

Abstract
Aims
Retention of low-density lipoprotein (LDL) cholesterol beneath the arterial endothelium initiates an inflammatory response culminating in atherosclerosis. Since the overlying endothelium is healthy and intact early on, it is likely that LDL passes through endothelial cells by transcytosis. However, technical challenges have made confirming this notion and elucidating the mechanisms of transcytosis difficult. We developed a novel assay for measuring LDL transcytosis in real time across coronary endothelial cell monolayers; we used this approach to identify the receptor involved.
Methods and results
Murine aortas were perfused ex vivo with LDL and dextran of a smaller molecular radius. LDL (but not dextran) accumulated under the endothelium, indicating that LDL transcytosis occurs in intact vessels. We then confirmed that LDL transcytosis occurs in vitro using human coronary artery endothelial cells. An assay was developed to quantify transcytosis of DiI-LDL in real time using total internal reflection fluorescence microscopy. DiI-LDL transcytosis was inhibited by excess unlabelled LDL, while degradation of the LDL receptor by PCSK9 had no effect. Instead, LDL colocalized partially with the scavenger receptor SR-BI and overexpression of SR-BI increased LDL transcytosis; knockdown by siRNA significantly reduced it. Excess HDL, the canonical SR-BI ligand, significantly decreased LDL transcytosis. Aortas from SR-BI-deficient mice were perfused ex vivo with LDL and accumulated significantly less sub-endothelial LDL compared with wild-type littermates.
Conclusion
We developed an assay to quantify LDL transcytosis across endothelial cells and discovered an unexpected role for SR-BI. Elucidating the mechanisms of LDL transcytosis may identify novel targets for the prevention or therapy of atherosclerosis.
Keywords: Endothelium, Transcytosis, LDL, SR-BI, Atherosclerosis
1. Introduction
One of the earliest stages in the pathogenesis of atherosclerosis is the deposition of low-density lipoprotein (LDL) cholesterol in the sub-endothelial intima. Remarkably, how circulating LDL traverses the endothelial cell layer remains poorly understood.
In principle, permeability across the endothelium could occur between cells (paracellular transport) or through the cytoplasm of individual cells (transcytosis).1 Early electron microscopic studies of rat arteries perfused with LDL demonstrated its internalization into cellular vesicles and its targeting to either lysosomes or the basolateral membrane of the cell.2 Importantly, LDL was never observed to be penetrating intercellular junctions. Furthermore, the size of LDL3 precludes paracellular passage without the opening of inter-endothelial junctions,4 yet an intact endothelium is observed overlying early atherosclerotic lesions.5 Thus, a number of investigators have suggested that LDL crosses the endothelium by transcytosis.6–8 Others have instead suggested that LDL exits the vasculature at areas of damaged or dividing endothelium, implicating the paracellular route.9,10 However, the rate of endothelial mitosis is vanishingly low (<0.05%),11 and more importantly, there is scant to no in vivo evidence of endothelial apoptosis or denudation early on in human atherosclerosis.5
The alternative route of endothelial transcytosis has been best characterized for the serum protein albumin; receptors on the apical plasmalemma mediate its internalization into membrane invaginations known as caveolae.12 After scission from the surface by a process requiring the GTPase mechanoenzyme dynamin, albumin-bearing vesicles make their way through the cytoplasm and undergo exocytosis at the basolateral cell surface.13 A major constituent of caveolae is the protein caveolin-1, the depletion of which prevents the formation of caveolae and ablates albumin transcytosis.14 Thus, it is intriguing to note that aortic rings from caveolin-1 knockout mice demonstrate a >50% reduction in LDL internalization6 while cav1−/−/ApoE−/− mice are protected from atherosclerosis (compared with ApoE−/− mice) despite having increased plasma LDL.15 This uncoupling of elevated plasma LDL levels and the development of atherosclerosis suggests a defect in the transit of LDL across the endothelial monolayer. This observation is all the more remarkable, because an increase in paracellular leak has been described in caveolin-1 knockout mice,16 implying that the paracellular route cannot account for the accumulation of sub-endothelial LDL. Indeed, overexpression of caveolin-1 specifically in the endothelium caused accelerated atherosclerosis,17,18 although transcytosis of LDL was not examined.
Thus, the route by which LDL crosses the endothelium is controversial, but the data favour transcytosis. It is an important issue since the transcellular transport of LDL is likely to be tightly regulated and amenable to therapeutic manipulation, in contrast to paracellular diffusion. However, there is a paucity of literature on LDL transcytosis and its regulation,7,19 likely due to technical difficulties in its study. Traditional experimental approaches using transwells are problematic, because manipulation of endothelial monolayers often introduces intercellular gaps, confounding analysis of transcytosis.1 Even a basic question like which receptor or receptors are involved is unanswered.
In this work, we report a novel assay that allows LDL transcytosis by single coronary artery endothelial cells to be quantified in real time. Using this approach, we describe an unexpected role for SR-BI.
2. Methods
2.1. Cell culture
In vitro experiments were performed using primary human coronary artery endothelial cells (HCAECs; Lonza, Canada) used in passages 6–9 and serum-starved for 3 h prior to experiments. While different batches of primary cells were used over the course of the project, cells from the same batch were used in individual experiments (e.g. control vs. an experimental condition).
2.2. Transwell transcytosis assay
Biotinylated LDL (Intracel, MD) was added to the upper chamber (50 µg/mL) of a transwell containing confluent HCAECs. Transwells (0.4 µm pore polyester, Costar, Corning, NY, USA) were incubated at 4°C for 10 min to allow LDL binding but not internalization. The transwell was then transferred to a fresh plate and was washed twice with cold PBS. Fresh media were added and cells were incubated at 37°C for 2 h with or without platelet-activating factor (PAF, 10 µM; Sigma-Aldrich, ON, Canada) or fluorescein-Na (1 µg/mL, MW 376 Da; Sigma-Aldrich).
