

Abstract
Currently, most methods for detecting gene–gene interactions (GGIs) in genome-wide association studies are divided into SNP-based methods and gene-based methods. Generally, the gene-based methods can be more powerful than SNP-based methods. Some gene-based entropy methods can only capture the linear relationship between genes. We therefore proposed a nonparametric gene-based information gain method (GBIGM) that can capture both linear relationship and nonlinear correlation between genes. Through simulation with different odds ratio, sample size and prevalence rate, GBIGM was shown to be valid and more powerful than classic KCCU method and SNP-based entropy method. In the analysis of data from 17 genes on rheumatoid arthritis, GBIGM was more effective than the other two methods as it obtains fewer significant results, which was important for biological verification. Therefore, GBIGM is a suitable and powerful tool for detecting GGIs in case–control studies.
Introduction
Genome-wide association studies (GWAS) have rapidly become a popular and powerful tool for human disease-associated gene discovery.1 Many single-SNP-based GWAS methods have emerged for several years.1, 2 However, due to the huge number of human genome SNPs (hundreds of thousands or millions in a test), the statistical power and efficiency of these methods are limited.3 In addition, human complex diseases are generally caused by the combined effect of multiple genes, and a single SNP is difficult to explain the pathogenesis of diseases.4, 5 The detection of interactions between genes is important to gain a better understanding of the genetic mechanisms of human complex diseases.
Large numbers of SNP–SNP interactions (SSIs) detecting methods appeared in recent years. In a case–control study, one form of SSI (or named co-associations) was epistasis, which was introduced ~100 years ago. These SSIs are associated with gene–gene interactions (GGIs). The GGIs (or gene–gene epistasis) are often characterized to be functional, compositional and statistical.6 The statistical definition of epistasis was first given by Fisher7 and developed further by Cockerham8 and Kempthorne,9 whereby the epistasis effect is considered as a deviation from additive genetic effects.10 Currently, popular SSIs detecting methods are based primarily on statistics,11, 12, 13 data mining,14, 15, 16 machine learning17, 18 and so on.19, 20 Statistical methods contain logistic regression model,11 the information entropy model;12, 13 data mining methods contain dimensionality reduction method,14 Bayesian method15, 16 and so on; machine learning methods are based on a tree and random forests17, 18 and so on.
We take only one SNP in a gene as a basic research unit, and only take into account the interactions between SNPs in these SSI methods. However, a gene that contains many SNPs should be the basic research unit, so the SSIs have limitations and cannot fully interpret the GGIs.21 We can use multiple SNPs in each gene (gene-based GGI methods), and these methods come with a potential increase of power.
There are some gene-based GGI detecting methods, such as canonical correlation-based U-statistic model,21 sparse canonical correlation analysis model,22 kernel canonical correlation-based U-statistic model (KCCU),23, 24 kernel regression model (KR),25 partial least squares path model (PLSPM and mPLSPM)26, 27 and so on. However, most of these methods can only reflect the linear relationship between two genes, and cannot reflect the nonlinear relationship. KCCU method can reflect nonlinear relationship between two genes, and is a useful and classic method.
Information entropy28, 29 is used to measure the uncertainty. The greater the uncertainty variables, the greater the entropy. The SNP-based entropy methods (SBEM) had been used to detect SSIs.12, 13 We proposed a gene-based information gain method (GBIGM), which is based on the entropy and information gain theory and views all SNPs in a gene for detecting GGIs in case–control studies. For a gene, we defined an information gain rate by comparing the entropy of the data with and without a gene's information. We consider IGR as a measure of genetic contribution for disease for this gene. While considering two genes, the IGR can be determined by comparing the joint entropy and individual entropies and we use it as a measure for epistasis. After comparing GBIGM with KCCU and SBEM both in the simulated and real data, we found that GBIGM can detect several epistasis types, and it is a valid and powerful gene-based method for detecting GGIs.
Materials and methods
Simulation data
Get the template data
The template data in simulation experiments are taken from HapMap.30, 31 In this study, we randomly select two gene regions,23 PARP1 and KRAS. PARP1 is located on chromosome 1, including seven SNPs (rs7537552, rs7537636, rs10495278, rs9287011, rs12090413, rs12092786 and rs12093044). KRAS is located on chromosome 12, including five SNPs (rs3924649, rs12307733, rs7980769, rs11836162 and rs11047882).
