

Abstract
In contrast to blood capillaries, lymphatic capillaries in peripheral tissues are composed of a single-cell layer of lymphatic endothelial cells (LECs) without a covering of mural cells. However, in lymphatic malformations, the enlarged lymphatic vessels were covered with mural cells. This study aimed to understand the molecular mechanism of differences between human dermal lymphatic endothelial cells (HDLECs) and human umbilical vein endothelial cells (HUVECs) and to determine the changes of LECs in the pathological condition of lymphatic malformation. Results showed that HDLECs exhibited lower expression of endothelial proteins, including VE-cadherin and CD31, than HUVECs; HDLECs also showed higher expression of mesenchymal proteins, including α-SMA, SM22α, calponin, and epithelial mesenchymal transition-related transcription factor Slug, than HUVECs. Likewise, HDLECs displayed higher permeability and weaker recruitment of SMCs than HUVECs; HDLECs also exhibited low PDGF-BB expression. TGF-β2 treatment and FGF2 depletion enhanced mesenchymal marker expression with increased permeability and reduced SMC recruitment. By contrast, Slug depletion in HDLECs enhanced VE-cadherin expression, inhibited α-SMA expression, decreased permeability, and enhanced PDGF-BB expression. These results suggested that HDLECs were in a mesenchymal status, which contributed to their functions and might determine their identities. Our data also revealed that miR143/145 was implicated in the mesenchymal status of HDLECs. In lymphatic malformations (LMs) treated with OK-432 sclerotherapy, immunohistochemistry results showed that Prox1 expression was reduced and mural cell investment was increased; these results indicated that LECs lost their mesenchymal status after OK-432 treatment was administered. The decreased mesenchymal status of LECs in LMs may induce dilated vessel constriction, which could be the mechanism of OK-432 sclerotherapy.
Keywords: Lymphatic endothelial cell, endothelial-mesenchymal transition, lymphatic malformation, permeability, sclerotherapy
Introduction
The lymphatic system is vital in tissue fluid homeostasis, biomacromolecule transport, immune surveillance, and lipid absorption [1,2]. This system is composed of a hierarchal vessel network with blind-ended lymphatic capillaries in peripheral tissues, pre-collecting lymphatic vessels, and collecting lymphatic vessels. In contrast to blood capillaries, lymphatic capillaries are composed of a single-cell layer of endothelial cells without a covering of mural cells [3,4]. Lymphatic capillaries facilitate the entry of immune cells and transport of macromolecules in the interstitial fluid. In addition, lymphatic endothelial cells (LECs) exhibit phenotype and functions distinct from blood endothelial cells (BECs) [3,4]. For instance, LECs are interconnected with one another by discontinuous button-like junctions with few expressed junction proteins; by contrast, BECs are interconnected with one another in a compact zip-like pattern [5]. LECs originate from BECs during embryonic development, and this process is initiated by homeobox transcription factor Prox1 [6,7]. Prox1, combined with VEGF-C/VEGFR-3, determines and maintains the unique identity and terminally differentiated state of LECs [7]. However, LECs exhibit plasticity because these cells can de-differentiate into endothelial cells with characteristics similar to BECs or can trans-differentiate into fibroblast-like cells [8].
Impairment in lymphatic system development and function were found in various lymphatic-related diseases, including lymphedema and tumor metastasis. Lymphatic malformations (LMs), previously known as lymphangiomas or cystic hygromas, are classified as one of low-flow vascular malformations, which are characterized by multiple cystic spaces lined with lymphatic endothelium of varying sizes [5,9]. Based on the classification of vascular anomalies introduced by Mulliken and Glowacki [10], LMs do not exhibit evident proliferation; LMs are also considered as a disorder of the vascular system but differ from hemangioma, which exhibits increased aberrant cell division with rapid growth [11]. Histological studies have compared normal lymphatic capillaries with microcystic LM tissues and revealed that the latter are full of thin-walled, irregularly shaped, and enlarged lumen, which is lined with LYVE-1-positive LECs and covered with smooth muscle actin (SMA)-positive cells [5,9]. However, the origin and the functions of these SMA-positive cells remain unclear. As such, the origins and the functions of SMA-positive cells should be determined to understand the mechanisms of LM development to establish novel therapeutics.
In this study, the mesenchymal status of lymphatic endothelial cells (HDLECs) was compared with that of blood endothelial cells (HUVECs) on the basis of expression levels of endothelial proteins and mesenchymal markers. The mesenchymal status of HDLECs was also reinforced and suppressed with treatment of TGF-β2 and depletion of Slug, a master transcription factor in epithelial/endothelial mesenchymal transition (EMT/EnMT) to further identify the precise role of mesenchymal status of HDLECs on permeability and mural cell recruitment. The expressions of Prox1 in LECs and α-SMA-positive cells in LMs were also determined to reveal the status of LECs in LMs.
Material and methods
Regents and antibodies
Culture media and buffers were purchased from Sciencell (Carlsbad, CA, USA). Primary antibodies against VE-cadherin were purchased from Cell Signaling Technology (Danvers, MA, USA). Primary antibodies against α-smooth muscle actin (α-SMA) were purchased from Epitomics (Burlingame, CA, USA). Primary antibodies against Prox1 were purchased from Proteintech (Chicago, IL, USA). Primary antibodies against LYVE1 were obtained from Abcam (Cambridge, MA, USA). Primary antibodies against α-Tubulin were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). For small-interfering RNA (siRNA)-mediated depletion, siRNA sequences against human Slug (5’-CAGACCCATTCTGATGTAAAG-3’) were cloned into pBRsi-hU6 (Genechem, Shanghai, China) lentiviral vector systems (Slug shRNA). The negative control siRNA (NC shRNA) and lentiviral vector package were provided by Genechem (Shanghai, China). The RNA interference efficiency has been confirmed in our previous study.
Cell culture
HUVECs were isolated from human umbilical cord veins according to our previous research [12]. HDLECs were purchased from Sciencell (Carlsbad, CA, USA). HDLECs and HUVECs were cultured in endothelial cell medium (ECM; Sciencell, Carlsbad, CA, USA) supplemented with 20% fetal bovine serum, SingleQuot (Cambrex Bio Science), and penicillin-streptomycin mixture. Human smooth muscle cells (SMCs) were purchased from Sciencell and cultured in DMEM supplemented with 20% fetal bovine serum. Only cells in passages 2-7 were used in the present study.
