed studies was performed using PubMed. Search terms included (i) mitochondria or mitochondrial; (ii) aging, ageing, older adults or elderly; and (iii) reactive oxygen species, mitochondria dynamics, mitochondrial proteostasis, cytosol, mitochondrial-associated membranes, redox homeostasis, electron transport chain, electron transport chain efficiency, epigenetic regulation, DNA heteroplasmy.Results: The importance of mitochondrial biology as a trait d’union between the basic biology of aging and the pathogenesis of age-related diseases is stronger than ever, although the emphasis has moved from reactive oxygen species production to other aspects of mitochondrial physiology, including mitochondrial biogenesis and turnover, energy sensing, apoptosis, senescence, and calcium dynamics. Conclusions: Mitochondria could play a key role in the pathophysiology of aging or in the earlier stages of some events that lead to the aging phenotype. Therefore, mitochondria will increasingly be targeted to prevent and treat chronic diseases and to promote healthy aging.
Key Words: Mitochondria, Aging, Lifespan
Mitochondria are ubiquitous, intracellular organelles involved in metabolic processes essential for production of the high-energy molecule adenosine triphosphate. About 1.5 billion years ago, mitochondria and eukaryotic cells established a symbiotic relationship, striking a deal that involved a full package of biological tasks, including energy production but also cellular homeostasis, iron processing, heme and steroid synthesis, apoptosis, and probably an array of biological tasks still to be uncovered. Mammalian mitochondria emerge and function through the co-ordinated action of two genomes: (a) Multiple copies of bacterial-like mitochondrial DNA (mtDNA) that encode for 22 transfer RNAs, 2 ribosomal RNAs, and 13 proteins implicated in oxidative phosphorylation, and (b) the nuclear genome (nDNA) that encodes for about another 1,500 structural and functional proteins. Evidence is emerging that defects in mitochondrial function may lead to chronic diseases and organ involvement with very heterogeneous clinical manifestations. In this review, we focus on neuromuscular defects and other phenotypes that are considered part of normal aging (1).
In Harman’s original free radical theory of aging, which was first proposed in 1978, reactive oxygen species (ROS) generated during aerobic metabolism can damage the mtDNA and proteins, thereby leading to deterioration of cells, tissues and organs and, eventually, the entire body. Over the years, the free-radical theory of aging has been repeatedly challenged and refuted by a “tsunami” of data showing that the mitochondrial function is a key to successful aging and aging phenotypes in ways the Harman’s theory never anticipated. As a result, the idea that mitochondria could play a central role in aging and age-related diseases has been increasingly emphasized. The purpose of this review is to scan the ever-growing literature on mitochondria from the perspective of aging research and try to identify priority questions that should be addressed in future research.
Mitochondria and ROS Production
The mtDNA is particularly susceptible to oxidative damage because of its proximity to free radical sources and the relative lack of a protein scaffold. It is generally believed that ROS generated during oxidative metabolism cause accumulation of mtDNA damage and mutations leading to loss of fidelity in newly synthetized proteins which ultimately impacts on mitochondria physiology, and may lead to premature senescence and aging (2,3). In particular, clonal accumulation of mtDNA deletion mutations is most important in fast twitch glycolytic (type II) muscle fibers, which are the fibers predominantly lost with aging (4–6). Premature muscle aging has also been observed in mice expressing a defective mtDNA polymerase gamma (7), whereas a mouse model of targeted overexpression of the human catalase gene in mitochondria exhibits a reduction in the age-dependent loss of skeletal muscle function and an improvement in Ca2+ signaling in myocytes (8).
Progressive mitochondrial dysfunction is considered a hallmark of aging (9,10), and impaired mitochondrial function causes an accelerated aging phenotype, which is particularly evident in high energy demanding tissues such as brain, heart, and skeletal muscle, and in kidney and liver, two organs with essential metabolic roles (5,11–13) (Figure 1). High levels of ROS cause macromolecular damage, whereas low levels of mitochondrial ROS production enhance systemic defense mechanisms by inducing an adaptive, hormetic response (known as “mitohormesis”) that confers better stress resistance and life-span extension in various animal models. Indeed, induction of mild mitohormesis in muscle promotes longevity in Drosophila via systemic repression of insulin signaling and preservation of mitochondrial and muscle function (14). Similarly, metformin may prolong life span through similar mechanism (15). Thus, it appears that the production of low levels of ROS may activate compensatory longevity-promoting signaling pathways. Excessive ROS toxicity is avoided through a powerful antioxidant system that scavenges ROS as they are generated. It is noteworthy that superoxide is scavenged by three superoxide dismutase isozymes: the cytosolic SOD1 (Cu2+Zn2+-SOD), mitochondrial SOD2 (Mn2+-SOD), and extracellular SOD3. Deficiency of both SOD1 and SOD2 leads to oxidative stress, but only SOD1 deficiency causes an accelerated sarcopenic phenotype in mice (16). A mainstream theory to explain mitohormesis is connected to uncoupling. Mitochondria generate energy by creating a proton gradient across their inner membrane. Some proteins of the inner mitochondrial membrane, such as the uncoupling proteins and the adenine translocator (17,18), can leak protons down the electrochemical gradient into the matrix and dissipate energy as heat while minimizing ROS production. Incidentally, brown fat uses this mechanism to produce heat in response to cold temperatures. Of significance, mild uncoupling of mitochondrial respiration has been implicated in life-span extension through its effects on metabolic rate and ROS production (19,20). Mitochondrial superoxide production increases with replicative age in cultured human fibroblasts and it is considered to be one of the major contributing factors to replicative senescence (21). This replicative senescence is delayed by mild mitochondrial uncoupling and is accompanied by deregulation of Ca2+-dependent signaling (22).
Figure 1.