Disease models
A disease model is a model that expresses the relationship between the gene and the disease.32 Disease models are usually divided into two types, namely, single-site and multi-locus disease models. In single-site disease models, the disease is linked to only one locus, and in multi-locus model the disease is related to multiple loci.
This paper concentrated on the interaction between two genes, so we generate case–control data with two-locus disease model. There are eight two-locus disease models when generating simulation data using gs2.0 software,33 as showed in Supplementary Figure 1.
In Supplementary Figure 1, α is the baseline value, which indicates the odds of disease when the two SNP's joint genotype is aabb. θ is the growth leading to illness when other genotype with respect to aabb.
Generate case–control data sets
In this study, we utilize all these eight two-locus disease models in gs2.0 program to generate simulated case–control data. In the simulation, one SNP in each gene is randomly selected to set the parameter in generating case–control data, and other SNPs are generated using the information of linkage disequilibrium. The parameter settings are given in Table 1.
Table 1. The parameter settings in simulation using gs2.0.
| Odds ratio | Sample size | Prevalence rate | |
|---|---|---|---|
| Negative data sets | 1.0 | 1000∼5000 | 0.1 |
| Positive data sets I | 1.2∼2.2 | 2000 | 0.1 |
| Positive data sets II | 1.6 | 1000∼5000 | 0.1 |
| Positive data sets III | 1.6 | 2000 | 0.01∼0.2 |
In case of the null hypothesis H0 (to generate negative data sets), there is no interaction between genes, then the odds ratio (OR) was set to be 1.0. We set same sample size for cases and controls (N cases and N controls, N is set to be 1000, 2000, 3000, 4000 and 5000) and prevalence rate to be 0.1.
In the case of the alternative hypothesis H1 (to generate positive data sets), to test the effects of OR, sample size and prevalence rate respectively in GGIs detecting, we set three scenarios to perform experiments. In the first scenario, we set OR to be 1.2, 1.4, 1.6, 1.8, 2.0 and 2.2, sample sizes to be 2000 and prevalence rate to be 0.1. In the second scenario, we set OR to be 1.6, sample size N to be 1000, 2000, 3000, 4000 and 5000 and prevalence rate to be 0.1. In the third scenario, we set OR to be 1.6, sample size N to be 2000 and prevalence rates to be 0.01, 0.05, 0.10, 0.15 and 0.20. For each parameter setting, we performed the experiment 100 times.
Rheumatoid arthritis data set
We apply our method to a rheumatoid arthritis (RA) data set (GSE39428).34 The data set contains 266 cases (RA) and 163 health controls. Genotyping is performed using a custom-designed Illumina 384-SNP VeraCode microarray (Illumina, San Diego, CA, USA) to determine possible associations of genes to RA. After pretreatment, we obtain 381 SNPs encoding 17 genes.
GBIGM
In a case–control study, we assume that there are Ncase cases and Ncontrol controls. For arbitrary two genes G1 and G2, there are k SNPs in gene G1 and t SNPs in gene G2. The framework of the GBIGM is described as follows.
The detailed description is as follows.