Clinical samples and immunofluorescence
Fifteen LM samples were collected after clinical surgical resections at the Hospital of Stomatology, Wuhan University (including 10 samples without any treatment, and 5 cases with OK-432 treatment). The study was approved from the review board of the Ethics Committee of Hospital of Stomatology, Wuhan University. The procedures for human tissues were performed according to the National Institutes of Health guidelines. Briefly, the specimens were fixed in buffered 4% paraformaldehyde and embedded in paraffin. After cut into slices, the tissues were dewaxed in xylene, rehydrated in ethanol and double-distilled water, antigen retrieved by microwave. After eliminating endogenic peroxidase with 3% hydrogen peroxide, the sections were incubated with 10% goat serum for 20 min, and antibodies at 4°C overnight. Then after washing, the sections were incubated with horseradish peroxidase-conjugated secondary antibodies followed by detections and staining with hematoxylin.
Immunofluorescence analysis for cells
The localization of VE-cadherin, Prox1 and α-SMA was detected by indirect immunofluorescence analysis. In brief, HDLECs and HUVECs were grown on glass coverslips with indicated treatment. Then, cells were washed with PBS, fixed in buffered 4% paraformaldehyde and blocked in 10% goat serum for 1 h at room temperature. Then cells were incubated with the primary antibodies overnight at 4°C followed by incubation with DyLight488-conjugated secondary antibody and DyLight549-conjugated secondary antibody (1:400; Jackson Lab, West Grove, PA, USA) for 1 h at room temperature. The nuclei were stained with DAPI, and the coverslips were mounted on a microscope slide with embedding medium (Invitrogen, Carlsbad, CA, USA). The cells were observed and photographed with a fluorescence microscope (Leica).
Real-time quantitative PCR
Isolation of total RNA, synthesis of cDNA and real-time quantitative PCR were carried out as described previously [13]. Briefly, total RNA was isolated with TRIzol reagent (Invitrogen). Aliquots (1 μg) of RNA were reverse transcribed to cDNA (20 μL) with oligo (dT) and M-MuLV reverse transcriptase (Invitrogen). One-fifth of the cDNA was used as a template for PCR using SYBR Premix Ex Taq™ (Perfect Real Time) kits (Takara, Kyoto, Japan) in an ABI 7500 real-time PCR system (Applied Biosystems, Foster City, CA, USA). GAPDH was selected as an internal control for each experiment. The primer nucleotide sequences for PCR are presented in Supplementary.
Permeability assay
Flux of FITC-conjugated dextran (FITC-dextran, 10 kDa, Invitrogen) across endothelial cells monolayers was used to measure the paracellular permeability. Briefly, endothelial cells (2 × 104) were seeded l in 300 μl medium into polycarbonate 24-well transwell chambers (0.4 μm; Corning, Tewksbury, MA, USA). Cells were incubated with FITC-dextran (0.1 mg/ml) in HBSS buffer for 60 min. Thereafter, the amount of tracer penetrating through the cell monolayer into the lower chamber was measured by a fluorometer (Turner) at an excitation wavelength of 494 nm and an emission wavelength of 521 nm, the integrity of monolayer was confirmed by methylene blue staining (Supplementary Figure 2).
Mural cells migration assays
The recruitment of SMCs was measured by detecting the migration ability of SMCs using Transwell system (8 μm; Corning). Endothelial cells (HDLECs or HUVECs; 5 × 105) were cultured in the lower chamber, while SMCs (5 × 105) were suspended in 100 μl of serum-deprived DMEM and seeded into the upper chambers. After incubation at 37°C for 12 h, the migrated cells were fixed and stained with crystal violet, and then photographed and counted.
Western blot analysis
The proteins were collected in M-PER (Pierce, Rockford, IL, USA), and the concentration was estimated using the BCA assay (Pierce). 30 μg of protein was separated on 10% SDS-polyacrylamide gels and then electroblotted on PVDF membranes (Roche Applied Science, Germany). The blots were blocked overnight with 5% non-fat dry milk and incubated with primary antibodies at dilutions recommended by the suppliers. Immunoblots were detected by HRP-conjugated secondary antibody (Pierce) using a chemiluminescence kit (Pierce) and photographed.
Statistical analysis
All values are expressed as the mean ± SEM of three independent experiments. Data analyses were conducted using Graphpad prime 5.01 (GraphPad Software, La Jolla, CA, USA). One-way ANOVA and the Student-Newman-Keuls test were used for statistical analysis. P < 0.05 was considered statistically significant.
Results
Mesenchymal status of HDLECs compared with HUVECs
We determined the distinctions between LECs and BECs because of the differences in structure and components between lymphatic capillaries and blood capillaries. In this study, the expressions of endothelial and lymphatic markers, as well as mesenchymal proteins in HDLECs and HUVECs, were investigated. VE-cadherin was highly expressed in HUVECs, and this high expression was localized around the margin of the cells in a zip-like pattern; by contrast, VE-cadherin was poorly expressed in HDLECs (Figure 1A). Likewise, immunofluorescence revealed that α-SMA expression was higher in HDLECs than in HUVECs. Real-time PCR assay results revealed that the expressions of endothelial markers, including VE-cadherin and CD31, were higher in HDLECs than in HUVECs; by contrast, the expressions of mesenchymal proteins, including α-SMA, smooth muscle 22α (SM22α), calponin, and fibroblast specific protein-1 (FSP1), were higher in HDLECs than in HUVECs. Furthermore, the expressions of Prox1 and podoplanin were higher in HDLECs than in HUVECs (Figure 1C-G). Western blot revealed similar results. Related transcription factors, such as Twist, Snai1/2, Zeb1/2, and others, are critical in EMT/EnMT [14]. In this study, real-time PCR and Western blot analysis results confirmed that Slug was highly expressed in HDLECs (Figure 1F and 1G). These results suggested that HDLECs were more mesenchymal than HUVECs.
Figure 1.
