

Abstract
Large tumor suppressor 1 and 2 (Lats1/2) regulate centrosomal integrity, chromosome segregation and cytokinesis. As components of the centralspindlin complex, the kinesin-like protein CHO1 and its splicing variant MKLP1 colocalize with chromosome passenger proteins and GTPases and regulate the formation of the contractile ring and cytokinesis; however, the regulatory mechanisms of CHO1/MKLP1 remain elusive. Here, we show that Lats1/2 phosphorylate Ser716 in the F-actin-interacting region of CHO1, which is absent in MKLP1. Phosphorylated CHO1 localized to the centrosomes and midbody, and the actin polymerization factor LIM-kinase 1 (LIMK1) was identified as its binding partner. Overexpression of constitutively phosphorylated and non-phosphorylated CHO1 altered the mitotic localization and activation of LIMK1 at the centrosomes in HeLa cells, leading to the inhibition of cytokinesis through excessive phosphorylation of Cofilin and mislocalization of Ect2. These results suggest that Lats1/2 stringently control cytokinesis by regulating CHO1 phosphorylation and the mitotic activation of LIMK1 on centrosomes.
Keywords: centrosome, CHO1, cytokinesis, Lats1, LIMK1, phosphorylation
Introduction
Cytokinesis, the process by which a single cell divides into 2 daughter cells following mitosis, occurs in 4 ordered steps, as follows: positioning of the division plane by the central spindle, cleavage furrow ingression, midbody formation, and abscission.1,2 Dysregulation of these steps leads to centrosome amplification and the generation of multinucleated cells, including tetraploid cells, which can develop into malignant tumors.
The centralspindlin complex comprising the kinesin-like motor protein KIF23 (also known as Kinesin-6 or CHO1/MKLP1 in mammals) and the Rho family GTPase-activating protein Cyk4 (also known as MgcRacGAP) is essential for the formation and maintenance of the central spindle and midbody during late mitosis (For a review see ref. 3). KIF23 proteins are conserved and regulate cytokinesis in various species.4,5 MKLP1 is an alternatively spliced form of CHO16,7 that lacks the filamentous actin (F-actin)-binding region (FABR) encoded by exon 18.8 Binding of CHO1 to F-actin is required for the terminal stage of cytokinesis but not microtubule bundling or midbody formation.9 CHO1/MKLP1 use plus end-directed motor activity to transport Cyk4 and Ect2, a guanine nucleotide exchange factor, to the equatorial cortex for cleavage furrow ingression, thereby inducing contractile ring formation.10,11 Inhibition of MKLP1 or CHO1 causes severe defects in central spindle organization, midbody formation and cytokinesis completion; 8,12,19 however, the unique functions of the CHO1 FABR are poorly understood.
During formation of the contractile ring, Ect2 and guanine nucleotide exchange factor H1 promote actin polymerization by regulating the Rho-GTP cycle (For a review see ref. 1). Dual specificity LIM-kinase 1 (LIMK1) inhibits the actin depolymerization factor Cofilin via phosphorylation at S3; in the terminal step of cytokinesis, Cofilin is dephosphorylated and activated by Slingshot, and localizes at the midbody until the completion of abscission.13,14 Overexpression of LIMK1 induces the formation of multinucleated cells because Slingshot is unable to dephosphorylate excessive levels of phosphorylated Cofilin.13,15 LIMK1 is also activated during early mitosis and regulates precise spindle positioning.14,15
In vertebrate somatic cells, disruption of the centrosome leads to cytokinesis failure caused by a loss of precise spindle positioning, and the acentrosomal daughter cells are arrested at the next G1 phase after mitosis.16 Moreover, the mother centriole is transiently repositioned to the midbody, leading to the completion of cell division.17 Cep55 is dissociated from the centrosome by Erk2/Cdk1-dependent phosphorylation and subsequently locates to the midbody, thereby regulating mitotic exit and cytokinesis.18 Despite these findings, the roles of centrosomes in cytokinesis are not fully understood.
Large tumor suppressors 1 and 2 (Lats1/2) are Ser/Thr kinases in the Hippo-signaling pathway that control organ size and tumor formation by negatively regulating Yap and Taz,19 and prevent tetraploidization by activating the p53 tumor suppressor.20 The mitotic kinase Aurora-A phosphorylates Lats2 at centrosomes (and spindle poles) and in the nucleus of HeLa-S3 cells, resulting in its re-localization to the central spindle, where it interacts with Lats1. In turn, Lats1 activates Aurora-B to prevent chromosome mis-segregation and cytokinesis failure. 21,22 Lats2 is essential for maturation of the centrosomes, formation of the mitotic spindle, and the completion of cytokinesis in mouse embryo fibroblasts (MEFs).23 Moreover, Lats1 localizes to the mitotic apparatus and regulates mitotic progression and cytokinesis.24,25,26 Lats1 inhibits the kinase activity of LIMK126 and a recent study showed that extra centrosomes caused by cytokinesis failure activate the Hippo pathway.27 These results suggest that Lats1/2 coordinately regulate centrosomal integrity, chromosome segregation and cytokinesis during mitosis; however, it remains unclear how they coordinate centrosomal integrity to ensure successful cytokinesis.
Here, we show that phosphorylation of CHO1 at S716 by Lats1 and Lats2 regulate its centrosomal localization, and that phosphorylated CHO1 interacts with and activates LIMK1 during early mitosis. Constitutive overexpression of phosphomimetic and non-phosphorylated mutants of CHO1 caused cytokinesis failure by excessive phosphorylation of Cofilin and mislocalization of Ect2, respectively, suggesting that the Lats1/2-CHO1-LIMK1 axis on the centrosome controls cytokinesis fidelity.
Results
Lats1 and Lats2 phosphorylate the CHO1 FABR during mitosis
CHO1 harbors a putative Lats1/2-binding motif (H-x-R-x-x-pS/pT; where H = His, R = Arg, pS/pT = phosphorylated Ser or Thr, and x = arbitrary residue) in its FABR (Fig. 1A), whereas MKLP1 lacks this region.8 The NCBI protein database includes sequences for human and mouse CHO1 (NP_612565 and NP_077207, respectively), as well as human MKLP1 (NP_004847), but not mouse MKLP1. However, an RT-PCR analysis identified MKLP1 expression in MEFs (Fig. S1A). Furthermore, a DNA sequence analysis revealed a high degree of homology between mouse and human CHO1 (identity of full length, 88.8%; similarity of full length, 94.5%; identity of exon 18, 82.7%; and similarity of exon 18, 90.4%). To investigate the functions of CHO1 and MKLP1, we examined whether S717 in the consensus Lats1/2-binding motif of mouse CHO1 (MmCHO1) is phosphorylated by Lats1/2. Kinase assays using GST-fused wild-type (WT) and a non-phosphorylatable S717A mutant of MmCHO1692-796 revealed that Lats1 and Lats2 predominantly phosphorylated S717 (S716 in human CHO1) in vitro (Fig. 1B). To confirm that S716 of human CHO1 is phosphorylated by Lats1 and Lats2, we generated a phospho-specific antibody against this residue (anti-pS716S717) (Fig. S1B). The expression level of 6Myc-tagged CHO1-pS716S717 was higher in HeLa-S3 cells treated with the microtubule depolymerizer nocodazole than non-treated asynchronous cells (Fig. S1C), suggesting that phosphorylation of this residue is enhanced during mitosis. Since MKLP1 lacks the S716 (S717 in mouse) residue of CHO1, anti-pS716S717 did not recognize exogenous 6Myc-tagged MKLP1 (Fig. 1C).
Figure 1.

(See previous page). Large tumor suppressors (Lats)1/2 phosphorylate CHO1-S716S717 during mitosis. (A) The primary structures of human and mouse CHO1 and human MKLP1. CC, coiled-coil domain. The Lats1/2 consensus sequences and phosphorylation sites are underlined and bold, respectively. (B) In vitro assays performed in the presence of [γ-32P] ATP using vector alone (control) or immunoprecipitated wild-type (WT) or kinase dead (KD) 6Myc-tagged Lats1 or Lats2, along with the expression of 3Flag-tagged Mob1A as a Lats1/2 activator. GST-fused truncated fragments (amino acids 692–796) of WT and S717A (SA) mutated MmCHO1-WT were used as substrates. (C) Immunoblot analyses of HeLa-S3 cells transfected with 6Myc-tagged vector alone, full-length MKLP1, or WT or S717A (SA) mutant MmCHO1. The cells were synchronized at the M phase by treatment with nocodazole. Asterisks indicate non-specific bands. (D) Immunoblot analyses of HeLa-S3 cells treated with mimosine (Mim), thymidine (Thy), taxol (Tax), nocodazole (Noc), or a thymidine single block-and-release (Thy+R10h). Control cells were asynchronous (Asy). The arrow indicates endogenous CHO1-pS716S717. For peptide competition assays, the anti-pS716S717 antibody was pre-incubated with phosphorylated (lanes 7–12) or non-phosphorylated (lanes 13–18) CHO1 antigen peptides. Asterisks indicate non-specific bands. Mcm2 is a marker of the G1 and S phases, whereas Aurora-A is a marker of the M phase. (E) Protein phosphatase (PPase) assay showing immunoblot analyses of endogenous CHO1-pS716S717 in HeLa-S3 cells synchronized with Tax, Noc, or Thy+R10h. The cell extracts were treated with or without 200 U of λPPase and PPase inhibitors. (F) Immunoblot analyses of HeLa-S3 cells transfected with siRNAs against Lats1 (#3509) or Lats2 (#581), and synchronized at mitosis by a thymidine single block-and-release (10 h). A GL2 (siRNA against firefly luciferase) was used as a negative control. The ratios of the band intensities of CHO1-pS716S717 to that in GL2 cells are shown.
