

Abstract
Purpose: The protein encoded by sphingomyelin phosphodiesterase 1, acid lysosomal (SMPD1) is a lysosomal acid sphingomyelinase. While there are increasing evidences to suggest that lysosomal enzyme defects and Parkinson’s disease (PD) have strong associations, and recently, SMPD1 p.L302P (c.T911C, NM_000543) was found to be a risk factor for PD in Ashkenazi Jewish ancestry population, we try to investigate the possible association between SMPD1 p.L302P and sporadic PD in ethnic Chinese population. Methods: 455 sporadic PD and 476 health controls were included in our study. SMPD1 p.L302P (c.T911C) was genotyped by matrix-assisted laser desorption/ionization-time-of-flight mass spectrometry (MALDI-TOF MS) and the results were confirmed by Sanger sequencing. Results: Our results showed that none of 455 sporadic PD and 476 health controls carried p.L302P. All of the 931 subjects’ genotypes were wild type TT. Our data indicated that in an ethnic Chinese population, p.L302P did not appear to be enriched in sporadic PD, and p.L302P may not be a risk factor for Chinese sporadic PD. And combine our data with the results from previous studies, we found that all of the 2,268 participants of Chinese population carrying no p.L302P. Conclusions: We could make a conclusion that p.L302P may not be common events for Chinese population. Sequencing of SMPD1 gene to find additional novel rare variants in the SMPD1 gene in diverse populations is needed.
Keywords: SMPD1, p.L302P, Parkinson’s disease, ethnic Chinese population, SNP
Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disorder, clinically characterized by resting tremor, bradykinesia, rigidity, postural instability and a positive response to dopamine replacement therapy. It is estimated that by 2030, the number of PD patients will get to between 8.7 and 9.3 million [1]. So, it is important to elucidate the etiology of PD and take proper measures to make early diagnosis and therapy of it. Although the mechanism of PD still remains unclear, recent studies suggested that a number of genes influence PD susceptibility [2-4].
Recently, Gan-Or et al [5] conducted a study with candidate gene approach to explore the possible association of founder mutations in the lysosomal storage disorder genes HEXA, SMPD1, and MCOLN1 (causing Tay-Sachs, Niemann-Pick A, and mucolipidosis type IV diseases, respectively) with PD in Ashkenazi Jewish ancestry population. A total of 938 PD and much more controls were included in the study. The results showed that SMPD1 p.L302P (L304P, c.T911C, NM_000543) was strongly associated with a highly increased risk for PD (odds ratio = 9.4, 95% confidence interval = [3.9-22.8], P<0.0001), as 9/938 patients with PD were carriers of this mutation compared to only 11/10,709 controls. This indicated that SMPD1 p.L302P is a novel risk factor for PD, just as GBA mutations, with small Minor Allele Frequency (MAF) but large odd ratios [6].
To investigate whether the SMPD1 p.L302P is a susceptible factor for sporadic PD in Chinese population, we performed an association study in ethnic Chinese PD patients and healthy controls.
Materials and methods
Patients and controls
The present study comprised 455 ethnic Chinese patients with PD and 476 healthy controls matched for age, gender, ethnicity and area of residence. All patients were consecutively recruited from the Department of Neurology of Xiangya Hospital (Central South University, State Key Laboratory of Medical Genetics of China) and the Neurodegenerative Disorders Research Center. A standard clinical neurological examination was performed for all patients and a diagnosis of idiopathic PD was made according to the United Kingdom PD Brain Bank Criteria [7]. None of the 455 patients had positive family history or carrying GBA mutations [8]. There are 235 males and 220 females patients. The average age at onset was 55.45 ± 10.38 years (range, 14-75 years). 476 healthy controls that are matched for age, gender, ethnicity and area of residence included 238 males and 238 females; average age at examination was 58.38 ± 12.09 years (range, 15-85 years). The control subjects were healthy people from the Xiangya Hospital Medical Center and healthy volunteers with no history of neurodegenerative diseases. A standard clinical neurological examination was performed on all control subjects and a diagnosis of possible idiopathic PD was excluded according to the United Kingdom PD Brain Bank Criteria. There was no statistically significant difference in the age and gender between patients and controls (P>0.05, using Chi-square test). This study protocol was approved by the Ethics Committee of Central South University and written informed consent was obtained from all subjects.
Genotype analysis
DNA was extracted from peripheral blood using standard techniques. And c.T911C (p.L302P) was genotyped by matrix-assisted laser desorption/ionization-time-of-flight mass spectrometry (MALDI-TOF MS) using the Mass Array system (Sequenom) as described [9]. For quality control, the positive and negative controls (with no DNA) were included on every 96-well assay plate. Briefly, locus-specific polymerase chain reaction and detection primers were designed using the MassArray Assay Design 3.0 software (Sequenom). Approximately 10 ng of genomic DNA was used to genotype each sample. The sample DNAs were amplified by primers flanking the targeted sequence, followed by dephosphorylation and allele-specific primer extension. Cleaned extension products were loaded into a 384-format Spectro-Chip and subjected to MALDI-TOF mass spectrometry. Finally, the resultant data were processed and analyzed by the Sequenom Mass Array Typer software (Sequenom). In order to confirm the results, 50 patients and 50 controls were randomly selected for Sanger sequencing.