To measure the amount of LDL flux, 100 µL of media was taken from the lower chamber and analysed by ELISA.1 The aliquot of media was added along with 50 µL of 1 µg/mL of horseradish peroxidase–conjugated sheep anti-LDL (Bioss, Burlington, ON, Canada) to a 96-well plate coated in streptavidin (Thermo Scientific, Waltham, MA, USA) at room temperature for 2 h with shaking. The plate was washed with buffer (25 mmol/L Tris, pH 7.2, 150 mmol/L NaCl, 0.05% Tween-20, 0.1% gelatin), then 50 µL of 3,3′,5,5′-tetramethylbenzidine (Thermo Scientific) was added to the well and incubated at room temperature for 15 min. Fifty microlitres of 2 mol/L sulfuric acid was added, and quantification of LDL was determined by measuring absorbance at 450 nm with a ThermoMax Microplate Reader (Molecular Devices, Sunnyvale, CA, USA). A standard curve using known quantities of LDL was used to determine the sensitivity of the assay.
2.3. Total internal reflection fluorescence transcytosis assay
Total internal reflection fluorescence (TIRF) microscopy images were acquired on an Olympus cell TIRF Motorized Multicolor TIRF module mounted on an Olympus IX81 microscope (Olympus, Hamburg, Germany). Samples were imaged using a ×150/1.45 objective with 561 nm excitation lasers and Volocity acquisition software. Unless otherwise indicated, the penetration depth, or the distance into the basal aspect of the cell that is visible by TIRF microscopy, was set at 110 nm. For each cell, 150 TIRF images were acquired at a frame rate of 6.67 per second for a constant duration of ∼22 s. At least 10 randomly selected cells were imaged in each experimental replicate. DiI-labeled human native (non-oxidized) LDL (2 µg/mL) with or without unlabelled LDL or high-density lipoprotein (HDL; Intracel, MD) was added to confluent cells seeded on 25 mm glass coverslips. Cells were put at 4°C to allow binding; after 10 min, cells were rinsed in cold PBS and fresh media (HEPES-buffered RPMI, i.e. HPMI)20 were added followed by immediate imaging with a heated (37°C) stage using a standard cell chamber (Life technologies, Catalog no. A7816). In some experiments, cells were pre-treated with Dyngo4A (30 µM; Abcam, Cambridge) for 30 min.
Quantification of the transcytotic events was performed in a blinded fashion using a vesicular detection and tracking algorithm custom-written and automated in Matlab.21 Using these scripts, images are filtered for noise and local background and individual vesicles identified based on their size (9–36 pixels2) and circularity (>0.2). The tracks of moving vesicles are then identified using a tracking algorithm that identifies vesicle tracks based on a maximum-probability assessment of how closely potential tracks resemble free and superdiffusive Brownian diffusion.22 The resulting tracks are analysed for the speed of vesicular movement, the duration the vesicle is present in the TIRF field, and the degree to which the particles' movement deviates from free Brownian diffusion (γ). Vesicles undergoing fusion with the plasma membrane are identified as those having a γ significantly less than that of an equivalent model population undergoing Brownian diffusion, typically 0 < γ < 0.873,23 and which undergo a decrease in fluorescence intensity over the last two time points of their tracks equivalent to a drop of at least 2.5 SDs of the vesicular intensity over the entire period the vesicle has been tracked. The assay overcomes the limitations of transwell and microscopy assays, as it differentiates actual fusion (transcytotic) events from both diffusion across endothelial junctions and transient interactions of vesicles with the basolateral plasma membrane.23 Under control conditions, cells performed 10–30 transcytosis events during the period of observation.
2.4. Colocalization assay and immunostaining
Cells were incubated with DiI-LDL (2 µg/mL) at 37°C for 10 min and then washed twice with cold PBS. Cells were then fixed with 4% paraformaldehyde (PFA), permeabilized, and blocked. Cells were incubated with an antibody to SR-BI (Santa Cruz Biotechnology, Dallas, Texas, USA) for 1 h followed by incubation with a goat anti-rabbit secondary antibody tagged with Alexa Fluor 488 (Jackson ImmunoResearch, West Grove, PA, USA). Z-stack images were then acquired using a spinning disc confocal microscope (Zeiss Axiovert 200 M microscope; ×60 objective, numerical aperture 1.35). After setting a threshold to exclude background, colocalization of LDL with endogenous SR-BI was calculated using Volocity version 6.0.1 and reported using Manders' coefficient. Further colocalization analysis was performed using ImageJ software (NIH) with the JACoP and Bioformats plugins installed.24,25 The images were cropped to each individual cell and a Pearson correlation coefficient measured for each cell. To determine whether the degree of colocalization was significant, each cell's image was subject to Costes’ analysis24 in which the image is randomized and the Pearson coefficient was calculated for the randomized image. Costes' analysis was performed 100 times per image to produce a statistically representative sample of non-interacting molecules. In separate experiments, 50-fold excess unlabelled LDL was added at the same time as DiI-LDL to demonstrate the specificity of colocalization with SR-BI.
For immunostaining, cells were grown to confluency on coverslips and then fixed with 4% PFA, permeabilized, and blocked. Cells were incubated with antibodies to either occludin or VE-cadherin (Santa Cruz) for 1h, followed by incubation with a goat anti-mouse Alexa Fluor 488 secondary (occludin) or a rabbit anti-goat Alexa Fluor 488 secondary (VE-Cadherin) (Jackson ImmunoResearch). Cells were stained with DAPI, mounted on slides, and imaged by spinning disk confocal microscopy on a Leica DMI6000B system (Wetzlar, Germany). Images were acquired with a z-stack interval of 0.2 µm.
For the time-lapse z-axis imaging, cells grown on coverslips were incubated with DiI-LDL (2 µg/mL) at 4°C for 10 min to allow membrane binding. A total of 20 µg/mL fluorescein lectin (Vector Labs, Burlington, ON, Canada) was added to the media during membrane binding to identify the plasma membrane. At Time 0, cells were rinsed three times with PBS, and then a subset of coverslips was immediately fixed in 4% PFA. The other group was treated with serum-free media and placed in the incubator at 37°C for 30 min, then washed 3× with PBS and fixed. For this time point, 20 µg/mL lectin was added during the final 10 min of LDL uptake followed by fixation. Cells were stained with DAPI, mounted on slides, and imaged by spinning disk confocal microscopy (Zeiss Axiovert 200 M). Images were acquired with a z-stack interval of 0.3 µm. LDL localization was quantified using ImageJ by first measuring the total LDL signal (red) in the image after background subtraction. The proportion of LDL that was intracellular was then calculated by subtracting the LDL signal in contact with the apical membrane [as identified by the lectin (green)] and dividing the remainder (the intracellular signal) by the total signal. Images and quantification are representative of four independent experiments; in each experiment, 10 high-power fields were acquired per condition.