The level of endogenous CHO1-pS716S717 was markedly higher in mitotic HeLa-S3 cells treated with taxol (a microtubule stabilizer), nocodazole, or a thymidine single block-and-release, than those in asynchronous cells or cells treated with mimosine or thymidine without release (Fig. 1D, lanes 1–6). The intensities of the CHO1-pS716S717 bands were decreased by pre-incubation of the antibody with its target phosphorylated peptide, but not non-phosphorylated peptide (Fig. 1D, lanes 7–18). The addition of lambda protein phosphatase to extracts of cells treated with taxol, nocodazole or a thymidine single block-and-release abolished the bands detected by anti-pS716S717, and this effect was prevented by the concomitant addition of phosphatase inhibitors (Fig. 1E). These results indicate that phosphorylation of CHO1-S716S717 occurs during both normal mitotic progression and after activation of the spindle assembly checkpoint.
In HeLa-S3 cells synchronized at mitosis by a thymidine single block-and-release, knockdown of Lats1, Lats2 or CHO1 using small interfering RNAs (siRNAs) reduced the level of CHO1-pS716S717, suggesting that Lats1 and Lats2 phosphorylate CHO1 during mitotic progression (Figs. 1F and S1D).
CHO1-pS716S717 localizes to the centrosome during mitosis
CHO1 localizes to the central spindle during late metaphase and is concentrated at the midbody during cytokinesis.12 In synchronized HeLa-S3 cells, CHO1-pS716S717 localized to the centrosomes and nucleus during interphase, and the signals became stronger during prophase. During metaphase and anaphase, CHO1-pS716S717 is mostly localized to the centrosomes (Figs. 2A, i–vi and S1E), which is distinct from the well-characterized mitotic localizations of CHO1 and MKLP1. Immunostaining with an antibody against a different region of the FABR of CHO1 showed a similar localization pattern (Fig. 2B). In a previous study, ectopically overexpressed CHO1 localized to the central spindle during anaphase,12 suggesting that the antibodies used here were unable to recognize endogenous CHO1 on the central spindle, which is present at this region at considerably lower levels than MKLP1. Both phospho-and non-phospho-S716S717 signals were identified at the midbody (Flemming body) during cytokinesis (Fig. 2A and B). The centrosomal localization of CHO1-pS716S717 in HeLa-S3 cells, confirmed by co-immunostaining of γ-tubulin (Fig. 2C), was decreased by disruption of LATS1 or LATS2 genes by programmable nucleases (Figs. 2D and S5A). A similar effect was observed following knockdown of CHO1/MKLP1 (Fig. 2E) and in a competition assay using phosphorylated S716S717 peptide (Fig. 2C), suggesting that Lats1/2 are responsible for the centrosomal phosphorylation of CHO1-S716S717. The CHO1-pS716S717 signals also colocalized slightly with phalloidin-staining at the centrosomes in mitotic HeLa-S3 cells (Fig. S1E).
Figure 2.
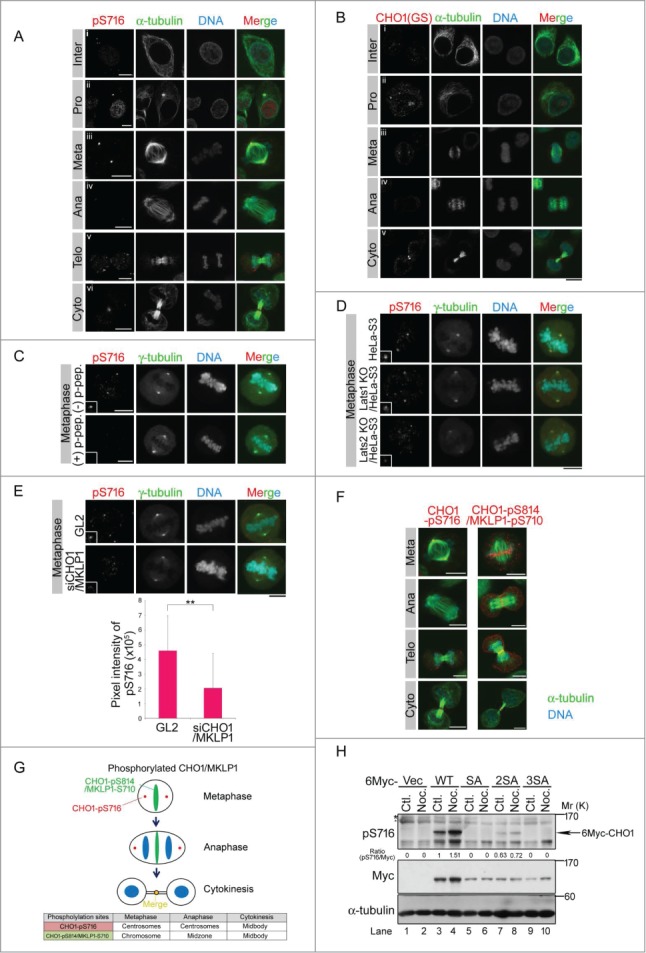
CHO1-pS716S717 localizes to centrosomes during mitosis. (A, B) Subcellular localizations of CHO1-pS716S717 (A) and CHO1 (B) in synchronized HeLa-S3 cells. Anti-CHO1[GS] is CHO1-specific antibody that recognizes the F-actin binding regions (FABR). (C) Peptide competition assay in which the anti-pS716S717 antibody was pre-incubated with phosphorylated (+) or non-phosphorylated (−) CHO1-S716S717 peptides prior to immunostaining. (D) Subcellular localization of CHO1-pS716S717 in metaphase large tumor suppressor (Lats)1 KO/HeLa-S3, Lats2 KO/HeLa-S3, and parental HeLa-S3 cells synchronized at mitosis by a thymidine single block-and-release (10 h). (E) Subcellular localization of metaphase CHO1-pS716S717 in HeLa-S3 cells that were transfected with siRNAs against the common 3′-UTR of CHO1 and MKLP1 and synchronized at mitosis by a thymidine single block-and-release (10 h). The GL2 siRNA was used as a negative control. The graph shows the pixel intensity of pS716 signals at the centrosome. Data represent the mean ± SD of n = 3 experiments. (F) Localizations of the pS716S717 and pS814S807 versions of CHO1 and/or MKLP1 in HeLa-S3 cells undergoing mitosis. A–F. Scale bar, 10 μm. (G) Overview of the subcellular localizations of CHO1-pS716S717 and CHO1-pS814S807/MKLP1-pS710 during the cell cycle. (H) Immunoblot analyses of exogenous CHO1-pS716S717 in HeLa-S3 cells that were transfected with 6Myc-tagged full-length MmCHO1-WT (wild type),-SA (S717A),-2SA (S805A, S807A) or-3SA (S717A, S805A, S807A), and treated with (Noc.) or without (Ctl.) nocodazole (80 ng/ml) for 18h.
Phosphorylation of MKLP1-S710 by an unidentified kinase generates a binding site for the 14-3-3 protein, which inhibits centralspindlin clustering, whereas phosphorylation of S708 by Aurora-B kinase during the anaphase to telophase transition inhibits the binding of 14-3-3 to MKLP1 and promotes centralspindlin clustering.28,29 S708 and S710 of human MKLP1 correspond to S812 and S814 of human CHO1 (S805 and S807 in mouse), respectively. We predicted that CHO1-S814S807 and MKLP1-S710 are another Lats1/2 phosphorylation site in CHO1 and MKLP1, because the peripheral sequence of CHO1-S814S807 and MKLP1-pS710 includes a putative Lats1/2 phosphorylation site. To characterize the distribution in HeLa-S3 cells, we raised a phospho-specific antibody against human CHO1-pS814. Examination of the anti-CHO1-pS814S807 antibody showed that it recognized not only CHO1-pS814/MKLP1-pS710 but also CHO1-pS812/MKLP1-pS708 (Fig. S2A) faintly and the corresponding phosphorylated sites of mouse CHO1 (Fig. S2B). Consistent with a previous study,30 CHO1-pS814/MKLP1-pS710 was also phosphorylated by Aurora-B but not Aurora-A in vitro (Fig. S2B). However, phosphorylation of endogenous CHO1-pS814/MKLP1-pS710 in nocodazole-treated mitotic cells was not inhibited by treatment with the Aurora-B inhibitor hesperadin (Fig. S2C), suggesting that Aurora-B is not solely responsible for phosphorylation of these residues in vivo. Indeed, CHO1-pS814/MKLP1-pS710 was phosphorylated by Lats2 but not Lats1 in vitro (Fig. S2B). Taken together, these results suggest that phosphorylation of CHO1-S716S717 and -S814S807 are regulated by Lats1 and Lats2, respectively.
