

Abstract
Although every organism shares some common features of replication, this process varies greatly among eukaryotic species. Current data show that mathematical models of the organization of origins based on possibility theory may be applied (and remain accurate) in every model organism i.e. from yeast to humans. The major differences lie within the dynamics of origin firing and the regulation mechanisms that have evolved to meet new challenges throughout the evolution of the organism. This article elaborates on the relations between chromatin structure, organization of origins, their firing times and the impact that these features can have on genome stability, showing both differences and parallels inside the eukaryotic domain.
Keywords: dormant origins, mathematical models of replication, origin competence, origin efficiency, origin firing, origin licensing, replication timing
Abbreviations
- APC
anaphase promoting complex
- ATR
ataxia telangiectasia mutated and Rad3-related kinase
- ARS
autonomously replicating sequences
- Cdc45
cell division control protein 45
- Cdc6
cell division control protein 6
- CDK
cyclin-dependent kinase
- CDT
C-terminus domain
- Cdt1
chromatin licensing and DNA replication factor 1
- CEN
centromere
- C-Frag
chromosome fragmentation
- CFSs
chromosome fragile sites
- Chk1
checkpoint kinase 1
- CIN
chromosome instability
- Clb2
G2/mitotic-specific cyclin Clb2
- CMG
Cdc45-MCM-GINS complex
- Dbf4
protein Dbf4 homolog A
- Dpb11
DNA replication regulator Dpb11
- DCR
Ddb1-Cu14a-Roc1 complex
- DDK
Dbf-4-dependent kinase
- Dfp1
Hsk1-Dfp1 kinase complex regulatory subunit Dfp1
- DSBs
double strand breaks
- E2F
E2F transcription factor
- EL
early to late origins transition
- ETG1
E2F target gene 1/replisome factor
- Fkh
fork head domain protein
- GCN5
histone acetyltransferase GCN5
- GINS
go-ichi-ni-san
- LE
late to early origins transition
- MCM2–7
minichromosome maintenance helicase complex
- NDT
N-terminus domain
- ORC
origin recognition complex
- ORCA
origin recognition complex subunit A
- ori
origin
- p53
tumor suppressor protein p53
- PCC
premature chromosome condensation
- PCNA
proliferating cell nuclear antigen
- Rif1
replication timing regulatory factor 1
- RO
replication origin
- RPD3
histone deacetylase 3
- RTC
replication timing control
- SCF
Skp1-Cullin-F-Box ligase
- SIR
sulfite reductase
- Sld2
replication regulator Sld2
- Sld3
replication regulator Sld3
- Swi6
chromatin-associated protein swi6
- Taz1
telomere length regulator taz1
- YKU70
yeast Ku protein.
Introduction
DNA replication prior to cell division ensures genome stability and as such is the most crucial process during the cell cycle. It enables cells to duplicate as rapidly as possible and use the least amount of energy and resources. Its accuracy depends on the effectiveness of DNA control and repair mechanisms.
Replication starts at particular locations called replication origins (ROs), which must undergo a specific sequence of binding events before the S phase. At the end of mitosis, before mitotic spindle disassembly, the pre-recognition complexes (pre-RCs) assemble at replication origins. First, the origin recognition complexes (ORCs) bind to potential assembly sites. At the onset of the G1 phase, ORCs, Cdc6 and Cdt1 act together to load minichromosome maintenance (MCM) 2–7 helicase complexes. Current data suggest that ORC binds first and then recruits Cdc6 and Cdt1,1-2 then pairs of ring-shaped MCM2–7 hexamers assemble around DNA).3-4 This initiation step of DNA replication is also referred to as licensing or pre-RC assembly.
The initiation itself is directly controlled by Dbf4-dependent kinase (DDK) and cyclin-dependent kinase (CDK) which are sequentially activated during the transition from G1 to S-phase. The first protein to act is DDK phosphorylating MCM2–7 subunits, which allows loading of Cdc45 and Sld3, followed by the phosphorylation of Sld2 and Sld3 by CDK, thus leading to the recruitment of GINS together with some additional factors (including DNA polymerases) and the assembly of a replisome capable of initiation of DNA synthesis. Afterwards, this Cdc45-MCM-GINS (CMG) complex slides ahead of the replication fork in order to unwind the DNA and grant access to the single-stranded template.5-6 This very sequence of events is common for all active origins, with the timing and efficiency of initiation making the difference.