2.5. siRNA and overexpression
To overexpress SR-BI, cells were transfected with SR-BI-GFP26 or GFP alone using Fugene HD (Promega, Madison, WI, USA) in accordance with the manufacturer's instructions. For transcytosis measurement, only cells expressing SR-BI-GFP or GFP alone were imaged by TIRF.
For SR-BI depletion, SR-BI siRNA (Qiagen, Valencia, CA, USA; Hs SCARB1 7, Cat. No. S102777201) or scrambled control siRNA (Qiagen) was transfected into cells using HiPerfect (Qiagen). Transcytosis was quantified 36 h later. Knockdown was confirmed by western blot. Lysates were analysed by SDS–PAGE using 12% polyacrylamide gel. Proteins were transferred to nitrocellulose membranes and then blocked and probed overnight at 4°C with the primary antibody. Blots were washed in PBS with Tween 20, and blots were incubated with horseradish peroxidase–conjugated secondary antibodies for 1 h, washed, and then visualized by enhanced chemiluminescence (Amersham, GE Healthcare, Mississauga, ON, Canada).
2.6. PCSK9 and LDLR
HEK293 cells grown in Dulbecco's Modified Eagle Medium (DMEM) were transiently transfected with a plasmid vector encoding human PCSK927 using FuGENE® HD Transfection Reagent according to manufacturer's instructions. DMEM was changed 12 h after transfection; 24 h later, cell culture supernatants were harvested and incubated with confluent primary HCAEC monolayers for a minimum of 6 h. Treated monolayers were then analysed for LDL transcytosis or harvested for immunoblotting as described above. Antibodies for LDLR and SR-BI were from Santa Cruz.
2.7. Ex vivo assay
Mouse work was performed in accordance with the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, seventh edition, 2011) and was approved by the Animal Care and Use Committee of the local government authorities (AUP# 896.18 and #542). Aortic arches were isolated from male C57BL/6 male mice (age 8–12 weeks, Figure 1), or male B6;129S2-Scarb1tm1Kri/J (SR-BI−/−) and wild-type (WT) littermate control mice (11–14 weeks old, Figure 7), all purchased from Jackson Laboratories (Bar Harbor, MN, USA). Mice were fed a standard chow diet and were given free access to food and water. Mice were sacrificed by isoflurane overdose, and then the chest cavity was opened. Three millilitres of 10 U/mL heparin were injected into the left ventricle, and the inferior vena cava was cut for exsanguination. The mouse was placed on a dissecting microscope stage, and the aorta was carefully cleaned and the branches off the arch were tied in situ. The aorta was then removed by cutting above the heart and before the renal branches and then mounted on a silicone-coated dish for cannulation. Vessels were cannulated with PE50 tubing and perfused for 3 h at a rate of 1 mL/h using MOPS [3-(N-morpholino)propanesulfonic acid] buffer containing biotinylated dextran (70 kDa, 100 µg/mL), as well as LDL (0.71 mg/mL) or PAF (50 nmol/L) (see Figure 1). In some experiments (Figure 7), perfusion was performed with biotinylated LDL (Alpha Diagnostics International, San Antonio, TX, USA) to increase the sensitivity of the assay and the ability to detect a difference between knockout and wild-type vessels. Vessels were then flushed with MOPS for 15 min, followed by perfusion with 4% PFA for 15 min. Vessels were subsequently immersed in 4% PFA for 15 min and then cryofixed.
Figure 1.

LDL accumulates in the ascending aorta by transcytosis. (A) Aortas from C57BL/6 mice were perfused ex vivo with LDL simultaneously with biotinylated dextran of a smaller molecular radius. Sequential sections, which were stained with Nile Red or streptavidin-Cy3 (to detect cholesterol and dextran, respectively), demonstrate that LDL accumulates in the sub-endothelial intima in the absence of dextran (inset). Arrows indicate cholesterol deposits (in red); nuclei are stained with DAPI (blue). Size bars, 10 μm. (B) No cholesterol is detected in vessels not perfused with LDL. Size bar, 13 μm. (C) As a positive control, aortas were perfused with dextran alone (i.e. no LDL) in the absence (top panels) or presence (bottom panels) of 50 nM PAF, which induces inter-endothelial gaps. Dextran (in red) accumulated in PAF-treated vessels (inset); nuclei are in blue. Arrows indicate the endothelium; dotted lines indicate the vessel wall. Size bars, 80 μm.
Figure 7.
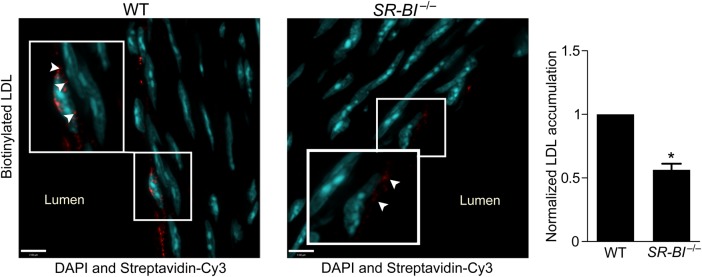
Deficiency of SR-BI reduces LDL transcytosis ex vivo. Aortas from SR-BI-deficient mice and wild-type (WT) littermates were perfused ex vivo with biotinylated LDL (25 µg/mL). Serial sections were stained with streptavidin-Cy3, and z-stack images were then acquired by confocal microscopy under identical microscope settings. Nuclei are stained with DAPI (blue). All perfusion, sectioning, staining, and image analysis were performed as four separate experiments, each containing one WT and one SR-BI−/− mouse. Images were quantified using ImageJ for Cy3 (red) fluorescence; all signal above background was measured. For each experiment, the same background correction was applied to images from both the knockout and matching wild-type aorta. Results are presented as the red fluorescence intensity in a SR-BI−/− aorta normalized to its WT equivalent. More punctae of LDL are visible in WT vs. SR-BI−/− vessels (inset, arrowheads). Quantification is shown at right. *P < 0.01 by one-sample t-test, four mice per group.
For the perfusion experiment with LDL and dextran (Figure 1), the ascending aorta was sectioned sequentially and stained using Nile Red and streptavidin-Cy3 (Sigma); Nile Red was chosen as it detects both free and esterified cholesterol and is more sensitive than Oil Red O.28 Vessels were then stained with DAPI and imaged by spinning disk confocal microscopy (Zeiss Axiovert 200 M microscope; Zeiss, Jena, Germany).