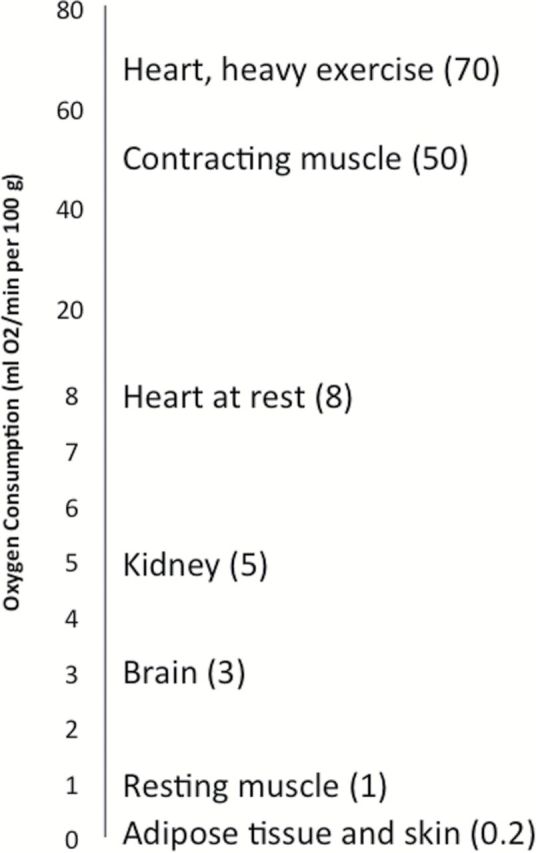
Tissue-specific oxygen consumption rate. Impaired mitochondrial function may cause an accelerated aging phenotype mainly in high energy demanding tissues such as brain, heart, and skeletal muscle, and in kidney and liver, two organs with essential metabolic roles.
Mitochondria Dynamics
Mitochondria constantly remodel their architecture through alternated asymmetric division (fission) and fusion that allow morphological transitions and adaptation to different functional situations (23). Fission is coordinated with DNA replication and is essential for mitochondrial duplication and biogenesis, which is a requirement for cell division and strongly connected to cell cycle. Fission is also an essential phase of mitochondria autophagy (mitophagy), which allows recycling of sections of mitochondria that has become dysfunctional or damaged (24). Fusion is the process by which mitochondria become interconnected. Through fusion, damaged mitochondria may acquire undamaged genetic material and maintain functionality, including the resynthesis of essential proteins. This alternate process of fusion and fission enables mitochondria to form a constantly changing tubular network, essential for health in most eukaryotic cells. Not surprisingly, mitochondrial dynamics are affected by fuel availability, ie, fuel excess stimulates fission and nutrient deficiency stimulates fusion (25). Studies have shown that an imbalance in this process contributes to the loss of mitochondrial homeostasis in sarcopenic muscles. In particular, aging is associated with the upregulation of mitochondrial fusion in skeletal muscle, possibly in order to cope with increased levels of mtDNA mutations (26). Defects in mitochondria maintenance and turnover because of impaired biogenesis and/or defective removal are thought to contribute to the pathogenesis of complex diseases not classically considered to involve mitochondria, including cancer, cardiovascular disease, and neurodegenerative diseases (27,28).
Mitophagy is required to guarantee cell survival (29) and is controlled by the mammalian target of rapamycin (mTOR), the adenosine monophosphate-activated protein kinase (AMPK), and forkhead box transcription factor 3 (FOXO3) pathways (30,31). Downregulation of mTOR stimulates mitophagy and upregulation of AMPK activates mitochondrial biogenesis, oxidative metabolism, and resistance to oxidative stress (32). This is consistent with the notion that by modulating mitochondrial activity, mTOR regulates both resting oxygen consumption and oxidative capacity. Interestingly, mTOR activity affects life span in many organisms.
It has been shown that aging is associated with defective autophagy that leads to the accumulation of dysfunctional mitochondria, oxidative stress, and inflammation. Studies have suggested that elevated autophagy activity preserves bioenergetics in centenarians (33), whereas defective mitophagy is associated with sarcopenia (34,35).
As indicated previously, mitochondrial quantity and function continuously adapt to meet the energy demands of the cell through a fine balance between mitophagy, mitochondrial biogenesis, and regulation of oxidative metabolism. The tilt up or down in this balance is mainly controlled by the peroxisome-proliferator–activated receptor γ coactivator 1α (PGC-1α), a pleiotropic transcriptional regulator that, among other functions, stimulates mitochondrial biogenesis, affects mitochondrial dynamics, modulates oxidative phosphorylation by interacting with the nuclear factors, erythroid 2-like 1 (NRF1) and 2 (NRF2), controls mitochondrial genome copy number through the transcription factor TFAM and affects lipid metabolism (36,37). There is some evidence that the age-associated decline of PGC-1α is the primary cause for the reduction of mitochondrial number and volume and the increase in apoptosis observed with aging and in some chronic diseases. Of significance, these steps are proposed to be part of the causal pathway to sarcopenia and frailty (38,39). A number of studies found that mitochondrial biogenesis is impaired with aging and is associated with a loss of mitochondrial content and function in energetically demanding tissues, such as skeletal muscle, brain, and heart (40,41). However, evidence for a central role of PGC-1α in driving such decline in humans is scant and testing this hypothesis remains an important area of research. The importance of PGC-1α to aging is also underlined by recent evidence of an interaction with tumor suppressor p53 and telomerase (42,43). In fact, telomere dysfunction has been associated with impaired mitochondrial biogenesis and function, decreased gluconeogenesis, cardiomyopathy, and increased ROS production (44). PGC-1α expression is finely tuned in response to a multitude of triggers, which may be viewed as metabolic sensors. Cell exposure to stressors, such as hydrogen peroxide (H2O2) or ischemia, generally induce PGC-1α expression (38). AMPK and SIRT1 are two metabolic sensors that directly affect PGC-1α activity through phosphorylation and deacetylation (45,46). It is widely believed that PGC-1α activity accounts for many of the beneficial effects of physical activity and dietary interventions in experimental animals (47). Studies have shown that a chronic inactivation of AMPK is associated with a decrease in mitochondrial biogenesis in aged animals, and that activation of AMPK may represent a therapeutic target to improve mitochondrial dysfunction with aging (48,49).
Mitochondria and Cytosol Relationship
Mitochondria are integrated in metabolic pathways, which include cytosolic protein synthesis and the intracellular network of membranes. The majority of mitochondrial proteins are translated in the cytoplasm and then imported, processed, and assembled in the mitochondrial matrix. Adequate proteostasis is maintained by chaperones that repair misfolded proteins and proteases that remove proteins irreversibly damaged (50).
Mitochondrial Proteostasis
Aging is associated with increased protein oxidative damage and an imbalance in proteostasis due to impairment of the ubiquitin-proteasome system (51). Such imbalance causes the accumulation of misfolded proteins that trigger a mitochondrial-specific unfolded protein response (UPRmt), which increases the production of chaperones and proteases in the attempt to re-establish proteostasis equilibrium. UPRmt has been implicated in life-span extension in worms, flies, and mice, suggesting a conserved role in the long-term maintenance of cellular homeostasis (52). Of note, many age-related neurodegenerative diseases, such as Parkinson’s disease and Alzheimer’s disease, involve the accumulation of misfolded proteins in both the endoplasmic reticulum (ER) and mitochondria (53). Recently, it has been suggested that augmenting mitochondrial stress signaling through the modulation of nicotinamide adenine dinucleotide (NAD+) levels can activate FOXO signaling, UPRmt and extend longevity (54). In this context, Lon protease appears as a key enzyme in aging and in situations of chronic stress (55). Lon participates in the degradation of oxidized proteins within the mitochondrial matrix induced by acute stress. Studies in mice suggest that Lon protease downregulation reduces the ability of the aging mitochondria to respond to stress (56,57).