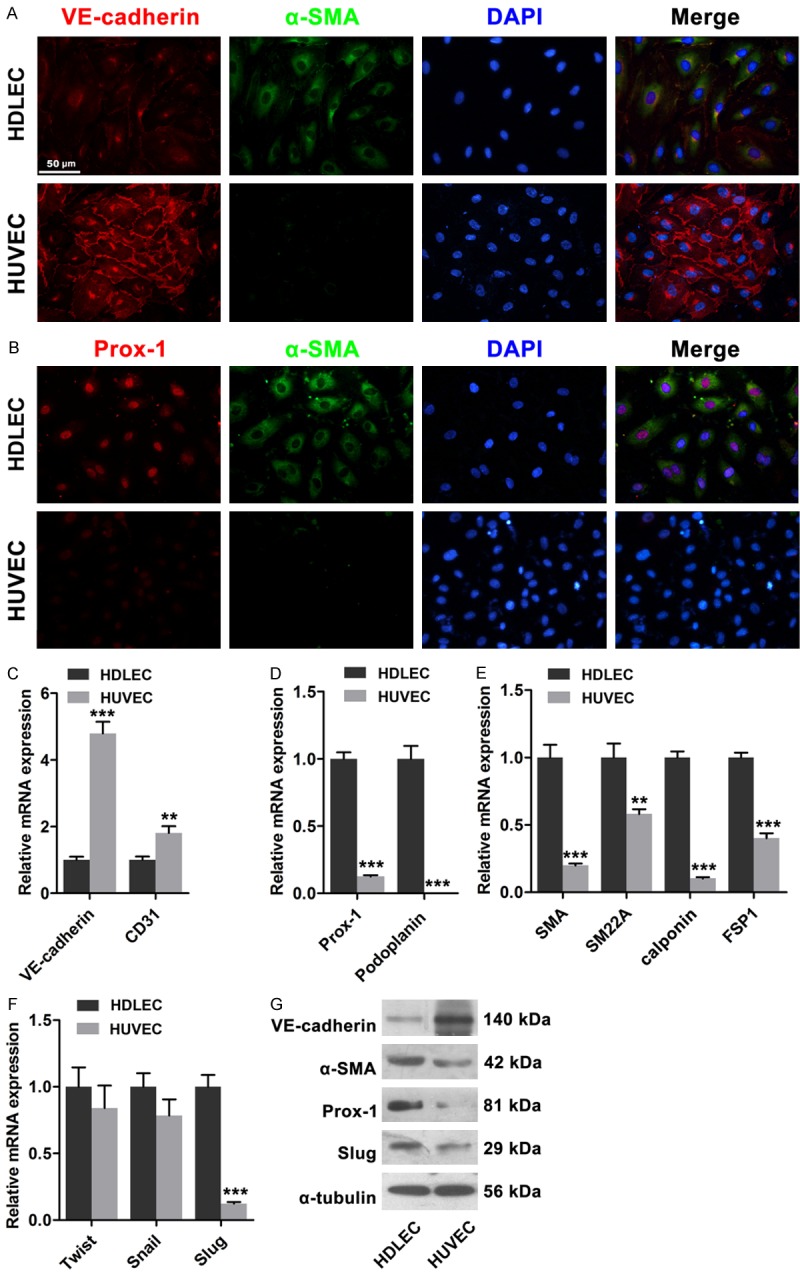
Mesenchymal status of HDLECs compared to HUVECs. A. Immunofluorescence showed lower expression of VE-cadherin and higher expression of α-SMA in HDLECs compared to HUVECs. B. Immunofluorescence showed higher expression of Prox1 in HDLECs. C. Endothelial markers VE-cadherin and CD31 were lower in HDLECs. D. Lymphatic endothelial markers Prox1 and Podoplanin were higher in HDLECs. E. Mesenchymal proteins including α-SMA, SM22α, SM and FSP-1 were higher in HDLECs. F. Real time PCR assays revealed the expression of Twist, Snail and Slug in HDLECs and HUVECs. G. Western blots showed the expression levels of VE-cadherin, α-SMA, Prox-1 and Slug in HDLECs compared to HUVECs. α-Tubulin was used as loading control. All data are presented as mean ± SE from three different experiments with duplicate. *, P < 0.05; **, P < 0.01 versus HDLECs.
Differences in functions related to permeability and SMC recruitment between HDLECs and HUVECs
To investigate the permeability and the ability of HDLECs and HUVECs to attract mural cells, we established permeability assay using a Transwell (0.4 μm) system by adding FITC-labeled dextrans in the upper chamber and by detecting them in the lower chamber (Figure 2A) and a Transwell system with a pore size of 8 μm to evaluate SMCs recruitment. HDLECs showed higher permeability than HUVECs, as indicated by more dextrans detected in the lower chambers (Figure 2B). The mRNA expression level of PDGF-BB, which has been reported as one of the most effective chemotaxins to attract pericytes or smooth muscle cells, was then determined by real-time PCR assay. The mRNA expression of PDGF-BB was lower in HDLECs than in HUVECs (Figure 2C). Furthermore, endothelial cell-induced migration of human SMCs was evaluated using a Transwell system with a pore size of 8 μm (Figure 2D). More SMCs migrated across the membrane when these SMCs were co-cultured with HUVECs, not with HDLECs (Figure 2E). Therefore, HDLECs exhibited higher permeability and weaker recruitment ability than HUVECs.
Figure 2.