Firing of the origins is controlled by multiple pathways and – along with licensing – is linked with epigenetic and genome stability systems. Both the licensing and origin activation processes vary throughout eukaryotic species. For instance in S. cerevisiae yeast, licensing is limited to specific parts of chromosomes, while in X. laevis frog embryo binding proteins can theoretically be attached at any location in the genome.7 Moreover, a few of the licensed origins will be activated and fired – approximately 90% of origins remain dormant and are replicated passively.8-12
In order to prevent re-replication of some segments of DNA, eukaryotic organisms divide replication into 2 distinct steps and ensure that the ability to license new origins is shut down before entering the S phase.1-2,13 The range of mechanisms involved vary between different organisms or cell types.1,14 In yeasts, CDKs phosphorylate pre-RC complexes, promoting their inactivation, while in animal cells inhibition of Cdt1 by geminin and its degradation dependent on a proliferating cell nuclear antigen (PCNA).
Moreover, the recent data suggest that even a minor alteration in the DNA replication process (noticed as “replication stress”) can be the cause of genome instability.15 DNA replication concurs with processes such as transcription or chromatin remodeling which provides an opportunity of inducing endogenous replication stress on several levels that can act locally as a discrete pause sites or globally by altering replication dynamics (i.e., altering origin firing, or slowing fork progression). The authors distinguished 4 main types of replication stress (from ‘inappropriate firing of replication origins’, through ‘obstacles to fork progression’ and ‘unbalanced replication during the depletion of dNTP pools’ right up to the mentioned above ‘interference between the replication and transcription programs’) and showed that depending on the stress stimulus intensity and its action time, one can observe either (i) only slight retardation of the cell cycle course when the replication stress is low, or (ii) catastrophic genome instability when it is high. Additionally, massive deregulation of DNA replication program due to high level of replication stress may cause senescence, genome chaos, cell death or tumorigenesis.13,15–18 To sum up, a low level of replication stress causes minor changes in cell fate enabling survival, while a higher stress level may lead to cell adaptation via genetic material changes. Genome variations are present in cells in patients suffering from many diseases e.g. Alzheimer disease or autism.19 Such alterations of the genetic material raise the probability of somatic evolution.20 However, the massive DNA replication missbehavior can lead to genome chaos, genome instability and - in this context - can be perceived as relevant to cancer biology.
In this article we present a comprehensive review of current knowledge about the structure of replication origins and their behavior throughout a cell cycle, along with an analysis of their organization supported by mathematical models.
Spatial Organization of Replication Origins
The eukaryotic genome is replicated by hundreds or thousands of replication forks, formed as shown in (Fig. 1). An adequate number of origins licensed before entering the S phase is important here, especially that forks may slow or even stall irreversibly in some conditions (because of exo- or endogenous replicative stress, e.g., on encountering damaged DNA). The mechanism of irreversible stalling is still unclear – it may be associated with replisome proteins disassembling from DNA,21 but certainly 2 adjacent stalled forks are a grave obstacle for a cell as a new origin cannot be licensed de novo between 2 stalled forks during the S phase. To overcome this issue, cells license many more origins than they normally use.11,22-24
Figure 1.
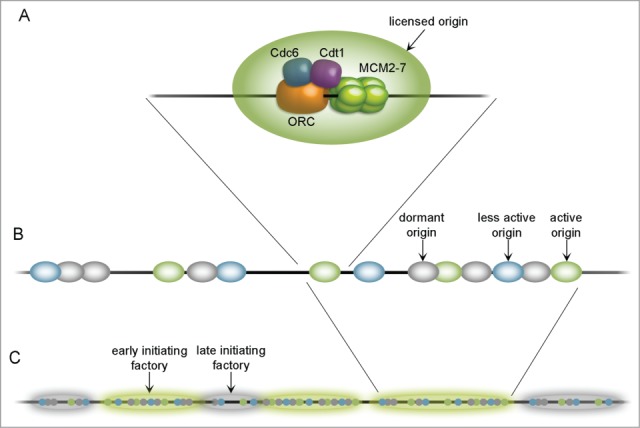
Organization of origins throughout the chromosome. A fully licensed replication origin (A) consists of core factors: ORC, Cdc6, Cdt1 and MCM 2–7 double hexamer, assembled on a DNA strand. Origins tend to group in clusters (B) containing highly efficient (active; green), less efficient (less active; blue) and dormant (gray) units. Together with other replication proteins they create replication factories (C) which may initiate earlier (highlighted green) or later (highlighted gray). Replication factories are said to contain approximately 20 licensed origins.