For the perfusion experiment using aortas from SR-BI−/− and wild-type (WT) littermate animals (Figure 7), the aortic arch was sequentially sectioned between the second and third branches at a thickness of 5 μm. Slides were air-dried for 2 h before staining. OCT medium was removed by submerging slides in PBS for 20 min. Slides were stained with 20 μL of Streptavidin-Cy3 (1:1000) in PBS for 30 min followed by 3× wash in PBS. The arch was then stained with DAPI followed by mounting with coverslips. Image acquisition was done by confocal microscopy on a Leica DMI6000 B microscope with settings kept constant between groups. A total of 30 images were randomly acquired from each vessel at a z-stack interval of 0.2 μm. All perfusion, sectioning, staining, and image analysis were performed as four separate experiments, each containing one WT and one SR-BI−/− mouse. Images were quantified using ImageJ for Cy3 (red) fluorescence; all signal above background was measured. For each experiment, the same background correction was applied to images from both the knockout and matching wild-type aorta. Results are presented as the red fluorescence intensity normalized to wild type.
2.8. Statistics
Statistical analysis was performed by use of GraphPad Prism software (GraphPad Prism 5.0; GraphPad Software Inc., La Jolla, CA, USA), and a combination of Student's t-tests and one-sample t-tests was used. Data are presented as mean ± SEM. All experiments were performed at least on four different occasions.
3. Results
3.1. LDL crosses the endothelium by transcytosis ex vivo
We first wanted to establish that LDL transcytosis occurs in intact arteries. Aortas were isolated from C57BL/6 mice, perfused with LDL and biotinylated dextran (of a smaller molecular radius than LDL), and sequential sections were then stained with Nile Red and Streptavidin-Cy3 to detect accumulation of cholesterol and dextran, respectively. Perfusion with LDL for several hours resulted in sub-endothelial cholesterol deposits (Figure 1A) that were absent in control vessels perfused without LDL (Figure 1B). No intimal dextran was observed despite its smaller size whether perfused at the same time as LDL (right-hand panel, Figure 1A) or when alone in the perfusate (Figure 1C). This was not due to impaired retention of the dextran, since aortas perfused with PAF, a known inducer of endothelial leak,29 demonstrated an obvious increase in accumulation of intimal dextran compared with control (Figure 1C). Taken together, these data suggest that LDL reached the sub-endothelial intima by transcytosis.
3.2. LDL transcytosis is performed by primary HCAECs in vitro
Primary HCAECs were grown in culture and exhibited a typical cobblestone morphology under phase contrast microscopy. In addition, they displayed continuous rings of tight junctions, as demonstrated by immunofluorescence for occludin, as well as adherens junctions, as shown by plasmalemmal staining for VE-cadherin (Figure 2A); these suggest that cell polarity is maintained. When confluent monolayers of these cells were pulsed with fluorophore-tagged LDL, we observed the progression of the LDL from the apical membrane downwards over time (Figure 2B). To confirm that actual LDL transcytosis occurs in HCAECs, we adapted our published assay for albumin transcytosis.1 Briefly, HCAECs seeded on transwells were exposed to biotin-LDL at 4°C, a temperature at which binding occurs but internalization is inhibited. Cells were rinsed to remove unbound LDL and placed at 37°C to allow transcytosis to begin; the binding and rinsing step is performed to decrease confounding from paracellular leak. Passage of LDL across the endothelial monolayer was measured using a highly sensitive ELISA (Figure 3A). As a control, we repeated the transcytosis assay in the presence of excess unlabelled LDL and found that permeability to biotin-LDL was significantly attenuated (Figure 3B) consistent with competition for binding sites. To prove that the assay is not affected by endothelial gaps, we added PAF to the cells. As expected, PAF significantly increased endothelial permeability to fluorescein (Figure 3C); however, it had no effect on the flux of membrane-bound LDL (Figure 3D).
Figure 2.

Primary HCAECs in culture retain endothelial characteristics and internalize LDL in a polarized fashion. (A) Coronary artery endothelial cells exhibit cobblestone morphology under phase contrast microscopy (left panel), as well as continuous rings of occludin (middle) and VE-cadherin (right panel) as determined by immunostaining. Images are z-stack projections and are representative of >5 independent experiments; nuclei are stained with DAPI (blue). Size bar is 19 µm. (B) Confluent HCAECs were pulsed with DiI-conjugated LDL and then rinsed to remove unbound ligand. After incubation at 37°C, monolayers were fixed at progressive time points and imaged by confocal microscopy. The apical membrane is labelled with a fluorescein-tagged lectin (green). Long images are side views (yz-axis) of separate cells with corresponding xy images shown at left for reference. xy images were taken 5 slices from the top of the cell in both cases, and white arrows denote vesicles containing LDL. Note the apical localization of LDL (red) at time zero (indicated by colocalization with the green lectin) followed by its movement towards the basal membrane (dotted line) 30 min later. Quantification is shown at right. Scale bar represents 3 µm, nuclei are stained with DAPI (blue); n = 4 independent experiments; *P < 0.001 by Student's t-test for 0 vs. 30 min.
Figure 3.

LDL transcytosis is performed by HCAECs in vitro. (A) Standard curve for ELISA for biotin-LDL showing linearity and high sensitivity; y-axis is optical density at 450 nm (see Methods). (B) Transwells coated with confluent HCAECs were allowed to bind biotin-LDL and then rinsed to remove unbound ligand. Two hours later, LDL in the lower chamber of the transwell was measured by ELISA. Addition of 50-fold excess unlabelled LDL (Comp) reduced biotin-LDL transcytosis, *P < 0.05 by Student's t-test. n = 7 experiments. (C and D) The transwell assay for LDL transcytosis is not affected by paracellular leak. Administration of PAF (10 µM) increased the permeability of endothelial monolayers to Na-fluorescein (C) but did not affect LDL transcytosis (D) compared with untreated cells (Ctrl); *P < 0.05 by one-sample t-test, n = 6 experiments.