CHO1-pS814 and MKLP1-pS710 localized to the aligned chromosomes during metaphase, moved to the central spindle, and then concentrated at the Flemming body during cytokinesis, which was similar to the S708/S710-diphosphorylated MKLP1 in a previous report28 (Figs. 2F and S3A). The mitotic CHO1-pS814/MKLP1-pS710 signal was decreased markedly following knockout of Lats2 in HeLa-S3 cells (Fig. S3B). The mitotic signals of CHO1-pS814/MKLP1-pS710 disappeared by peptide competition assay (Fig. S3C). The mitotic localization patterns of the phosphorylated CHO1 proteins are summarized in Figure 2G. A competition assay using non-phosphorylated target peptides specifically recognized CHO1-pS814/MKLP1-pS710 bands (Fig. S2D). Knockdown of CHO1/MKLP1 using small siRNAs reduced the level of CHO1-pS814/MKLP1-pS710 (Fig. S2E). Although CHO1-S814S807 was phosphorylated during mitosis, the corresponding S710 residue of MKLP1 remained phosphorylated throughout the cell cycle (Fig. S2D), even after inhibition of Aurora-B (Fig. S2C), which is consistent with a previous report.28 These results suggest that, during mitotic progression and/or exit, CHO1-pS716S717 plays a role that is distinct from CHO1-pS814S807 and MKLP1 -pS710 at spatially independent areas.
To examine the relationship between CHO1-pS716S717 and -pS812S805 and -pS814S807, HeLa-S3 cells were transfected with 3 different non-phosphorylated MmCHO1 mutants in which the residues corresponding to S716, S812 and S814 of human CHO1 were replaced with alanine as follows: SA (S717A), 2SA (S805A and S807A) and 3SA (S717A, S805A, and S807A). In nocodazole-treated transfected cells, S716S717 was phosphorylated in CHO1-WT but not CHO1-SA. Notably, the signal of S716S717 was diminished in CHO1-2SA and -3SA, suggesting that phosphorylation of this residue is supported by phosphorylation of S812S805 and S814S807 (Fig. 2H). In kinase assays using Aurora-A or Aurora-B, the levels of phosphorylated CHO1-S716S717 were similar in cells expressing CHO1-WT and CHO1-SA, suggesting that neither kinase could phosphorylate CHO1-S716S717 directly (Fig. S2F). It is likely that phosphorylation of CHO1-S716S717 is regulated mainly by Lats1 and Lats2, which may be supported by phosphorylation of S812S805 and S814S807.
CHO1 interacts with LIMK1 in a pS716S717-dependent manner
To identify interacting partners of CHO1, lysates of HeLa-S3 cells expressing 6Myc-tagged full-length CHO1-WT were immunoprecipitated with an anti-Myc antibody and analyzed by mass spectrometry. Among the candidates identified, we focused on LIMK1, which was derived from the band that displayed the highest specificity of binding to CHO1 but not MKLP1 (Fig. S3D).
To determine whether phosphorylation of CHO1-S716S717 is required for the interaction between CHO1 and LIMK1, we co-expressed 3Flag-tagged full-length LIMK1-WT with 6Myc-tagged full-length MKLP1 or CHO1-WT, -SA (non-phosphorylatable), -SD (phosphomimetic), -2SA, or -3SA in HeLa-S3 cells (Fig. 3A). An immunoprecipitation (IP) assay confirmed that CHO1-WT and CHO1-SD bound to LIMK1, but CHO1-SA did not (Fig. 3B), suggesting that the CHO1-LIMK1 interaction depends on S716S717 phosphorylation. Since CHO1-S716S717 was not phosphorylated in CHO1-2SA and -3SA (Fig. 2H), the interaction of LIMK1 with these proteins, as well as MKLP1, was limited (Fig. 3B). These results suggest that phosphorylation of CHO1-S716S717 is important for binding to LIMK1.
Figure 3.

CHO1-pS716S717 interacts with LIM-kinase 1 (LIMK1). (A) Schematic illustrations of phosphorylated wild-type (WT) and mutant CHO1 (SA, SD, 2SA, 3SA). (B) Immunoblot analyses showing the requirement for pS716S717 for the physical interaction of CHO1 with LIMK1. HeLa-S3 cells were co-transfected with 3Flag-LIMK1 and a 6Myc-tagged MKLP1 or full-length MmCHO1-WT, -SA, -SD, -2SA, or -3SA. (C) Overview of the CHO1 and LIMK1 deletion mutants and their interactions. (D) Immunoblot analyses of lysates of HeLa-S3 cells co-expressing 3Flag-LIMK1 and 6Myc-full-length MmCHO1 or the indicated deletion mutants, showing the physical interaction of full-length LIMK1 with the stalk domain of CHO1. (E) Immunoblot analyses of lysates of HeLa-S3 cells co-expressing 3Flag-CHO1 and 6Myc-tagged full-length LIMK1 or the indicated deletion mutants, showing the physical interaction of full-length CHO1 with the LIM domain of LIMK1.
CHO1 has 3 domains, as follows (Fig. 3C): the motor domain possesses a kinesin-like motor activity that is directed toward the plus end of microtubules; the stalk domain is essential for dimerization of CHO1 and its interaction with Cyk4 and RhoGAP, which regulates proper localizations of these proteins at the midzone and the microtubule-bundling activity to disassemble the contractile ring; and the tail domain contains 2 protein-binding domains (a FABR and an Arf6-binding domain) and a nuclear localization signal, which contribute to midbody matrix stabilization and nuclear localization, respectively.9,31 To determine which domain interacts with LIMK1, HeLa-S3 cells were co-transfected with 3Flag-tagged full-length LIMK1-WT and 6Myc-tagged full-length or individual domains of CHO1. IP and immunoblotting analyses revealed that LIMK1 interacted with the stalk domain and full-length CHO1, but not the motor and tail domains (Fig. 3D).
LIMK1 comprises LIM domains, a PDZ domain and a kinase domain (Fig. 3C). The LIM domain interacted with full-length CHO1, although an interaction between full-length CHO1 and full-length LIMK1 was not observed (Fig. 3E).
Taken together, these results suggest that the stalk domain of CHO1 physically associates with the LIM domain of LIMK1, and the interaction is dependent on phosphorylation of CHO1-S716S717, -S812S805 and -S814S807.
Lats1/2 and CHO1 regulate the centrosomal localization and activity of LIMK during mitosis
A previous study revealed that LIMK1 phosphorylated at T508 (pT508), which is an activating phosphorylation site phosphorylated by Rho family small GTPases, and their downstream kinases, such as PAK and ROCK, colocalizes with γ-tubulin at centrosomes during prophase to early telophase, and then translocates partially to the cleavage furrow during late mitosis and cytokinesis.15,32 To determine whether the subcellular localization of LIMK1 correlates with the interaction of CHO1, we examined the effect of knockdown of CHO1 on the localization of LIMK1-pT508 to centrosomes. Unlike that of γ-tubulin, the signal intensity of LIMK1-pT508 was lower in CHO1 knockdown cells than control cells (Fig. 4A–C). The signal intensities of both LIMK1-pT508 and γ-tubulin were decreased following knockdown of Lats1 and/or Lats2 (Fig. 4D–F), suggesting that Lats1/2 affect the regulation of centrosomal LIMK1. Compared with that in control cells, the level of phosphorylated Cofilin (pS3) was also lower in CHO1 knockdown cells and Lats1 or Lats2 knockdown cells that were synchronized at mitosis by a thymidine single block-and-release (Figs. 4G and H). By contrast, in asynchronous (almost non-mitotic) cells, the level of Cofilin-pS3 was increased by depletion of Lats1 and/or Lats2 (Fig. 4I), which is consistent with the previous finding that Lats1 interacts directly with and inhibits the kinase activity of LIMK1.26 Because the total level of LIMK1 was not affected by Lats1/2 knockdown (Fig. 4H), a reduction in the centrosomal level of LIMK1-pT508 may be responsible for changing the localization or activity of the protein. Overall, these results indicate that Lats1/2 and CHO1 regulate the localization of LIMK1-pT508 on the centrosome.
Figure 4.

(See previous page). Large tumor suppressor (Lats)1/2 and CHO1 regulate the centrosomal localization and activity of LIM-kinase (LIMK)1 during mitosis. (A) Centrosomal localization of LIMK1-pT508 in metaphase HeLa-S3 cells transfected with a control (GL2) or CHO1/MKLP1-specific siRNA and synchronized by a thymidine single block-and-release (10 h). Scale bar, 10 μm. (B, C) The signal intensities of LIMK1-pT508 (B) and γ-tubulin (C) on centrosomes in CHO1/MKLP1 knockdown cells during metaphase. Data represent the mean ± SD of n = 3 independent experiments (30 cells per experiment). (D) Centrosomal localization of phosphorylated LIMK1-pT508 and γ-tubulin in metaphase HeLa-S3 cells transfected with a control (GL2), Lats1-or Lats2-specific siRNA. Lats2 knockout cells (KO/HeLa-S3) were also transfected with Lats1 siRNA. The cells were synchronized by a thymidine single block-and-release (10 h). Scale bar, 10 μm. (E, F) The signal intensities of LIMK1-pT508 (E) and γ-tubulin (F) on the centrosomes in Lats1-, Lats2-, and Lats1/2-depleted cells during metaphase. Data represent the mean ± SD of n = 3 independent experiments (30 cells per experiment). (G) Immunoblot analyses of Cofilin-pS3 in HeLa-S3 cells transfected with a control (GL2) or CHO1/MKLP1-specific siRNA and synchronized by a thymidine single block-and-release (10 h). The ratio of the amount of Cofilin-pS3 in CHO1/MKLP1 knockdown cells to that in control cells is shown. (H) Immunoblot analyses of Cofilin-pS3 in HeLa-S3 cells transfected with a control (GL2), Lats1-, or Lats2-specific siRNA and synchronized by a thymidine single block-and-release (10 h). (I) Immunoblot analyses of Cofilin-pS3 in asynchronous parental HeLa-S3 cells, Lats1 KO/HeLa-S3 cells, Lats2 KO/HeLa-S3 cells, and Lats2 KO/HeLa-S3 cells transfected with Lats1 siRNA.