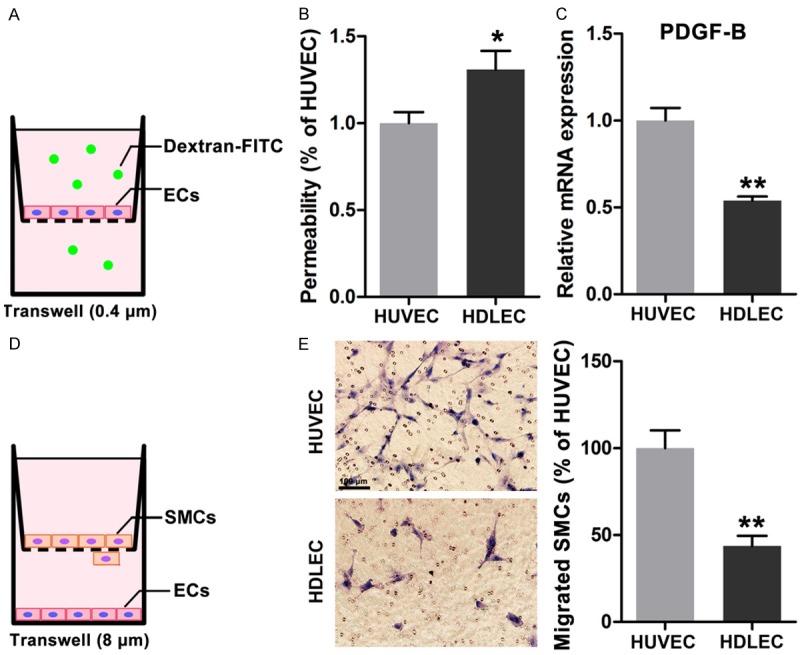
Different permeability and recruitment ability of HDLECs compared to HUVECs. A. Diagram of permeability assays of endothelail cells. B. Higher permeability was found in HDLECs. C. Less expression of PDGF-BB in HDLECs compared to HUVECs. D. Diagram of recruitment ability of endothelial cells by measuring the migrated SMCs in co-culture system. E. More SMCs were migrated across the membrane in co-culture system with HUVECs. All data are presented as mean ± SE from three different experiments with duplicate. *, P < 0.05; **, P < 0.01 versus HUVECs.
Enhanced mesenchymal status of HDLECs increased their permeability but decreased their recruitment ability
On the basis of our findings, we speculated that functional differences between HDLECs and HUVECs may be attributed to distinct mesenchymal status. Therefore, we enhanced the mesenchymal status of HDLECs by inducing EnMT according to published protocols [8]. After TGF-β2 treatment was administered for 48 h under FGF2-depleting conditions, VE-cadherin expression was significantly downregulated, whereas α-SMA expression was markedly increased (Figure 3A). mRNA and protein expressions of VE-cadherin, α-SMA, and SM22α were then determined. Our results revealed that mRNA and protein expressions of VE-cadherin were significantly reduced; by contrast, mRNA and protein expressions of α-SMA and SM22α were markedly increased; these results are consistent with those in previous studies [8]. The permeability of TGF-β2-treated HDLECs was determined and the results showed that more dextrans were detected, suggesting increased permeability and enhanced mesenchymal status of HDLECs. The mRNA expression of PDGF-BB was reduced and the ability to recruit SMCs was impaired (Figure 3E and 3F). In summary, increased mesenchymal status of HDLECs could increase permeability and impair mural cell recruitment.
Figure 3.
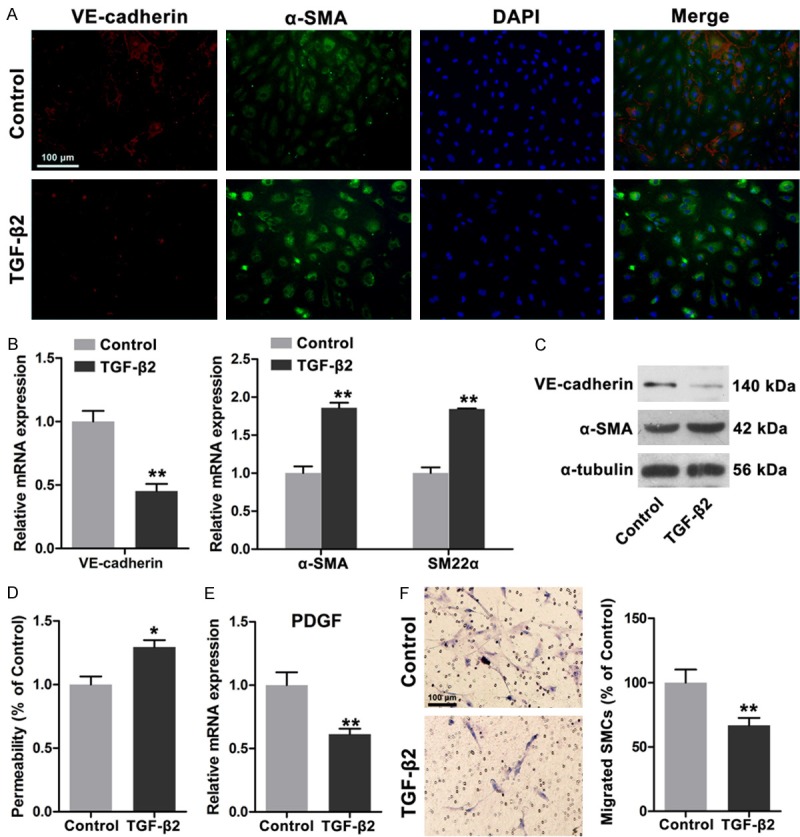
TGF-β2 induced endothelial mesenchymal transition of HDLECs with increased permeability and weakened recruitment of SMCs. A. Immunofluorescence showed decreased VE-cadherin and increased α-SMA in HDLECs after TGF-β2 treatment (20 ng/ml for 48 h). B. Real time PCR assays revealed decreased VE-cadherin expression and increased α-SMA and SM22α expression. C. Western blots showed the decreased VE-cadherin and increased α-SMA in HDLECs with TGF-β2 treatment. D. Increased permeability was observed in HDLECs after TGF-β2 treatment. E. Decreased PDGF-BB expression in HDLECs after TGF-β2 treatment. F. HDLECs treated with TGF-β2 showed weakened ability to recruit SMCs. All data are presented as mean ± SE from three different experiments with duplicate. *, P < 0.05; **, P < 0.01 versus control group.