Mitochondrial-associated membranes and mitochondrial lipids
Accumulating evidence indicates that the ER and mitochondrial outer membrane establish a tight interplay through mitochondrial-associated membranes (MAMs) (58). MAMs are important signaling sites connecting the ER and mitochondria with functional roles during apoptosis, autophagy, Ca2+ transport, inflammation, and lipid synthesis and mitochondrial dynamic events. The ER is a major lipid-synthesizing organelle, and lipids that are constituents of the mitochondrial outer membrane and inner membrane are partially supplied by the ER through MAMs and then modified within mitochondria, although some of the lipids, such as cardiolipin, are specifically synthesized in mitochondria. Cardiolipin is a phospholipid exclusively found in the inner mitochondrial membrane, where it plays an important structural role in the organization and assembly of the electron transport chain respiratory complexes (59,60) and guarantees the impermeability of this membrane. Cardiolipin peroxidation and depletion occur with aging and in a variety of pathological conditions and are associated with energy deficiency (61,62).
It has been suggested that the benefits of caloric restriction (CR) in healthspan and life span in mice may be mediated by a decrease in long-chain n-3 polyunsaturated fatty acid (PUFA) in the mitochondrial membrane. Such decrease in PUFA alters the permeability and/or the proton leak across inner mitochondrial membrane, thereby decreasing the sensitivity to lipid peroxidation. The increased susceptibility to lipid oxidative damage and downstream protein and genome toxicity affects both aging and life span (63,64). Liver mitochondrial membranes of long-lived species show a lower level of free radical production and a lower degree of unsaturation of fatty acids which, in turn, may contribute to slowing down the aging process (65). However, lifelong treatment with the beta-blocker drug atenolol decreases membrane fatty acid unsaturation and oxidative stress in heart and skeletal muscle mitochondria without changing mice longevity (66). In contrast, diets containing a low proportion of PUFAs and high amount of monounsaturated and saturated fats may maximize life span in animals maintained on CR (67).
Mitochondria and apoptosis
An imbalance between proliferation and cell death has been linked to many age-related diseases. Mitochondria could play a key role in the initiation and amplification of cell death, as fragmentation, remodeling, and permeabilization of the mitochondrial outer membrane are events that contribute to the initiation of apoptosis. Excessive mitochondrial Ca2+ uptake may trigger mitochondrial dysfunction and apoptosis through the permeabilization of the mitochondrial outer membrane (68,69). Although the mechanisms of apoptosis are not fully understood, an increase in mitochondrial permeability transition pore opening, which is modulated by anti-apoptotic proteins such as Bax/Bak and Bcl-2, and the release of cytochrome c are critical triggers in mitochondrial-regulated apoptosis (70). In reperfused aging cardiomyocytes, cell death and mitochondrial permeability transition pore opening are increased despite attenuated ROS burst and mitochondrial calcium overload (71). Apoptosis mediates the pro-longevity response to mitochondrial ROS in Caenorhabditis elegans (72). Mitochondria change morphology and assume a characteristic punctiform shape in apoptotic cells, suggesting that this organelle plays an important role in the process that eventually leads to cell death (73,74).
Mitochondria respiratory chain efficiency
Four multiprotein complexes (complexes I to IV) form the mitochondrial respiratory chain (electron transport chain) components, which are connected by two electron transporters: coenzyme Q (CoQ) and cytochrome c. With the exception of complex II and the electron transporters, the respiratory chain complexes are built by the contribution of both mtDNA and nDNA genomes. This co-ordination is highly regulated and responds not only to energy requirements but also to cellular and mitochondria homeostasis. High-energy electrons move from either NADH to oxygen through complex I to complex III and complex IV or FADH2 to oxygen through complex II—complex III—complex IV to generate the mitochondrial membrane potential that drives adenosine triphosphate biosynthesis by complex V. Complexes are not randomly distributed throughout the inner mitochondrial membrane but rather rigidly organized in a super-assembly architecture that ensures efficiency and adaptability (75). The assembly and stability of mitochondria supercomplexes are key to efficient electron flux from reduced coenzymes to molecular oxygen, and to prevent superoxide generation (76–78). Alterations of the electron transport chain components’ assembly due to aging and/or chronic diseases may lead to higher electron leakage and pathological increase in ROS production. For example, a defective supercomplex organization and subsequent mitochondrial dysfunction may be implicated in cardiac failure (79,80). Mechanisms involved in the destabilization of supercomplexes with aging are unknown, but a failure in lipid-protein interactions (ie, cardiolipin with complex I) has been described (81,82). A central role in cellular homeostasis is played by CoQ, which is a central component of the “respirasome.” CoQ is synthesized by a highly regulated multiprotein complex that involves at least 10 nDNA-encoded proteins. CoQ is essential for beta-oxidation of fatty acids, pyrimidine nucleotide synthesis, and mitochondrial permeability transition pore regulation (83).
Recent studies have shown that abnormally elevated resting metabolic rate is a risk factor for mortality and is highly correlated with the cellular levels of cytochrome oxidase, and mitochondrial content and/or efficiency (84,85). Although the mechanism for this association is still a matter of speculation, a compelling hypothesis is that higher resting metabolic rate could be an indicator of higher protein turnover rates in those mitochondria that remain undamaged and need to compensate for the accumulation of dysfunctional mitochondria. If this hypothesis is correct, resting metabolic rate abnormally elevated for the age of an individual could be used as a good clinical indicator of mitochondrial dysfunction associated with aging, and perhaps even as a biomarker of unsuccessful aging.