Phosphorylation of CHO1-S716S717 is required for the anchoring and activation of LIMK1 on centrosomes
Next, we determined the impact of Lats1/2-mediated phosphorylation of CHO1 on the centrosomal localization of LIMK1-pT508 during metaphase. Overexpression of CHO1-WT in HeLa-S3 cells (Fig. S4A) increased the signal intensity of LIMK1-pT508 but not γ-tubulin on the centrosomes (Fig. 5A–C). Overexpression of CHO1-SD, a constitutive phosphomimetic form of the protein, increased the signal intensity of LIMK1-pT508 even further, whereas overexpression of non-phosphorylatable CHO1-SA only modestly increased the signal intensity than CHO1-WT (Figs. 5A and B). LIMK1-T508 is phosphorylated by itself and Aurora-A; in turn, LIMK1-pT508 phosphorylates Aurora-A at T288, an activating phosphorylation site, thereby forming a positive feedback loop to regulate mitotic spindle morphology.33 The signal intensity of Aurora-A-pT288 on the centrosomes during the G2 phase and metaphase was increased significantly by overexpression of CHO1-SD (Figs. 5D, E, S5B, and S5C), and immunostaining with an anti-α-tubulin antibody revealed that spindle formation was also augmented, suggesting hyperactivation of Aurora-A (Fig. 5F and G). However, although the signal intensity of γ-tubulin was slightly lower in cells expressing CHO1-SA than in CHO1-SD cells (Fig. 5A and C), the signal intensities of Aurora-A-pT288 at the centrosomes and α-tubulin at the mitotic spindle were not decreased by overexpression of CHO1-SA (Fig. 5D–G). During anaphase, the centrosomal signals of LIMK1-pT508 in CHO1-WT and -SA cells declined gradually to their steady levels, whereas those in CHO1-SD cells were maintained at a high level (Fig. 5H and I). Indeed, when we examined the effect of CHO1 (WT, SA, and SD) on the kinase activity of LIMK1 in an in vitro kinase assay containing [γ-32P] ATP, the kinase activity of 3Flag-LIMK1 was increased in the cells stably expressing the CHO-SD mutant (Fig. S6). Since LIMK1-pT508 phosphorylates Cofilin-S3 to induce contractile ring assembly, the defused signals of Cofilin-pS3 in metaphase cells were predictably enhanced by the overexpression of CHO-SD (Fig. 5J).
Figure 5.

(See previous page). Phosphorylation of CHO1 at S716S717 is required for the anchoring and activation of LIM-kinase 1 (LIMK1) on the centrosome. (A) Centrosomal localization of LIMK1-pT508 and γ-tubulin in metaphase HeLa-S3 cells stably expressing vector alone or 6Myc-tagged CHO1-WT, -SA (S717A), or -SD (S717D). The cells were synchronized by a thymidine single block-and-release (10 h). Scale bar, 10 μm. The insets show enlarged images of signals at the centrosome. (B, C) The signal intensities of LIMK1-pT508 (B) and γ-tubulin (C) on centrosomes in metaphase HeLa-S3 cells stably expressing the constructs described in (A). Data represent the mean ± SD of n = 3 independent experiments (30 cells per experiment). (D, E) The signal intensities of Aurora-A-pT288 on centrosomes in G2 phase (D) and metaphase (E) HeLa-S3 cells stably expressing the constructs described in (A). Cells were synchronized by a thymidine single block-and-release (10 h). Data represent the mean ± SD of n = 3 independent experiments (30 cells per experiment). (F) The mitotic spindle in metaphase HeLa-S3 cells stably expressing the constructs described in (A). Cells were synchronized by a thymidine single block-and-release (10 h) and stained with anti-α-tubulin. Scale bar, 10 μm. (G) The percentages of the cells described in (F) with a mitotic spindle during metaphase. The cells were classified according to the α-tubulin signal intensity. Data represent the mean ± SD of n = 3 experiments. (H, I) The centrosomal localization of LIMK1-pT508 in anaphase HeLa-S3 cells stably expressing the constructs described in (A). In (I), the data represent the mean ± SD of n = 3 independent experiments (30 cells per experiment). (J) The level of Cofilin-pS3 in metaphase HeLa-S3 cells stably expressing the constructs described in (A). Cells were synchronized by a thymidine single block-and-release (10 h). Scale bar, 10 μm.
Overall, these results suggest that phosphorylation of CHO1-S716S717 is necessary for the anchoring and activation of LIMK1 on centrosomes; however, it may not be essential for the activation of centrosomal Aurora-A and subsequent spindle formation.
Phosphorylation of CHO1-S716 is required for cytokinesis
CHO1 and MKLP1 are essential for the completion of cytokinesis. Ectopic expression of a CHO1 mutant lacking the FABR (amino acids 696–787) does not rescue cytokinesis failure caused by siRNA-mediated knockdown of CHO1 completely,9 suggesting that the regulation of cytokinesis by CHO1 is dependent on its FABR. Therefore, we performed a rescue assay to determine whether ectopically expressed CHO1-WT overcomes the cytokinesis failure phenotypes caused by knockdown of CHO1 in HeLa-S3 cells. Overexpression of CHO1-WT suppressed the increased number of multinucleated cells (including binucleated cells) caused by knockdown of CHO1/MKLP1 (Figs. 6A and S1D). Consistent with previous reports, these findings suggest that cytokinesis completion requires both MKLP1 and CHO1. Notably, overexpression of the CHO1-SA and -SD mutants increased the numbers of multinucleated cells, whereas overexpression of CHO1-WT did not (Fig. 6C). As expected, because they were unable to support the phosphorylation of S716S717 (Fig. 2H), overexpression of CHO1-2SA and -3SA increased the number of multinucleated cells (Figs. 2H and 6C). These results indicate that both unphosphorylated and constitutively phosphorylated CHO1-S716 prevent the completion of cytokinesis and normal nuclear envelope formation, suggesting that the precise temporal and spatial regulation of this residue is essential.
Figure 6.
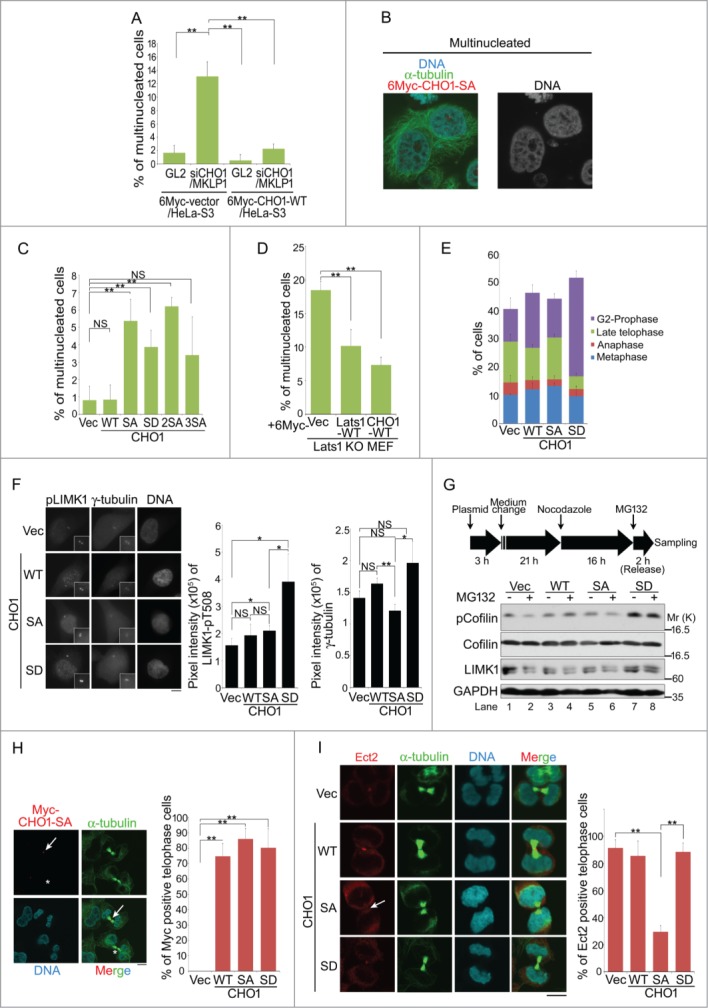
Phosphorylation of CHO1 at S716S717 is required for cytokinesis. (A) The percentages of multinucleated cells in HeLa-S3 populations stably expressing 6Myc-vector or 6Myc-CHO1-WT (wild type) that were transfected with a control (GL2) or CHO1/MKLP1-specific siRNA. The cells were fixed 72 h after transfection. (B) Representative images of immunostained multinucleated cells (including binucleated cells). (C) The percentages of multinucleated cells in a HeLa-S3 population stably expressing 6Myc-tagged vector alone or CHO1-WT, -SA, -SD, -2SA, or -3SA. (D) The percentages of multinucleated cells in Lats1-KO mouse embryo fibroblasts (MEFs) transfected with 6Myc-tagged vector alone, large tumor suppressor (Lats)1-WT, or CHO1-WT. The cells were fixed 72 h after transfection. (E) The percentages of HeLa-S3 cells stably expressing 6Myc-tagged vector alone, CHO1-WT, -SA, or -SD at each stage of the cell cycle. The cells were synchronized by a thymidine single block-and-release (10 h). (F) The centrosomal localization of LIM-kinase 1 (LIMK1)-pT508 in G2 phase HeLa-S3 cells stably expressing 6Myc-tagged vector alone or CHO1-WT, -SA, or -SD. The cells were synchronized by a thymidine single block-and-release (10 h). Scale bar, 10 μm. The bar graphs show the intensities of the LIMK1-pT508 (center) and γ-tubulin (right) signals on the centrosomes in the G2 phase. Data represent the mean ± SD of n = 3 independent experiments (30 cells per experiment). (G) Immunoblot analyses of Cofilin-pS3 in HeLa-S3 cells transiently expressing 6Myc-tagged vector alone or CHO1-WT, -SA, or -SD. Transfected HeLa-S3 cells were treated with nocodazole (80 ng/ml) for 16 h prior to releasing for 2 h with (+) or without (−) MG132 (5 nM). (H) Midbody localization (arrow) of ectopic 6Myc-CHO1-SA in HeLa-S3 cells undergoing cytokinesis. The asterisks indicate mislocalization of CHO1-SA. Scale bar, 10 μm. The bar graph shows the percentages of cells expressing 6Myc-tagged vector alone or CHO1-WT, -SA or -SD with midbody localization of the exogenous protein during late telophase. (I) Midbody localization of Ect2 during cytokinesis in HeLa-S3 cells stably expressing 6Myc-tagged vector alone or CHO1-WT, -SA, or -SD. The arrow indicates mislocalization of Ect2. Scale bar, 10 μm. The bar graph shows the percentages of cells undergoing cytokinesis with midbody Ect2 signals. (B–E, H, I) Data represent the mean ± SD of n = 3 experiments (more than 100 cells per experiment).