Step 1: Compute the entropy H0 of initial data set
In the initial data set D, the samples are divided either cases or controls, and p(case) is the proportion of cases in D, which is calculated
 |
H(•) is defined as a classic entropy function, and the entropy H0 of D can be defined as
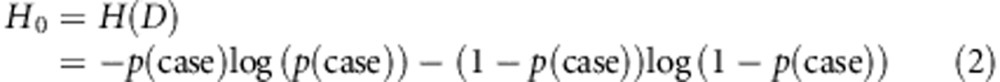 |
Step 2:
(1) Compute the conditional entropy H1 and information gain rate ΔR1 for gene G1
The k SNPs in gene G1 are quantified as X,
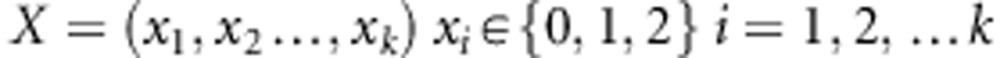 |
The conditional entropy H1, information gain IG(D\X) and information gain rate ΔR1 for gene G1 are defined as follows:
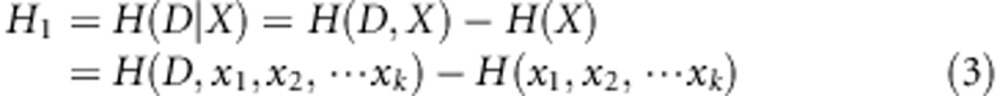 |
 |
 |
Here the conditional entropy H1 of D conditioned on X can be calculated as the difference between joint entropy of D and X and entropy of X. For any D and X, H(D\X)≤H(D) constant sets up. So the information gain IG(D\X) describes the difference value of entropies conditioned on X or not, which can reflect the importance of X. The information gain rate ΔR1 is a normalized information gain.
(2) Compute the conditional entropy H2 and information gain rate ΔR2 for gene G2
The t SNPs in gene G2 are quantified as Y,
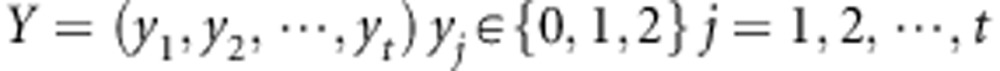 |
Similarly, the conditional entropy H2, information gain IG(D\Y) and information gain rate ΔR2 for gene G2 are defined as follows:
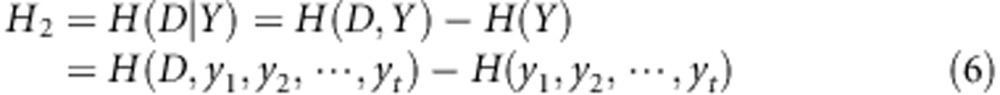 |
 |
 |
Step 3: Compute conditional entropy H1,2 and information gain rate ΔR1,2 for gene G1 and G2
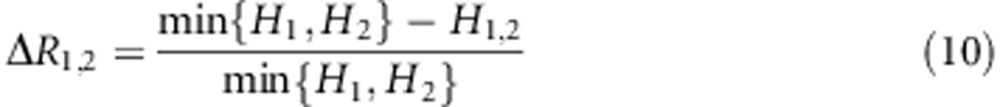 |
ΔR1,2 is the normalized difference between the entropy conditioned on X and Y and the entropy conditioned on X or Y (the minimum between entropy conditioned on X and entropy conditioned on Y). It represents the normalized information gain while considering both X and Y and considering only X or Y. Therefore, the larger the ΔR1,2, the larger probability of epistasis between X and Y.
In the initial data, we calculate statistics ΔR1,2 and denote it as ΔR1,22.
Step 4: Perform relabeling and generate new data set
As the model does not assume any distribution of the data, it is difficult to use conventional parametric test for significant inference. In this study, we use a displacement detection test method (Permutation)35, 36 for detecting significant GGIs. In the permutation test, we relabel the samples to generate a new random case and control groups, then recalculate the statistic, construct the empirical distribution and finally estimate P-values.
Step 5: Repeat steps (2) to (4) m times
For a given number (m) of permutation times, repeat steps (2) to (4) m times. We will obtain m statistics ΔR1,2, and we denote them as 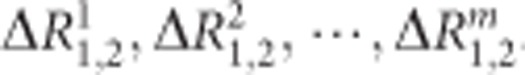 .
.
Step 6: Estimate P-value
Define the null hypothesis, alternative hypothesis and significance level.
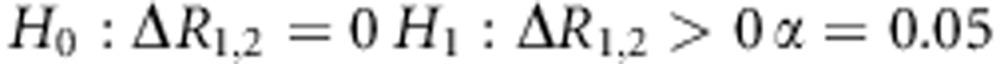 |
While we perform the permutation, the random samples are following the null hypothesis H0. Therefore, according to m statistics from random permutation samples, we can get the experience sampling distribution (empirical distribution) for the statistics ΔR1,2 following the null hypothesis H0.
We count the number of statistics ΔR1,2i that will be equal to or greater than ΔR1,20.