Mitochondria and redox homeostasis
The efficiency of mitochondrial function is maintained by a complex network of redox-sensors that alter energy production in response to changes in the cellular environment and levels of endogenous metabolites, such as Fe(II), succinate, and ROS. During aging, deregulation of various signaling pathways implicated in metabolism can induce mitochondrial dysfunction. The insulin-like growth factor 1 (IGF1), mTOR, AMPK, and sirtuin proteins act as sensors to detect slight changes in metabolites (eg, amino acids, adenosine monophosphate, and NAD+) that evoke rapid adaptive responses through signaling via FOXO, PGC-1α, and other regulatory proteins (86). Of note, CR mimetics and exercise promote longevity in animal models by impacting on these pathways (87,88). A chronic reduction of NAD+, its precursor nicotinamide, and/or adenosine triphosphate disrupts cellular bioenergetics and causes cumulative mitochondrial damage believed to be associated with aging (54). The lowering in NAD+ levels contributes to the mitochondrial decay associated with skeletal muscle aging by inducing a pseudohypoxic state that disrupts nuclear-mitochondrial communication (89), a process shown to be modulated by SIRT1 and hypoxia-inducible factor 1 (HIF-1α) (Figure 2). It follows that the pharmacological restoration of normal NAD+ and nicotinamide levels could possibly mitigate cellular bioenergetics dysfunction during aging and enhance the UPRmt response (90,91). In keeping with this hypothesis, the benefits of CR have been linked to the activation of NAD+-dependent sirtuins whose activity mediate the adaptive response to low energy by promoting the deacetylation of downstream protein targets, such as PGC-1α, FOXO, and p53.
Figure 2.

Signaling pathways implicated in metabolism and mitochondrial dysfunction during aging. A well-controlled balance between mitochondrial biogenesis and mitophagy ensures successful aging. Caloric restriction (CR), CR mimetics and exercise generate mild stress that result in elevated production of adenosine monophosphate (AMP), nicotinamide adenine dinucleotide, and/or ROS levels with subsequent activation of metabolic sensors, such as AMP kinase (AMPK) and the protein deacetylase SIRT1. Activated AMPK inhibits the insulin/IGF-1/mTOR signaling and triggers, along with SIRT1, the biogenesis of new mitochondria via PGC-1α-mediated transcriptional regulation. Modulation of tumor suppressor p53 promotes mitophagy by replacing defective mitochondria with new functionally competent mitochondria. Hence, the burden of disability in older persons may be dramatically reduce through preservation and improvement of mitochondrial quality.
Aging is apparently associated with disturbances in mitochondrial iron homeostasis, particularly its metabolism and storage. These defects have been associated with some age-related degenerative diseases with functional consequences involving the motor and cognitive performance levels (92,93). Cellular and mitochondrial iron accumulation increases with aging in some postmitotic tissues, such as skeletal muscle, brain, and peripheral nervous system, and this could generate ROS (94). Iron scavenging and removal might, therefore, prevent cellular and mitochondrial oxidative damage and attenuate age-related mitochondrial decay. Muscle inactivity is associated with disturbances in intracellular calcium homeostasis, accumulation of fatty acids and a reduced rate of mitochondrial protein import (95,96). These events increase the production of ROS in the muscle and as a consequence may lead to muscle atrophy.
Mitochondria and epigenetic regulation
Mitochondria may also play an important role in epigenetic regulation by providing numerous co-substrates produced in the tricarboxylic acid cycle (TCA cycle) that are required for epigenetic and transcriptional processes, such as histone modifications and chromatin remodeling (97). Methylation of nuclear DNA is a major component of the epigenetic system in mammalian cells and may strongly affect the regulation of mitochondrial function, since most of the mitochondrial proteins are encoded by nDNA. Because mitochondria are redox sensors, changes in the energy status in the mitochondria in response to alterations in the levels of metabolites, such as Fe(II), succinate, or even dietary changes, could be involved in epigenetic alterations with aging. This will produce changes in the expression of several genes that regulate mitochondrial metabolism and nuclear-encoded mitochondrial genes (called the “mitochondrial retrograde pathway”) (98). Salminen and colleagues (2014) proposed that mitochondria sense stressful conditions and react by shaping the epigenetic landscape of chromatin to promote survival or trigger a senescent phenotype (97).
Mitochondria and mtDNA heteroplasmy
The coexistence of wild-type and mutated copies of mtDNA, known as mtDNA heteroplasmy, may play a role in aging and in other age-related diseases. Somatic mtDNA mutations accumulate with aging in mammals and have been proposed to provide a survival advantage (3). There is significant accumulation of tissue-related and allele-related heteroplasmies during human aging (99). In fact, mtDNA heteroplasmy has been found in centenarians, suggesting that it could contribute to longevity and healthy aging (100,101). Interestingly, lower mtDNA copy number is associated with prevalent frailty and could be a significant predictor of all-cause mortality in humans (102).
Conclusions
Mitochondria are both the factory for cellular energy production and the hot spots, where different signals are integrated to produce homeostatic responses at the cellular level. It is not clear whether mitochondrial dysfunction is either a cause or a consequence of aged-associated diseases (or both), but increasing evidence suggests that energy balance is central to both successful aging and protection from age-related diseases. Numerous biological signals modulate mitochondrial function, and it is only when mitochondria are fine-tuned, healthy, and efficient that all their multiple and highly energy-demanding processes can occur normally (Figure 3). Some of the acute or chronic pathological conditions associated with aging might be a consequence of some stress-induced signaling events that occur within the mitochondria or converge to mitochondria. Mitochondria could play a key role in the pathophysiology of aging or in the earlier stages of some events that lead to the aging phenotype, such as sarcopenia. Thus, deficits in bioenergetics caused by a decline in mitochondrial function may impair normal cellular activities as we age and, as a consequence, compromise the cellular ability to adapt to various physiological stresses leading finally to weakness, frailty, and disability. There is also evidence of disease-specific mitochondrial dysfunctions that lead to further mitochondrial damage or to the exhaustion of reserves for compensatory biogenesis, and these outcomes lead to a vicious cycle of progressive tissue degeneration and loss of function.
Figure 3.

Factors affecting aged mitochondria. Numerous biological processes modulate mitochondrial function. Defects in mitochondria maintenance and turnover because of impaired biogenesis and/or defective removal are thought to contribute to the pathogenesis of complex diseases and aging. The assembly and maintenance of respiratory complexes that can sustain a high energetic flux without generating excessive ROS, and the quantity and quality of energetic fuel are essential to health and successful aging.
The assembly and maintenance of respiratory complexes that can sustain a high energetic flux without generating excessive ROS, and the quantity and quality of energetic fuel are essential to health and successful aging. Dysfunctional mitochondria have to be either repaired constantly or degraded in order to prevent cellular damage and maintain mitochondrial homeostasis. An imbalance in mitochondrial biogenesis and turnover can contribute to the aberrant increase in mitochondrial mass or content with paradoxical loss of function seen during aging and in several age-related diseases. Therefore, a good equilibrium between mitochondrial biogenesis and mitophagy is required in order to maintain cellular homeostasis and the epigenetic and proteomic adaptability changes that promote survival under stress conditions. A deregulation of this equilibrium could be detrimental and lead to the accumulation of mitochondrial damage that may trigger apoptosis during aging. This is consistent with studies showing that most interventions that prolong life span evoke decreases in the activity of nutrient signaling pathways and activate metabolic sensors, such as sirtuins, cellular redox homeostasis, and mitochondrial biogenesis pathways.