Decreased mesenchymal status by Slug depletion in HDLECs decreased permeability and enhanced SMC recruitment
Slug triggers EnMT by suppressing VE-cadherin transcription [13,15]. Our previous data demonstrated that Slug is strongly expressed in HDLECs compared with that in HUVECs. Therefore, we investigated whether or not Slug suppression affects the functions of HDLECs, including permeability and mural cell recruitment. After the cells were transfected with shRNA lentivirus against Slug, HDLECs showed a significantly reduced Slug expression at mRNA and protein levels. Likewise, VE-cadherin expression was increased at mRNA and protein levels. Mesenchymal proteins, including α-SMA and SM22α, were remarkably reduced (Figure 4A and 4B). Prox1, which is a critical transcription factor of LEC differentiation and maintenance, was also downregulated after Slug was depleted in HDLECs (Figure 4A and 4B). Immunofluorescence results also indicated that α-SMA and Prox1 expressions were decreased (Figure 4C). Permeability was also evaluated, and the results revealed that Slug depletion decreased the permeability of HDLECs, Thus, the integrity of the endothelial monolayer was increased (Figure 4D). Moreover, the mRNA expression of PDGF-BB was increased after Slug interference occurred (Figure 4E). SMC migration also increased, as detected by Transwell migration assays (Figure 4F); this result suggested that the recruitment ability of HDLECs was enhanced by Slug depletion. These results further indicated that suppressed mesenchymal status by targeting Slug in HDLECs could decrease permeability and enhance mural cell recruitment.
Figure 4.
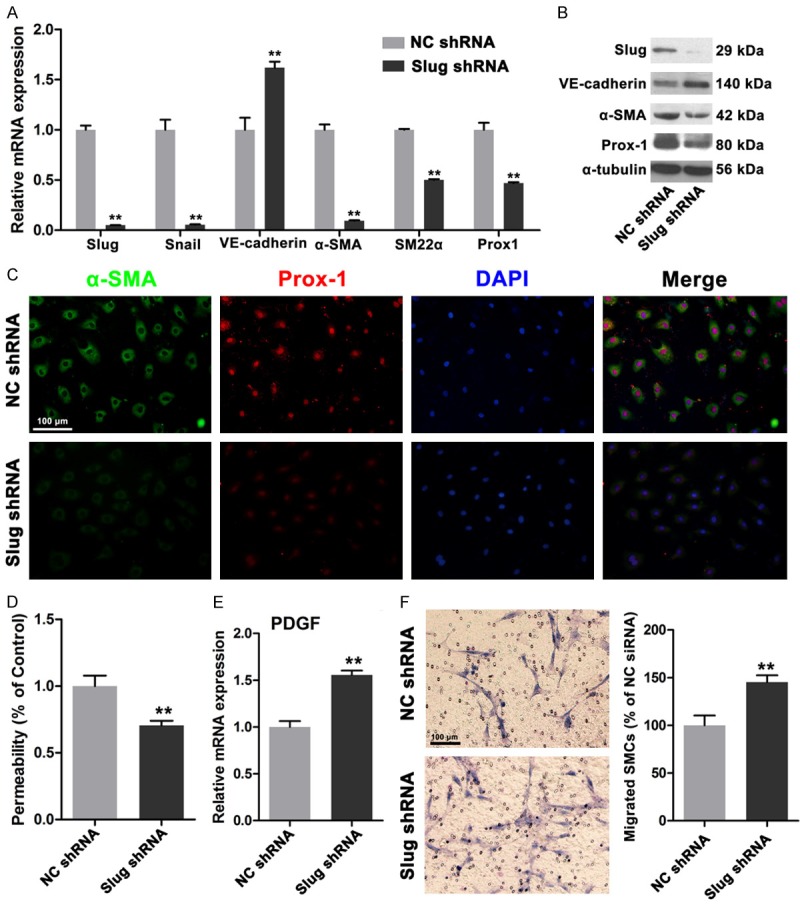
Depletion of Slug reversed the mesenchymal status of HDLECs. A. Real time PCR assays showed depletion of Slug increased the expression of VE-cadherin and decreased α-SMA, SM22α and Prox1 expressions. B. Western blots revealed the increased VE-cadherin, decreased α-SMA, SM22α and Prox1 expressions in protein levels. C. Immunofluorescence showed decreased α-SMA and Prox1 in HDLECs with depletion of Slug. D. Decreased permeability of HDLECs with depletion of Slug. E. Increased PDGF-BB in HDLECs treated with Slug shRNA. F. HDLECs with depletion of Slug showed enhanced ability of SMCs recruitment. All data are presented as mean ± SE from three different experiments with duplicate. *, P < 0.05; **, P < 0.01 versus NC shRNA group.
miR-143/145 is involved in mesenchymal status of HDLECs
miR-143 and miR-145 are secreted by endothelial cells and then absorbed by smooth muscle cells to regulate fate and contractile phenotype [16]. In this study, the expression levels of miR-143 and miR-145 in HDLECs were compared with those in HUVECs. miR-143 and miR-145 expressions were higher in HDLECs than in HUVECs (Figure 5A). After TGF-β2 treatment was administered, miR-143 expression was significantly increased in HDLECs (Figure 5B). After Slug was depleted in HDLECs by lentivirus-mediated RNA interference, miR-143 and miR-145 expressions were reduced (Figure 5C). Considering the significant role of miR-143/145 gene cluster in the development and the phenotype formation of smooth muscle cells, we suggested that miR-143/145 might participate in mesenchymal status regulation of HDLECs.
Figure 5.
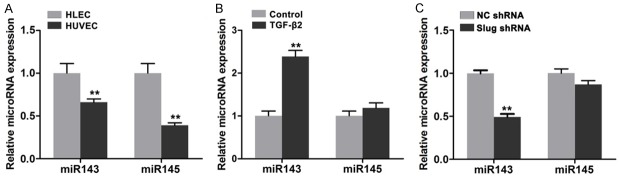
miR143/145 involved in the mesenchymal status of HDLECs. A. Higher expression of miR143 and miR145 in HDLECs compared to HUVECs. B. TGF-β2 treatment increased the expression of miR143 and miR145 in HDLECs. C. RNA interference against Slug reduced the expression of miR143 and miR145 in HDLECs. All data are presented as mean ± SE from three different experiments with duplicate. *, P < 0.05; **, P < 0.01.