Statistical analysis
The average age at onset of PD patients and average age of controls at examination were analyzed with SPSS 17.0 software (SPSS Inc., Chicago, IL, USA). The distribution difference of Male/Female in case and control groups was tested using the Chi-square test. A two-tailed test P-value of <0.05 was considered statistically significant.
Results
Our study included 455 sporadic PD and 476 health controls in Chinese population, and we used MALDI-TOF MS to determine the p.L302P (c.T911C) genotype which was also confirmed by Sanger sequencing, but none of them carried p.L302P. All of the 931 subjects’ genotypes were wild type TT.
Discussion
The etiology of PD is still elusive. However, there are growing evidences that genetics play an important role in the pathogenesis of PD. Recently, a lot of susceptible genes that have been reported by GWAS or candidate gene approach contributed to a high risk of getting PD [10,11]. These different susceptible genes could be classified into different pathways that could damage cell normal metabolisms. The cellular autophagy-lysosomal pathway (ALP) is one of the pathways that play an essential role in PD pathogenesis [12]. Excessive accumulation of aberrant proteins in the dopaminergic neurons cytoplasm may be caused by dysfunction of normal ALP pathway, which could ultimately results in cell death and the clinical manifestations of PD. And lysosomal enzymes including proteins encoded by GBA and SMPD1 play an important role in maintaining the ALP pathway normal functions [12,13]. Thus, when we are exploring the mechanisms of PD, we could easily pay attention to ALP pathway, and also the lysosomal enzymes including proteins encoded by GBA and SMPD1 could easily draw our attention.
p.L302P located in SMPD1 was identified by Gan-Or et al [5] study in a cohort of Ashkenazi Jewish PD patients. With the results that p.L302P multiplies by 9 the risk to develop PD, which provided additional evidences to the viewpoint that ALP pathway involves in PD pathogenesis. However, in our study, we did not replicate this variant’s enrichment in our 455 PD subjects. This is consistent to Wu et al [14] results (Table 1; Figure 1), which found no p.L302P in 1,139 participants (579 PD and 560 control subjects) in Chinese Taiwan population. Although p.L302P is rare, and p.L302P was found only in 9 of 938 patients with PD in Ashkenazi Jewish ancestry population, we could expect there should be about 4-5 patients carry this variant in our PD group if it has the similar role in increasing the risk of PD. But we did not detect the postulated enrichment. A larger sample size study will be of great interest. But, the negative results also suggested possible ethnic-specific effect of this variant, just like that MAPT H1H2 could contribute to substantial risk of PD in Caucasia, but little in Asia, and vice versa when considering LRRK2 G2385R and R1628P [15,16]. Recently, Foo et al [17] did not detect p.L302P in 198 Chinese populations individuals (98 PD, and 98 healthy controls), either (Table 1; Figure 1); however, R591C another rare variant locating in SMPD1 was identified to increase the risk of PD (P=0.009) in their study. This may also prove the ethnic-specific effect of the gene variants, and sequencing of SMPD1 gene to find additional novel rare variants in the SMDP1 gene is needed.
Table 1.
Summary of published literature on associations between variants locating in SMPD1 and PD
| Variants | Population | Allele frequency in PD | Allele frequency in controls | P | OR (95% CI) | Ref |
|---|---|---|---|---|---|---|
| p.L302P | Ashkenazi Jewish | 9/938 | 11/10,709 | P<0.0001 | 9.4 (3.9-22.8) | [5] |
| Taiwanese | 0/579 | 0/560 | - | - | [13] | |
| Singaporean and Mainland Chinese | 0/99 | 0/99 | - | - | [16] | |
| Mainland Chinese | 0/455 | 0/476 | - | - | Our study | |
| p.R591C | Singaporean and Mainland Chinese | 2/806 | 0/7,481 | P=0.009 | - | [16] |
| p.P533L, | Singaporean and Mainland Chinese | 17/806 | 90/7,481 | P=0.047 | 1.76 (1.05-2.97) | [16] |
| p.P332R | Singaporean and Mainland Chinese | 10/806 | 134/7,481 | P=0.322 | 0.69 (0.36-1.32) | [16] |
| p.Y500H | Singaporean and Mainland Chinese | 1/806 | 5/7,481 | P=0.459 | 1.86 (0.22-15.9) | [16] |
| p.R496L | Ashkenazi Jewish | 4/938 | 52/10,709 | P=1.0 | - | [5] |
| c.996delC | Ashkenazi Jewish | 5/938 | 29/10,709 | P=0.3 | - | [5] |
| p.G508R | Singaporean and Mainland Chinese | 232/806 | 1,944/7,481 | P=0.120 | 1.13 (0.97-1.31) | [16] |
OR: odds ratios; CI: confidence interval; PD: Parkinson’s disease.
