

Abstract
It remains poorly understood how the haematopoietic stem/progenitor cells (HSPC) are attracted to their niches and the functional consequences of such interaction. In the present study, we show that the cell cycle regulator cyclin A1 in association with vascular endothelial growth factor receptor 1 (VEGFR1), is required for HSPC and their niches to maintain their function and proper interaction. In the absence of cyclin A1, the HSPC in the BM are increased in their frequency and display an increased migratory and homing ability. Concomitantly, the ability of the endosteal and central BM niche zones to attract and home the wild-type HSPC is significantly reduced in cyclin A1-null mice as compared to the wild-type controls. The impaired proliferation and homing of HSPC in the BM of cyclin A1-null mice are attributed to the increased density of microvessels in the endosteal and central BM niche zones, which is associated with the increased VEGFR1 expression. Thus, modulation of cyclin A1 and VEGFR1 in HSPC and their niches may provide new insights into therapeutic approaches.
Keywords: bone marrow transplantation, cyclin A1, haematopoietic stem and progenitor cells, homing and migration, VEGFR1
Introduction
In adult BM, HSPC fulfill the function in self-renewal, proliferation and differentiation during homeostasis and after external injury. HSPC are not randomly distributed in the BM, but rather reside in specialized microenvironments referred to as niches.1-5 Both HSPC and their niches are necessary to generate the cells that are needed to replenish the blood and immune systems.1,5-7 At least 2 niche zones for HSPC in the BM anatomical regions have been described: the endosteal and the central BM niche zones.5,8-10
It is known that HSPC are located near bone surfaces in the endosteal niche zone or in perivascular sites adjacent to endothelium of blood vessels in the central BM zone.7,8 The presence of osteoblastic signaling in the endosteal zone, but the absence of this signaling in the central BM zone appears to be the major functional difference between these 2 BM zones.6 Further, HSPC preferentially and consistently trans-endothelially migrate and home to the endosteal zone of the BM of recipient mice after transplantation.9
The endosteal niche zone is the region along bone surfaces which are enriched in bone cells and microvessels; the central BM niche zone is the region excluded from the bone surfaces and is enriched with marrow vascularity and blood vessels.11,12 Each niche zone is characterized by specific cell types including endothelial cells, osteoblasts and mesenchymal stromal cells, all of which have been described.11,13 A recent study reports that HSPC preferentially migrate and home to the endosteal niches of recipient mice after transplantation.14 However, it remains unknown which types of niche cells may attract HSPC to preferentially home to the endosteal niche zones. Increasing evidence suggests that the endothelial cells promote HSPC homing via specific adhesion molecules to migrate close to bone on a competitive basis.11 VEGFR1 is expressed by the endothelial cells of blood vessels and contributes to homing and dormancy of HSPC.7,15-17 VEGFR1 plays an important role in vascular niches.17–20 Since the marrow vessels expressing VEGFR1 form a network that connect the bone and marrow, which in turn create specific environments for HSPC.11 It is thus interesting to investigate whether VEGFR1 may play an important role in regulation of interaction of HSPC with their BM niches.
The mammalian cell cycle regulator cyclin A1 is specifically expressed in male germ cells and in BM cells.21-23 Cyclin A1 expression has also been detected in endothelial and osteoblast cell lines.24,25 Cyclin A1 is highly expressed in leukemia, cancers of testes, breast, lung and prostate.26-30 Further, cyclin A1 is specifically expressed in acute myeloid leukemic stem cells, and it is of interests to design targeted immunotherapy to selectively target leukemic stem cells that overexpress cyclin A1.31,32 Targeted overexpression of cyclin A1 in early myeloid cells results in pathogenesis of acute myeloid leukemia in transgenic mice.33 Perturbations in niche signaling have been shown to enhance the stem cell mobilization and change the cellular compositions in BM niches, which are associated with pathological conditions such as myeloproliferative disorders.34
In the present study we show that cyclin A1 is associated with VEGFR1, and has an important function in regulating proliferative, migratory and homing ability of HSPC, and their interaction with the endosteal and central BM niches. Cyclin A1 and VEGFR1 play an important role in regulation of microvessels of the endosteal and central BM niche zones, Our study also provides a molecular basis for a potential therapeutic opportunity to protect HSPC and their niches for bone marrow transplantation.
Results
Loss of cyclin A1 function increases HSPC numbers in endosteal and central BM zones
Cyclin A1 expression was detected in stem cell enriched fraction of mouse BM: Lin−c-Kit+Sca1+(LSK)-IL7α− cells designated as LSK-HSPC, and in Lin−Sca-1+lymphoid progenitors (Fig. 1A). Cyclin A1 protein expression was detected in endothelial cells of perivascular vessels, which expressed CD31, a cell surface marker for blood vessels (Fig. 1B). Immunohistochemical analysis further confirmed that cyclin A1 was expressed in the endothelial cells and osteoblasts within the BM micro-anatomical regions: the proximal/distal epiphyses and metaphysic zones (Fig. 1B, C and Fig. S1). To study cyclin A1 function in haematopoietic cells and their BM niches in vivo, we established mice (Ccna1−/−) in which Ccna1allelesencoding for cyclin A1 were deleted. We assessed the number and frequency of different subpopulations of haematopoietic cells in the BM of cyclin A1-deficient mice (Ccna1−/−) and age-matched wild-type controls (Ccna1+/+) by FACS analysis (Fig. 1D and E). The absolute numbers and frequency of the stem cell-enriched LSK-HSPC were significantly increased in the BM of Ccna1−/− mice by 49% and 33% compared with that of Ccna1+/+ mice (Fig. 1F, mean frequency of LSK-HSPC in Lin−cells per Ccna1+/+ mouse = 0.84%; mean LSK-HSPC per Ccna1−/− mouse = 1.11%, difference = 0.27%; 95% CI = 0.08% to 0.13%, n = 19 mice of each genotype, p = 0.003). The frequency of LSK-HSPC-enriched Lin− cells in the BM of Ccna1−/− mice was higher than that of Ccna1+/+ mice (mean Lin− cells per Ccna1+/+ mouse = 39.58%, mean Lin− cells per Ccna1−/− mouse = 48.68%; difference = 4.1%; 95% CI: 7.3 to 6.78%; n = 25 mice of each genotype, p = 0.01) (Fig. S2). While the frequency of the more differentiated lineages including myeloid progenitors (MP), the lymphoid progenitors (LP) and the common lymphoid progenitors (CLP) in Ccna1−/− mice was similar to that in Ccna1+/+ mice (Fig. 1F). There was no significant difference in the frequency of LSK-HSPC, MP and LP in the spleen between Ccna1−/− and Ccna1+/+ mice (Fig. 1G). These data suggest that cyclin A1 may play a role in maintaining proper numbers of HSPC in the BM.
Figure 1.
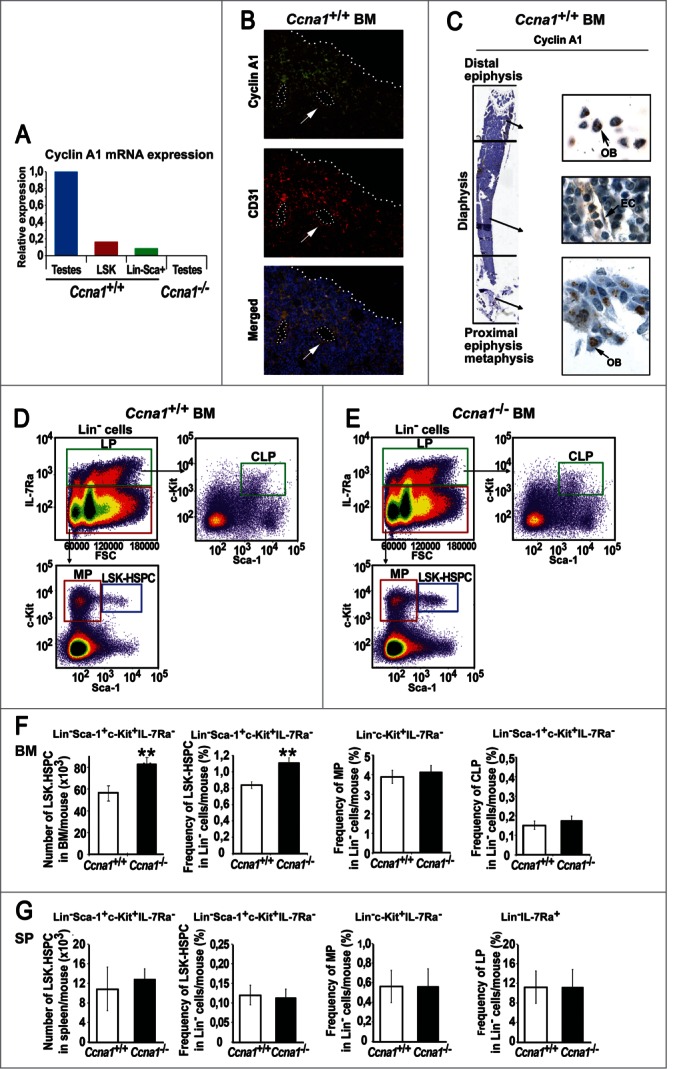
Loss of cyclin A1 function results in the increased numbers of HSPC in the BM of Ccna1−/− mice. (A) Expression of cyclin A1 mRNA in sorted Lin−Sca-1+c-Kit+ HSPC (LSK), Lin−Sca-1+ lymphoid progenitors, testis tissues (testes) from Ccna1+/+ mice and testis tissues (testes) from Ccna1−/− mice was determined using semi-quantitative RT-PCR. Relative expression from 3 independent experiments is shown. (B) Representative photographs show the distribution of cyclin A1 in endothelial cells of perivascular blood vessels that are stained positive for CD31, as determined by immunofluorescence analysis. Antibody against cyclin A1 was conjugated with Alexa Fluor 488 (green) and antibody to CD31 was conjugated Alexa Fluor 594 (red), 4′,6-Diamidino-2-phenylindole (DAPI) showing the nucleus staining is in blue. Cells that are co-stained with cyclin A1 and CD31 are indicated as “Merge.” (C) Representative pictures of the femur long bone of a Ccna1+/+ mouse, stained with antibody against cyclin A1. The micro-anatomic zones including proximal, distal epiphyses, metaphysis and diaphysis regions are indicated. Osteoblasts (OB) and endothelial cells (EC) are indicated. (D and E) Representative FACS plots of isolated BM Lin− cells from Ccna1+/+ and Ccna1−/− mice are stained and sorted with the cell surface markers as indicated. (F) Total number and frequency of subpopulations of BM cells per mouse that were quantified by FACS analysis are shown in the graphs. Data represent mean values + SEM (n = 19 pairs of mice from each genotype). (G) Total numbers and frequency of subpopulations of haematopoietic cells from spleen (SP) per mouse that are quantified by FACS analysis are shown. Data represent mean values + SEM (n = 3 pairs of mice from each genotype). The statistically significance is indicated by “*.” One “*” indicates that P ≤ 0.05, Two “**” indicates that P ≤ 0.01.