Knockout and knockdown of Lats1 cause cytokinesis failure and increase the number of multinucleated cells.26 To determine whether CHO1 is responsible for Lats1-mediated cytokinesis, we generated Lats1−/− mice by targeting exon 5, which encodes part of the kinase domain, and isolated MEFs (Lats1-KO MEFs) (Mukai and Nojima, unpublished data). Consistent with previous reports, Lats1-KO MEFs had an augmented population of multinucleated cells (Fig. S4B). Overexpression of CHO1-WT or Lats1-WT in Lats1-KO MEFs suppressed the formation of multinucleated cells (Fig. 6D), suggesting that Lats1 and CHO1 cooperatively regulate cytokinesis.
A cell-cycle analysis revealed that CHO1-SA cells progressed normally toward late telophase and formed the midbody, but failed to undergo abscission, whereas a large proportion of the CHO1-SD cells exhibited prolonged arrest at the G2 phase or prophase. Compared with control cells, a smaller percentage of CHO1-SD cells were at the late telophase stage with the midbody (Fig. 6E), presumably as a consequence of premature mitotic exit without cytokinesis (so-called ‘mitotic slippage’). Furthermore, compared with that in control cells, the centrosomal LIMK1-pT508 signal was markedly higher in CHO1-SD cells during the G2 phase (Fig. 6F), which is consistent with the results obtained for the metaphase and anaphase cells (Fig. 5B and I). Because constitutive hyper-phosphorylation of Cofilin by LIMK1 promotes the formation of multinucleated cells by preventing cytokinesis completion,15 we examined the phosphorylation of Cofilin-S3 in CHO1-expressing HeLa-S3 cells that were synchronized at either the metaphase/anaphase transition by treatment with the proteasome inhibitor MG132 after nocodazole arrest-and-release, or at a later stage of mitosis by nocodazole arrest-and-release only. Cells expressing CHO1-SD exhibited constitutively hyperphosphorylated Cofilin during metaphase/anaphase and onward, whereas cells expressing vector alone or CHO1-WT or -SA did not (Fig. 6G; see also Fig. 5J). These results suggest that CHO1-SD promotes tight binding and excessive activation of LIMK1 on centrosomes, thereby inducing arrest at the G2 phase or prophase through hyperactivation of the LIMK1-Aurora-A positive feedback loop and the subsequent failure of cytokinesis caused by hyper-phosphorylation of Cofilin.
Although overexpression of CHO1-SA did not promote LIMK1 activity and Cofilin phosphorylation during mitosis, it eventually caused cytokinesis failure (Figs. 5B, J and 6C). Like MKLP1, CHO1 is a kinesin-like motor protein that translocates to the central spindle during anaphase and the midbody during late telophase. Because CHO1-SA and -SD also localized to the midbody, it is unlikely that the phosphorylation of S716S717 directly affects their motor activities (Fig. 6H). To determine why overexpression of CHO1-SA caused cytokinesis failure, we focused on the regulation of other components of the centralspindlin complex, including HsCyk4 and Ect2, which colocalize with CHO1/MKLP1 at the midbody and regulate actomyosin contractility. Ect2 was mislocalized in CHO1-SA mutant cells (Fig. 6I). Meanwhile, although phosphorylation of HsCyk4 (pS387) by Aurora-B is essential for cytokinesis completion,34 both HsCyk4-pS387 and Aurora-B localized normally to the midbody in CHO1-SA cells as well as -WT, -SD and control cells undergoing cytokinesis (Fig. S4C and S4D). These results indicate that the midbody is intact in CHO1-SA mutant cells, suggesting that mislocalization of Ect2 is responsible for cytokinesis failure in these cells.
Taken together, these results suggest that phosphorylation of CHO1-S716S717 by Lats1 and Lats2 is required for anchoring and activation of LIMK1 at centrosomes to regulate Cofilin phosphorylation during early mitosis, and for the recruitment of Ect2 to the midbody during late mitosis, thereby controlling both the execution (advance preparation) and completion of cytokinesis.
Discussion
The results presented here demonstrate that Lats1 and Lats2 phosphorylate S716S717 in the FABR of CHO1 during mitosis; this event is supported by phosphorylation of S812S805 and S814S807 by Aurora-B and Lats2, respectively, and CHO1 proteins phosphorylated at these sites were differentially located during metaphase and anaphase. Because it was not observed that centrosomal CHO1 is phosphorylated at S814, the initial phosphorylation of these residues may trigger S716 phosphorylation to promote localization of CHO1 to the centrosomes, and may subsequently be abolished by phosphatases. During late mitosis, phosphorylation of MKLP1-S708 (corresponding to CHO1-S812) prevents its interaction with the 14-3-3 protein and promotes clustering and stable localization of the centralspindlin complex at the central spindle.28 Phosphorylation of S716 may inhibit the translocation of CHO1 from the centrosomes to the central spindle during early mitosis because S814 of centrosomal CHO1 proteins is unphosphorylated throughout the cell cycle (Fig. S3A). Localization of CHO1-pS814 at the central spindle is similar to the well-known localizations of MKLP1 and Lats2-pS380. 28,22
LIMK1 was identified as a mitotic association partner of CHO1-pS716. Activated LIMK1-pT508 phosphorylates Cofilin during the early stages of mitosis (prometaphase and metaphase) to induce actomyosin contractility, and the levels of phosphorylated LIMK1 and Cofilin are decreased during the late stages of mitosis (telophase and cytokinesis) to allow cytokinesis completion.15 Overexpression of LIMK1 induces cytokinesis failure and the production of multinucleated cells; therefore, it may function as a rheostat-like molecule that is stringently regulated to ensure successful cytokinesis.
Based on the results presented here, we propose that centrosomal CHO1 is a key regulator of mitosis that activates LIMK1 by anchoring it to the centrosome in a Lats1/2-dependent manner during early mitosis (Fig. 7). The Lats1/2-CHO1-LIMK1 axis may regulate cytokinesis initiation by phosphorylating Cofilin on centrosomes and distributing it throughout the cytoplasm. In addition to its well-characterized role in mitotic spindle organization, the centrosome may also function as a pivotal site of signal transduction to regulate cytokinesis initiation. However, because loss or disruption of centrosomes in vertebrate cells does not always prevent cytokinesis (abscission), but rather induces cell-cycle arrest of the daughter cells at the subsequent G1 phase,16 the centrosomes may play a non-essential role in efficient signal transduction of the Lats1/2-CHO1-LIMK1 axis.
Figure 7.
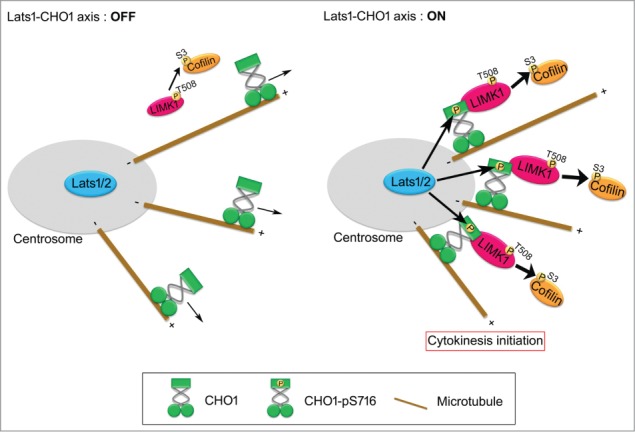
A model for the role of Lats1/2-CHO1-LIMK1 axis in preparation of cytokinesis initiation on centrosome. When Lats1/2-CHO1 axis is activated, LIMK1 is anchored to centrosomes by S716-phosphorylated CHO1, which in turn efficiently activates LIMK1 and promotes phosphorylation of Cofilin for preparation of cytokinesis initiation (right panel). If Lats1/2-CHO1 axis is not activated, CHO1 does not localize at centrosome, thereby decreasing the activating phosphorylation level and activation of LIMK1 to phosphorylate Cofilin (left panel). Eventually, dysregulation of Lats1/2-CHO1 axis fails to initiate cytokinesis.