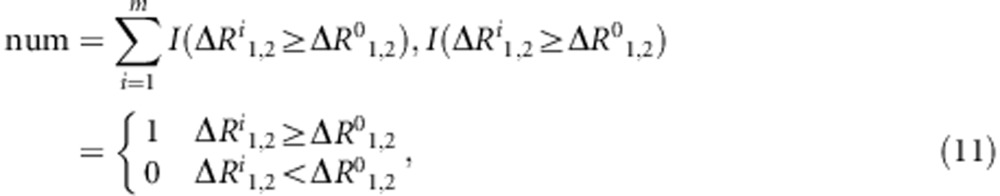 |
here /(·) is an indicator function.
Then the P-value can be estimated as
 |
SBEM
The SBEM has been used to detect SSIs.12, 13 We use one SNP as a representative in a gene each time, then calculate the entropy-based statistic and estimate the significance similar to GBIGM; at last we select the most significant SSI result as the significance of GGI. So SBEM is a univariate SNP method and GBIGM is a multiple SNPs method. The detailed description of SBEM can be found in the paper of Dong et al.12
KCCU
The kernel canonical correlation-based U-statistic model (KCCU)23, 24 can reflect nonlinear relationship between two genes and is a useful and classic method. In KCCU, the maximum kernel canonical coefficient of the two genes is taken as a measure of GGI in cases and controls. Let the genotyped data of case–control study be 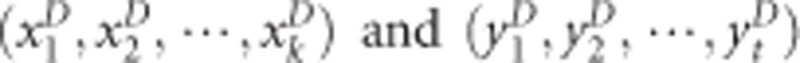 for gene G1 and gene G2 for cases, and
for gene G1 and gene G2 for cases, and 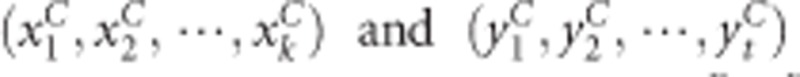 for controls. The maximum kernel canonical coefficient krD between
for controls. The maximum kernel canonical coefficient krD between 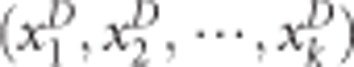 and
and 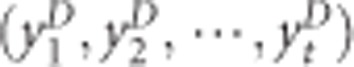 obtained through kernal canonical correlation analysis (KCCA) could be considered as a measurement of gene-based GGI in cases and krc between
obtained through kernal canonical correlation analysis (KCCA) could be considered as a measurement of gene-based GGI in cases and krc between 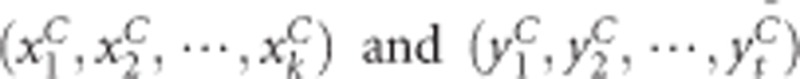 be a measurement of GGI in controls. After a transformation to krD and krc analogous to Fisher's simple correlation coefficient transformation, we can obtain a KCCU statistic. The detailed description of KCCU can be found in the paper of Yuan et al.23
be a measurement of GGI in controls. After a transformation to krD and krc analogous to Fisher's simple correlation coefficient transformation, we can obtain a KCCU statistic. The detailed description of KCCU can be found in the paper of Yuan et al.23
Evaluation indexes
To test the effectiveness of GBIGM, we select power and false positive rate (Type I error probability) pa as evaluation indexes, and perform a comparative analysis with SBEM12 and KCCU.23
Power
The power of a statistical test is the probability that it correctly rejects the null hypothesis when the null hypothesis is incorrect (the alternative hypothesis is true). In this study, we performed the simulation m (100) times. The power is the frequency of rejection of the null hypothesis case in the positive data sets (the alternative hypothesis H1 is true) under a certain significance level (α=0.05). As the total numbers of tests are different among SBEM, GBIGM and KCCU, we use 2 different formulas to calculate the power.