Most of previous theoretical studies have not concerned the importance of origin placement. Yet, a large number of licensed origins, distributed properly along chromosomal DNA, is essential to complete replication within a reasonable period of time. Thus it may be expected that the replication origins have been evolutionarily selected so that the replication time is minimized and there is also some kind of a security tool to act under stressful conditions (excess number of origins).
Origin Competence, Efficiency and Dormancy
There are various parameters used by scientists to describe replication behavior. Origin efficiency and competence are distinct populational measures that are typically determined by statistical methods. Competence, briefly, is the ability of a particular licensed origin to successfully activate. With activation comes another factor – activation timing. Some of the origins are activated early in the S phase, while others tend to be initiated later. There is an interrelation between replication timing and the efficiency of adjacent ROs – late (or less efficient) origins are often passively replicated by their earlier (or more efficient) neighbors. These two parameters may be generalized to earlier ROs being more efficient and the converse.25-26 Timing and efficiency can also describe the probability of origin firing – early timing reflects a high chance of RO activation early in the S phase, and high efficiency depicts probability of consistent firing across the entire replication process. Hence it has been suggested that timing and efficiency may be dictated by some fundamental mechanisms.27
Dormant origins are fully licensed and are capable of being initiated, but remain inactive until their firing is necessary. This feature shows that the number of activated ROs may vary according to some conditions. Cells with a reduced number of licensed origins still replicate successfully if not exposed to exogenous replicative stress from the ‘supply’ origin loci.23-24 This implies that dormancy of ROs is regulated in order for origins to become active only when necessary. An analysis of metazoan cells reveals the existence of ‘factories’ – subnuclear foci with high local concentrations of enzymes required for DNA replication. Each factory contains up to 20 replication forks. The timing system of origin firing appears to be simple – it is most likely that one fork fires from a single RO cluster, although its choice is random. Activation of dormant origins starts when a fork slows or stalls, which is a simple response to this stochastic event. Fiber analysis of DNA replication in metazoan cells shows clusters of 2–10 neighboring ROs being activated nearly at the same time, and groups of clusters from different regions of chromatin activating replication origins in different subperiods of the S phase (Fig. 1 B–C).28-29
As mentioned earlier, the majority of ROs remain dormant. When a progressing replication forks reach inactive MCM2–7 complex, then MCM2–7 becomes displaced from DNA, while the origin stays unlicensed and is replicated passively (Fig. 2B-D).22-23 But if 2 converging replication forks stall and there is an inactive licensed RO between them, it can be activated to finish replication of the endangered site, thus preventing loss of some parts of the genome. Dormancy may mainly be the effect of the relative inefficiency of some ROs; however, the number and distribution of licensed origins appear to be the key to successful replication. Replication may fail at chromosome ends, when a replication fork stalls and there is no other licensed distal origin, or if 2 neighboring forks stall within a chromosome (Fig. 2).
Figure 2.
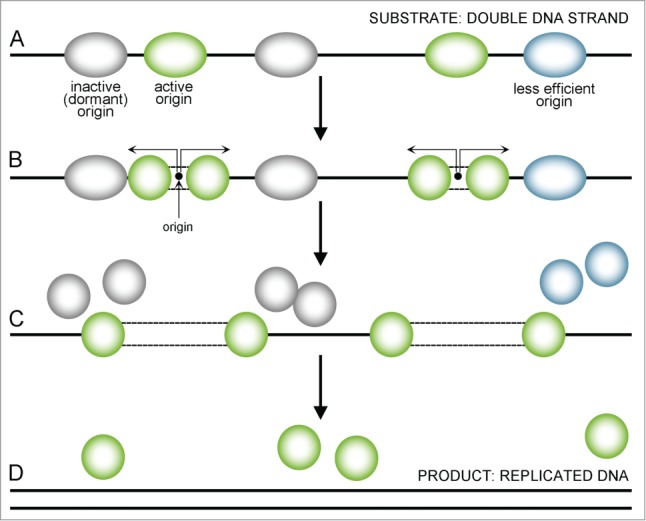
A sequence presenting uninterrupted replication. Replication starts from multiple sites (A) with the most active origins (green) firing earlier and the less active (less efficient) ones being delayed (blue) or dormant (gray). When an active origin starts replication, a double MCM2–7 complex divides and single MCM2–7 helicases move forward in both directions, replicating the DNA strand (B, indicated by arrows). Less efficient (blue) or dormant (gray) origins that fail to activate disassemble when the replication forks reach them and are replicated passively (C). When replication forks reach the end of a chromosome or another fork comes from the other side, they also get dismantle – the final ‘product’ of this process is replicated DNA (D).