3.3. Development of a novel assay to study LDL transcytosis
While the transwell assay suggests that LDL transcytosis occurs in vitro, the method is cumbersome and not amenable to the molecular dissection of transcytosis. Transwell assays are affected by edge effects, low transfection efficiency (particularly in primary cells), and the potential of constant confounding from intercellular gaps. To overcome these issues, we developed a novel assay using TIRF microscopy.21 This technique selectively visualizes the basal (bottom) surface of cells (e.g. 100–150 nm or so), excluding overlying noise from the rest of the cytosol. DiI-labeled (fluorescent) LDL is added apically to confluent endothelial monolayers, and the fusion of LDL-containing vesicles with the basolateral membrane is observed in real time (see Supplementary material online, Movie S1 and Figure 4A and B). Quantification of fusion events is performed in a blinded and automated fashion. We validated this new method in a number of ways. First, to prove that the assay is not confounded by endocytic traffic (i.e. vesicles moving in and out of the TIRF zone), we reduced the penetration depth of the TIRF laser to 75 nm; as expected, this had no effect on the number of transcytosis events detected (Figure 4C). As with the transwell method, the TIRF assay was not affected by an increase in paracellular leak induced by PAF (Figure 4D). In addition, an excess of unlabelled ligand caused an almost four-fold decrease in LDL transcytosis (Figure 4E), consistent with a receptor-mediated process rather than simple diffusion across paracellular gaps. Finally, incubation with Dyngo4A,30 a specific chemical inhibitor of the GTPase dynamin (Figure 4F), essentially abrogated the process.
Figure 4.

Novel assay to study LDL transcytosis. (A) Schematic of the assay. DiI-LDL is added to confluent endothelial monolayers and is detected by TIRF microscopy as it enters the bottom of the cell. (B) Still images from the Supplemental Video. Top panel depicts initial image, bottom panel is 150 ms later. Blue arrowhead denotes LDL-containing vesicle that disappears due to exocytosis; white arrowheads show vesicles that do not fuse. (C) As a control for endocytic traffic, the penetration depth (PD) of the TIRF laser was decreased and exocytic events were quantified. Changing the PD of the TIRF laser had no effect; n = 4. (D) The TIRF assay is not affected by paracellular leak. Endothelial monolayers were treated with PAF as in Figure 2, and transcytosis events were quantified; there was no significant difference between control (ctrl) and PAF-treated cells, n = 5. (E) Addition of 50-fold excess unlabelled LDL at the same time as DiI-LDL reduced transcytosis, *P < 0.01 by one-sample t-test, n = 4. (F) Pre-incubation for 30 min with Dyngo4A (30 µM), a specific inhibitor of the GTPase dynamin, blocked LDL transcytosis; *P = 0.001 by one-sample t-test, n = 4 experiments.
3.4. Degradation of the LDLR by PCSK9 does not decrease LDL transcytosis
We then wished to take advantage of this assay to identify which receptor was capable of mediating LDL transcytosis. To determine whether the high-affinity LDL receptor (LDLR) is required, we exposed coronary endothelial cells to exogenous proprotein convertase subtilisin/kexin type 9 (PCSK9)27 which mediates degradation of the receptor. As expected, PCSK9-treated cells demonstrated almost complete loss of LDLR (Figure 5A); of note, levels of another cell surface receptor, SR-BI, were unchanged. Under these conditions, LDL transcytosis was not affected (Figure 5B).
Figure 5.

Degradation of LDLR by PCSK9 does not decrease LDL transcytosis. Confluent human coronary artery endothelial monolayers were treated with media from PCSK9-expressing or untransfected (control) HEK293 cells. (A) PCKS9 degrades LDLR but not SR-BI; α-actinin is the loading control; blots are representative of five independent experiments and quantification is to their right; *P < 0.01 by one-sample t-test for LDLR levels in control vs. PCSK9-treated cells. (B) Treatment with PCSK9 (as in A) was followed by measurement of LDL transcytosis using the TIRF assay. Degradation of LDLR did not affect LDL transcytosis, n = 5.
3.5. SR-BI mediates LDL transcytosis across HCAECs
Since LDLR does not mediate LDL transcytosis, we turned our attention elsewhere. The scavenger receptor SR-BI was described some time ago to bind native LDL,31 but the physiological significance of this interaction is unknown. Interestingly, SR-BI is expressed in coronary endothelium and is capable of bona fide endocytosis.32 We observed partial colocalization of SR-BI and DiI-LDL (Pearson's coefficient R2: 0.228 ± 0.029; Costes' R2: 0.0025 ± 0.0009; Manders' coefficient = 0.577 ± 0.073; Figure 6A), which was >100-fold higher than that expected by chance. Addition of 50-fold excess unlabelled LDL essentially abrogated the colocalization. Overexpression of SR-BI resulted in a 50% increase in LDL transcytosis (Figure 6B), while even incomplete knockdown by siRNA had the opposite effect, resulting in a decrease in transcytosis by ∼40% (Figure 6C and D). Lastly, addition of excess HDL, the canonical ligand for SR-BI, significantly attenuated transcytosis of LDL (Figure 6E), suggesting competition for the receptor.
Figure 6.

SR-BI mediates LDL transcytosis. (A) Coronary artery endothelial cells were incubated with DiI-LDL then fixed and immunostained for SR-BI. Z-stack images were then acquired using a spinning disk confocal microscope. DiI-LDL colocalized partially with SR-BI (arrows). Pearson coefficient R2: 0.228 ± 0.029; Costes' R2: 0.0025 ± 0.0009; Manders' coefficient = 0.577 ± 0.073. Addition of 50-fold unlabelled LDL essentially ablated colocalization of DiI-LDL and SR-BI (Pearson R2 = 0.125 ± 0.015; Manders' coefficient = 0.107 ± 0.025; P < 0.001 by Student's t-test for comparison of the Manders' coefficients, P = 0.01 by Student's t-test for Pearson's coefficients); n = 8 experiments with DiI-LDL alone and n = 4 with excess unlabelled LDL. White scale bar is 3 µm. (B) Cells transfected with SR-BI-GFP exhibited increased LDL transcytosis compared with cells transfected with GFP alone (Control), as measured by TIRF, *P < 0.01 by one-sample t-test, n = 4 experiments. (C) Cells were transfected with siRNA to SR-BI or scrambled control (Control) followed by western blotting. Levels of LDLR were probed as a control for off-target effects while α-actinin was the loading control. (D) Even partial depletion of SR-BI by siRNA caused a significant drop in LDL transcytosis compared with scrambled control (scRNA) as measured by TIRF; *P < 0.05 by one-sample t-test, n = 5 experiments. (E) Addition of 50-fold unlabelled HDL at the same time as DiI-LDL reduced LDL transcytosis, *P < 0.05 by one-sample t-test, n = 4 experiments.