It is known that HSPC are located in the central BM zone and the endosteal zone within the BM, and both of the niche zones are enriched with perivascular blood vessels.11,12 As mentioned above, cyclin A1 expression was detected in endothelial cells of perivascular blood vessels and in osteoblasts of the bone surfaces in the BM. We next assessed whether loss of cyclin A1 function my affect the frequency of LSK-HSPC residing in the BM niche zones. Using flow cytometry, the frequencies of LSK-HSPC harvested from the endosteal and central BM niche zones in Ccna1−/− and Ccna1+/+ mice were assessed (Fig. 2A). The frequency of LSK-HSPC in both endosteal zone and central BM zone in Ccna1−/− mice was increased by 31% and 26% as compared with that of Ccna1+/+ mice (Fig. 2B–E, mean frequency of LSK-HSPC in Lin- cells from the endosteal zone/per Ccna1+/+ mouse = 0.78%; mean frequency of LSK-HSPC in Lin- cells from the endosteal zone/per Ccna1−/− mouse = 1.02%, difference = 0.24; 95% CI = 0.13 to 0.24%, p = 0.018; mean frequency of LSK-HSPC in Lin- cells from the central BM zone /per Ccna1+/+ mouse = 0.94%; mean frequency of LSK-HSPC in Lin- cells from the central BM zone /per Ccna1−/− mouse = 1.19%, difference = 0.24; 95% CI = 0.14 to 0.18%, p = 0.01). This data show that cyclin A1 plays a role in maintaining LSK-HSPC numbers within the endosteal and vascular niches.
Figure 2.
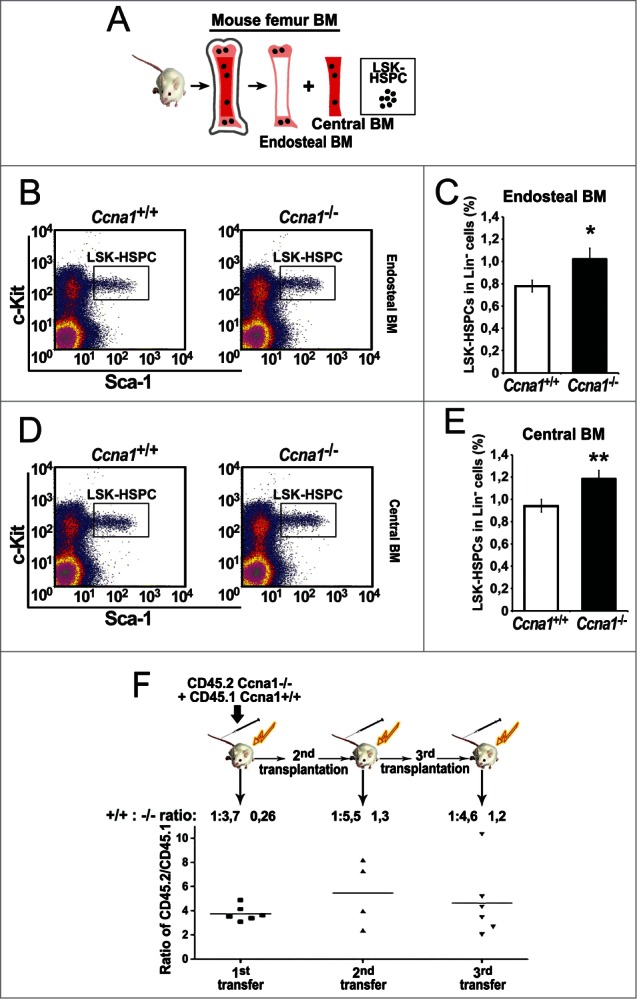
The frequency of HSPC is increased in the endosteal and central BM regions from Ccna1−/− compared with that of litter-mate Ccna1+/+ mice. Ccna1−/− HSPC display increased proliferation rate compares with that of Ccna1+/+ HSPC. (A) schematic illustration of the procedures to isolate and sort LSK-HSPCs from endosteal and central BM regions. (B and D) Representative FACS raw data from the assessment of LSK-HSPCs in the endosteal vs. central BM zones from Ccna1−/− and Ccna1+/+ littermate controls are shown. (C and E) The frequency of LSK-HSPCs within Lin− population in the endosteal regions in (C) and in the central BM region in (E) from Ccna1−/− and Ccna1+/+ mice is shown. Mean frequency of LSK-HSPC in the endosteal zone per Ccna1+/+ mouse = 0.78%, mean frequency of LSK-HSPC in the endosteal zone per Ccna1−/− mouse = 1.02%, difference = 0.24, 95% CI = 0.13 to 0.24%, p = 0.018, mean frequency of LSK-HSPC in the central BM zone per Ccna1+/+ mouse = 0.94%, mean frequency of LSK-HSPC per Ccna1−/− mouse = 1.19%, difference = 0.24, 95% CI = 0.14 to 0.18%, p = 0.01. (n = 3 mice of each genotype). (F) A cartoon depicting the procedures of assessing the competitive repopulation ability of Ccna1−/− BM using serial BM transplantations is shown in the upper panel. The competitive transplantation assay is shown in the lower panel. The recipients from the primary transplantation were found to have a mixed percentage of donor chimeras, with an average ratio of CD45.2:CD45.1 being 3.7:1 (+0.26) at 4 months post-transplantation. The ratios of CD45.2:CD45.1 for secondary: 5.5:1 (+1.3) and tertiary rounds of serial transplantation: 4.6 (+1.2) are indicated. (n = 4–6 recipients per round of transplantation). Data represent average CD45.2:CD45.1 ratios + SEM.
We next asked if the observed differences in HSPC between Ccna1−/− and Ccna1+/+ mice may be accounted for the altered proliferation of HSPC or their susceptibility to apoptosis. Cell cycle analysis using short-term BrdU incorporation in vivo assay was performed. There was a higher proportion of mitotic cells, suggesting that the increased frequency of LSK-HSPC in Ccna1−/− BM may be associated with the increased proliferation (Fig. S3A). We next assessed whether the increased frequency of LSK-HSPC may be related to the apoptosis status. We assessed apoptosis status in the LSK-HSPC, MP, LP and Lin− cells from Ccna1−/− and Ccna1+/+ mice under steady-state conditions. The subpopulations were isolated and labeled with antibodies against the phenotypic cell surface markers in combination with Annexin-V and 7AAD staining followed by FACS analysis. There was no significant difference between genotypes in the detection of early apoptosis (Annexin-V+/7AAD-) vs. late apoptosis and necrosis (Annexin-V+/7AAD+) in LSK-HSPC, MP, LP and Lin− cells between genotypes (Figure S3B–E). Further, mRNA expression of the second A-type cyclin, Ccna2, encoding cyclin A2 remained unchanged in Ccna1−/− mice suggesting that cyclin A2 does not compensate for loss of cyclin A1 (Supplementary Fig. 4). Thus, cyclin A1 function is important for maintaining the proper number and frequency of HSPC in the specialized BM niche zones.
Cyclin A1-deficient HSPC show an increased ability to proliferate in irradiated host BM after long-term transplantation
To further investigate whether loss of cyclin A1 gene indeed affected the number and frequency of HSPC, we performed long-term competitive bone marrow transplantation assays. BM cells from Ccna1−/− (CD45.2) were mixed with wild-type competitor CD45.1 BM cells and transplanted into lethally irradiated recipients (CD45.1) (Fig. 2F). The proportion of Ccna1−/− BM cells was 2-fold higher than wild-type cells in the recipients at 4 months post-transplantation (Fig. 2F). When the HSPC from these mice was used for a secondary transplantation into irradiated recipients, the number of Ccna1−/− BM cells was again 2-fold higher than the wild-type cells in the irradiated recipients after 4 months of secondary transplantation (Fig. 2F). Upon tertiary transplantation, the number of Ccna1−/− BM cells once again displayed 2-fold increase relative to the wild-type cells (Fig. 2F). These data suggest that Ccna1−/− BM cells retained the feature of higher frequency of HSPC over the wild-type after reconstitution experiments.