Increased α-SMA-positive cells coverage in human LM after OK-432 treatment
OK-432 is a lyophilized mixture of low-virulence group A Streptococcus pyogenes Su strain incubated with a low concentration of benzyl penicillin, which is commonly used for sclerotherapy to treat LM [17]. Enlarged lymphatic vessels lined with thin LYVE1-positive LECs were found in untreated LMs (Figure 6A). Around the vessels, α-SMA-positive cells were found in close contact with endothelial cells. After the cystic spaces were treated with OK-432, the enlarged vessels were constricted and α-SMA-positive cell recruitment was increased. Immunofluorescence results suggested that the lined LECs of LMs treated with OK-432 showed a decreased expression of Prox1, a specific marker protein that determines the identity and the phenotype of LECs, compared with those of untreated LMs (Figure 6B). In addition, the mRNA expression of PDGF-BB in LMs (n=3) was compared with that in skins (n=3). As shown in Supplementary Figure 1A, the mRNA expression of PDGF-BB was much higher than that in skins. The ratio of SMC/LEC in untreated and OK-432 treated LMs (n=3) was compared (Supplementary Figure 1B). In summary, LECs that line the cyst of LMs showed a weakened mesenchymal status but with increased α-SMA-positive cell coverage after OK-432 sclerotherapy was administered; this result might reduce the size of cysts in LMs, thereby relieving symptoms.
Figure 6.
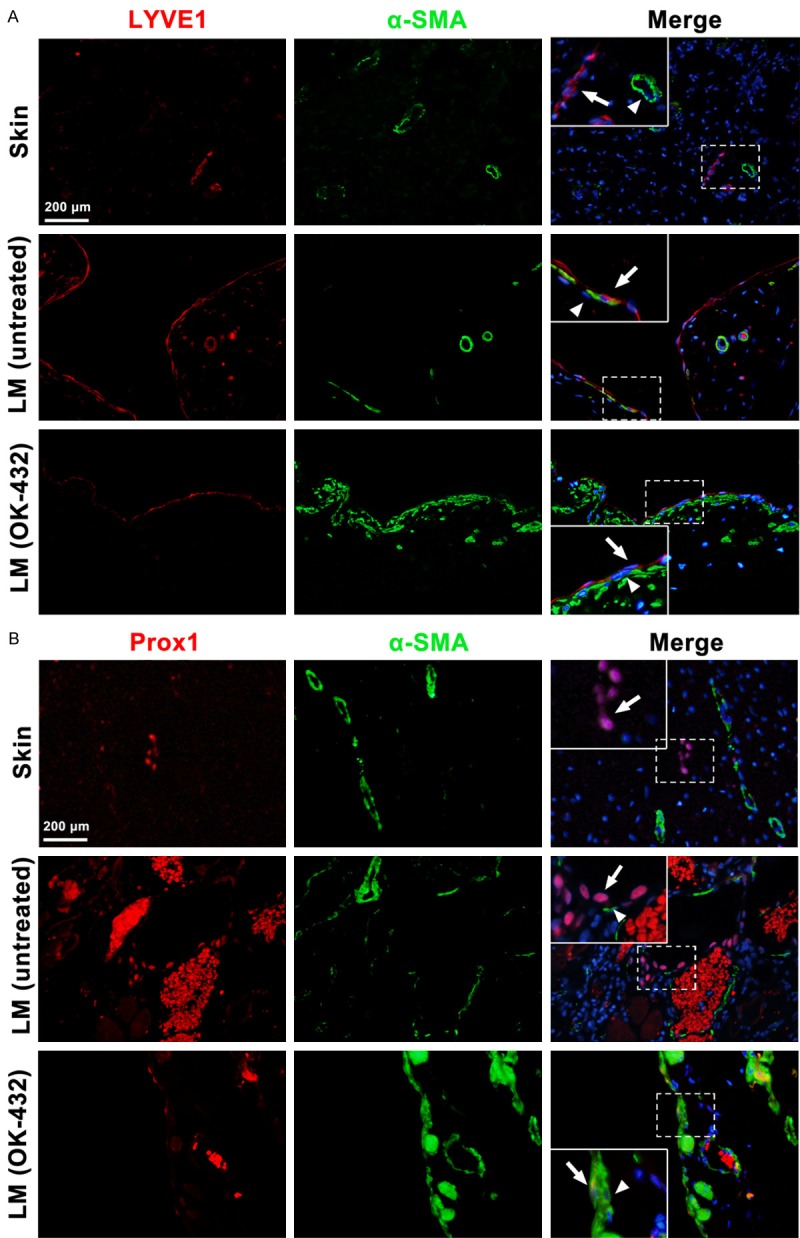
Increased investment of α-SMA positive mural cells and decreased expression of Prox1 in LMs after OK-432 sclerotherapy. A. Immunofluorescence showed increased α-SMA positive cells recruitment in the enlarged lymphatic vessels in LMs after OK-432 sclerotherapy. B. Immunofluorescence revealed weakened expression of Prox1 in LECs lined in the dilated vessels in LMs after OK-432 sclerotherapy. The arrows showed the lymphatic endothelial cells, the triangle pointed the recruited mural cells.
Discussion
We provided evidence of the mesenchymal status of HDLECs by comparing the different expressions of endothelial and mesenchymal proteins. Our results revealed the identity and the functions of HDLECs, including permeability of endothelial cell layer and mural cell recruitment. Moreover, the mesenchymal status of HDLECs was regulated by Slug, a classical EMT/EnMT-related transcription factor. Immunohistochemistry and immunofluorescence results suggested that LECs in LMs may lose mesenchymal status and be covered with an increased number of α-SMA-positive mural cells after sclerotherapy was administered; thus, sclerotherapy might be used to treat LMs effectively.
In embryonic development, LECs were derived from vein endothelial cells [18,19]. However, the characteristics of LECs differ from those of BECs. Primary lymphatic vessels, which are considered as lymphatic capillaries, are blind-ended structures that lack basal lamina and mural cells [4]. Only a single-cell layer of overlapping ECs constitutes these lymphatic capillaries. These overlapping ECs are interconnected with one another by specialized discontinuous button-like junctions with few intercellular tight junctions or adherent junctions; compared with BECs, this structure exhibits enhanced permeability and efficiently collects interstitial fluid, macromolecules, and immune cells [4]. VE-cadherin, not CD31, is necessary to maintain tight junction integrity of lymphatic capillaries [20]. Therefore, we initially analyzed VE-cadherin expression in LECs and BECs under culture conditions. Our results showed that VE-cadherin expression was considerably higher in BECs than in LECs. Permeability of endohtelial cells was then analyzed, and our results showed that the permeability was higher in LEC monolayer than in BEC monolayer. EnMT, which was first addressed in diseases, such as pulmonary fibrosis, kidney fibrosis, and cancers, is a process by which endothelial cells lose endothelial characteristics but gain mesenchymal or fibroblastic features, as represented by the downregulation of VE-cadherin, CD31, CD34, and other endothelial molecules and by the upregulation of SMA, SM22α, FSP-1, and mesenchymal proteins [8,21-23]. Considering that VE-cadherin was poorly expressed in HDLECs, we investigated the mesenchymal marker proteins in HDLECs. Mesenchymal proteins, including α-SMA, SM22α, calponin, and FSP-1, were highly expressed in HDLECs compared with HUVECs. The EMT/EnMT-related transcription factor Slug was highly expressed in HDLECs. On the basis of these findings, we described mesenchymal status as the condition at which mesenchymal proteins are highly expressed in HDLECs.