Because we did not detect stable interactions of Lats1/2 with CHO1 (data not shown), Lats1/2 likely dissociates from CHO1 immediately after phosphorylation. Lats1 interacts with LIMK1 and inhibits its kinase activity toward Cofilin; 26 however, this function of Lats1 is likely distinct from its role in the Lats1/2-CHO1-LIMK1 pathway because it does not require intact kinase activity.26 Since the activation of LIMK1 and phosphorylation of Cofilin are dispensable for the final step of cytokinesis, inactive Lats1 located away from the centrosomes may interact with LIMK1 and inhibit the phosphorylation of Cofilin at the midbody during abscission. In support of this idea, both CHO1 and Lats1 lacking the kinase domain interacted with LIMK1, whose LIM domain influenced its mitotic phosphorylation and activation (Fig. 3C and 3E).15 These results suggest that the Lats1/2-CHO1 axis activates LIMK1 at centrosomes and inactive Lats1 inhibits LIMK1 at the midbody. LIMK1 interacted with the stalk domain of CHO1, which is also present in MKLP1 and contains the S812 and S814 sites. Since phosphorylation of S812 and S814 did not occur in centrosomes (Fig. 2F), the interaction of LIMK1 with CHO1 that is dependent on phosphorylation of these sites may be required for loading CHO1/MKLP1 onto the central spindle during late mitosis.
The mechanism by which CHO1 but not MKLP1 is retained at the centrosomes throughout the cell cycle is unknown. Centrosome-associated tyrosine-phosphorylated cortactin promotes F-actin-driven centrosome separation. 35 Since CHO1 contains a FABR, it may bind tightly to centrosome-associated F-actin; indeed, faint colocalization of CHO1 with F-actin was observed (Fig. S1E). S307 and T508 of LIMK on centrosomes (and spindle poles) are phosphorylated by Aurora-A, and inhibition of this kinase causes mislocalization of phosphorylated LIMK1. Aurora-A is also phosphorylated and activated by LIMK1, thereby forming a feedback loop to regulate mitotic spindle morphology.33 Constitutive phosphorylation of CHO1-S716 promotes Aurora-A activation and subsequent centrosome maturation (Fig. 5D–F); hence, it is likely that the Lats1/2-CHO1-LIMK1 axis coordinately regulates Aurora-A-mediated centrosome maturation and Cofilin-mediated cytokinesis.
In the Hippo-signaling pathway, actin depolymerization promotes the kinase activities of Lats1 and Lats2.36 The level of CHO1-pS716S717 was elevated following the treatment of HeLa-S3 cells with cytochalasin B (Fig. S4E), suggesting that the Lats1/2-CHO1-LIMK1 axis is activated by actin depolymerization. As upstream regulators of Lats1/2, Mst1/2 regulate cell proliferation and play a key role in centrosome duplication.37 Knockdown of Mst2 reduced the level of CHO1-pS716S717 (Fig. S4F), suggesting that some components of the Hippo pathway can regulate the Lats1/2-CHO1-LIMK1 axis. However, because Lats1/2 are regulated by other kinases such as Aurora-A, Chk1 and PKA during cell-cycle regulation and in the Hippo pathway,22,38,39 additional studies of the Lats1/2-CHO1-LIMK1 pathway are required.
Materials and Methods
Plasmids
The full-length mouse Cho1 sequence (Acc. No. NM_024245) was amplified from a 10T 1/2 cDNA library by PCR and cloned into the AscI and NotI sites of mammalian expression vectors (pCMV6myc and p3Flag) and a bacterial expression vector (pGST6P). A PCR-based method was used to construct full-length CHO1 mutants containing point mutations (S717A, S717D, S805A/S807A, and S717A/S805A/S807A), and the NsiI/NotI fragment of wild-type CHO1 was replaced with the corresponding fragment of mutant CHO1. The human LIM-kinase 1 (LIMK1) sequence (Acc. No. NM_002314) was amplified from a testis cDNA library by PCR and cloned into the AscI and NotI sites of the pCMV6myc and p3Flag vectors. Truncated forms of CHO1 (amino acids 1–425, 426–626, and 627–953) and LIMK1 (amino acids 1–147, 148–346, and 347–647) were generated by PCR and cloned into the AscI and NotI sites of the pCMV6myc vector. A truncated form of CHO1 (amino acids 692–796) was synthesized (by GenScript) and cloned into the AscI and NotI sites of pGST6P vector. Mouse Lats1 and Lats2 and human Mob1A were described previously. 23,25 All amplified sequences and mutations were confirmed by DNA sequencing.
Cell culture
HeLa-S3 and 293T cell lines were maintained as described previously.23 Lats1−/- MEFs were generated (Mukai and Nojima, unpublished data) and maintained as described previously. 23
Cell cycle synchronization and plasmid transfection
Cells were synchronized at the G1 phase, S phase, meta/anaphase, or prometaphase as described previously.22 For immunofluorescence staining, cells were synchronized at the S phase by a thymidine single block, released from the block, and then fixed with formaldehyde at the indicated mitotic stages. To assess the interaction of endogenous CHO1 and LIMK1 or γ-tubulin and pS716S717-CHO1 or pT508-LIMK1 in vivo, cells were synchronized at the S phase by a thymidine single block and released from the block for 10 h. Transient transfections of HeLa-S3 and 293T cells were performed using Lipofectamine (Invitrogen, 18324-020) and PLUS (Invitrogen, 11514-015) reagents, according to the manufacturer's instructions.
In vitro kinase assay
In vitro Lats1/2-kinase and Aurora-A kinase assays were performed as described previously.22 In vitro Aurora-B kinase assays were performed by incubating 1 μg of Aurora-B active kinase (Merck, D8NN055N-I) or 1 μg of GST-tagged Aurora-B-KD (K106R) with GST-tagged CHO1 for 30 min at 30°C in Aurora-B kinase buffer (50 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 1 mM EGTA, 1 mM DTT, 5 mM NaF, 0.05 mM Na3VO4, and 5 mM β-glycerophosphate) containing 20 μM ATP with or without [γ-32P] ATP. For the LIMK1 in vitro kinase assay, HeLa-S3 cells stably expressing 6Myc-tagged CHO1 (WT, SA, or SD) were transfected with 3Flag-tagged LIMK1 and then treated with nocodazole (80 ng/ml) for 16 h before being released into normal growth conditions for 3 h with MG132 (5 nM; CALBIOCHEM, 474790) and the phosphatase inhibitor okadaic acid (0.1 μM; Sigma, O4511). LIMK1 was immunoprecipitated with anti-Flag from these cell extracts and then incubated in LIM-kinase buffer (20 mM HEPES-NaOH, pH 7.2, 5 mM MgCl2, 5 mM MnCl2, 1 mM DTT, 10 mM NaF, 20 mM β-glycerophosphate, 1 mM PMSF, 10 μg/ml Leupeptin, and 2 μg/ml PepstatinA) containing 20 μM ATP with [γ-32P] ATP.
Antibodies
The anti-CHO1-pS716S717 polyclonal antibody was generated against phosphorylated S716 of human CHO1 (S717 in mouse). Briefly, rabbits were injected with the following KLH-conjugated phosphopeptide: LHRRSN(PO3H2)SCSSISV (pS716; this antigen sequence is identical to the flanking sequence of S717 in mouse CHO1). The antisera were affinity purified using a phospho-antigen peptide column. To eliminate the reaction of non-specific antibodies with unphosphorylated antigen peptide, the antibody preparation was passed through a non-phospho-CHO1-peptide (S716: LHRRSNSCSSISV) column. A novel polyclonal antibody against exon 18 (anti-CHO1[GS]), which encodes the F-actin binding region (FABR) of both human and mouse CHO1, was generated by injecting a rabbit with a KLH-conjugated peptide sequence (RQQEPGQSKTC) corresponding to the specific region of CHO1 (amino acids 749–759 and 750–760 in human and mouse, respectively). The anti-CHO1-S814S807 polyclonal antibody was newly generated against phosphopeptides RSR(PO3H2)SAGSRWVDHKP (pS814; pS807 in mouse). Similarly, the anti-Ect2 and anti-HsCyk4-pS387 polyclonal antibodies were generated against the RETDVSPFPPRKRP peptide and GLYRI(PO3H2)SGCDRTVKE phospho-peptide, respectively. The anti-CHO1-pS716S717, anti-CHO1[GS], anti-CHO1-S814S807, anti-Ect2, and anti-HsCyk4-pS387 antibodies were generated and purified by GenScript. Monoclonal antibodies against the following targets were used: α-tubulin (Sigma, T5168), γ-tubulin (Sigma, T6557), FLAG-tag (clone M2; Sigma, F3165), Myc-tag (clone PL14; MBL, M047-3), CHO1/MKLP1 (Abnova, B01), Aurora-B (AIM-1) (BD Bioscience, 611083), and Cofilin (clone E-8; Santa Cruz Biotechnology, sc-376476). Polyclonal antibodies against the following targets were used: Myc-tag (MBL, JM-3995-3), phosphorylated histone H3 (S10) (Upstate, #06-570), phospho-Aurora-A/AIK (Thr-288) (Cell Signaling Technology, #3091), CHO1/MKLP1 (Santa Cruz Biotechnology, sc-867), Cyclin B1(Santa Cruz Biotechnology, sc-752), LIMK1 (Santa Cruz Biotechnology, sc-8387), LIMK1 (Cell Signaling Technology, #3842), phospho-LIMK1 (Thr508/Thr505) (Cell Signaling Technology, #3841), phospho-Cofilin (Ser3) (Cell Signaling Technology, #3313), and Mst2 (Cell Signaling Technology, #3952). The anti-GST monoclonal antibody and anti-Lats1 rabbit, anti-Lats2, and anti-Aurora-A polyclonal antibodies have been described previously. 22,25
Dot blot analysis, immunoblotting and immunoprecipitation
Dot blotting, preparation of cell lysates, immunoblotting and immunoprecipitation (IP) were performed as described previously.22 Anti-EGFP and anti-FLAG polyclonal antibodies (Sigma, F7425) were used as negative control IgGs in Figure 3F. The ratios of the band intensities were determined using ImageJ software.