Increased proliferation capability of Ccna1−/− HSPC is associated with their increased ability to home to their host BM
It is suggested that the increased proliferation capability of HSPC is associated with their enhanced ability to migrate and home to the BM.35,36 We next investigated whether the increased proliferative capability of Ccna1−/− HSPC may impair their homing frequency after BM transplantation. Purified BM Lin− cells of Ccna1−/− mice and Ccna1+/+ mice were stained with a fluorescence dye carboxyfluorescein diacetate succinimidyl ester (CFSE) and were transplanted into lethally irradiated recipient mice (Fig. 3A). The frequency of transplanted donor HSPC that homed to the host BM 18 hours post-transplantation were sorted and assessed by FACS analysis (Fig. 3A). There was a 98% increase in frequency of donor Ccna1−/− HSPC that homed to the host BM, as compared with that of donor Ccna1+/+ HSPC (Fig. 3B, p = 0.012). The number of donor HSPC integrated into the spleen, thymus and lymph nodes was similar between the irradiated genotypes (Fig. S5). It is known that a small number of circulating HSPC after entering blood stream will periodically migrate to their BM niches.35,36 We also transplanted BM Lin− cells of Ccna1−/− mice and Ccna1+/+ mice into non-irradiated recipient mice in which the host BM niches and circulation systems remained intact (Fig. 3C). The Ccna1−/− HSPC also displayed an increased ability to enter into the non-irradiated host BM after 18 hours BM transplantation, and the homing frequency of Ccna1−/− HSPC was 43% higher than that of Ccna1+/+controls (Fig. 3D). The homing frequency index is significantly higher in Ccna1−/− HSPC relative to controls (p = 0.029). While the number of donor HSPC integrated into the spleen, thymus and lymph nodes of non-irradiated host mice was similar between genotypes (Fig. S6). Thus, Ccna1−/− HSPC display alterations in their ability of homing and mobilization. This suggests that cyclin A1 plays an important role in regulation of homing and mobilization of HSPC to their host BM after transplantation.
Figure 3.
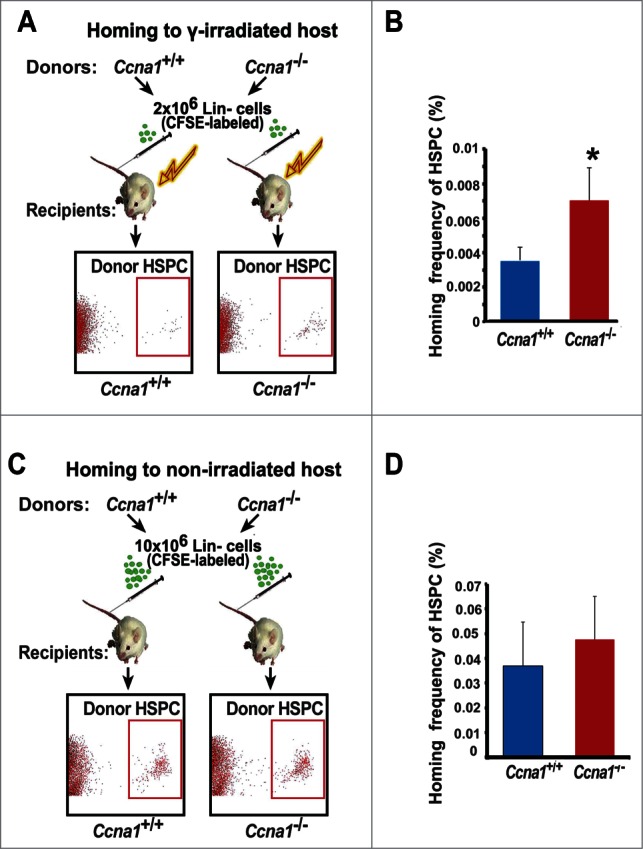
Ccna1−/− HSPC display increased homing and mobilization frequencies compared with that of Ccna1+/+ HSPC. (A, C) A cartoon of the homing assay procedure is depicted. The donor Lin− cells were purified from BM of the Ccna1−/− mice or Ccna1+/+ littermates and were labeled with CFSE dye. For experiments in (A), the recipient mice were lethally irradiated. For experiments in (C), the recipient mice were not treated with irradiation. Representative FACS plots show that donor HSPC are present in the recipient BM. (B and D) The percentage of homed CFSE-positive Ccna1−/− HSPC, referred as homing frequency relative to that of Ccna1+/+ controls in irradiated host BM (p = 0.012) in (B) and in non-irradiated host BM (p = 0.029) in (D) are shown. The data represent mean values + SEM (n = 3) for each genotype for each experiment. Three independent experiments were performed.
The absence of cyclin A1 reduces the ability of endosteal and endosteal zones to attract and home normal HSPC
A large body of evidence suggests that the number and frequency of HSPC are regulated by their specialized niches in the BM.1,5 We hypothesized that the altered frequency of HSPC in Ccna1−/− mice may be associated with alterations in their niches. To assess the function of the BM niches, we performed BM transplantation and assessed ability of the endosteal and central BM zones from Ccna1+/+ mice to host transplanted donor Ccna1+/+ HSPC (Fig. 4A). In agreement with a reported study,14 Ccna1+/+HSPC preferentially homed to the endosteal niches in the BM of recipient Ccna1+/+ mice after BM transplantation (Fig. 4B). However, the ability of the non-irradiated endosteal niches and the central BM niches in Ccna1−/− mice to attract and home Ccna1+/+ HSPC was significantly reduced compared to that of Ccna1+/+mice (Fig. 4C, for endosteal BM zone, p = 0.03, for central BM zone, p = 0.043). This suggests that loss of cyclin A1 results in a reduced ability of endosteal and central BM niche zones to interact with and host transplanted HSPC.
Figure 4.
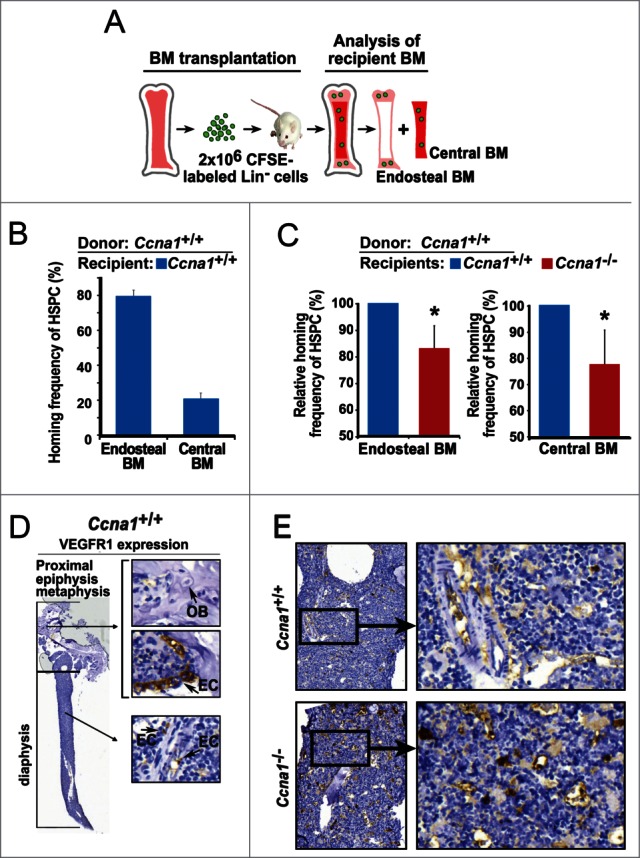
Loss of cyclin A1 function impairs ability of the endosteal and central BM niches to interact with donor Ccna1+/+ HSPC and increased VEGFR1 expression in the BM. (A) A cartoon of the homing assay procedure and assessment of homed Ccna1+/+ HSPC in the endosteal and central BM niches of recipient mice are depicted. (B) Homing frequency of Ccna1+/+ HSPC to the endosteal and central BM niches of recipient Ccna1+/+mice. n = 4 recipient mice for each genotype. Data represent mean values + SEM of 3 independent experiments. (C) Homing frequency of Ccna1+/+ HSPC to the endosteal and central BM niches of recipient Ccna1−/− mice and Ccna1+/+ littermates. n = 4 recipient mice for each genotype. Data represent mean values + SEM of 3 independent experiments. For endosteal BM zone, p = 0.03, for central BM zone, p = 0.043. (D) Representative microphotographs show the immunohistochemical analysis of femur bone sections from a Ccna1+/+ mouse using antibody against VEGFR1. VEGFR1-negative osteoblasts (OB) and VEGFR1-positive endothelial cells (EC) of the blood vessels are indicated by arrows. (E) Representative pictures show VEGFR1 expression in the BM samples from Ccna1−/− and Ccna1+/+ littermate.