3.6. Aortas from SR-BI-deficient mice accumulate less LDL during ex vivo perfusion
Finally, to validate our in vitro findings, we isolated the aortas from SR-BI-deficient mice and their wild-type littermates, followed by perfusion for 3 h with biotinylated LDL. Sequential sections of the vessels were then examined and quantified for intimal accumulation of LDL. Consistent with the cell culture experiments, deficiency of SR-BI caused a >40% reduction in aortic LDL accumulation (Figure 7). Thus, SR-BI contributes to LDL transcytosis both in vitro and ex vivo.
4. Discussion
Our study addresses the critical question of how LDL crosses the endothelium to accumulate in the intima, a necessary first step in the pathogenesis of atherosclerosis. We provide evidence in both intact vessels and in vitro that LDL can cross endothelial cells by transcytosis. This is consistent with the observation that early atherosclerotic lesions occur despite apparently healthy overlying endothelium.5
Elucidation of the molecular regulation of transcytosis is best determined in vitro, yet a major technical barrier is the difficulty in distinguishing it from paracellular leak, since manipulation of an endothelial monolayer by pharmacological or molecular approaches often perturbs barrier integrity.1
Accordingly, we devised a means of quantifying transcytosis by single cells in real time, reasoning that this would avoid confounding by paracellular leak or low transfection efficiency in primary cells. After validating this technique, we then sought to identify the receptor involved in LDL transcytosis.
While the best characterized receptor for LDL is its high affinity receptor (LDLR), our data suggest that it is unlikely to be required for LDL transcytosis; specifically, almost total degradation of LDLR by PCSK933 did not impair transcytosis. The lack of involvement of LDLR in transcytosis is perhaps unsurprising as it is known from human patients that mutations in LDLR causing marked elevations in circulating LDL nonetheless predispose to accelerated atherosclerosis.34
Instead, we present multiple lines of evidence indicating that SR-BI mediates transcytosis of LDL, in addition to its previously established roles in HDL metabolism35 and steroidogenesis.36 This finding was unexpected given that deficiency of SR-BI has been reported to cause accelerated atherosclerosis in mice.36,37 However, the phenotype of these animals is dominated by expression of the receptor in the liver37,38 and macrophages.39,40 Our data suggest that endothelial SR-BI has a more complex role and may contribute to early atherosclerotic lesions. This apparent duality of SR-BI is not without precedent, as data suggest that in such early lesions, SR-BI expression in bone marrow-derived cells is also pathogenic.41 It is also possible that the pro-atherogenic contributions of SR-BI in the arterial endothelium are nullified by the receptor's beneficial role in the liver and macrophages. Our study used multiple approaches to implicate SR-BI in vitro, followed by validation of these results ex vivo; further work will be required to confirm these observations in vivo. However, it is reassuring that our observation that LDL and HDL can compete for binding to SR-BI is mirrored by an earlier report by Rohrer et al.,35 who found that LDL could decrease binding of HDL to aortic endothelium. These findings suggest the possibility that very low-density lipoprotein (VLDL), another reported ligand for SR-BI,42 could also inhibit LDL transcytosis, but this remains to be established experimentally.
Our data imply that in addition to SR-BI, other receptors are likely to contribute to LDL transcytosis. The endothelium expresses other scavenger receptors including CD36, SR-AI/II, LOX-1, and SREC.43,44 Of these,45 only SR-BI and CD36 have been reported to bind native LDL31,46 and to be capable of performing bona fide receptor-mediated endocytosis.32,47 Intriguingly, the majority of both SR-BI and CD36 is found in lipid rafts or caveolae. Thus, the role of CD-36 in LDL transcytosis is the subject of ongoing work in our laboratory. Furthermore, although the receptor-mediated endocytosis of LDL by LDLR is well characterized,48 the relationship, if any, of endothelial retention of cholesterol to cholesterol transcytosis remains uncertain. We believe that our development of a dynamic and facile assay for quantifying LDL transcytosis by individual cells will greatly facilitate the elucidation of this important issue.
In closing, given that the deposition of sub-endothelial LDL is the first step in atherosclerosis, it is startling how little is known about the process. We have devised a novel assay to quantify LDL transcytosis; our data implicate transcytosis as an important route and point to SR-BI as a contributing receptor. This assay is likely to prove useful in the study of transcytosis of other ligands, including modified forms of LDL and other lipoproteins such as HDL.35 Further study into the mechanisms of LDL transcytosis may lead to new therapeutic approaches for the prevention and treatment of atherogenesis.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This work was supported by the Canadian Institutes of Health Research (102741 to N.G.S.); Canada Research Chair (216684 to N.G.S.); Foundation Leducq (13 CVD 03 to N.G.S.). S.M.A received MD/PhD funding from the CIHR and the McLaughlin Centre for Molecular Medicine.
Acknowledgements
The authors thank Johnny Su for assistance with densitometry and Michael Woodside and Paul Paroutis of the Imaging Facility at the Hospital for Sick Children for technical assistance with TIRF.
Conflict of interest: none declared.