Figure 1.
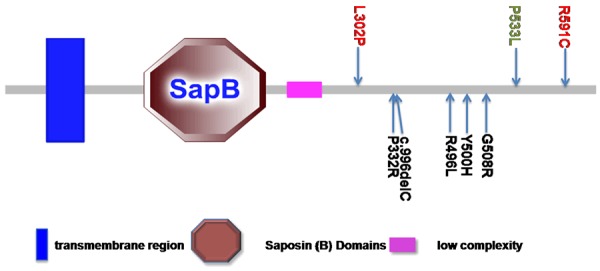
SMPD1 protein and the location of PD associated variants. Variants in red are identified with association to PD, variants in green are identified with potential association to PD, and variants in black are identified with no associations to PD. It is worthy of note that larger sample size of replication is needed to draw the definite conclusions. (http://smart.embl-heidelberg.de).
Conclusions
To our knowledge, this is the first study to assess the association of SMPD1 p.L302P variants in a cohort of ethnic Chinese PD patients and controls in Mainland China. Our data indicated that in an ethnic Chinese population, p.L302P did not appear to be enriched in sporadic PD, and p.L302P may not be a risk factor for Chinese sporadic PD. And combine our data with the results from Wu and Foo et al [14,17], we found that all of the 2,268 participants of Chinese population carrying no p.L302P. We could make a conclusion that p.L302P may not be common events for Chinese population. Sequencing of SMPD1 gene to find additional novel rare variants in the SMDP1 gene in diverse populations is needed.
Acknowledgements
We would like to thank all participants in this study. This work was supported by grant 2011CB510001 from the Major State Basic Research Development Program of China (973 Program) (to Drs. Bei-sha Tang), grants 81361120404, 81430023, 81130021, 81171198, 81371405 and 81571248 from the National Natural Science Foundation of China (to Drs. Bei-sha Tang and Ji-feng Guo et al), and grant 2014zzts077 from the Fundamental Research Funds for the Central Universities of Central South University (to Dr. Kai Li).
Disclosure of conflict of interest
None.
References
- 1.Dorsey ER, Constantinescu R, Thompson JP, Biglan KM, Holloway RG, Kieburtz K, Marshall FJ, Ravina BM, Schifitto G, Siderowf A, Tanner CM. Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology. 2007;68:384–386. doi: 10.1212/01.wnl.0000247740.47667.03. [DOI] [PubMed] [Google Scholar]
- 2.Spatola M, Wider C. Genetics of Parkinson’s disease: the yield. Parkinsonism Relat Disord. 2014;20(Suppl 1):S35–S38. doi: 10.1016/S1353-8020(13)70011-7. [DOI] [PubMed] [Google Scholar]
- 3.Bonifati V. Genetics of Parkinson’s disease--state of the art, 2013. Parkinsonism Relat Disord. 2014;20(Suppl 1):S23–S28. doi: 10.1016/S1353-8020(13)70009-9. [DOI] [PubMed] [Google Scholar]
- 4.Puschmann A. Monogenic Parkinson’s disease and parkinsonism: clinical phenotypes and frequencies of known mutations. Parkinsonism Relat Disord. 2013;19:407–415. doi: 10.1016/j.parkreldis.2013.01.020. [DOI] [PubMed] [Google Scholar]
- 5.Gan-Or Z, Ozelius LJ, Bar-Shira A, Saunders-Pullman R, Mirelman A, Kornreich R, Gana-Weisz M, Raymond D, Rozenkrantz L, Deik A, Gurevich T, Gross SJ, Schreiber-Agus N, Giladi N, Bressman SB, Orr-Urtreger A. The p. L302P mutation in the lysosomal enzyme gene SMPD1 is a risk factor for Parkinson disease. Neurology. 2013;80:1606–1610. doi: 10.1212/WNL.0b013e31828f180e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, Bar-Shira A, Berg D, Bras J, Brice A, Chen CM, Clark LN, Condroyer C, De Marco EV, Durr A, Eblan MJ, Fahn S, Farrer MJ, Fung HC, Gan-Or Z, Gasser T, Gershoni-Baruch R, Giladi N, Griffith A, Gurevich T, Januario C, Kropp P, Lang AE, Lee-Chen GJ, Lesage S, Marder K, Mata IF, Mirelman A, Mitsui J, Mizuta I, Nicoletti G, Oliveira C, Ottman R, Orr-Urtreger A, Pereira LV, Quattrone A, Rogaeva E, Rolfs A, Rosenbaum H, Rozenberg R, Samii A, Samaddar T, Schulte C, Sharma M, Singleton A, Spitz M, Tan EK, Tayebi N, Toda T, Troiano AR, Tsuji S, Wittstock M, Wolfsberg TG, Wu YR, Zabetian CP, Zhao Y, Ziegler SG. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med. 2009;361:1651–1661. doi: 10.1056/NEJMoa0901281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry. 1992;55:181–184. doi: 10.1136/jnnp.55.3.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun QY, Guo JF, Wang L, Yu RH, Zuo X, Yao LY, Pan Q, Xia K, Tang BS. Glucocerebrosidase gene L444P mutation is a risk factor for Parkinson’s disease in Chinese population. Mov Disord. 2010;25:1005–1011. doi: 10.1002/mds.23009. [DOI] [PubMed] [Google Scholar]