Recent studies found that some levels of hydrogen peroxide could have hormetic roles that promote healthspan and longevity, perhaps due to enhanced mitopaghy and proteostasis. In fact, some anti-aging drugs and CR mimetics, such as resveratrol and rapamycin, are also regulators or inducers of autophagy and mitochondrial biogenesis.
There is absolutely no doubt that mitochondrial dysfunction remains one of the enduring mainstream theories to explain aging and processes that lead to the development of aging phenotypes. Understanding which mitochondrial molecular pathways can alter homeostatic ROS levels may reveal whether or how free radicals could be involved both in aging and the delay of age-related diseases. Although poorly understood, the role of ROS on mitochondrial and cellular health is likely to be nonlinear, with positive physiological effects within a narrow range of concentrations. The emerging importance of mitochondrial physiology at the interface of aging and diseases suggests that mitochondrial dysfunction may be the shared mechanism by which the risk of developing chronic disease and multimorbidity increases as we age. This inter-relationship can be further complicated by the fact that many diseases may exacerbate mitochondrial damage, thus triggering a downward vicious cycle (Figure 4). Testing this hypothesis has important clinical implications and will require technological advances and extensive collaboration between basic scientists, epidemiologists and clinicians for the development of new intervention strategies that can reduce the burden of disability in older persons by preserving or improving mitochondrial function.
Figure 4.

Mitochondria as a key regulator in the pathophysiology of aging or in the earlier stages of some events that may lead to the aging phenotype. Even minor mitochondrial dysfunction when coupled with specific organ susceptibility may cause diseases, such as atherosclerosis, and also conditions that we still do not recognize as diseases, such as arterial stiffness or sarcopenia. The difference between the so-called “diseases” and these age-related conditions resides in our ignorance. What is clear is that some disease may sometime accelerate mitochondrial function decline and create a vicious cycle that leads to frailty and disability. For example, atherosclerosis of the femoral arteries may cause irreversible mitochondrial dysfunction in the leg muscle, further accelerating sarcopenia. If this hypothesis is correct, for example, early revascularization or treatment that protect mitocondrial integrity may substantial change the prognosis and clinical evolution of peripheral arterial disease. Indeed, integrating traditional pathophysiology and mitochondrial biology may increase our understanding of disease and help identify new therapeutic targets.
Funding
This research was supported by the Intramural Research Program of NIH, National Institute on Aging.
Conflict of Interest
There are no conflicts of interest to report.
References
- 1. DiMauro S, Schon EA, Carelli V, Hirano M. The clinical maze of mitochondrial neurology. Nat Rev Neurol. 2013;9(8):429–444. 10.1038/nrneurol.2013.126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Harman D. Free radical theory of aging: dietary implications. Am J Clin Nutr. 1972;25(8):839–843. [DOI] [PubMed] [Google Scholar]
- 3. Larsson N-G. Somatic mitochondrial DNA mutations in mammalian aging. Annu Rev Biochem. 2010;79:683–706. 10.1146/annurev-biochem-060408-093701 [DOI] [PubMed] [Google Scholar]
- 4. Wanagat J, Cao Z, Pathare P, Aiken JM. Mitochondrial DNA deletion mutations colocalize with segmental electron transport system abnormalities, muscle fiber atrophy, fiber splitting, and oxidative damage in sarcopenia. FASEB J. 2001;15(2):322–332. [DOI] [PubMed] [Google Scholar]
- 5. Bua E, Johnson J, Herbst A, et al. Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am J Hum Genet. 2006;79(3):469–480. 10.1086/507132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pak JW, Aiken JM. Low levels of mtDNA deletion mutations in ETS normal fibers from aged rats. Ann N Y Acad Sci. 2004;1019:289–293. [DOI] [PubMed] [Google Scholar]
- 7. Trifunovic A, Wredenberg A, Falkenberg M, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429(6990), 417–423. [DOI] [PubMed] [Google Scholar]
- 8. Umanskaya A, Santulli G, Xie W, Andersson DC, Reiken SR, Marks AR. Genetically enhancing mitochondrial antioxidant activity improves muscle function in aging. Proc Natl Acad Sci U S A. 2014;111(42), 15250–15255. 10.1073/pnas.1412754111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6), 1194–1217. 10.1016/j.cell.2013.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Newgard CB, Pessin JE. Recent progress in metabolic signaling pathways regulating aging and life span. J Gerontol A Biol Sci. 2014;69(1), S21–S27. 10.1093/gerona/glu058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Coen PM, Jubrias SA, Distefano G, et al. Skeletal muscle mitochondrial energetics are associated with maximal aerobic capacity and walking speed in older adults. J Gerontol A Biol Sci Med Sci. 2013;68(4), 447–455. 10.1093/gerona/gls196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Peterson CM, Johannsen DL, Ravussin E. Skeletal muscle mitochondria and aging: a review. J Aging Res. 2012;194821. 10.1155/2012/194821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sevini F, Giuliani C, Vianello D, et al. mtDNA mutations in human aging and longevity: controversies and new perspectives opened by high-throughput technologies. Exp Gerontol. 2014;56, 234–244. 10.1016/j.exger.2014.03.022 [DOI] [PubMed] [Google Scholar]