For SBEM, when testing the GGIs, we need to compute all the SSIs between the two genes. Therefore, we need to make a multiple testing adjustment. Here we made use of the Bonferroni adjustment method.37 Assume the significance level α=0.05, gene G1 has k SNPs, gene G2 has t SNPs, then there are a total of k × t SNP pairs to be tested. For each SNP pair, we simulate m times to get P-values. If the number of P-values less than α is m1, the power of SBEM can be calculated as follows:
 |
For GBIGM and KCCU, if the number of the P-values less than α is m2, the power can be calculated as follows:
 |
False positive rate P α
False positive rate Pα is the probability that it falsely rejects the null hypothesis when the null hypothesis is true, and it is also known as the probability of committing a Type I error. At a specific significance level (α=0.05), we perform the simulation 100 times with each different sample sizes when the null hypothesis is true. The observed Pα changes when the sample size changes from small to large. When Pα is stable near the significance level α, we consider that the sample size is large enough to obtain a robust test result.
Results
Power
The effect of OR
When OR value changes from 1.2 to 2.2, the power of GBIGM, SBEM and KCCU are indicated as Figure 1.
Figure 1.

The power comparison among three different GGIs detecting methods under different disease models and different OR values. The horizontal axis is the odd ratios ranging in 1.2 to 2.2 and the vertical axis is the power obtained from the three methods. There are 8 different disease models from a to h.
With the increasing OR value, the power of GBIGM, SBEM and KCCU are monotonously increasing under seven different genetic models except the recessive–recessive model. The power of GBIGM is significantly higher than the power of SBEM and KCCU under seven different genetic models except the recessive–recessive model. When the OR value reaches 1.8, under the five disease models of the dominant–dominant model, a special interaction model, multiplicative–multiplicative model, exclusive OR model and additive–additive model, the power of GBIGM has reached 80%, whereas the power of SBEM and KCCU are only about 40%. Under the recessive–recessive model, when the OR value gradually changes from 1.2 to 2.2, the power of the three GGIs detecting models are consistently below 20%, which indicates that these three GGIs detecting models are unsuitable when the disease data conform with the recessive–recessive model.
The effect of sample size
When sample size changes from 1000 to 5000, the power of GBIGM, SBEM and KCCU are shown in Figure 2.
Figure 2.

The power comparison among three different GGIs detecting methods under different disease models and different sample sizes. The horizontal axis is the sample size ranging in 1000 to 5000 and the vertical axis is the power obtained from the three methods. There are 8 different disease models from a to h.
With an increase in the sample size, the power of GBIGM, SBEM and KCCU are monotonously increasing under seven different genetic models except the recessive–recessive model. The power of GBIGM is significantly higher than the power of SBEM and KCCU under seven different genetic models except the recessive–recessive model. When the sample size reaches 4000, under the six disease models of the dominant–dominant model, a special interaction model, multiplicative–multiplicative model, exclusive OR model, addictive–addictive model and threshold model, the power of GBIGM have reached 80%, whereas the power of SBEM and KCCU are only about 40%. Under the recessive–recessive model, when the sample size gradually changes from 1000 to 5000, the power of the three GGIs detection model are consistently below 20%, which indicates that these three GGIs detecting models are unsuitable when the disease data conforms with the recessive–recessive model.
The effect of prevalence rate
When the prevalence rate changes from 0.01 to 0.20, the power of GBIGM, SBEM and KCCU are shown in Figure 3.
Figure 3.

The power comparison among three different GGIs detecting methods under different disease models and different prevalence rates. The horizontal axis is the prevalence rate ranging in 0.01 to 0.2 and the vertical axis is the power obtained from the three methods. There are 8 different disease models from a to h.
From the results, we can see that there are no significant correlations between the power and prevalence rate, which indicate these GGIs detecting methods are not influenced from the prevalence rate. In the recessive–recessive model, the addictive–addictive model, the multiplicative–multiplicative model and the exclusive model, the power of GBIGM are significantly higher than the power of SBEM and KCCU. There are no significant differences between the power of these three methods in other models.
From the comparison of the power of these methods under different disease models and parameter settings, we conclude that all of these three methods are unsuitable for detecting GGIs when the interactions are the recessive–recessive model type. In other disease types, GBIGM is significantly powerful than SBEM and KCCU.
False positive rates
Set significance level α=0.05, when the sample size gradually changes from 1000 to 5000, the false positive rate Pα of GBIGM are shown in Table 2.