Protein phosphatase assays
For protein phosphatase (PPase) assays, cells were lysed in modified TNE250 lysis buffer containing 5 nM MG132 but excluding phosphatase inhibitors (NaF, Na3VO4, and β-glycerophosphate). The lysates were incubated at 4°C for 30 min with or without 200 U of λ-PPase (New England BioLabs, P0753L) in the presence or absence of phosphatase inhibitors (1 mM NaF, 1 mM Na3VO4, 10 mM β-glycerophosphate, and 100 ng/ml okadaic acid).
Immunofluorescence staining
To collect mitotic cells, the cells were blocked at the S phase by the addition of 2.5 mM thymidine for 24 h, released, and then fixed 10 or 11 h later. Cells were incubated with the primary antibodies, followed by AlexaFluor 488-and/or 594-conjugated anti-rabbit/mouse IgG (Molecular Probes) as secondary antibodies in Tris buffered saline with Tween 20 containing 5% FBS. DNA was stained using Hoechst 33258 (Sigma, B-2883). The cells were observed using light fluorescence microscopy (model BX51; Olympus), confocal LSM (model FV10i; Olympus), and Fluoview software (Olympus). The signal intensities of phospho-LIMK1 at the centrosomes and midbody were calculated using MetaView software (Universal Imaging Ltd).
Competition assays
In competition assays, the primary antibodies (anti-pS716S717and anti-pS814S807) were pre-incubated with or without the corresponding phosphorylated or non-phosphorylated CHO1 peptides (10 μg for immunoblotting and 5 μg for immunofluorescence staining) at room temperature for 40 min.
Small interfering RNAs
The siRNA duplexes were transfected into growing HeLa-S3 cells using Lipofectamine 2000 reagent (Life Technologies), according to the manufacturer's instructions. The cells were fixed or lysed at the indicated times after transfection. The following siRNA duplexes were used: firefly luciferase (GL2) as a negative control, 5′-CGUACGCGGAAUACUUCGAdTdT-3′; Lats1-3509, 5′-ACUUUGCCGAGGACCCGAAdTdT-3′; Lats2-581, 5′-GUUCGGACCUUAUCAGAAAdTdT-3′; LIMK1, 5′-UGGCAAGCGUGGACUUUCAdTdT-3′; and 3′UTR of CHO1/MKLP1 5′-GCAGUCUUCCAGGUCAUCUdTdT-3′. The Mst2 siRNAs (#1–3) were purchased from OriGene (SR304635).
Disruption of the LATS1 and LATS2 genes in HeLa-S3 cells
The Crispr/Cas-9 system was used to generate the Lats1 knockout HeLa-S3 cell line (Lats1 KO/HeLa-S3), following a similar method described in a previous report.40 Briefly, HeLa-S3 cells were transfected with a pX330 vector (Addgene) containing a target sequence for the LATS1 gene and cultured in DMEM (Sigma, D5796) containing 5% FBS. A single cell was then cloned by limiting dilution (Mukai and Nojima, unpublished data). The Lats2 knockout HeLa-S3 cell line (Lats2 KO/HeLa-S3) was generated using the transcription activator-like effector nuclease system (Torigata and Nojima, unpublished data). Validation and confirmation of the targeting efficiency in each clonal line were performed by PCR, DNA sequencing and immunoblotting.
RT-PCR
Total RNAs were extracted from mouse embryo fibroblasts (MEFs) and HeLa-S3 cells using the RNeasy Mini Kit, according to the manufacturer's instructions (Qiagen). The cDNAs from MEFs or the mouse 10T 1/2 library were synthesized from 0.3 μg of RNA using the High-Capacity cDNA Archive Kit (Applied Biosystems). PCR was performed with 60 ng of cDNA, Taq polymerase supplemented with the PCR× Enhancer System (Takara), and the following primers: CHO1/MKLP1 forward, 5′-TTAGAAGCCAGGTTGCAAGG-3′; CHO1/MKLP1 reverse, 5′-GGCTTATGATCTACCCATCTG-3′; GAPDH forward, 5′-TCACCATCTTCCAGGAGCGAG-3′; and GAPDH reverse, 5′-GCTGTAGCCGTATTCATTGTC-3′. The following thermal cycling profile was used: initial denaturation at 95°C for 3 min; 35 cycles of 95°C for 30 s, 50°C for 30 s, and 72°C for 2 min; and then a final extension at 72°C for 5 min. The PCR products were subjected to agarose gel electrophoresis and ethidium bromide staining.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. Patrick Hughes for critically reading the manuscript; Dr. Kazunobu Saito of the Core Instrumentation Facility (RIMD, Osaka University) for technical assistance with peptide mapping by tandem mass spectrometry; and Dr. Yoshitaka Fujihara (RIMD, Osaka University) for technical advice regarding the Crispr/Cas-9 system.
Funding
This work was supported in part by the Kudo Scientific Foundation (to NY), by Grant-in-Aid for Japan Society for the Promotion of Science Fellows (to AO), and by Grants-in-Aid for Scientific Research (C to NY and B to HN) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
SUPPLEMENTAL MATERIAL
Supplemental data for this article can be accessed on the publisher's website.
Author Contributions
AO and SM performed all experiments and analyzed data; KT generated the Lats2 KO/HeLa-S3 cells; AO, NY and HN wrote the manuscript; and NY and HN conceived the experimental design.
References
- 1.Fededa JP, Gerlich DW. Molecular control of animal cell cytokinesis. Nat Cell Biol 2012; 14:440–47; PMID:22552143; http://dx.doi.org/ 10.1038/ncb2482 [DOI] [PubMed] [Google Scholar]
- 2.Normand G, King RW. Understanding cytokinesis failure. Adv Exp Med Biol 2010; 676:27–55; PMID:20687468; http://dx.doi.org/ 10.1007/978-1-4419-6199-0_3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Glotzer M. The 3Ms of central spindle assembly: microtubules, motors and MAPs. Nat Rev Mol Cell Biol 2009;10:9–20; PMID:19197328; http://dx.doi.org/ 10.1038/nrm2609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adams RR, Tavares AA, Salzberg A, Bellen HJ, Glover DM. pavarotti encodes a kinesin-like protein required to organize the central spindle and contractile ring for cytokinesis. Genes Dev 1998; 15:1483–94; PMID:9585508; http://dx.doi.org/ 10.1101/gad.12.10.1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Raich WB, Moran AN, Rothman JH, Hardin J. Cytokinesis and midzone microtubule organization in Caenorhabditis elegans require the kinesin-like protein ZEN-4. Mol Biol Cell 1998; 9:2037–49; PMID:9693365; http://dx.doi.org/ 10.1091/mbc.9.8.2037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nislow C, Lombillo VA, Kuriyama R, McIntosh JR. A plus-end-directed motor enzyme that moves antiparallel microtubules in vitro localizes to the interzone of mitotic spindles. Nature 1992; 359:543–7; PMID:1406973; http://dx.doi.org/ 10.1038/359543a0 [DOI] [PubMed] [Google Scholar]
- 7.Sellitto C, Kuriyama R. Distribution of a matrix component of the midbody during the cell cycle in Chinese hamster ovary cells. J Cell Biol 1988; 106:431–9; PMID:2448315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuriyama R, Gustus C, Terada Y, Uetake Y, Matuliene J. CHO1, a mammalian kinesin-like protein, interacts with F-actin and is involved in the terminal phase of cytokinesis. J Cell Biol 2002; 156:783–90; PMID:11877456; http://dx.doi.org/ 10.1083/jcb.200109090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matuliene J, Kuriyama R. Role of the midbody matrix in cytokinesis: RNAi and genetic rescue analysis of the mammalian motor protein CHO1. Mol Biol Cell 2004;15:3083–94; PMID:15075367; http://dx.doi.org/ 10.1091/mbc.E03-12-0888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nishimura Y, Yonemura S. Centralspindlin regulates ECT2 and RhoA accumulation at the equatorial cortex during cytokinesis. J Cell Sci 2006; 119:104–14; PMID:16352658; http://dx.doi.org/ 10.1242/jcs.02737 [DOI] [PubMed] [Google Scholar]
- 11.Yüce O, Piekny A, Glotzer M. An ECT2-centralspindlin complex regulates the localization and function of RhoA. J Cell Biol 2005; 170:571–82; PMID:16103226; http://dx.doi.org/ 10.1083/jcb.200501097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matuliene J, Kuriyama R. Kinesin-like protein CHO1 is required for the formation of midbody matrix and the completion of cytokinesis in mammalian cells. Mol Biol Cell 2002; 13:1832–45; PMID:12058052; http://dx.doi.org/ 10.1091/mbc.01-10-0504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaji N, Ohashi K, Shuin M, Niwa R, Uemura T, Mizuno K. Cell cycle-associated changes in Slingshot phosphatase activity and roles in cytokinesis in animal cells. J Biol Chem 2003; 278:33450–55; PMID:12807904; http://dx.doi.org/ 10.1074/jbc.M305802200 [DOI] [PubMed] [Google Scholar]