- 14. Owusu-Ansah E, Song W, Perrimon N. Muscle mitohormesis promotes longevity via systemic repression of insulin signaling. Cell. 2013;155(3), 699–712. 10.1016/j.cell.2013.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Martin-Montalvo A, Mercken EM, Mitchell SJ, et al. Metformin improves healthspan and lifespan in mice. Nat Commun. 2013;4:2192. 10.1038/ncomms3192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shi Y, Ivannikov MV, Walsh ME, et al. The lack of CuZnSOD leads to impaired neurotransmitter release, neuromuscular junction destabilization and reduced muscle strength in mice. PloS One. 2014;9(6), e100834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Azzu V, Brand MD. Degradation of an intramitochondrial protein by the cytosolic proteasome. J Cell Sci. 2010;123(Pt 4), 578–585. 10.1242/jcs.060004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Divakaruni AS, Brand MD. The regulation and physiology of mitochondrial proton leak. Physiology (Bethesda, MD). 2011; 26(3), 192–205. 10.1152/physiol.00046.2010 [DOI] [PubMed] [Google Scholar]
- 19. Amara CE, Shankland EG, Jubrias SA, Marcinek DJ, Kushmerick MJ, Conley KE. Mild mitochondrial uncoupling impacts cellular aging in human muscles in vivo. Proc Natl Acad Sci U S A. 2007;104(3), 1057–1062. 10.1073/pnas.0610131104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mookerjee SA, Divakaruni AS, Jastroch M, Brand MD. Mitochondrial uncoupling and lifespan. Mec Ageing Dev. 2010;131(7–8), 463–472. 10.1016/j.mad.2010.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lawless C, Jurk D, Gillespie CS, Shanley D, Saretzki G, von Zglinicki T, Passos JF. A stochastic step model of replicative senescence explains ROS production rate in ageing cell populations. PloS One. 2012;7(2), e32117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Passos JF, Saretzki G, Ahmed S, et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 2007;5(5), e110. 10.1371/journal.pbio.0050110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. López-Lluch G, Irusta PM, Navas P, de Cabo R. Mitochondrial biogenesis and healthy aging. Exp Gerontol. 2008;43(9), 813–819. 10.1016/j.exger.2008.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ding W-X, Yin X-M. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem. 2012;393(7), 547–564. 10.1515/hsz-2012-0119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rambold AS, Kostelecky B, Lippincott-Schwartz J. Fuse or die: shaping mitochondrial fate during starvation. Commun Integr Biol. 2011;4(6):752–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Joseph A-M, Adhihetty PJ, Wawrzyniak NR, et al. Dysregulation of mitochondrial quality control processes contribute to sarcopenia in a mouse model of premature aging. PloS One. 2013;8(7), e69327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rajawat YS, Hilioti Z, Bossis I. Aging: central role for autophagy and the lysosomal degradative system. Mech Ageing Dev. 2009;8(3), 199–213. 10.1016/j.arr.2009.05.001 [DOI] [PubMed] [Google Scholar]
- 28. Gouspillou G, Sgarioto N, Kapchinsky S, et al. Increased sensitivity to mitochondrial permeability transition and myonuclear translocation of endonuclease G in atrophied muscle of physically active older humans. FASEB J. 2014;28(4):1621–1633. 10.1096/fj.13-242750 [DOI] [PubMed] [Google Scholar]
- 29. Rodriguez-Hernandez A, Cordero MD, Salviati L, et al. Coenzyme Q deficiency triggers mitochondria degradation by mitophagy. Autophagy. 2009;5(1):19–32. [DOI] [PubMed] [Google Scholar]
- 30. Hirota Y, Kang D, Kanki T. The physiological role of mitophagy: new insights into phosphorylation events. Int J Cell Biol. 2012;(3):354914–354918. 10.1155/2012/354914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhao J, Brault JJ, Schild A, et al. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab. 2007;6(6):472–483. 10.1016/j.cmet.2007.11.004 [DOI] [PubMed] [Google Scholar]
- 32. Guarente L. Aging research-where do we stand and where are we going? Cell. 2014;159(1):15–19. 10.1016/j.cell.2014.08.041 [DOI] [PubMed] [Google Scholar]
- 33. Sgarbi G, Matarrese P, Pinti M, et al. Mitochondria hyperfusion and elevated autophagic activity are key mechanisms for cellular bioenergetic preservation in centenarians. Aging (Albany NY). 2014;6(4):296–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sandri M. Autophagy in skeletal muscle. FEBS Lett. 2010;584(7):1411–1416. 10.1016/j.febslet.2010.01.056 [DOI] [PubMed] [Google Scholar]
- 35. O’Leary MF, Vainshtein A, Iqbal S, Ostojic O, Hood DA. Adaptive plasticity of autophagic proteins to denervation in aging skeletal muscle. Am J Physiol Cell Physiol. 2013;304(5):C422–30. 10.1152/ajpcell.00240.2012 [DOI] [PubMed] [Google Scholar]
- 36. Kelly DP, Scarpulla RC. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004;18(4):357–368. [DOI] [PubMed] [Google Scholar]
- 37. Virbasius JV, Scarpulla RC. Activation of the human mitochondrial transcription factor A gene by nuclear respiratory factors: a potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc Natl Acad Sci U S A. 1994;91(4):1309–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Handschin C, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr Rev. 2006;27(7):728–735. 10.1210/er.2006-0037 [DOI] [PubMed] [Google Scholar]
- 39. Gouspillou G, Sgarioto N, Norris B, et al. The relationship between muscle fiber type-specific PGC-1alpha content and mitochondrial content varies between rodent models and humans. PLoS One. 2014;9(8):e103044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Short KR, Bigelow ML, Kahl J, et al. Decline in skeletal muscle mitochondrial function with aging in humans. Proc Natl Acad Sci U S A. 2005;102(15):5618–5623. 10.1073/pnas.0501559102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Conley KE, Jubrias SA, Esselman PC. Oxidative capacity and ageing in human muscle. J Physiol (Lond). 2000;526(Pt 1):203–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jaskelioff M, Muller FL, Paik J-H, et al. Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature. 2011;469(7328):102–106. 10.1038/nature09603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sahin E, Colla S, Liesa M, et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470(7334):359–365. 10.1038/nature09787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sahin E, DePinho RA. Axis of ageing: telomeres, p53 and mitochondria. Nat Rev Mol Cell Biol. 2012;13(6):397–404. 10.1038/nrm3352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jäger S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A. 2007;104(29):12017–12022. 10.1073/pnas.0705070104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1alpha. J Biol Chem. 2005;280(16):16456–16460. 10.1074/jbc.M501485200 [DOI] [PubMed] [Google Scholar]