In the consequence of MCM helicase DNA-triggered, 2 single-stranded DNA (ssDNA) strands are exposed. This state, though necessary, poses the risk of DNA being damaged and replication – compromised. The accumulation of ssDNA at replication forks is one of the hallmarks of replication stress.15 It may also expose them to endonucleases which will lead to post-replicative double strand breaks (DSBs).
Mathematical Review of Origin Location
The formation of replication factories is a consequence of replication itself. Analysis using a simple mathematical model reveals that origins characterized by low competence or large fluctuations in activation time tend to group together (Fig. 3C).30 This trend may reflect the fact that if origins either tend to take very long time activating or are likely to fail clustering them reduces the risk that large parts of the chromosome will not be replicated on time. On the other hand, when origins are highly competent and have well-defined and preferentially short activation time, maximal coverage and even distribution are favored as an optimal solution (Fig. 3). We already know that some parts of chromatin are unfavorable for origin placement, e.g. extensively transcribed regions, but the process of origin placement seems to be neither fully stochastic nor preferential.29
Figure 3.
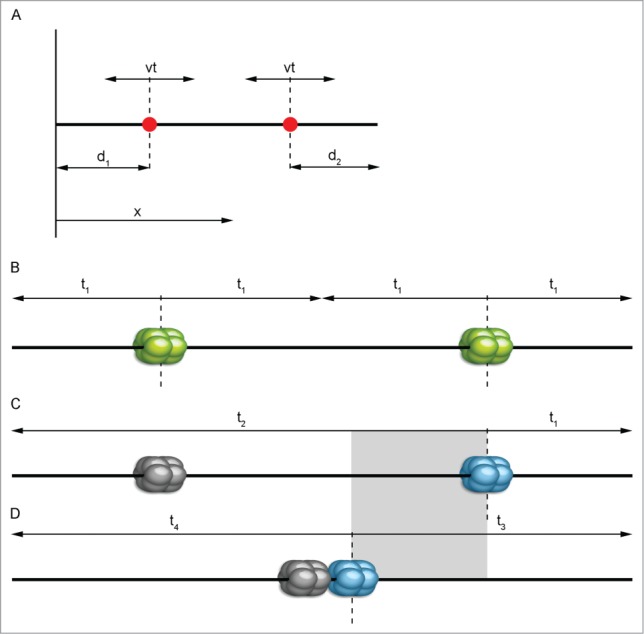
Graphical representation of the simplest mathematical replication model. A simple mathematical model (A) concerning only 2 origins (red dots) gives basic information about location trend. We may consider a chromosome as a line parallel to the x axis. Then, the position of a specific origin on this chromosome can be evaluated by its coordinate x with d1 and d2 being the distance from the nearest end of the chromosome. Replication forks travel from a fired origin in both directions with constant speed v, replicating DNA at time t (according to Karschau et al.30, modified). In the case of high replication origin activity (B), the most probably scenario is that both of them will activate successfully. Separate positioning further from the center toward the sides of a chromosome provides the shortest replication time (t1). If origins are low efficient (C and D), one of them is likely not to be activated and to remain dormant (gray-colored) and another one will have to pick up the replication of an entire chromosome. Positioning of low-active replication origins as if they were active will prolong replication time as seen in (C) by the amount of time marked with t2. The most advantageous solution here is grouping origins together near the center (D) – overall replication time will be shortened (t3 and t4). The gray rectangle shows the approximate difference in replication ending time between (C) and (D).