Loss of cyclin A1 resulted in an increase in micro-vessel densities accompanied with increased VEGFR1 expression in HSPC and their niche cells
To further investigate whether the altered function of the endosteal and central BM niches in Ccna1−/− mice may be due to the alterations in components that are essential for niche structure and function, we examined a panel of proteins that are important for extracellular matrix and vascularization Among a panels of proteins examined, VEGFR1 expression observed in the endothelial sinuses of the BM from Ccna1+/+ mice as determined by immunohistochemical analysis of multiple bone sections (Fig. 4D). The increased VEGFR1 expression was co-incident with a higher density of endothelial sinuses in the BM of Ccna1−/− mice (Fig. 4E). It has been shown that VEGFR1 is an important component expressed by the endothelial sinuses of the BM which support the homing and dormancy of HSPC.18 Haematopoietic progenitor cells (HPC) from mobilized peripheral blood display enhanced migration and marrow homing compared to steady-state bone marrow HPC. To determine whether VEGFR1 may be expressed in HSPC nearby the endothelial cells of blood vessels, we co-stained the BM sections of Ccna1+/+mice with antibodies against VEGFR1, CD31 and c-kit VEGFR1. It has been shown that the network of vessels interacts with haematopoietic cells in the BM.37 We observed that VEGFR1 was expressed in HSPC and their surrounding endothelial niche cells In the BM of Ccna1+/+ mice (Fig. 5A). VEGFR1 expression was increased in the BM cells of Ccna1−/− mice (Fig. 5A, B). We further applied confocal/multiphoton microscopy combined with immunostaining to visualize HSPC and microvessels that expressed VEGFR1. VEGFR1 expression in Ccna1−/− HSPC and endothelial niche cells from both endothelial and central BM niche zones was remarkably increased compared with that of Ccna1+/+ mice (Fig. 5C). Using confocal/multiphoton microscopy, we observed that both endosteal and central BM niche zones of Ccna1−/− mice displayed an 83.3% increased density of endothelial CD31-positive microvessels compared to that of Ccna1+/+ mice (Fig. 5D and E, p = 0.03). These data suggest that loss of cyclin A1 function is required for regulation of VEGFR1 expression and proper microvessel density in the BM niches.
Figure 5.
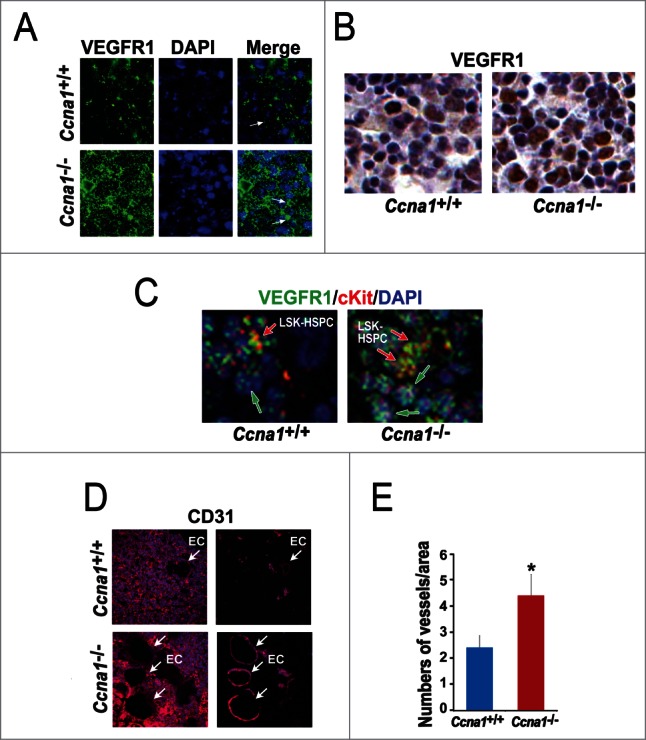
Loss of cyclin A1 function impairs VEGFR1 expression in HSPC and in endothelial cells of blood vessels in both endosteal and central BM niches, which is accompanied by the increased density of blood vessels. (A) Immunofluorescence staining of VEGFR1 in the BM cells of the central BM niches of Ccna1+/+ mice and Ccna1−/− mice. (B) Representative confocal microscopy pictures highlight both HSPC co-stained with antibodies to VEGFR1, c-kit and DAPI (in red, green and blue merged) and non-HSPC only stained with antibody to VEGFR1 and DAPI (in green and blue merged). (C) Similar areas of the BM shown in (B) are assessed by immunohistochemical analysis using antibody against VEGFR1. (D) Representative confocal microscopy pictures of analysis of the endosteal and central BM niches using antibody against CD31 in red. The areas of the BM of Ccna1−/− and Ccna1+/+ mice contain perivascular vessels are shown in the left panel. The vessels in the same area show in the left panel are highlighted and are shown in the right panel. (E) Quantification of the numbers of vessels in the total BM of the Ccna1−/− and Ccna1+/+ mice. The numbers of vessels per microscopic area (x40 amplification) were counted on multiple sections of the BM, p = 0.03 (n = 4 mice each genotype).
The increased VEGFR1 in the endosteal and central BM niches in Ccna1−/− mice is associated with the impaired proliferative capability of HSPC
Next, we wanted to assess whether the elevated level of VEGFR1 in endothelial niche cells of microvessels is responsible for the increased proliferative activity of Ccna1−/− HSPC. As shown in Figure 6A, Ccna1−/− HSPC expressing high level of VEGFR1 are in close contact with their surrounding endothelial niche cells in the BM of Ccna1−/− mice. We hypothesized that the increased proliferative activity in HSPC in the Ccna1−/− BM may be a direct consequence of the increased VEGFR1 expression due to cyclin A1-deficiency. We sorted CD31-positive endothelial cells and LSK-HSPC from the BM of Ccna1−/− mice using FACS (Fig. 6B). We blocked VEGFR1 production in the endothelial cells using neutralizing antibodies against VEGFR1 or matched isotype control. We then assessed whether blockage of VEGFR1 in the endothelial niche cells may affect LSK-HSPC proliferation in the co-culture of these 2 populations (Fig. 6B). After 48 hours of co-culture of LSK-HSPC with the endothelial cells in which VEGFR1 production was blocked, the frequency of LSK-HSPC was significantly reduced in the co-culture down to 23% as compared with that co-cultured with control niche cells (Fig. 6B–D; mean numbers of in control co-cultures = 100.67; mean numbers of LSK-HSPC in VEGFR1-depleted co-cultures = 77.67, difference = 23; 95% CI = 17.97 to 14.56; p = 0.03). This intriguing finding suggests that the increased proliferative activity in HSPC in the Ccna1−/− BM may be a direct consequence of the increased VEGFR1 expression due to cyclin A1-deficiency.
Figure 6.
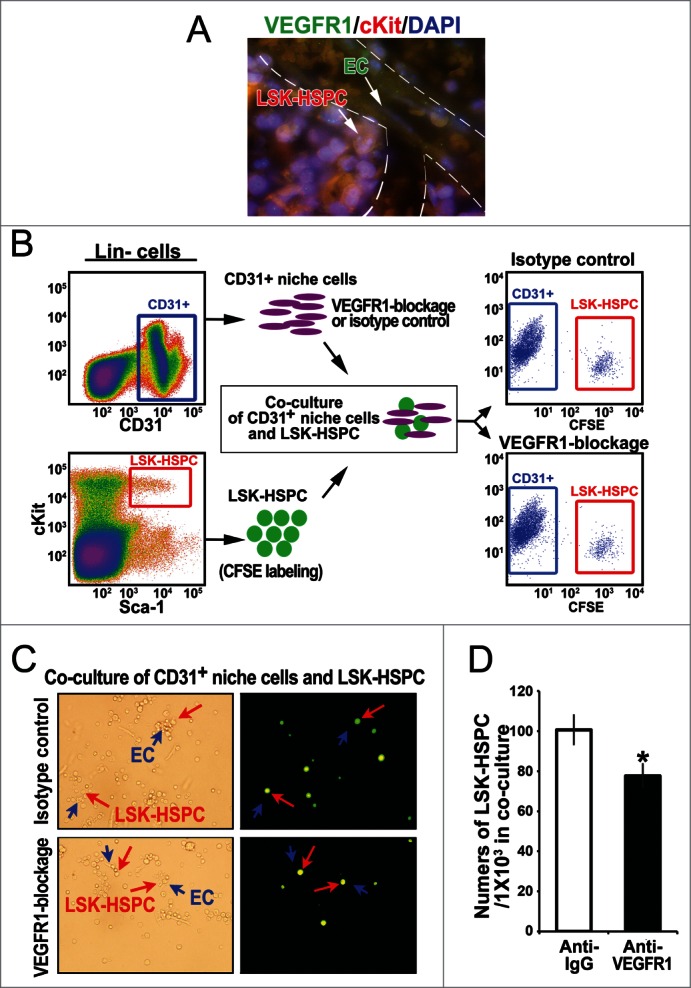
Altered VEGFR1 signaling in the endothelial niche cells is in part responsible for altered proliferation of LSK-HSPC in Ccna1−/− mice. (A) Representative pictures of immunofluorescence analysis of an area contains blood vessels in Ccna1−/− BM using antibodies against VEGFR1 (green), c-kit (red) and DAPI to highlight the nucleus. HSPC co-stained with anti-VEGFR1 and anti-c-kit and DAPI are shown in orange, while the endothelial cells of blood vessels positive only for VEGFR and not c-kit are shown in green, as indicated by arrows. (B) Representative FACS plots of sorted CD31-positive niche cells (CD31+) and sorted LSK-HSPC using FACS Aria sorters are shown in the left panel. A cartoon picture shows that CD31-positive niche cells co-culture with LSK-HSPC. Representative FACS plots of CD31-positive niche cells and LSK-HSPC after co-culture for 48 hours are shown in the right panel. (C) Representative microphotographs show the morphology of the cells after 48 hours of co-culture. The contrast microscopic images show LSK-HSPC in the co-culture with CD31-positive niche cells in the left panel. The immunofluorescence images of the same areas are shown. LSK-HSPC are stained in green and CD31-positive cells are unstained. The arrows indicate these cells at same position as shown in the left panel. LSK-HSPC are indicated by the red arrows and CD31-positive cells in close contact with LSK-HSPC are indicated by blue arrows. In the co-cultures, CD31-positive cells in which IgG isotype control or anti-VEGFR1 were used are indicated. (D) The relative frequency of LSK-HSPC in the co-culture with CD31-positive niche cells with or without VEGFR1 blockage.