- 9.Buetow KH, Edmonson M, MacDonald R, Clifford R, Yip P, Kelley J, Little DP, Strausberg R, Koester H, Cantor CR, Braun A. High-throughput development and characterization of a genomewide collection of gene-based single nucleotide polymorphism markers by chip-based matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Proc Natl Acad Sci U S A. 2001;98:581–584. doi: 10.1073/pnas.021506298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lill CM, Roehr JT, McQueen MB, Kavvoura FK, Bagade S, Schjeide BM, Schjeide LM, Meissner E, Zauft U, Allen NC, Liu T, Schilling M, Anderson KJ, Beecham G, Berg D, Biernacka JM, Brice A, DeStefano AL, Do CB, Eriksson N, Factor SA, Farrer MJ, Foroud T, Gasser T, Hamza T, Hardy JA, Heutink P, Hill-Burns EM, Klein C, Latourelle JC, Maraganore DM, Martin ER, Martinez M, Myers RH, Nalls MA, Pankratz N, Payami H, Satake W, Scott WK, Sharma M, Singleton AB, Stefansson K, Toda T, Tung JY, Vance J, Wood NW, Zabetian CP, Young P, Tanzi RE, Khoury MJ, Zipp F, Lehrach H, Ioannidis JP, Bertram L. Comprehensive research synopsis and systematic meta-analyses in Parkinson’s disease genetics: The PDGene database. PLoS Genet. 2012;8:e1002548. doi: 10.1371/journal.pgen.1002548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nalls MA, Plagnol V, Hernandez DG, Sharma M, Sheerin UM, Saad M, Simon-Sanchez J, Schulte C, Lesage S, Sveinbjornsdottir S, Stefansson K, Martinez M, Hardy J, Heutink P, Brice A, Gasser T, Singleton AB, Wood NW. Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet. 2011;377:641–649. doi: 10.1016/S0140-6736(10)62345-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharma N. Lysosomal enzyme defects and Parkinson disease. Neurology. 2013;80:1544–1545. doi: 10.1212/WNL.0b013e31828f1958. [DOI] [PubMed] [Google Scholar]
- 13.Trinh J, Farrer M. Advances in the genetics of Parkinson disease. Nat Rev Neurol. 2013;9:445–454. doi: 10.1038/nrneurol.2013.132. [DOI] [PubMed] [Google Scholar]
- 14.Wu RM, Lin CH, Lin HI. The p. L302P mutation in the lysosomal enzyme gene SMPD1 is a risk factor for Parkinson disease. Neurology. 2014;82:283. doi: 10.1212/WNL.0000000000000004. [DOI] [PubMed] [Google Scholar]
- 15.Wang C, Cai Y, Zheng Z, Tang BS, Xu Y, Wang T, Ma J, Chen SD, Langston JW, Tanner CM, Chan P. Penetrance of LRRK2 G2385R and R1628P is modified by common PD-associated genetic variants. Parkinsonism Relat Disord. 2012;18:958–963. doi: 10.1016/j.parkreldis.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 16.Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, Kawaguchi T, Tsunoda T, Watanabe M, Takeda A, Tomiyama H, Nakashima K, Hasegawa K, Obata F, Yoshikawa T, Kawakami H, Sakoda S, Yamamoto M, Hattori N, Murata M, Nakamura Y, Toda T. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet. 2009;41:1303–1307. doi: 10.1038/ng.485. [DOI] [PubMed] [Google Scholar]
- 17.Foo JN, Liany H, Bei JX, Yu XQ, Liu J, Au WL, Prakash KM, Tan LC, Tan EK. Rare lysosomal enzyme gene SMPD1 variant (p. R591C) associates with Parkinson’s disease. Neurobiol Aging. 2013;34:2813–2890. doi: 10.1016/j.neurobiolaging.2013.06.010. [DOI] [PubMed] [Google Scholar]