References
- 1.Armstrong SM, Khajoee V, Wang C, Wang T, Tigdi J, Yin J, Kuebler WM, Gillrie M, Davis SP, Ho M, Lee WL. Co-regulation of transcellular and paracellular leak across microvascular endothelium by dynamin and rac. Am J Pathol 2012;180:1308–1323. [DOI] [PubMed] [Google Scholar]
- 2.Vasile E, Simionescu M, Simionescu N. Visualization of the binding, endocytosis, and transcytosis of low-density lipoprotein in the arterial endothelium in situ. J Cell Biol 1983;96:1677–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bhalodkar NC, Blum S, Rana T, Kitchappa R, Bhalodkar AN, Enas EA. Comparison of high-density and low-density lipoprotein cholesterol subclasses and sizes in Asian Indian women with Caucasian women from the Framingham Offspring Study. Clin Cardiol 2005;28:247–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.von Eckardstein A, Rohrer L. Transendothelial lipoprotein transport and regulation of endothelial permeability and integrity by lipoproteins. Curr Opin Lipidol 2009;20:197–205. [DOI] [PubMed] [Google Scholar]
- 5.Williams KJ, Tabas I. The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol 1995;15:551–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frank PG, Pavlides S, Cheung MW, Daumer K, Lisanti MP. Role of caveolin-1 in the regulation of lipoprotein metabolism. Am J Physiol Cell Physiol 2008;295:C242–C248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bian F, Yang X, Zhou F, Wu PH, Xing S, Xu G, Li W, Chi J, Ouyang C, Zhang Y, Xiong B, Li Y, Zheng T, Wu D, Chen X, Jin S. C-reactive protein promotes atherosclerosis by increasing LDL transcytosis across endothelial cells. Br J Pharmacol 2014;171:2671–2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quest AF, Leyton L, Parraga M. Caveolins, caveolae, and lipid rafts in cellular transport, signaling, and disease. Biochem Cell Biol 2004;82:129–144. [DOI] [PubMed] [Google Scholar]
- 9.Cancel LM, Fitting A, Tarbell JM. In vitro study of LDL transport under pressurized (convective) conditions. Am J Physiol Heart Circ Physiol 2007;293:H126–H132. [DOI] [PubMed] [Google Scholar]
- 10.Lin SJ, Jan KM, Chien S. Role of dying endothelial cells in transendothelial macromolecular transport. Arteriosclerosis 1990;10:703–709. [DOI] [PubMed] [Google Scholar]
- 11.Cancel LM, Tarbell JM. The role of mitosis in LDL transport through cultured endothelial cell monolayers. Am J Physiol Heart Circ Physiol 2011;300:H769–H776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tiruppathi C, Song W, Bergenfeldt M, Sass P, Malik AB. Gp60 activation mediates albumin transcytosis in endothelial cells by tyrosine kinase-dependent pathway 10.1074/jbc.272.41.25968. J Biol Chem 1997;272:25968–25975. [DOI] [PubMed] [Google Scholar]
- 13.Oh P, McIntosh DP, Schnitzer JE. Dynamin at the neck of caveolae mediates their budding to form transport vesicles by GTP-driven fission from the plasma membrane of endothelium. J Cell Biol 1998;141:101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schubert W, Frank PG, Razani B, Park DS, Chow CW, Lisanti MP. Caveolae-deficient endothelial cells show defects in the uptake and transport of albumin in vivo. J Biol Chem 2001;276:48619–48622. [DOI] [PubMed] [Google Scholar]
- 15.Frank PG, Lee H, Park DS, Tandon NN, Scherer PE, Lisanti MP. Genetic ablation of caveolin-1 confers protection against atherosclerosis. Arterioscler Thromb Vasc Biol 2004;24:98–105. [DOI] [PubMed] [Google Scholar]
- 16.Schubert W, Frank PG, Woodman SE, Hyogo H, Cohen DE, Chow CW, Lisanti MP. Microvascular hyperpermeability in caveolin-1 (-/-) knock-out mice. Treatment with a specific nitric-oxide synthase inhibitor, L-NAME, restores normal microvascular permeability in Cav-1 null mice. J Biol Chem 2002;277:40091–40098. [DOI] [PubMed] [Google Scholar]
- 17.Fernandez-Hernando C, Yu J, Davalos A, Prendergast J, Sessa WC. Endothelial-specific overexpression of caveolin-1 accelerates atherosclerosis in apolipoprotein E-deficient mice. Am J Pathol 2010;177:998–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fernandez-Hernando C, Yu J, Suarez Y, Rahner C, Davalos A, Lasuncion MA, Sessa WC. Genetic evidence supporting a critical role of endothelial caveolin-1 during the progression of atherosclerosis. Cell Metab 2009;10:48–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun SW, Zu XY, Tuo QH, Chen LX, Lei XY, Li K, Tang CK, Liao DF. Caveolae and caveolin-1 mediate endocytosis and transcytosis of oxidized low density lipoprotein in endothelial cells. Acta Pharmacologica Sinica 2010;31:1336–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jaumouille V, Farkash Y, Jaqaman K, Das R, Lowell CA, Grinstein S. Actin cytoskeleton reorganization by Syk regulates Fcgamma receptor responsiveness by increasing its lateral mobility and clustering. Dev Cell 2014;29:534–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Azizi PM, Zyla RE, Guan S, Wang C, Liu J, Bolz SS, Heit B, Klip A, Lee WL. Clathrin-dependent entry and vesicle-mediated exocytosis define insulin transcytosis across microvascular endothelial cells. Mol Biol Cell 2015;26:740–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Becherer U, Pasche M, Nofal S, Hof D, Matti U, Rettig J. Quantifying exocytosis by combination of membrane capacitance measurements and total internal reflection fluorescence microscopy in chromaffin cells. PLoS One 2007;2:e505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crocker JC, Grier DG. Methods of digital video microscopy for colloidal studies. J Colloid Interface Sci 1996;179:298–310. [Google Scholar]
- 24.Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc 2006;224:213–232. [DOI] [PubMed] [Google Scholar]
- 25.Linkert M, Rueden CT, Allan C, Burel JM, Moore W, Patterson A, Loranger B, Moore J, Neves C, Macdonald D, Tarkowska A, Sticco C, Hill E, Rossner M, Eliceiri KW, Swedlow JR. Metadata matters: access to image data in the real world. J Cell Biol 2010;189:777–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neculai D, Schwake M, Ravichandran M, Zunke F, Collins RF, Peters J, Neculai M, Plumb J, Loppnau P, Pizarro JC, Seitova A, Trimble WS, Saftig P, Grinstein S, Dhe-Paganon S. Structure of LIMP-2 provides functional insights with implications for SR-BI and CD36. Nature 2013;504:172–176. [DOI] [PubMed] [Google Scholar]