Alterations in VEGFR1 signaling due to cyclin A1-deficiency may impair the interactions between HSPC and their niches
It has been shown that VEGFR1 signals may mediate migration of HSPC.16 Next, we hypothesize that the increased homing and migratory ability of Ccna1−/− HSPC in vivo may be a direct consequence of the increased VEGFR1 expression due to cyclin A1-deficiency. To this end, we first assessed the ability of sorted LSK-HSPC from Ccna1−/− and Ccna1+/+mice to migrate toward the BM chemo-attractants using migration assays. LSK-HSPC from Ccna1−/− mice displayed an increased migratory ability by 214% compared to that of Ccna1+/+ mice (Fig. 7A, mean percentage of migrated Ccna1+/+ LSK-HSPC = 16.21%; mean percentage of migrated Ccna1−/− LSK-HSPC = 50.96%, difference = 34.74; 95% CI = 18.32 to 42.88%, p = 0.016). Next, we blocked VEGFR1 production in LSK-HSPC sorted from the BM of Ccna1−/− and Ccna1+/+ mice. Blocking VEGFR1 signaling in Ccna1−/− LSK-HSPC significantly reduced their initially increased migration rate down to 52% compared to that of isotype controls (Fig. 7B, p = 0.038). Further, Ccna1−/− LSK-HSPC displayed a reduced ability to attach to fibronectin, a component of the BM niches, down to 99.9% to that observed in Ccna1+/+ LSK-HSPC (Fig. 7C). These data suggest that alteration in VEGFR1 signaling contributes to the altered homing and mobilization of Ccna1−/− HSPC and impairs ability of LSK-HSPC to interact with their niche components. Taken together, VEGFR1 may form a molecular network together with cyclin A1 to mediate the interaction between HSPC and their niches (Fig. 7D).
Figure 7.
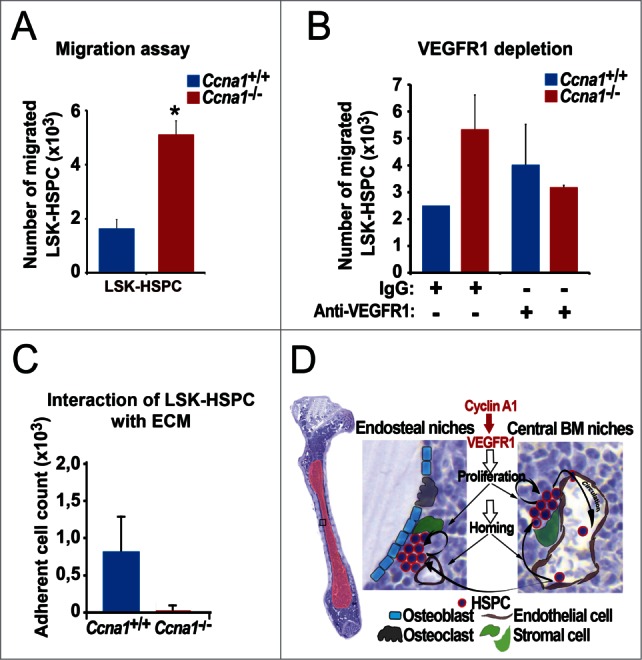
Altered VEGFR1 signaling in LSK-HSPC is in part responsible for altered migration and mobilization of LSK-HSPC in Ccna1−/− mice. (A) Migration assay to assess the migratory ability of sorted LSK-HSPCs cells from Ccna1−/− and Ccna1+/+ mice in in vitro is shown. Mean number of migrated Ccna1+/+ LSK-HSPC = 1621 per 10000 total cells applied in the migration assay; mean number of migrated Ccna1−/− LSK-HSPC = 5096, difference = 3475 cells, 95% CI = 1832 to 4288, p = 0.016. Data represent mean values + SEM from 3 independent experiments (n = 6 mice of each genotype). (B) Migration assay to evaluate the involvement of VEGFR1 in mediating the migratory ability of HSPCs from Ccna1−/ and Ccna1+/+ mice. For the blockage of VEGFR1 production, 1 µg/ml of anti-VEGFR1, or 1 µg/ml IgG antibody was used. p = 0.038. (C) Assessment of the interaction of LSK-HSPC from Ccna1−/ and Ccna1+/+ mice with fibronectin, an extracellular matrix component (ECM). The numbers of cells that are attached to the fibronectin-coated surface are shown. Data represent mean values + SEM from 2 independent experiments. (D) A schematic model to illustrate cyclin A1 function in the regulation of proliferation, homing and mobilization of HSPC and their interaction with the endothelial niche cells of perivascular vessels in the endosteal niches and central BM niches. Cyclin A1 regulates interaction of HSPC with their niches through VEGFR1. In both endosteal niches and central BM niches, endothelial niche cells of blood vessels that produce VEGFR1 may have a dominant and stimulatory influence on proliferation, migration and homing of HSPC under homeostatic conditions.
Discussion
In the present study, we show that cyclin A1 is an important regulatory factor that mediates interaction of HSPC with their niches in the BM. Loss of cyclin A1 function in HSPC results in: (i) an increase in numbers and frequency of HSPC in the BM; (ii) increased ability of HSPC to migrate and home to the host BM after BM transplantation; (iii) decreased ability of HSPC to interact with extra cellular matrix in the BM. Conversely, loss of cyclin A1 function in the endothelial niche cells results in: (i) increased level of VEGFR1 expression in the endothelial vessels; (ii) increased numbers of micro-vessels in the endosteal and central BM zones; (iii) decreased ability of the BM to engraft HSPC. Thus, loss of cyclin A1 function is associated with defects in HSPC behaviors such as migration, proliferation and regeneration in the BM.
Previous studies have shown that cyclin A1 is an important cell cycle regulator that is specifically expressed in male germ cells and in BM cells.21-23 However, expression pattern of cyclin A1 in HSPC vs. niche cells, and its function in haematopoietic system have not been reported. Our finding in the present study reveals that cyclin A1 is expressed in HSPC and the endothelial niche cells in the BM of mice. Cyclin A1 function is required for maintaining proper frequency and numbers of HSPC in the BM. The BM of Ccna1−/− mice contains significantly higher numbers of HSPC compared with the controls. When Ccna1−/− HSPC was used together with Ccna1+/+ HSPC for transplantation into irradiated recipients, the number of Ccna1−/− BM cells was 2-fold higher than Ccna1+/+ cells in the irradiated recipients after series of transplantations. The proportion of mitotic cells was increased within Ccna1−/− HSPC compared with that of Ccna1+/+ HSPC. It is suggested that higher numbers of HSPC is a result of their increased proliferative activity, and the active proliferating HSPC may be more prone to DNA damages and apoptosis induced by cytotoxic agents.43 Although we did not detect severe phenotype in haematopoietic system in Ccna1−/− mice under steady state condition. Our unpublished data show that Ccna1−/− mice suffer the early death compare with Ccna1+/+ mice after exposure to high-dose irradiation.
The homing and lodging of HSPC into the host BM are highly coordinated events, and HSPC preferentially home to the endosteal and vascular niches.38 It has been reported that several key adhesion molecules and chemokine receptors such as VLA-4 or CXCR4 which are expressed by HSPC mediate homing and lodging of HSPC, as these receptors are able to adhere and bind to their ligands that are present within the BM niches.14,11 In the present study, we show that Ccna1−/− HSPC display an increased ability to migrate and home to the irradiated/non-irradiated host BM as compare to Ccna1+/+ controls. It is likely that loss of cyclin A1 function leads to an alteration in the chemokine receptor signaling, thus alters the migration and homing rates of HSPC to the BM.
It has been shown that HSPC are preferentially maintained in vascular and perivascular domains of the BM sinusoidal network.14,38,39 Consistent with previous reported studies, we also show that HSPC are located adjacent to blood vessels in both endosteal and central BM zones. However, the vascular structure and microvessel density are altered in the BM of Ccna1−/− mice. We further show that the ability of the endosteal and central BM niches in Ccna1−/− mice to engraft Ccna1+/+ HSPC is significantly reduced compare to that of Ccna1+/+ mice. Since cyclin A1 is also expressed in endothelial niche cells of the vascular vessels in the BM, it is likely that loss of cyclin A1 function may also affect proliferation of endothelial niches cells, and leads to the altered microvessel density of the BM niches.
The niches for HSPC in different regions of the BM remain incompletely defined.3 It is believed that vascular volume in the BM region may have impact on the homing of HSPC.11 However, the factors that regulate vascular volume in the BM remain to be identified. In the present study, we observed that the increased microvessel density was co-incident of increased VEGFR1 expression in the endothelial niche cells in the Ccna1−/− BM. To define the functional relationship between cyclin A1 and VEGFR1 in regulating interactions of HSPC with their niches, we performed co-culture experiment in which purified Ccna1−/− CD31-positive endothelial cells were co-cultured with Ccna1−/− LSK-HSPC cells. We found that blockage of VEGFR1 production in the endothelial cells resulted into decreased numbers of Ccna1−/− LSK-HSPC in the co-culture as compared to that of the controls. This suggests that cyclin A1 regulates vessels density and interactions of HSPC with endothelial niche cells via VEGFR1-dependent signaling. It is known that VEGFR1 binds to several ligands including PlGF, VEGF-A, and VEGF-B.40 Further, circulating soluble VEGFR1 and VEGFR2 significantly attenuated VEGF-induced neovascularization.41,42 In the present study, VEGF expression level in BM HSPC and endothelial cells remains similar between Ccna1−/− mice and Ccna1+/+ mice as determined by immunohistochemical analysis and western blot analysis, however, VEGFR1 expression is significantly increased in both HSPC and endothelial cells in Ccna1−/− BM. It is possible that the increased production of VEGFR1 may be the causative factor that contributes to the alteration in microvessel density. Alternatively, the increased VEGFR1 expression may be the consequence of the altered proliferation of the endothelial cells caused by cyclin A1-deficiency. As summarized in Figure 7D, our finding provides new evidence that endothelial vessels are one of the important components of the BM niches, and cyclin A1 and VEGFR1 are involved in regulating proliferation and trafficking of HSPC between the endosteal niches and the central BM niches within the BM. Our findings support the hypothesis that specific molecular signaling from the endothelial vessels influence proliferation and migration of HSPC.43 Our previous reported studies showed that cyclin A1 promoted invasion of tumor cells by modulating VEGF expression.29,30 Our findings in the present study further highlights the functional importance of cyclin A1 and VEGFR1 signaling in hematopoiesis and may provide new insights into therapeutic approaches. Future studies may be conducted to further address the mechanisms on how cyclin A1 and VEGFR1 are interconnected in haematopoietic system.