Analysis of stochasticity in the licensing of fixed and well-known origin loci in S. cerevisiae provides an equational explanation of this phenomenon, with DNA modeled as a one-dimensional length and assuming that there are only 2 loci in the considered region (Fig. 3).30
At first thought, for the 2 origin sites considered, one could expect that the most convenient configuration is one with isolated loci. However if the origins have a significant chance of failing, it would mean that one part of the chromosome would have to wait until the replication fork from the other site reaches it, which would extend the replication time. Therefore, being grouped together near the center is a huge advantage for origins of low competence. Likewise, this model holds true for the extended analysis, where more real models of chromosomes with many loci are investigated. Algorithmical simulations performed for groups of more than 2 origins also point to the conclusion that clusters of many highly competent origins are unfavorable. There is indeed a transition in the optimal configuration of origins (from isolated to grouped) connected with lower competence of the locus and longer of activation time needed. This phenomenon is confirmed by comparing theoretical data with experimental findings for S. cerevisiae and X. laevis (Fig. 3).30
This simplified model, modified further to take into account the timing of origin firing, reveals more information about the parameters indicating origin placement. The expanded analyses were done in the same way for the 2-origin model of unit length and for groups of origins, using realistic parameters of what is known about X. laevis. Using X. laevis as a source is more relevant as, unlike in yeast, any of its loci are able to become an origin. Despite that, biologists have found only more or less equally-spaced groups of 5–10 pMCMs separated about 10kb apart.10,31
Data show that optimal locations of origins are not constant and may be presented as a function of standard deviation (its symbol is σ and it show how spread out the origins are). A sharp transition can be observed at σ = 0.25. Above this, it is best to have 2 origins placed in the middle of the investigated segment, but minor differences occur for σ < 0.25 compared to the previous model (where firing time has not been considered). Next, this formula may be implicated for a full-chromosome model of X. laevis which has approximately 1/1.5 pMCM/kb.10 σ in both X. laevis and S. cerevisiae is 6–10 min long, and the S phase is about 20 min long. In that case, 64 pMCMs were distributed on a 100 kb artificial chromosome either uniformly or randomly. The results indicate that the best replication timing is achieved when 8 groups of 8 origins are present (about 1.1 min advantage over using random loci). This structure is also convenient for protection against fluctuations between rounds of the cell cycle (single initiation event is enough for replication fork activation).
Timing is a major issue for actively proliferating cells, such as early embryos, but for many cell types time of replication may be prolonged by delays due to the activation of cell cycle checkpoints. This possibility is greatly advantageous if we consider issues such as fork stalling or DNA damages that may happen during replication. Another algorithmical theory takes into account the probability of fork stalling and replication failure and may be applied for cells without restrictions of the duration of replication.32 If we consider a region of the genome between 2 neighboring ROs (denoted by a symbol ‘D’ after Newman et al.32), there is a small probability of a fork stalling at each one of the base pairs within this area. To maintain unreplicated DNA within ‘D’ after firing or inactivating (after passive replication) all of the origins, both of the forks shall enter ‘D’ from left and right side and both shall stall before meeting (double fork stall; see also Fig. 4a'). The origin placement of forks is irrelevant in this case because of no time restrictions. Analyses made using elementary probability theory show that there is a very low chance for double fork stall within ‘D’ because the average distance that a fork can travel before being stalled is larger than the typical distance between 2 origins.
Figure 4.
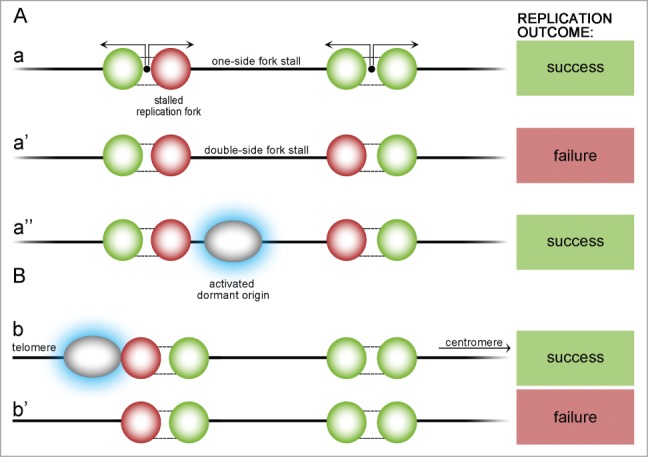
Possible scenarios of origin stalling within a chromosome and at a telomeric region. Stall events may occur either within the chromosome (A) or at the telomeric region (B). One-side stall event (a) does not endanger replication if it happens within the chromosome – DNA will be replicated by another active fork. If, unlikely, active forks stall on 2 sides of yet unreplicated DNA (a'), part of the DNA between them cannot be replicated and replication partially fails, but if there is a dormant origin between (a”), it is activated (blue highlight) and can finish the replication. At the telomeric chromosome end there must be a dormant ‘emergency’ origin that become activated in case of stalling (b). If not, DNA from a telomeric region cannot be replicated (b').