- 14.Kaji N, Muramoto A, Mizuno K. LIM kinase-mediated cofilin phosphorylation during mitosis is required for precise spindle positioning. J Biol Chem 2008; 283:4983–92; PMID:18079118; http://dx.doi.org/ 10.1074/jbc.M708644200 [DOI] [PubMed] [Google Scholar]
- 15.Amano T, Kaji N, Ohashi K, Mizuno K. Mitosis-specific activation of LIM motif-containing protein kinase and roles of cofilin phosphorylation and dephosphorylation in mitosis. J Biol Chem 2002; 277:22093–102; PMID:11925442; http://dx.doi.org/ 10.1074/jbc.M201444200 [DOI] [PubMed] [Google Scholar]
- 16.Khodjakov A, Rieder CL. Centrosomes enhance the fidelity of cytokinesis in vertebrates and are required for cell cycle progression. J Cell Biol 2001; 153:237–42; PMID:11285289; http://dx.doi.org/ 10.1083/jcb.153.1.237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Piel M, Nordberg J, Euteneuer U, Bornens M. Centrosome-dependent exit of cytokinesis in animal cells. Science 2001; 291:1550–3; PMID:11222861; http://dx.doi.org/ 10.1126/science.1057330 [DOI] [PubMed] [Google Scholar]
- 18.Fabbro M, Zhou BB, Takahashi M, Sarcevic B, Lal P, Graham ME, Gabrielli BG, Robinson PJ, Nigg EA, Ono Y, et al.. Cdk1/Erk2-and Plk1-dependent phosphorylation of a centrosome protein, Cep55, is required for its recruitment to midbody and cytokinesis. Dev Cell 2005; 9: 477–88; PMID:16198290; http://dx.doi.org/ 10.1016/j.devcel.2005.09.003 [DOI] [PubMed] [Google Scholar]
- 19.Pan D. The hippo signaling pathway in development and cancer. Dev Cell 2010; 19:491–505; PMID:20951342; http://dx.doi.org/ 10.1016/j.devcel.2010.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aylon Y, Michael D, Shmueli A, Yabuta N, Nojima H, Oren M. A positive feedback loop between the p53 and Lats2 tumor suppressors prevents tetraploidization. Genes Dev 2006; 20:2687–700; PMID:17015431; http://dx.doi.org/ 10.1101/gad.1447006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Toji S, Yabuta N, Hosomi T, Nishihara S, Kobayashi T, Suzuki S, Tamai K, Nojima H. The centrosomal protein Lats2 is a phosphorylation target of Aurora-A kinase. Genes Cells 2004; 9:383–97; PMID:15147269; http://dx.doi.org/ 10.1111/j.1356-9597.2004.00732.x [DOI] [PubMed] [Google Scholar]
- 22.Yabuta N, Mukai S, Okada N, Aylon Y, Nojima H. The tumor suppressor Lats2 is pivotal in Aurora A and Aurora B signaling during mitosis. Cell Cycle 2011; 10: 2724–36; PMID:21822051; http://dx.doi.org/ 10.4161/cc.10.16.16873 [DOI] [PubMed] [Google Scholar]
- 23.Yabuta N, Okada N, Ito A, Hosomi T, Nishihara S, Sasayama Y, Fujimori A, Okuzaki D, Zhao H, Ikawa M, et al.. Lats2 is an essential mitotic regulator required for the coordination of cell division. J Biol Chem 2007; 29:19259–71; PMID:17478426; http://dx.doi.org/ 10.1074/jbc.M608562200 [DOI] [PubMed] [Google Scholar]
- 24.Nishiyama Y, Hirota T, Morisaki T, Hara T, Marumoto T, Iida S, Makino K, Yamamoto H, Hiraoka T, Kitamura N, et al.. A human homolog of Drosophila warts tumor suppressor, h-warts, localized to mitotic apparatus and specifically phosphorylated during mitosis. FEBS Lett 1999; 459:159–65; PMID:10518011; http://dx.doi.org/ 10.1016/S0014-5793(99)01224-7 [DOI] [PubMed] [Google Scholar]
- 25.Yabuta N, Mukai S, Okamoto A, Okuzaki D, Suzuki H, Torigata K, Yoshida K, Okada N, Miura D, Ito A, et al.. N-terminal truncation of Lats1 causes abnormal cell growth control and chromosomal instability. J Cell Sci 2013; 126: 508–20; PMID:23230145; http://dx.doi.org/ 10.1242/jcs.113431 [DOI] [PubMed] [Google Scholar]
- 26.Yang X, Yu K, Hao Y, Li DM, Stewart R, Insogna KL, Xu T. LATS1 tumour suppressor affects cytokinesis by inhibiting LIMK1. Nat Cell Biol 2004; 6:609–17; PMID:15220930; http://dx.doi.org/ 10.1038/ncb1140 [DOI] [PubMed] [Google Scholar]
- 27.Ganem NJ, Cornils H, Chiu SY, O'Rourke KP, Arnaud J, Yimlamai D, Théry M, Camargo FD, Pellman D. Cytokinesis failure triggers hippo tumor suppressor pathway activation. Cell 2014;158:833–48; PMID:25126788; http://dx.doi.org/ 10.1038/nrc3824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Douglas ME, Davies T, Joseph N, Mishima M.. Aurora B and 14-3-3 coordinately regulate clustering of centralspindlin during cytokinesis. Curr Biol 2010; 20:927–33; PMID:20451386; http://dx.doi.org/ 10.1016/j.cub.2010.03.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Joseph N, Hutterer A, Poser I, Mishima M. ARF6 GTPase protects the post-mitotic midbody from 14-3-3-mediated disintegration. EMBO J 2012; 31:2604–14; PMID:22580824; http://dx.doi.org/ 10.1038/emboj.2012.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guse A, Mishima M, Glotzer M. Phosphorylation of ZEN-4/MKLP1 by aurora B regulates completion of cytokinesis. Curr Biol 2005; 15:778–86; PMID:15854913; http://dx.doi.org/ 10.1016/j.cub.2005.03.041 [DOI] [PubMed] [Google Scholar]
- 31.Mishima M, Kaitna S, Glotzer M. Central spindle assembly and cytokinesis require a kinesin-like protein/RhoGAP complex with microtubule bundling activity. Dev Cell 2002; 2:41–54; PMID:11782313; http://dx.doi.org/ 10.1016/S1534-5807(01)00110-1 [DOI] [PubMed] [Google Scholar]
- 32.Chakrabarti R, Jones JL, Oelschlager DK, Tapia T, Tousson A, Grizzle WE. Phosphorylated LIM kinases colocalize with gamma-tubulin in centrosomes during early stages of mitosis. Cell Cycle 2007; 6:2944–52; PMID:18000399; http://dx.doi.org/ 10.4161/cc.6.23.4957 [DOI] [PubMed] [Google Scholar]
- 33.Ritchey L, Ottman R, Roumanos M, Chakrabarti R. A functional cooperativity between Aurora A kinase and LIM kinase1: implication in the mitotic process. Cell Cycle 2012; 11: 296–309; PMID:22214762; http://dx.doi.org/ 10.4161/cc.11.2.18734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Minoshima Y, Kawashima T, Hirose K, Tonozuka Y, Kawajiri A, Bao YC, Deng X, Tatsuka M, Narumiya S, May WS Jr, et al.. Phosphorylation by aurora B converts MgcRacGAP to a RhoGAP during cytokinesis. Dev Cell 2003; 4:549–60; PMID:12689593; http://dx.doi.org/ 10.1016/S1534-5807(03)00089-3 [DOI] [PubMed] [Google Scholar]
- 35.Wang W, Chen L, Ding Y, Jin J, Liao K. Centrosome separation driven by actin-microfilaments during mitosis is mediated by centrosome-associated tyrosine-phosphorylated cortactin. J Cell Sci 2008; 121:1334–43; PMID:18388321; http://dx.doi.org/ 10.1242/jcs.018176 [DOI] [PubMed] [Google Scholar]
- 36.Wada K, Itoga K, Okano T, Yonemura S, Sasaki H. Hippo pathway regulation by cell morphology and stress fibers. Development 2011; 138:3907–14; PMID:21831922; http://dx.doi.org/ 10.1242/dev.070987 [DOI] [PubMed] [Google Scholar]
- 37.Mardin BR, Lange C, Baxter JE, Hardy T, Scholz SR, Fry AM, Schiebel E. Components of the Hippo pathway cooperate with Nek2 kinase to regulate centrosome disjunction. Nat Cell Biol 2010; 12:1166–76; PMID:21076410; http://dx.doi.org/ 10.1038/ncb2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim M, Kim M, Lee S, Kuninaka S, Saya H, Lee H, Lee S, Lim DS. cAMP/PKA signalling reinforces the LATS-YAP pathway to fully suppress YAP in response to actin cytoskeletal changes. EMBO J 2013; 32:1543–55; PMID:23644383; http://dx.doi.org/ 10.1038/emboj.2013.102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suzuki H, Yabuta N, Okada N, Torigata K, Aylon Y, Oren M, Nojima H. Lats2 phosphorylates p21/CDKN1A after UV irradiation and regulates apoptosis. J Cell Sci 2013; 126:4358–68; PMID:23886938; http://dx.doi.org/ 10.1242/jcs.125815 [DOI] [PubMed] [Google Scholar]
- 40.Mashiko D, Fujihara Y, Satouh Y, Miyata H, Isotani A, Ikawa M. Generation of mutant mice by pronuclear injection of circular plasmid expressing Cas9 and single guided RNA. Sci Rep 2013; 3:3355; PMID:24284873; http://dx.doi.org/ 10.1038/srep03355 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.