Materials and Methods
Mice and PCR genotyping
Mice homozygous for a targeted disruption of the Ccna1 gene 44 were generated. Ccna1−/−mice and Ccna1+/+ mice in C57BL/6 inbred background were established and used. Genotyping to identify mutant or wild type alleles was done by polymerase chain reaction (PCR)-based analysis as previously described.38 Both females and males at 12–14 weeks of age were used. All procedures were performed under ethical permission and were in accord with guidelines of the Swedish Regional Animal Care and Well-fare Committee and the Institutional Animal Care and Use Committee of Columbia University Medical Center.
Real-time RT-PCR and cDNA amplification
To examine the expression of Ccna1 in the sorted mouse BM populations, total cellular RNA was isolated from total BM, LSK-HSPC or Lin−Sca-1+ cells and subjected to RT-PCR analysis. PCR with primers specific for full-length mouse Ccna1 (forward primer: 5′-CATATGATGCATCGCCAGAGCTCCA-3′, reverse primer: 5′-CCCGGGTCACTGCAGGGGAAGAACTA-3′) (Invitrogen, Carlsbad, CA, USA). Thermocycling programs consisted of 94°C for 2 min followed by 31 cycles of 94°C for 30 sec, 58°C for 30 sec and 68°C for 2 min and 68°C for 10 min. Quantitative PCR was performed with a 7300 Real Time PCR System (Applied Biosystems, Carlsbad, CA, USA). PCR products of cyclin A1 of approximately 1.2 kb in size were excised and sequenced. All assays were run in either duplicate or triplicate for each individual sample and repeated.
Purification of subpopulations by fluorescence-activated cell sorting (FACS)
BM was isolated from tibias and femurs. To deplete lineage-positive (Lin+) cells, the EasySep Mouse Haematopoietic Progenitor Enrichment Cocktail Kit (StemCell Technologies, Vancouver, British Columbia, Canada) was used according to the manufacturer's protocol. In brief, Lin+ cells were specifically labeled with dextran-coated magnetic microparticles using biotinylated antibodies against cell surface antibodies including anti-CD4, CD8, CD3, CD11b, CD19, Mac1, CD45R/B220, GR1 and TER119, and bispecific Tetrameric Antibody Complex (TAC). Populations of LSK-HSPC (Lin−IL-7Ra−Sca-1+c-kit+, LSK) were isolated by flow cytometric cell sorting (FACS) as detailed below. Isolated Lin− cells from BM and spleen were further stained with phycoerythrin–Cy7 (PE-Cy)-conjugated anti-Sca-1, phycoerythrin (PE)-conjugated anti-c-kit/CD117, Alexa Fluor 647-conjugated anti-IL-7Rα cocktail and fluorescin isothiocyanate (FITC)-conjugated Annexin V and 7AAD (BD PharMingen of BD Biosciences, San Jose, CA, USA). The myeloid progenitors (MP): Lin−IL-7Ra−Sca1−c-Kit+ population, lymphoid progenitors (LP): Lin−IL-7Ra+ cells, and common lymphoid progenitors (CLP): Lin−IL-7Ra+sca-1+c-Kit+ cells were also sorted and isolated on FACS Aria sorters (Becton Dickinson, Mountain View, CA, USA). The data were analyzed using FCS Express software (DeNovo Software, Los Angeles, CA, USA). The total number of HSPC per mouse was calculated based on the frequency of this population within the Lin- cells as described.45
Competitive BM transplantation assay
Competitive repopulation assays were modified from previously described assays by Szilvassy et al.40 Age- and sex-matched B6.SJL CD45.2 Ccna1−/− mice were used as a source of competitor marrow cells. Age- and sex-matched B6.SJL CD45.1 Ccna1+/+ recipient mice (Taconic, Germantown, NY, USA) were lethally irradiated with 2 doses of 5.5 Gy administered 3 hour apart using a 137Cs source at a dose rate of 1Gy/min. A total of 2 × 106 BM cells were injected into the tail vein of recipient mice at a ratio of 2:1 CD45.2 Ccna1−/−: CD45.1 Ccna1+/+. Four months post transplantation, recipients were sacrificed and BM was harvested for a second round of transplantation. Secondary transplant recipients were monitored for 4 months, sacrificed and then BM was harvested for a third round of competitive transplants. Tertiary recipients were monitored for 4 months and then sacrificed. Four to 6 recipients were used for each round of transplantation. Beginning 8 weeks after transplantation, donor contribution to overall hematopoiesis was determined by using 100 µl of peripheral blood obtained from the retro-orbital plexus. After red blood cell lysis with EL lysis solution (Qiaqen, Sollentuna, Sweden), donor and competitor hematopoiesis were analyzed by co-staining cells with FITC-conjugated CD45.1 (Ccna1+/+) and PECy7-conjugated CD45.2 (Ccna1−/−) antibodies.
BM homing assays
The donor BM Lin− cells were obtained from Ccna1−/− mice and wild-type littermate controls. In order to monitor the donor cells, the cells were labeled with carboxyfluorescein diacetate, succinimidyl ester (CFSE) (Invitrogen, Stockholm, Sweden) according to the manufacturer's instructions. For BM transplantation to irradiated recipients, recipient mice were lethally irradiated with 2 doses of 5 Gy administered 2 hour apart using a 137Cs source at a dose rate of 1Gy/min. After irradiation, the recipient mice received 2 × 106 of donor Lin− cells labeled with CFSE via intravenous tail injection. The recipient mice were euthanized for tissue isolation at either 6 or 18 hours post-transplantation. For BM transplantation to non-irradiated recipients, 1 × 107donor BM Lin− cells labeled with CFSE were transplanted to mice via tail vein administration. The recipient mice were euthanized after 18 hours. Homing efficiency is defined as the ratio of either CFSE+ events in the recipient BM divided by the total number of transplanted donor cells. In both assays, BM, spleen, thymus and lymph node cells were removed from recipient mice at the time points mentioned. CFSE–positive mononuclear single cell suspensions were prepared from the tissues and were stained with Annexin V/7AAD. The proportion of the donor cells labeled with CFSE and cell viability were assessed by flow cytometry on a CyAn ADP instrument (Beckman Coulter, Miami, FL, USA).
Immunofluorescence and immunohistochemical analysis
Bones (femur) were collected and fixed overnight in zinc formalin (Histolab Inc., Gothenborg, Sweden), decalcified in Formic Acid Bone Decalcifier (Histolab Inc., Gothenborg, Sweden) for 48 hours, dehydrated, and embedded in wax (Histolab Inc.,). After deparaffinization, rehydration and epitope unmasking, sections were stained with polyclonal antibodies against cyclin A1 at 1:250, C-kit at 1:200, or VEGFR1 at 1:500 (Santa Cruz Biotechnology Inc., California, USA) and c-Kit at 1:100 (BioLegend, San Diego, CA, USA). Secondary anti-rabbit conjugated to Alexa Fluor 488 (Invitrogen, Stockholm, Sweden) and anti-donkey conjugated to Rhodamine (Chemicon International Inc., Temecula, CA, USA) or anti-goat conjugated to FITC antibodies (Invitrogen, Stockholm, Sweden) and 4′,6-Diamidino-2-phenylindole counterstain (SERVA Electrophoresis GmbH, Heidelberg, Germany) was used to visualize cell nuclei. The slides were examined under an Olympus AX70 fluorescent microscope and Zeiss LSM 700 confocal microscope (Carl Zeiss Microscopy GmbH, Jena, Germany). For immunohistochemical analysis, the staining procedure was performed using a semiautomatic staining machine (Ventana ES, Ventana Inc., Tucson, AZ).
Cell cycle analysis
Cell cycle analysis of LSK-HSPC was performed in an in vivo assay, mice were injected with 1 mg (200 µl) of 5-bromodeoxyuridine (BrdU) (Sigma-Aldrich, St. Louis, MO, USA) intraperitoneally (IP) 50 min prior to being sacrificed and BM was harvested as described above. BM cells were stained with linage-specific and LSK-specific antibodies as described above. Subsequent staining with FITC-conjugated BrdU and PerCP-conjugated Annexin V and 7-AAD was performed according to the BrdU Flow Kit Staining Protocol (BD Biosciences, San Jose, CA, USA). The cells were then subsequently subjected to cell cycle analysis on a BD LSR II (Becton Dickinson, Mountain View, CA, USA) or FACS Calibur (Becton Dickinson, Mountain View, CA, USA). The data was assessed using FlowJo or FCS Express software (DeNovo Software, Los Angeles, CA, USA). Cell cycle was also performed in an in vitro assay. 1 × 104 of isolated LSK-HSPC cells of Ccna1−/− mice and wild-type littermates were washed with PBS and fixed in 70% ice-cold ethanol at −20°C overnight. Fixed cells were centrifuged at 400×g and washed with PBS. The cells were stained with 50 µg/ml propidium iodide (Sigma-Aldrich, Stockholm, Sweden), 0.1% Tritin X-100 (Sigma-Aldrich, Stockholm, Sweden), 100 µg/ml RNAse-A (AppliChem, Kongens Lyngby, Danmark) for 40 min at RT in the dark, and the PI-elicited fluorescence of individual cells was measured using flow cytometry (FACS Calibur, Becton Dickinson, Mountain View, CA, USA). The data was assessed using FCS Express software (DeNovo Software, Los Angeles, CA, USA).