Table 2. False positive rate of the GBIGM in different sample sizes.
|
Sample size |
|||||
|---|---|---|---|---|---|
| Model | 1000 | 2000 | 3000 | 4000 | 5000 |
| Dominant–dominant model | 0.02 | 0.05 | 0.05 | 0.05 | 0.03 |
| Recessive–dominant model | 0.04 | 0.04 | 0.06 | 0.05 | 0.04 |
| Recessive–recessive model | 0.05 | 0.04 | 0.04 | 0.04 | 0.04 |
| A special interaction model | 0.07 | 0.05 | 0.05 | 0.06 | 0.04 |
| Additive–additive model | 0.80 | 0.78 | 0.78 | 0.77 | 0.76 |
| Multiplicative–multiplicative model | 0.04 | 0.05 | 0.04 | 0.06 | 0.05 |
| Exclusive OR model | 0.08 | 0.06 | 0.04 | 0.06 | 0.04 |
| Threshold model | 0.09 | 0.08 | 0.06 | 0.05 | 0.08 |
When the sample size changes from 1000 to 5000 gradually, false positive rate Pα are stable nearby the significance level α=0.05 under seven different genetic models except the additive–additive model. It indicates that GBIGM is unsuitable for detecting GGIs when the interaction is the additive–additive model type. The results confirmed the stability of the model in most disease models.
GBIGM does not depend on any statistical distribution or model, so it could detect both the linear relationship and the nonlinear relationship between two genes. Integrate the power analysis and false positive rate analysis, we believe that GBIGM we proposed is a valid and robust method for detecting GGIs under six disease models except additive–additive model and recessive–recessive model.
Applications in RA data
In these data, the genes and SNP numbers in these genes are shown in Table 3. The numbers of SNPs in genes vary from 2 to 128. We applied GBIGM, SBEM and KCCU in this data set for detecting GGIs related to RA.
Table 3. The description of genes and SNP numbers in RA data.
| Gene | Chromsome | SNP number |
|---|---|---|
| PADI1 | 1 | 3 |
| PADI2 | 1 | 7 |
| PADI4 | 1 | 5 |
| PADI6 | 1 | 6 |
| PRKD3 | 2 | 7 |
| GC | 4 | 12 |
| GLRX | 5 | 2 |
| CDSN | 6 | 5 |
| PSORS1C1 | 6 | 14 |
| TXNDC5 | 6 | 128 |
| CA1 | 8 | 38 |
| BUB3 | 10 | 3 |
| SORBS1 | 10 | 10 |
| VDR | 12 | 19 |
| SERPINA1 | 14 | 5 |
| PCSK6 | 15 | 74 |
| DNAH9 | 17 | 43 |
In GBIGM and KCCU, we set the significance level α=0.05, and obtained 5 (3.68%) and 76 (55.88%) significant GGIs, respectively. In SBEM, for each gene–gene pair, we took the minimum SNP–SNP P-value as the gene–gene P-value, and we obtained 123 (90.44%) significant GGIs at the significance level α=0.05 or 74 (54.41%) significant GGIs at the significance level α=0.001. The detailed results are presented in Supplementary Table 1. As the total number of gene–gene pair is 136, we obtained too many significant results when using SBEM and KCCU. The GBIGM method we proposed can significantly decrease the number of significant results, which is very important for the biological verification.
Of the five significant GGIs detected by GBIGM, they were also detected in KCCU, and the ranks of P-values were 33, 38, 24, 10 and 13. Especially, in these 17 genes, there was only one gene (PADI4) confirmed to be related to RA by OMIM.38, 39 We detected a potential interaction between PADI4 and BUB3 in GBIGM (P-value=0.038, rank 4) and KCCU (P-value=9.62E–13, rank 10), but not in SBEM at the significance level α=0.001 (P-value=0.034, rank 118). The ranks of these five significant GGIs in the three different methods are shown in Table 4. Therefore, the gene-based methods, especially the GBIGM method we proposed, have the potential to be more powerful than the SNP-based methods.