- 27.Poirier S, Mayer G, Benjannet S, Bergeron E, Marcinkiewicz J, Nassoury N, Mayer H, Nimpf J, Prat A, Seidah NG. The proprotein convertase PCSK9 induces the degradation of low density lipoprotein receptor (LDLR) and its closest family members VLDLR and ApoER2. J Biol Chem 2008;283:2363–2372. [DOI] [PubMed] [Google Scholar]
- 28.Fowler SD, Greenspan P. Application of Nile red, a fluorescent hydrophobic probe, for the detection of neutral lipid deposits in tissue sections: comparison with oil red O. J Histochem Cytochem 1985;33:833–836. [DOI] [PubMed] [Google Scholar]
- 29.Huang Q, Wu M, Meininger C, Kelly K, Yuan Y. Neutrophil-dependent augmentation of PAF-induced vasoconstriction and albumin flux in coronary arterioles. Am J Physiol 1998;275:H1138–H1147. [DOI] [PubMed] [Google Scholar]
- 30.McCluskey A, Daniel JA, Hadzic G, Chau N, Clayton EL, Mariana A, Whiting A, Gorgani NN, Lloyd J, Quan A, Moshkanbaryans L, Krishnan S, Perera S, Chircop M, von Kleist L, McGeachie AB, Howes MT, Parton RG, Campbell M, Sakoff JA, Wang X, Sun JY, Robertson MJ, Deane FM, Nguyen TH, Meunier FA, Cousin MA, Robinson PJ. Building a better dynasore: the dyngo compounds potently inhibit dynamin and endocytosis. Traffic 2013;14:1272–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Calvo D, Gomez-Coronado D, Lasuncion MA, Vega MA. CLA-1 is an 85-kD plasma membrane glycoprotein that acts as a high-affinity receptor for both native (HDL, LDL, and VLDL) and modified (OxLDL and AcLDL) lipoproteins. Arterioscler Thromb Vasc Biol 1997;17:2341–2349. [DOI] [PubMed] [Google Scholar]
- 32.Pagler TA, Rhode S, Neuhofer A, Laggner H, Strobl W, Hinterndorfer C, Volf I, Pavelka M, Eckhardt ER, van der Westhuyzen DR, Schutz GJ, Stangl H. SR-BI-mediated high density lipoprotein (HDL) endocytosis leads to HDL resecretion facilitating cholesterol efflux. J Biol Chem 2006;281:11193–11204. [DOI] [PubMed] [Google Scholar]
- 33.Peterson AS, Fong LG, Young SG. PCSK9 function and physiology. J Lipid Res 2008;49:1595–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karayan L, Qiu S, Betard C, Dufour R, Roederer G, Minnich A, Davignon J, Genest J Jr. Response to HMG CoA reductase inhibitors in heterozygous familial hypercholesterolemia due to the 10-kb deletion (‘French Canadian mutation’) of the LDL receptor gene. Arterioscler Thromb 1994;14:1258–1263. [DOI] [PubMed] [Google Scholar]
- 35.Rohrer L, Ohnsorg PM, Lehner M, Landolt F, Rinninger F, von Eckardstein A. High-density lipoprotein transport through aortic endothelial cells involves scavenger receptor BI and ATP-binding cassette transporter G1. Circ Res 2009;104:1142–1150. [DOI] [PubMed] [Google Scholar]
- 36.Trigatti B, Rayburn H, Vinals M, Braun A, Miettinen H, Penman M, Hertz M, Schrenzel M, Amigo L, Rigotti A, Krieger M. Influence of the high density lipoprotein receptor SR-BI on reproductive and cardiovascular pathophysiology. Proc Natl Acad Sci USA 1999;96:9322–9327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huszar D, Varban ML, Rinninger F, Feeley R, Arai T, Fairchild-Huntress V, Donovan MJ, Tall AR. Increased LDL cholesterol and atherosclerosis in LDL receptor-deficient mice with attenuated expression of scavenger receptor B1. Arterioscler Thromb Vasc Biol 2000;20:1068–1073. [DOI] [PubMed] [Google Scholar]
- 38.Varban ML, Rinninger F, Wang N, Fairchild-Huntress V, Dunmore JH, Fang Q, Gosselin ML, Dixon KL, Deeds JD, Acton SL, Tall AR, Huszar D. Targeted mutation reveals a central role for SR-BI in hepatic selective uptake of high density lipoprotein cholesterol. Proc Natl Acad Sci USA 1998;95:4619–4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang W, Yancey PG, Su YR, Babaev VR, Zhang Y, Fazio S, Linton MF. Inactivation of macrophage scavenger receptor class B type I promotes atherosclerotic lesion development in apolipoprotein E-deficient mice. Circulation 2003;108:2258–2263. [DOI] [PubMed] [Google Scholar]
- 40.Covey SD, Krieger M, Wang W, Penman M, Trigatti BL. Scavenger receptor class B type I-mediated protection against atherosclerosis in LDL receptor-negative mice involves its expression in bone marrow-derived cells. Arterioscler Thromb Vasc Biol 2003;23:1589–1594. [DOI] [PubMed] [Google Scholar]
- 41.Van Eck M, Bos IS, Hildebrand RB, Van Rij BT, Van Berkel TJ. Dual role for scavenger receptor class B, type I on bone marrow-derived cells in atherosclerotic lesion development. Am J Pathol 2004;165:785–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Van Eck M, Hoekstra M, Out R, Bos IS, Kruijt JK, Hildebrand RB, Van Berkel TJ. Scavenger receptor BI facilitates the metabolism of VLDL lipoproteins in vivo. J Lipid Res 2008;49:136–146. [DOI] [PubMed] [Google Scholar]
- 43.Dhaliwal BS, Steinbrecher UP. Scavenger receptors and oxidized low density lipoproteins. Clin Chim Acta 1999;286:191–205. [DOI] [PubMed] [Google Scholar]
- 44.Rhainds D, Brissette L. The role of scavenger receptor class B type I (SR-BI) in lipid trafficking. defining the rules for lipid traders. Int J Biochem Cell Biol 2004;36:39–77. [DOI] [PubMed] [Google Scholar]
- 45.Terpstra V, van Amersfoort ES, van Velzen AG, Kuiper J, van Berkel TJ. Hepatic and extrahepatic scavenger receptors: function in relation to disease. Arterioscler Thromb Vasc Biol 2000;20:1860–1872. [DOI] [PubMed] [Google Scholar]
- 46.Calvo D, Gomez-Coronado D, Suarez Y, Lasuncion MA, Vega MA. Human CD36 is a high affinity receptor for the native lipoproteins HDL, LDL, and VLDL. J Lipid Res 1998;39:777–788. [PubMed] [Google Scholar]
- 47.Brundert M, Heeren J, Merkel M, Carambia A, Herkel J, Groitl P, Dobner T, Ramakrishnan R, Moore KJ, Rinninger F. Scavenger receptor CD36 mediates uptake of high density lipoproteins in mice and by cultured cells. J Lipid Res 2011;52:745–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goldstein JL, Brown MS. The LDL receptor. Arterioscler Thromb Vasc Biol 2009;29:431–438. [DOI] [PMC free article] [PubMed] [Google Scholar]