Apoptosis assay
For measuring the apoptosis, purified Lin− cells from BM or the spleen were co-stained with cell surface markers, Annexin-V conjugated with fluorescin isothiocyanate (FITC) and 7-AAD dye. Subsequently, the stained cells were subjected to FACS analysis for the determination of early and late apoptotic populations in LSK-HSPC and lineage-specific subpopulations. The flow cytometric analysis was performed using CyAn ADP (Beckman Coulter, Miami, FL, USA) or FACS Aria cell analyzers (Becton Dickinson, Mountain View, CA, USA).
Cell migration assay and antibody blockage
LSK-HSPC sorted by FACS (1 × 104 cells) were suspended in 300 µL of serum-free IMDM medium (PAA laboratories, Pasching, Austria). The cells then were then placed into the trans-well migration chambers using Cultrex Transwell Cell Migration Assays (Trevigen Inc., Gaithersburg, MD, USA) were used by following the manufacturer's instructions. IMDM media supplemented with 20% serum was used as chemo-attractant in the bottom chamber. The chambers were incubated at 37°C in a presence of 5% CO2 in a humidified environment for 18 hours. The cells that migrated through the transwells were stained with calcein-AM and the fluorescence intensity was measurement on a Tecan Infinite-200 microplate reader (Tecan Group Ltd, Männedorf, Switzerland) at 485 nm excitation and 520 nm emission. The proportion of migrated cells was calculated using standard curves. For the blockage of VEGFR1 production, antibody-based neutralization was performed, and 200 ng/ml of anti-VEGFR1, or equal amount of IgG antibody was used as previously described.46
The effect of cyclin A1-deficiency on the interaction of LSK HSPC with extracellular matrix fibronectin
A 96-well plate was coated overnight at 4°C with Fibronectin (AMS Biotechnology, Abingdon, UK) diluted in Dulbecco's PBS at final concentration 50 μg/ml in volume 100 μL. As negative controls, wells were incubated with Dulbecco's PBS only. After coating, the wells were washed 3 times with 1× PBS and 2% BSA in 1x PBS were added to the wells and incubated for 1 hour at 37°C. 1 × 104 LSK HSPC were used for adherent assays. The recombinanat fibronectin at final concentration 50 μg/ml was used to coat the wells (AMS Biotechnology, Abingdon, UK). The adhered cells were then washed and stained with Calcein-AM for measuring the adherence on a Tecan Infinite-200 microplate reader (Tecan Group Ltd, Männedorf, Switzerland) at 485 nm excitation and 520 nm emissions.
VEGFR blockage in CD31-positive niche cells and co-culture of niche cells with LSK-HSPC
LSK-HSPC or CD31-positive niche cells were sorted by FACS. For the blockage of VEGFR1 protein in CD31-positive cells, antibody-based neutralization was performed, and 200 ng of anti-VEGFR1, or IgG antibody was used as previously described.47 5 × 104 CD31-positive cells were plated together with 1 × 104 CFSE-labeled LSK-HSPC in IMDM medium in 96-well plates. The cells were cultured for 48 to 72 hours followed by FACS and cell counting analyses.
Statistical analysis
Statistical analysis was performed using the paired Student t test (Graphpad Software, San Diego, CA) and the Mann-Whitney U test; a P value of <0.05 was considered significant. The p values used for comparison of variables between groups of patients were based on the Student's t-test with 95% confidence intervals.
All procedures are in accordance with the Helsinki declaration of 1975.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. Boris Reizis for advice on the bone marrow transplantation assays and Elise Nilsson and Per-Anders Bertilsson for technical help. We thank Dr. Anders Bredberg for his help to analyze and interpret data.
Funding
This work was supported by the grants from the Swedish Cancer Society, The Swedish National Research Council, The Swedish Children Foundation, Malmö Hospital Cancer Foundation, Malmö Hospital Foundation and Gunnar Nilsson Cancer Foundation. Crafoord Foundation to JLP and a grant from the NIH (R01 HD034915) to DJW and support for LEB from training grant T32 DK007647. The support of the Russian Government Program of Competitive growth of Kazan Federal University to RM is acknowledged.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Lymperi S, Ferraro F, Scadden DT. The HSC niche concept has turned 31. Has our knowledge matured? Ann N Y Acad Sci 2010; 1192:12-8; PMID: 20392212; http://dx.doi.org/ 10.1111/j.1749-6632.2009.05223.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature 2014; 505:327-34; PMID: 24429631; http://dx.doi.org/ 10.1038/nature12984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morrison SJ, Spradling AC. Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell 2008; 132:598-611; PMID: 18295578; http://dx.doi.org/ 10.1016/j.cell.2008.01.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Scadden DT. The stem-cell niche as an entity of action. Nature 2006; 441:1075-9; PMID: 16810242; http://dx.doi.org/ 10.1038/nature04957 [DOI] [PubMed] [Google Scholar]
- 5.Xie Y, Yin T, Wiegraebe W, He XC, Miller D, Stark D, Perko K, Alexander R, Schwartz J, Grindley JC, et al.. Detection of functional haematopoietic stem cell niche using real-time imaging. Nature 2009; 457:97-101; PMID:19052548; http://dx.doi.org/ 10.1038/nature07639 [DOI] [PubMed] [Google Scholar]
- 6.Ding L, Morrison SJ. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 2013; 495:231-5; PMID: 23434755; http://dx.doi.org/ 10.1038/nature11885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wilson A, Trumpp A. Bone-marrow haematopoietic-stem-cell niches. Nat Rev Immunol 2006; 6:93-106; PMID: 16491134; http://dx.doi.org/ 10.1038/nri1779 [DOI] [PubMed] [Google Scholar]
- 8.Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, Martin RP, Schipani E, Divieti P, Bringhurst FR, et al.. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 2003; 425:841-6; PMID: 14574413; http://dx.doi.org/ 10.1038/nature02040 [DOI] [PubMed] [Google Scholar]
- 9.Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 2005; 121:1109-21; PMID: 15989959; http://dx.doi.org/ 10.1016/j.cell.2005.05.026 [DOI] [PubMed] [Google Scholar]
- 10.Zhang J, Niu C, Ye L, Huang H, He X, Tong WG, Ross J, Haug J, Johnson T, Feng JQ, et al.. Identification of the haematopoietic stem cell niche and control of the niche size. Nature 2003; 425:836-41; PMID: 14574412; http://dx.doi.org/ 10.1038/nature02041 [DOI] [PubMed] [Google Scholar]
- 11.Bianco P. Bone and the hematopoietic niche: a tale of two stem cells. Blood 2011; 117:5281-8; PMID: 21406722; http://dx.doi.org/ 10.1182/blood-2011-01-315069 [DOI] [PubMed] [Google Scholar]
- 12.Yin T, Li L. The stem cell niches in bone. J Clin Invest 2006; 116:1195-201; PMID: 16670760; http://dx.doi.org/ 10.1172/JCI28568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garrett RW, Emerson SG. Bone and blood vessels: the hard and the soft of hematopoietic stem cell niches. Cell Stem Cell 2009; 4:503-6; PMID: 19497278; http://dx.doi.org/ 10.1016/j.stem.2009.05.011 [DOI] [PubMed] [Google Scholar]
- 14.Ellis SL, Grassinger J, Jones A, Borg J, Camenisch T, Haylock D, Bertoncello I, Nilsson SK. The relationship between bone, hemopoietic stem cells, and vasculature. Blood 2011; 118:1516-24; PMID: 21673348; http://dx.doi.org/ 10.1182/blood-2010-08-303800 [DOI] [PubMed] [Google Scholar]
- 15.Forsberg EC, Smith-Berdan S. Parsing the niche code: the molecular mechanisms governing hematopoietic stem cell adhesion and differentiation. Haematologica 2009; 94:1477-81; PMID: 19880773; http://dx.doi.org/ 10.3324/haematol.2009.013730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA, et al.. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005; 438:820-7; PMID: 16341007; http://dx.doi.org/ 10.1038/nature04186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kopp HG, Hooper AT, Avecilla ST, Rafii S. Functional heterogeneity of the bone marrow vascular niche. Ann N Y Acad Sci 2009; 1176:47-54; PMID: 19796232; http://dx.doi.org/ 10.1111/j.1749-6632.2009.04964.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bonig H, Priestley GV, Oehler V, Papayannopoulou T. Hematopoietic progenitor cells (HPC) from mobilized peripheral blood display enhanced migration and marrow homing compared to steady-state bone marrow HPC. Exp Hematol 2007; 35:326-34; PMID: 17258081; http://dx.doi.org/ 10.1016/j.exphem.2006.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dimmeler S, Zeiher AM. Akt takes center stage in angiogenesis signaling. Circ Res 2000; 86:4-5; PMID: 10625297; http://dx.doi.org/ 10.1161/01.RES.86.1.4 [DOI] [PubMed] [Google Scholar]