- 47. Li L, Muhlfeld C, Niemann B, et al. Mitochondrial biogenesis and PGC-1alpha deacetylation by chronic treadmill exercise: differential response in cardiac and skeletal muscle. Basic Res Cardiol. 2011;106(6):1221–1234. 10.1007/s00395-011-0213-9 [DOI] [PubMed] [Google Scholar]
- 48. McCarty MF. Chronic activation of AMP-activated kinase as a strategy for slowing aging. Med Hypotheses. 2004;63(2):334–339. [DOI] [PubMed] [Google Scholar]
- 49. Reznick RM, Zong H, Li J, et al. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab. 2007;5(2):151–156. 10.1016/j.cmet.2007.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Baker MJ, Tatsuta T, Langer T. Quality control of mitochondrial proteostasis. Cold Spring Harb Perspect Biol. 2011;3(7). 10.1101/cshperspect.a007559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Maharjan S, Oku M, Tsuda M, Hoseki J, Sakai Y. Mitochondrial impairment triggers cytosolic oxidative stress and cell death following proteasome inhibition. Sci Rep. 2014;4:5896. 10.1038/srep05896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jensen MB, Jasper H. Mitochondrial proteostasis in the control of aging and longevity. Cell Metab. 2014;20(2):214–225. 10.1016/j.cmet.2014.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bernales S, Soto MM, McCullagh E. Unfolded protein stress in the endoplasmic reticulum and mitochondria: a role in neurodegeneration. Front Aging Neurosci. 2012;4:5. 10.3389/fnagi.2012.00005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mouchiroud L, Houtkooper RH, Moullan N, et al. The NAD(+)/Sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell. 2013;154(2):430–441. 10.1016/j.cell.2013.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ngo JK, Pomatto LCD, Davies KJA. Upregulation of the mitochondrial Lon Protease allows adaptation to acute oxidative stress but dysregulation is associated with chronic stress, disease, and aging. Redox Biol. 2013;1(1):258–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bota DA, Van Remmen H, Davies KJA. Modulation of Lon protease activity and aconitase turnover during aging and oxidative stress. FEBS Lett. 2002;532(1–2):103–106. [DOI] [PubMed] [Google Scholar]
- 57. Ngo JK, Davies KJA. Mitochondrial Lon protease is a human stress protein. Free Radic Biol Med. 2009;46(8):1042–1048. 10.1016/j.freeradbiomed.2008.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rizzuto R, Pinton P, Carrington W, et al. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998;280(5370):1763–1766. [DOI] [PubMed] [Google Scholar]
- 59. Birk AV, Chao WM, Bracken C, Warren JD, Szeto HH. Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br J Pharmacol. 2014;171(8):2017–2028. 10.1111/bph.12468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Szeto HH. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br J Pharmacol. 2014;171(8):2029–2050. 10.1111/bph.12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Szeto HH, Birk AV. Serendipity and the discovery of novel compounds that restore mitochondrial plasticity. Clin Pharmacol Ther. 2014;96(6):672–683. 10.1038/clpt.2014.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Liu S, Soong Y, Seshan SV, Szeto HH. Novel cardiolipin therapeutic protects endothelial mitochondria during renal ischemia and mitigates microvascular rarefaction, inflammation, and fibrosis. Am J Physiol Renal Physiol. 2014;306(9):F970–F980. 10.1152/ajprenal.00697.2013. [DOI] [PubMed] [Google Scholar]
- 63. Pamplona R, Barja G, Portero-Otin M. Membrane fatty acid unsaturation, protection against oxidative stress, and maximum life span: a homeoviscous-longevity adaptation? Ann N Y Acad Sci. 2002;959:475–490. [DOI] [PubMed] [Google Scholar]
- 64. Chen Y, Hagopian K, McDonald RB, et al. The influence of dietary lipid composition on skeletal muscle mitochondria from mice following 1 month of calorie restriction. J Gerontol A Biol Sci Med Sci. 2012;67(11):1121–1131. 10.1093/gerona/gls113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pamplona R, Prat J, Cadenas S, et al. Low fatty acid unsaturation protects against lipid peroxidation in liver mitochondria from long-lived species: the pigeon and human case. Mech Ageing Dev. 1996;86(1): 53–66. [DOI] [PubMed] [Google Scholar]
- 66. Gomez A, Sanchez-Roman I, Gomez J, et al. Lifelong treatment with atenolol decreases membrane fatty acid unsaturation and oxidative stress in heart and skeletal muscle mitochondria and improves immunity and behavior, without changing mice longevity. Aging Cell. 2014;13(3):551–560. 10.1111/acel.12205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. López-Domínguez JA, Ramsey JJ, Tran D, et al. The influence of dietary fat source on life span in calorie restricted mice. J Gerontol A Biol Sci Med Sci. 2014;1–8. 10.1093/gerona/glu177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Van Vliet AR, Verfaillie T, Agostinis P. New functions of mitochondria associated membranes in cellular signaling. Biochim Biophys Acta. 2014;1843(10):2253–2262. 10.1016/j.bbamcr.2014.03.009. [DOI] [PubMed] [Google Scholar]
- 69. Takeuchi A, Kim B, Matsuoka S. The destiny of Ca(2+) released by mitochondria. J Physiol Sci. 2015;65(1):11–24. 10.1007/s12576-014-0326-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mitochondrial Control of Apoptosis Signaling Pathway—Web Cell Signaling http://www.cellsignal.com/contents/science-pathway-research-apoptosis/mitochondrial-control-of-apoptosis-signaling-pathway/pathways-apoptosis-control Acceesed March 2015.