Table 4. The ranks of the five significant GGIs in the results of three methods.
|
Rank |
||||
|---|---|---|---|---|
| Gene 1 | Gene 2 | GBIGM | KCCU | SBEM |
| GC | PRKD3 | 1 | 33 | 53 |
| PADI1 | PRKD3 | 2 | 38 | 1 |
| CDSN | SERPINA1 | 3 | 24 | 86 |
| BUB3 | PADI4 | 4 | 10 | 118 |
| PADI6 | SERPINA1 | 5 | 13 | 81 |
Discussion
The GBIGM we proposed used the information gain as a statistic to detect the interactions between genes in case–control studies. As a nonparametric method, our model could detect both the linear relationship and the nonlinear relationship between two genes. Compared with SBEM and KCCU in the simulated data, the power of GBIGM proposed are larger than the others in the most cases. The method we proposed is stable to the sample size through the test of false positive rates. It is a suitable and powerful tool for detecting GGIs for most disease models except recessive–recessive model (other methods are also not suitable) and additive–additive model (high false positive rates). Compared with the other methods, GBIGM can obtain fewer significant results, and it is important for biologists to perform biological verification. We built an online analysis platform of GBIGM for the scientists using (http://nclab.hit.edu.cn/GBIGM).
Acknowledgments
This work was supported in part by the National Natural Science Foundation of China (61271346, 61300116, 61172098, 61402132 and 91335112), Specialized Research Fund for the Doctoral Program of Higher Education of China (20112302110040), Fundamental Research Funds for the Central Universities (HIT.KISTP.201418), Natural Science Foundation of Heilongjiang Province (QC2013C063) and the Fund of Heilongjiang Education Department (12531298 and 12531299).
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary Material
References
- Balding DJ: A tutorial on statistical methods for population association studies. Nat Rev Genet 2006; 7: 781–791. [DOI] [PubMed] [Google Scholar]
- Zheng G, Meyer M, Li W, Yang Y: Comparison of two-phase analyses for case-control genetic association studies. Stat Med 2008; 27: 5054–5075. [DOI] [PubMed] [Google Scholar]
- Visscher PM, Hemani G, Vinkhuyzen AA et al: Statistical power to detect genetic (co)variance of complex traits using SNP data in unrelated samples. PLoS Genet 2014; 10: e1004269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardon LR, Bell JI: Association study designs for complex diseases. Nat Rev Genet 2001; 2: 91–99. [DOI] [PubMed] [Google Scholar]
- Maher B: Personal genomes: the case of the missing heritability. Nature 2008; 456: 18–21. [DOI] [PubMed] [Google Scholar]
- Phillips PC: Epistasis—the essential role of gene interactions in the structure and evolution of genetic systems. Nat Rev Genet 2008; 9: 855–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RA: The correlation between relatives on the supposition of Mendelian inheritance. Trans R Soc Edinb 1918; 52: 35. [Google Scholar]
- Cockerham CC: An extension of the concept of partitioning hereditary variance for analysis of covariances among relatives when epistasis is present. Genetics 1954; 39: 859–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempthorne O: The correlation between relatives in a random mating population. Proc R Soc Lond B Biol Sci 1954; 143: 102–113. [PubMed] [Google Scholar]
- Cordell HJ: Epistasis: what it means, what it doesn't mean, and statistical methods to detect it in humans. Hum Mol Genet 2002; 11: 2463–2468. [DOI] [PubMed] [Google Scholar]
- Schwender H, Ickstadt K: Identification of SNP interactions using logic regression. Biostatistics 2008; 9: 187–198. [DOI] [PubMed] [Google Scholar]
- Dong C, Chu X, Wang Y et al: Exploration of gene-gene interaction effects using entropy-based methods. Eur J Human Genet 2008; 16: 229–235. [DOI] [PubMed] [Google Scholar]
- Kang G, Yue W, Zhang J, Cui Y, Zuo Y, Zhang D: An entropy-based approach for testing genetic epistasis underlying complex diseases. J Theor Biol 2008; 250: 362–374. [DOI] [PubMed] [Google Scholar]
- Ritchie MD, Hahn LW, Roodi N et al: Multifactor-dimensionality reduction reveals high-order interactions among estrogen-metabolism genes in sporadic breast cancer. Am J Human Genet 2001; 69: 138–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu JS: Bayesian inference of epistatic interactions in case-control studies. Nat Genet 2007; 39: 1167–1173. [DOI] [PubMed] [Google Scholar]