To clarify the relationship between LEC mesenchymal status and functions, including permeability and mural cell recruitment ability, we moderated the mesenchymal status of HDLECs by enhancing EnMT or by suppressing EnMT. In a previous study [8], TGF-β2 with depleted FGF2 can induce EnMT in HDLECs, as indicated by an increase in α-SMA and other mesenchymal proteins and by a continuous decrease in VE-cadherin. We successfully suppressed HDLEC mesenchymal status, decreased α-SMA, and increased VE-cadherin by reducing Slug expression. We found that increased mesenchymal status in HDLECs impaired integrity and mural cell recruitment ability and vice versa. Slug depletion also decreased Prox1 expression, suggesting the possible interaction between these transcription factors. Prox1, the main regulatory factor of the identity and the phenotype of LECs [3], functions as a binary switch, which can suppress BEC identity but can promote and maintain LEC identity [24]. Switching off Prox1 activity can sufficiently initiate a reprogramming cascade, leading to the dedifferentiation of LECs to BECs [24]. Slug may control the fate of LECs by regulating Prox1 expression. Slug can directly bind to the promoter of VE-cadherin, thereby inducing and maintaining the mesenchymal status of endothelial cells; this process might be another mechanism controlling the identity of LECs [15].
Foxc2–/– mice show abnormal lymphatic vascular patterning with increased pericyte investment of lymphatic vessels, agenesis of valves, and lymphatic dysfunction, which are similar to the appearance of LMs [25]. The loss of Foxc2 increases the expressions of PDGF-BB, endoglin, and collagen IV of LECs; this increased expression further contributes to mural cell recruitment and lamina establishment that may be accounted for the interaction between ECs and SMCs [25]. Foxc2 expressed in cancer cells promotes EMT [26-28]; Foxc2 is also necessary to maintain mesenchymal phenotype after EMT is induced in breast cancers [26]. This result is similar to that observed in Foxc2 in LECs; in particular, Foxc2 is a crucial factor that determines and maintains LEC identity [29,30]. Another crucial fate-determining factor of LECs, Prox1 functions as a positive moderator of EMT by inhibiting E-cadherin via miR-9 [30]. Therefore, Prox1 cooperated with Foxc2 to determine the mesenchymal status of LECs in a similar manner. LECs are among the few differentiated cell types that likely lose endothelial identity but gain a mesenchymal phenotype. These dedifferentiated ECs require a constant expression of certain genes, such as Prox1 and Foxc2, to maintain phenotypic identity. In LMs, LECs might lose mesenchymal status acompanied with increased PDGF-BB expression and mural cell recruitment. LMs are independent from the lymphatic or venous system [9] with accumulating fluids that enlarge the lymphatic vessels; as a result, cystic morphological characteristics were observed. The loss of mesenchymal status of LECs in LMs may result in increased pressure because mechanical stimulus could initiate the maturity of endothelial progenitor cells and induce vascular differentiation of stem cells.
In summary, HDLECs displayed higher expression of mesenchymal proteins than BECs, indicating mesenchymal status. Mesenchymal proteins were involved in increased permeability and weakened mural cell recruitment. Mesenchymal status suggested that HDLECs were in poorly differentiated with high plasticity; nevertheless, these characteristics could be changed under pathological conditions, such as high pressure in LMs. Under such conditions, LECs lost their mesenchymal status and matured to a BEC-like status with increased integrity and mural recruitment. This disease could be treated by inducing the trans-differentiation from LECs to BEC via intralesional injection of sclerosing agents.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (81170977, 81371159) to YF Zhao, and Specialized Research Fund for the Doctoral Program of Higher Education (20130141130006) to YF Zhao.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Alitalo K, Tammela T, Petrova TV. Lymphangiogenesis in development and human disease. Nature. 2005;438:946–953. doi: 10.1038/nature04480. [DOI] [PubMed] [Google Scholar]
- 2.Tammela T, Alitalo K. Lymphangiogenesis: Molecular mechanisms and future promise. Cell. 2010;140:460–476. doi: 10.1016/j.cell.2010.01.045. [DOI] [PubMed] [Google Scholar]
- 3.Chen H, Griffin C, Xia L, Srinivasan RS. Molecular and cellular mechanisms of lymphatic vascular maturation. Microvasc Res. 2014;96:16–22. doi: 10.1016/j.mvr.2014.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oliver G, Srinivasan RS. Endothelial cell plasticity: how to become and remain a lymphatic endothelial cell. Development. 2010;137:363–372. doi: 10.1242/dev.035360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perkins JA, Manning SC, Tempero RM, Cunningham MJ, Edmonds JL Jr, Hoffer FA, Egbert MA. Lymphatic malformations: current cellular and clinical investigations. Otolaryngol Head Neck Surg. 2010;142:789–794. doi: 10.1016/j.otohns.2010.02.025. [DOI] [PubMed] [Google Scholar]
- 6.Hong YK, Detmar M. Prox1, master regulator of the lymphatic vasculature phenotype. Cell Tissue Res. 2003;314:85–92. doi: 10.1007/s00441-003-0747-8. [DOI] [PubMed] [Google Scholar]
- 7.Makinen T, Norrmen C, Petrova TV. Molecular mechanisms of lymphatic vascular development. Cell Mol Life Sci. 2007;64:1915–1929. doi: 10.1007/s00018-007-7040-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ichise T, Yoshida N, Ichise H. FGF2-induced Ras-MAPK signalling maintains lymphatic endothelial cell identity by upregulating endothelial-cell-specific gene expression and suppressing TGFbeta signalling through Smad2. J Cell Sci. 2014;127:845–857. doi: 10.1242/jcs.137836. [DOI] [PubMed] [Google Scholar]