- 71. Fernandez-Sanz C, Ruiz-Meana M, Castellano J, et al. Altered FoF1 ATP synthase and susceptibility to mitochondrial permeability transition pore during ischaemia and reperfusion in aging cardiomyocytes. Thromb Haemost. 2015;113(3), 441–451. 10.1160/TH14-10–0901. [DOI] [PubMed] [Google Scholar]
- 72. Yee C, Yang W, Hekimi S. The intrinsic apoptosis pathway mediates the pro-longevity response to mitochondrial ROS in C. elegans. Cell. 2014;157(4), 897–909. 10.1016/j.cell.2014.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hiona A, Sanz A, Kujoth GC, et al. Mitochondrial DNA mutations induce mitochondrial dysfunction, apoptosis and sarcopenia in skeletal muscle of mitochondrial DNA mutator mice. PLoS One. 2010;5(7):e11468. 10.1371/journal.pone.0011468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Seo AY, Joseph A-M, Dutta D, Hwang JCY, Aris JP, Leeuwenburgh C. New insights into the role of mitochondria in aging: mitochondrial dynamics and more. J Cell Sci. 2010;123(Pt 15):2533–2542. 10.1242/jcs.070490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Lapuente-Brun E, Moreno-Loshuertos R, Acin-Perez R, et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science. 2013;340(6140):1567–1570. 10.1126/science.1230381. [DOI] [PubMed] [Google Scholar]
- 76. Kitazoe Y, Kishino H, Hasegawa M, Matsui A, Lane N, Tanaka M. Stability of mitochondrial membrane proteins in terrestrial vertebrates predicts aerobic capacity and longevity. Genome Biol Evol. 2011;3:1233–1244. 10.1093/gbe/evr079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Maranzana E, Barbero G, Falasca AI, Lenaz G, Genova ML. Mitochondrial respiratory supercomplex association limits production of reactive oxygen species from complex I. Antioxid Redox Signal. 2013;19(13):1469–1480. 10.1089/ars.2012.4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lenaz G, Genova ML. Supramolecular organisation of the mitochondrial respiratory chain: a new challenge for the mechanism and control of oxidative phosphorylation. Adv Exp Med Biol. 2012;748:107–144. 10.1007/978-1-4614-3573-0_5. [DOI] [PubMed] [Google Scholar]
- 79. Rosca MG, Vazquez EJ, Kerner J, et al. Cardiac mitochondria in heart failure: decrease in respirasomes and oxidative phosphorylation. Cardiovasc Res. 2008;80(1):30–39. 10.1093/cvr/cvn184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Frenzel M, Rommelspacher H, Sugawa MD, Dencher NA. Ageing alters the supramolecular architecture of OxPhos complexes in rat brain cortex. Exp Gerontol. 2010;45(7–8):563–572. 10.1016/j.exger.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 81. Petrosillo G, Matera M, Moro N, Ruggiero FM, Paradies G. Mitochondrial complex I dysfunction in rat heart with aging: critical role of reactive oxygen species and cardiolipin. Free Radic Biol Med. 2009;46(1):88–94. 10.1016/j.freeradbiomed.2008.09.031. [DOI] [PubMed] [Google Scholar]
- 82. Paradies G, Paradies V, De Benedictis V, Ruggiero FM, Petrosillo G. Functional role of cardiolipin in mitochondrial bioenergetics. Biochim Biophys Acta. 2014;1837(4):408–417. 10.1016/j.bbabio.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 83. López-Lluch G, Rodriguez-Aguilera JC, Santos-Ocana C, Navas P. Is coenzyme Q a key factor in aging? Mech Ageing Dev. 2010;131(4):225–235. 10.1016/j.mad.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 84. Crescenzo R, Bianco F, Mazzoli A, Giacco A, Liverini G, Iossa S. Alterations in proton leak, oxidative status and uncoupling protein 3 content in skeletal muscle subsarcolemmal and intermyofibrillar mitochondria in old rats. BMC Geriatr. 2014;14:79. 10.1186/1471-2318-14-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Fabbri E, An Y, Schrack JA, et al. Energy metabolism and the burden of multimorbidity in older adults: results from the Baltimore longitudinal study of aging. J Gerontol A Biol Sci Med Sci. 2014;1–7. 10.1093/gerona/glu209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Houtkooper RH, Williams RW, Auwerx J. Metabolic networks of longevity. Cell. 2010;142(1):9–14. 10.1016/j.cell.2010.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hepple RT, Baker DJ, McConkey M, Murynka T, Norris R. Caloric restriction protects mitochondrial function with aging in skeletal and cardiac muscles. Rejuvenation Res. 2006;9(2):219–222. 10.1089/rej.2006.9.219. [DOI] [PubMed] [Google Scholar]
- 88. Ruas JL, White JP, Rao RR, et al. A PGC-1α isoform induced by resistance training regulates skeletal muscle hypertrophy. Cell. 2012;151(6):1319–1331. 10.1016/j.cell.2012.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Gomes AP, Price NL, Ling AJY, et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155(7):1624–1638. 10.1016/j.cell.2013.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Mendelsohn AR, Larrick JW. Partial reversal of skeletal muscle aging by restoration of normal NAD(+) levels. Rejuvenation Res. 2014;17(1):62–69. 10.1089/rej.2014.1546. [DOI] [PubMed] [Google Scholar]
- 91. Prolla TA, Denu JM. NAD+ deficiency in age-related mitochondrial dysfunction. Cell Metab. 2014;19(2):178–180. 10.1016/j.cmet.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 92. Rotig A, de Lonlay P, Chretien D, et al. Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat Genet. 1997;17(2):215–217. 10.1038/ng1097-215. [DOI] [PubMed] [Google Scholar]
- 93. Isaya G. Mitochondrial iron-sulfur cluster dysfunction in neurodegenerative disease. Front Pharmacol. 2014;5:29. 10.3389/fphar.2014.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Kotzbauer PT, Truax AC, Trojanowski JQ, Lee VM-Y. Altered neuronal mitochondrial coenzyme A synthesis in neurodegeneration with brain iron accumulation caused by abnormal processing, stability, and catalytic activity of mutant pantothenate kinase 2. J Neurosci. 2005;25(3):689–698. 10.1523/jneurosci.4265-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Weiss N, Andrianjafiniony T, Dupré-Aucouturier S, Pouvreau S, Desplanches D, Jacquemond V. Altered myoplasmic Ca(2+) handling in rat fast-twitch skeletal muscle fibres during disuse atrophy. Pflugers Arch. 2010;459(4):631–644. 10.1007/s00424-009-0764-x. [DOI] [PubMed] [Google Scholar]
- 96. Bhattacharya A, Lustgarten M, Shi Y, et al. Increased mitochondrial matrix-directed superoxide production by fatty acid hydroperoxides in skeletal muscle mitochondria. Free Radic Biol Med. 2011;50(5):592–601. 10.1016/j.freeradbiomed.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Salminen A, Kaarniranta K, Hiltunen M, Kauppinen A. Krebs cycle dysfunction shapes epigenetic landscape of chromatin: novel insights into mitochondrial regulation of aging process. Cell Signal. 2014;26(7):1598–1603. 10.1016/j.cellsig.2014.03.030. [DOI] [PubMed] [Google Scholar]
- 98. Shaughnessy DT, McAllister K, Worth L, et al. Mitochondria, Energetics, Epigenetics, and Cellular Responses to Stress. Environ Health Perspect. 2014;1221271–1278. 10.1289/ehp.1408418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Li M, Schroder R, Ni S, Madea B, Stoneking M. Extensive tissue-related and allele-related mtDNA heteroplasmy suggests positive selection for somatic mutations. Proc Natl Acad Sci U S A. 2015;112(8), 2491–2496. 10.1073/pnas.1419651112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Giuliani C, Barbieri C, Li M, et al. Transmission from centenarians to their offspring of mtDNA heteroplasmy revealed by ultra-deep sequencing. Aging. 2014;6(6), 454–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Rose G, Romeo G, Dato S, et al. Somatic point mutations in mtDNA control region are influenced by genetic background and associated with healthy aging: a GEHA study. PloS One. 2010;5(10), e13395. 10.1371/journal.pone.0013395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Ashar FN, Moes A, Moore AZ, et al. Association of mitochondrial DNA levels with frailty and all-cause mortality. J Mol Med (Berl). 2015;93(2), 177–186. 10.1007/s00109-014-1233-3. [DOI] [PMC free article] [PubMed] [Google Scholar]