- Jiang X, Barmada MM, Visweswaran S: Identifying genetic interactions in genome-wide data using Bayesian networks. Genet Epidemiol 2010; 34: 575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Liu CT, Zhang M, Zhang H: A forest-based approach to identifying gene and gene gene interactions. Proc Natl Acad Sci USA 2007; 104: 19199–19203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz DF, Konig IR, Ziegler A: On safari to Random Jungle: a fast implementation of random forests for high-dimensional data. Bioinformatics 2010; 26: 1752–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo CL, Liew MJ, Mohamad MS, Salleh AH: A review for detecting gene-gene interactions using machine learning methods in genetic epidemiology. Biomed Res Int 2013; 2013: 432375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upstill-Goddard R, Eccles D, Fliege J, Collins A: Machine learning approaches for the discovery of gene-gene interactions in disease data. Brief Bioinform 2013; 14: 251–260. [DOI] [PubMed] [Google Scholar]
- Peng Q, Zhao J, Xue F: A gene-based method for detecting gene-gene co-association in a case-control association study. Eur J Human Genet 2010; 18: 582–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waaijenborg S, Zwinderman AH: Sparse canonical correlation analysis for identifying, connecting and completing gene-expression networks. BMC Bioinformatics 2009; 10: 315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Z, Gao Q, He Y et al: Detection for gene-gene co-association via kernel canonical correlation analysis. BMC Genet 2012; 13: 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson NB, Jenkins GD, Larson MC et al: Kernel canonical correlation analysis for assessing gene-gene interactions and application to ovarian cancer. Eur J Human Genet 2014; 22: 126–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson NB, Schaid DJ: A kernel regression approach to gene-gene interaction detection for case-control studies. Genet Epidemiol 2013; 37: 695–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Yang X, Yuan Z et al: A PLSPM-based test statistic for detecting gene-gene co-association in genome-wide association study with case-control design. PLoS One 2013; 8: e62129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Zhao J, Yuan Z, Zhang X, Ji J, Xue F: A powerful latent variable method for detecting and characterizing gene-based gene-gene interaction on multiple quantitative traits. BMC Genet 2013; 14: 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon CE: A mathematical theory of communication. Bell Syst Tech J 1948; 27: 45. [Google Scholar]
- Shannon CE, Weaver W: The Mathematical Theory of Communication. Univ of Illinois Press: Champaign, IL, USA, 1949. [Google Scholar]
- Thorisson GA, Smith AV, Krishnan L, Stein LD: The International HapMap Project Web site. Genome Res 2005; 15: 1592–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International HapMap CInternational HapMap C, Frazer KA International HapMap C, Ballinger DG et al: A second generation human haplotype map of over 3.1 million SNPs. Nature 2007; 449: 851–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Reich J: A complete enumeration and classification of two-locus disease models. Hum Hered 2000; 50: 334–349. [DOI] [PubMed] [Google Scholar]
- Li J, Chen Y: Generating samples for association studies based on HapMap data. BMC Bioinformatics 2008; 9: 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett T, Wilhite SE, Ledoux P et al: NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res 2013; 41: D991–D995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Q, Soerensen M, Kruse TA, Christensen K, Christiansen L: A novel permutation test for case-only analysis identifies epistatic effects on human longevity in the FOXO gene family. Aging Cell 2013; 12: 690–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry KJ, Johnston JE, Mielke PWJr: Analysis of trend: a permutation alternative to the F test. Percept Mot Skills 2011; 112: 247–257. [DOI] [PubMed] [Google Scholar]
- Dunn OJ: Multiple comparisons among means. J Am Statist Assoc 1961; 56: 52–64. [Google Scholar]
- Schorderet DF: Using OMIM (On-line Mendelian Inheritance in Man) as an expert system in medical genetics. Am J Med Genet 1991; 39: 278–284. [DOI] [PubMed] [Google Scholar]
- McKusick VA: Mendelian inheritance in man and its online version, OMIM. Am J Hum Genet 2007; 80: 588–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.