- 20.Kobayashi H, Butler JM, O'Donnell R, Kobayashi M, Ding BS, Bonner B, Chiu VK, Nolan DJ, Shido K, Benjamin L, et al.. Angiocrine factors from Akt-activated endothelial cells balance self-renewal and differentiation of haematopoietic stem cells. Nat Cell Biol 2010; 12:1046-56; PMID: 20972423; http://dx.doi.org/ 10.1038/ncb2108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ekberg J, Holm C, Jalili S, Richter J, Anagnostaki L, Landberg G, Persson JL. Expression of cyclin A1 and cell cycle proteins in hematopoietic cells and acute myeloid leukemia and links to patient outcome. Eur J Haematol 2005; 75:106-15; PMID: 16004607; http://dx.doi.org/ 10.1111/j.1600-0609.2005.00473.x [DOI] [PubMed] [Google Scholar]
- 22.Sweeney C, Murphy M, Kubelka M, Ravnik SE, Hawkins CF, Wolgemuth DJ, Carrington M. A distinct cyclin A is expressed in germ cells in the mouse. Development 1996; 122:53-64; PMID: 8565853 [DOI] [PubMed] [Google Scholar]
- 23.Yang R, Nakamaki T, Lubbert M, Said J, Sakashita A, Freyaldenhoven BS, Spira S, Huynh V, Muller C, Koeffler HP. Cyclin A1 expression in leukemia and normal hematopoietic cells. Blood 1999; 93:2067-74; PMID: 10068680 [PubMed] [Google Scholar]
- 24.Bao L, Odell AF, Stephen SL, Wheatcroft SB, Walker JH, Ponnambalam S. The S100A6 calcium-binding protein regulates endothelial cell-cycle progression and senescence. FEBS J 2012; 279:4576-88; PMID: 23095053; http://dx.doi.org/ 10.1111/febs.12044 [DOI] [PubMed] [Google Scholar]
- 25.Yu S, Sharma R, Nie D, Jiao H, Im HJ, Lai Y, Zhao Z, Zhu K, Fan J, Chen D, et al.. ADAR1 ablation decreases bone mass by impairing osteoblast function in mice. Gene 2013; 513:101-10; PMID: 23123729; http://dx.doi.org/ 10.1016/j.gene.2012.10.068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liao C, Li SQ, Wang X, Muhlrad S, Bjartell A, Wolgemuth DJ. Elevated levels and distinct patterns of expression of A-type cyclins and their associated cyclin-dependent kinases in male germ cell tumors. Int J Cancer 2004; 108:654-64; PMID: 14696091; http://dx.doi.org/ 10.1002/ijc.11573 [DOI] [PubMed] [Google Scholar]
- 27.Shames DS, Girard L, Gao B, Sato M, Lewis CM, Shivapurkar N, Jiang A, Perou CM, Kim YH, Pollack JR, et al.. A genome-wide screen for promoter methylation in lung cancer identifies novel methylation markers for multiple malignancies. PLoS Med 2006; 3:e486; PMID: 17194187; http://dx.doi.org/ 10.1371/journal.pmed.0030486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Syed Khaja AS, Dizeyi N, Kopparapu PK, Anagnostaki L, Harkonen P, Persson JL. Cyclin A1 modulates the expression of vascular endothelial growth factor and promotes hormone-dependent growth and angiogenesis of breast cancer. PLoS One 2013; 8:e72210; PMID: 23991063; http://dx.doi.org/ 10.1371/journal.pone.0072210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wegiel B, Bjartell A, Ekberg J, Gadaleanu V, Brunhoff C, Persson JL. A role for cyclin A1 in mediating the autocrine expression of vascular endothelial growth factor in prostate cancer. Oncogene 2005; 24:6385-93; PMID: 16007189 [DOI] [PubMed] [Google Scholar]
- 30.Wegiel B, Bjartell A, Tuomela J, Dizeyi N, Tinzl M, Helczynski L, Nilsson E, Otterbein LE, Harkonen P, Persson JL. Multiple cellular mechanisms related to cyclin A1 in prostate cancer invasion and metastasis. J Natl Cancer Inst 2008; 100:1022-36; PMID: 18612129; http://dx.doi.org/ 10.1093/jnci/djn214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ekberg J, Landberg G, Holm C, Richter J, Wolgemuth DJ, Persson JL. Regulation of the cyclin A1 protein is associated with its differential subcellular localization in hematopoietic and leukemic cells. Oncogene 2004; 23:9082-9; PMID: 15489899; http://dx.doi.org/ 10.1038/sj.onc.1208090 [DOI] [PubMed] [Google Scholar]
- 32.Ochsenreither S, Majeti R, Schmitt T, Stirewalt D, Keilholz U, Loeb KR, Wood B, Choi YE, Bleakley M, Warren EH, et al.. Cyclin-A1 represents a new immunogenic targetable antigen expressed in acute myeloid leukemia stem cells with characteristics of a cancer-testis antigen. Blood 2012; 119:5492-501; PMID: 22529286; http://dx.doi.org/ 10.1182/blood-2011-07-365890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liao C, Wang XY, Wei HQ, Li SQ, Merghoub T, Pandolfi PP, Wolgemuth DJ. Altered myelopoiesis and the development of acute myeloid leukemia in transgenic mice overexpressing cyclin A1. Proc Natl Acad Sci U S A 2001; 98:6853-8; PMID: 11381140; http://dx.doi.org/ 10.1073/pnas.121540098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lane SW, Scadden DT, Gilliland DG. The leukemic stem cell niche: current concepts and therapeutic opportunities. Blood 2009; 114:1150-7; PMID: 19401558; http://dx.doi.org/ 10.1182/blood-2009-01-202606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bhattacharya D, Czechowicz A, Ooi AG, Rossi DJ, Bryder D, Weissman IL. Niche recycling through division-independent egress of hematopoietic stem cells. J Exp Med 2009; 206:2837-50; PMID: 19887396; http://dx.doi.org/ 10.1084/jem.20090778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Massberg S, Schaerli P, Knezevic-Maramica I, Kollnberger M, Tubo N, Moseman EA, Huff IV, Junt T, Wagers AJ, Mazo IB, et al.. Immunosurveillance by hematopoietic progenitor cells trafficking through blood, lymph, and peripheral tissues. Cell 2007; 131:994-1008; PMID: 18045540; http://dx.doi.org/ 10.1016/j.cell.2007.09.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hooper AT, Butler JM, Nolan DJ, Kranz A, Iida K, Kobayashi M, Kopp HG, Shido K, Petit I, Yanger K, et al.. Engraftment and reconstitution of hematopoiesis is dependent on VEGFR2-mediated regeneration of sinusoidal endothelial cells. Cell Stem Cell 2009; 4:263-74; PMID: 19265665; http://dx.doi.org/ 10.1016/j.stem.2009.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lo Celso C, Fleming HE, Wu JW, Zhao CX, Miake-Lye S, Fujisaki J, Côté D, Rowe DW, Lin CP, Scadden DT. Live-animal tracking of individual haematopoietic stem/progenitor cells in their niche. Nature 2009; 457:92-6; PMID: 19052546; http://dx.doi.org/ 10.1038/nature07434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 2012; 481, 457-62; PMID: 22281595; http://dx.doi.org/ 10.1038/nature10783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yancopoulos GD, Davis S, Gale NW Rudge JS, Wiegand SJ, Holash J. Vascular-specific growth factors and blood vessel formation. Nature 2000; 407:242-8; PMID: 11001067; http://dx.doi.org/ 10.1038/35025215 [DOI] [PubMed] [Google Scholar]
- 41.Jacobi J, Tam BY, Wu G, Hoffman J, Cooke JP, Kuo CJ. Adenoviral gene transfer with soluble vascular endothelial growth factor receptors impairs angiogenesis and perfusion in a murine model of hindlimb ischemia. Circulation 2004; 110:2424-9; PMID: 15477417; http://dx.doi.org/ 10.1161/01.CIR.0000145142.85645.EA [DOI] [PubMed] [Google Scholar]
- 42.Chan CK, Chen CC, Luppen CA, Kim JB, DeBoer AT, Wei K, Helms JA, Kuo CJ, Kraft DL, Weissman IL. Endochondral ossification is required for haematopoietic stem-cell niche formation. Nature 2009; 457:490-4; PMID: 19078959; http://dx.doi.org/ 10.1038/nature07547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yin T, Li L. The stem cell niches in bone. J Clin Invest 2006; 116:1195-201; PMID: 16670760; http://dx.doi.org/ 10.1172/JCI28568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu D, Matzuk MM, Sung WK, Guo Q, Wang P, Wolgemuth DJ. Cyclin A1 is required for meiosis in the male mouse. Nat Genet 1998; 20:377-80; PMID: 9843212; http://dx.doi.org/ 10.1038/3855 [DOI] [PubMed] [Google Scholar]
- 45.Yilmaz OH, Valdez R, Theisen BK, Guo W, Ferguson DO, Wu H, Morrison SJ. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 2006; 441:475-82; PMID: 16598206; http://dx.doi.org/ 10.1038/nature04703 [DOI] [PubMed] [Google Scholar]
- 46.Szilvassy SJ, Humphries RK, Lansdorp PM, Eaves AC, Eaves CJ. Quantitative assay for totipotent reconstituting hematopoietic stem cells by a competitive repopulation strategy. Proc Natl Acad Sci U S A 1990; 87:8736-40; PMID: 2247442; http://dx.doi.org/ 10.1073/pnas.87.22.8736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wegiel B, Ekberg J, Talasila KM, Jalili S, Persson JL. The role of VEGF and a functional link between VEGF and p27Kip1 in acute myeloid leukemia. Leukemia 2009; 23:251-61; PMID: 18987662; http://dx.doi.org/ 10.1038/leu.2008.300 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.