- 9.Ramashankar , Prabhakar C, Shah NK, Giraddi G. Lymphatic malformations: A dilemma in diagnosis and management. Contemp Clin Dent. 2014;5:119–122. doi: 10.4103/0976-237X.128689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mulliken JB, Glowacki J. Hemangiomas and vascular malformations in infants and children: a classification based on endothelial characteristics. Plast Reconstr Surg. 1982;69:412–422. doi: 10.1097/00006534-198203000-00002. [DOI] [PubMed] [Google Scholar]
- 11.Boscolo E, Bischoff J. Vasculogenesis in infantile hemangioma. Angiogenesis. 2009;12:197–207. doi: 10.1007/s10456-009-9148-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zou HX, Jia J, Zhang WF, Sun ZJ, Zhao YF. Propranolol inhibits endothelial progenitor cell homing: a possible treatment mechanism of infantile hemangioma. Cardiovasc Pathol. 2013;22:203–210. doi: 10.1016/j.carpath.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 13.Zhang W, Chen G, Ren JG, Zhao YF. Bleomycin induces endothelial mesenchymal transition through activation of mTOR pathway: a possible mechanism contributing to the sclerotherapy of venous malformations. Br J Pharmacol. 2013;170:1210–1220. doi: 10.1111/bph.12355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 15.Lopez D, Niu G, Huber P, Carter WB. Tumor-induced upregulation of Twist, Snail, and Slug represses the activity of the human VE-cadherin promoter. Arch Biochem Biophys. 2009;482:77–82. doi: 10.1016/j.abb.2008.11.016. [DOI] [PubMed] [Google Scholar]
- 16.Cordes KR, Sheehy NT, White MP, Berry EC, Morton SU, Muth AN, Lee TH, Miano JM, Ivey KN, Srivastava D. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature. 2009;460:705–710. doi: 10.1038/nature08195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Joshi SR, Comer BS, McLendon JM, Gerthoffer WT. MicroRNA Regulation of Smooth Muscle Phenotype. Mol Cell Pharmacol. 2012;4:1–16. [PMC free article] [PubMed] [Google Scholar]
- 18.Pichol-Thievend C, Hogan BM, Francois M. Lymphatic vascular specification and its modulation during embryonic development. Microvasc Res. 2014;96:3–9. doi: 10.1016/j.mvr.2014.07.011. [DOI] [PubMed] [Google Scholar]
- 19.Yang Y, Garcia-Verdugo JM, Soriano-Navarro M, Srinivasan RS, Scallan JP, Singh MK, Epstein JA, Oliver G. Lymphatic endothelial progenitors bud from the cardinal vein and intersomitic vessels in mammalian embryos. Blood. 2012;120:2340–2348. doi: 10.1182/blood-2012-05-428607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baluk P, Fuxe J, Hashizume H, Romano T, Lashnits E, Butz S, Vestweber D, Corada M, Molendini C, Dejana E, McDonald DM. Functionally specialized junctions between endothelial cells of lymphatic vessels. J Exp Med. 2007;204:2349–2362. doi: 10.1084/jem.20062596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hashimoto N, Phan SH, Imaizumi K, Matsuo M, Nakashima H, Kawabe T, Shimokata K, Hasegawa Y. Endothelial-mesenchymal transition in bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 2010;43:161–172. doi: 10.1165/rcmb.2009-0031OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Potenta S, Zeisberg E, Kalluri R. The role of endothelial-to-mesenchymal transition in cancer progression. Br J Cancer. 2008;99:1375–1379. doi: 10.1038/sj.bjc.6604662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 24.Johnson NC, Dillard ME, Baluk P, McDonald DM, Harvey NL, Frase SL, Oliver G. Lymphatic endothelial cell identity is reversible and its maintenance requires Prox1 activity. Genes Dev. 2008;22:3282–3291. doi: 10.1101/gad.1727208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Petrova TV, Karpanen T, Norrmen C, Mellor R, Tamakoshi T, Finegold D, Ferrell R, Kerjaschki D, Mortimer P, Yla-Herttuala S, Miura N, Alitalo K. Defective valves and abnormal mural cell recruitment underlie lymphatic vascular failure in lymphedema distichiasis. Nat Med. 2004;10:974–981. doi: 10.1038/nm1094. [DOI] [PubMed] [Google Scholar]
- 26.Hollier BG, Tinnirello AA, Werden SJ, Evans KW, Taube JH, Sarkar TR, Sphyris N, Shariati M, Kumar SV, Battula VL, Herschkowitz JI, Guerra R, Chang JT, Miura N, Rosen JM, Mani SA. FOXC2 expression links epithelial-mesenchymal transition and stem cell properties in breast cancer. Cancer Res. 2013;73:1981–1992. doi: 10.1158/0008-5472.CAN-12-2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu B, Han SM, Tang XY, Han L, Li CZ. Overexpressed FOXC2 in ovarian cancer enhances the epithelial-to-mesenchymal transition and invasion of ovarian cancer cells. Oncol Rep. 2014;31:2545–2554. doi: 10.3892/or.2014.3119. [DOI] [PubMed] [Google Scholar]
- 28.Ren YH, Liu KJ, Wang M, Yu YN, Yang K, Chen Q, Yu B, Wang W, Li QW, Wang J, Hou ZY, Fang JY, Yeh ET, Yang J, Yi J. De-SUMOylation of FOXC2 by SENP3 promotes the epithelial-mesenchymal transition in gastric cancer cells. Oncotarget. 2014;5:7093–7104. doi: 10.18632/oncotarget.2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seo S, Fujita H, Nakano A, Kang M, Duarte A, Kume T. The forkhead transcription factors, Foxc1 and Foxc2, are required for arterial specification and lymphatic sprouting during vascular development. Dev Biol. 2006;294:458–470. doi: 10.1016/j.ydbio.2006.03.035. [DOI] [PubMed] [Google Scholar]
- 30.Wu X, Liu NF. FOXC2 transcription factor: a novel regulator of lymphangiogenesis. Lymphology. 2011;44:35–41. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.