The stall rate may slightly vary at different locations on chromosomes, but the scale of these variations is much smaller than the median stalling distance of replication forks, therefore it does not affect analysis significantly. What is more interesting, chromosome fragile sites were thought to be large domains of increased fork stalling probability. Research shows that these sites are indeed regions containing a shortage of active ROs – regions where the calculated double stall events are highly plausible. Avoidance of double stalls, as a factor, suggests that ROs should be spaced regularly rather than randomly. Examination of S. cerevisiae origins clearly reveals that in vivo they are distributed uniformly with only some minor deviations, which meets all of the above calculations.30-32 Theoretically, for an infinite chromosome with randomly-placed origins, the calculated ratio of the standard deviation of origin separation divided by the mean (R) equals 1. For finite strands R < 1 and this factor reflects some degree of randomness.32 All of the 16 yeast chromosomes have an R lower than that given by random distribution which indicates that their origin spatial distribution is below randomness. Similar conclusions were arrived at after comparison of 3 other yeast species – Kluyveromyces lactis, Lachancea kluyveri and Kluyveromyces waltii (recently named Lachancea waltii). R values of all above considered species are similar, but this is rather because of evolutionary pressure to distribute ROs in a regular manner, and not due to maintaining origin positions. More distant relatives of S.cerevisiae – fission yeast S. pombe – also exhibited non-random RO placement. A higher R value may be dictated by different organization of origins caused by a looser DNA sequence consensus.33-34 Origin distribution in S. pombe resembles more that of metazoans. K. lactis, L. kluyveri and L. waltii have the largest mean inter-origin distance and, interestingly, the lowest R values – this may be a reflection of evolution compensating larger spacing with a more even distribution. Therefore, minimization of the double stall threat must have had an impact on RO positioning throughout the yeast genome, however, more variations are present in inter-origin spacing which may suggest an evolutionary aim to decrease error possibility despite difficulties in creating a perfect order of origins.
Double fork stalls are more likely to occur between widely-spaced origins (Fig. 4A). Despite a large number of RO, there may be a situation where a separation larger than the standard deviation appears, thus increasing the probability of a double stall in a particular region. In S. cerevisiae the largest gaps between neighboring origins were 90.1, 88.5 and 79.3 kb. Theoretical simulations of randomly distributed origins provided an average largest gap of 169 ± 31 kb (the minimum value obtained in simulations is 116 kb) and calculated frequency of double stalled forks is 4.6%. These values are significantly higher than those observed in vivo in yeast and are another confirmation of an evolutionary-favored limit to the maximum separation between origins. These data were confirmed by experimental work on S. cerevisiae with 5 efficient origins deleted. The chromosome with an origin-less region of approximately 160 kb (close to the calculated maximum gap) had a 3 times higher loss rate compared to the correct value.35 This overall protection against fork stalling does not reflect dormancy or activity of origins, but rather the total number of licensed ROs.
Telomeres are critical regions for a linear eukaryotic genome – if one replication fork stalls at a place beyond the furthest RO, there is no other fork to finish DNA replication (Fig. 4B). Each one of the 16 S. cerevisiae chromosomes has the closest origin placed on average 404 ± 273 bp away from the chromosome end. Hence the largest distance from the end of the chromosome to the nearest origin is 730 bp across all 32 telomeric ends. It is still far less than the general distance between origins in the chromosome body. Supporting experimental evidence also shows that for a chromosome with a 160 kb origin-less telomeric region, the rate of loss is more than 20 times higher than in the same region within a chromosome body.35 Yet, this effect appears to be smaller than theoretically predicted, which may imply that some additional mechanism exists to enhance telomeric replication. It still consistently confirms the thesis of origin distribution being determined by the risk of stall events as one of major factors.
Gindin et al.36 provide a new interesting mechanical model of predicting DNA replication in human cells. This chromatin structure-based algorithm considers only factors that have an impact on replication timing. Interestingly, in this model the fork collision mechanism is highly important, while region density in DNase-HS or even separation of replication into licensing and initiation (or origin firing) proved to be insignificant. The factor that dominates the timing program in human cells is the selection of the pre-RC location assembly. This model predicts replication timing even with every pre-RC complex indicating constant probability of subsequent initiation. This is a well-known mechanism in yeast (and described earlier) but not yet tested in metazoan cells. In summary, the human timing model is able to predict replication time and provides cell-specific timing patterns. This data strongly support the stochasticity of the licensing process which is determined by chromatin structure.
Another new study by Li et al.37 reveals information that the replication rate obtained from a single cell responded to the behavior of cell populations, and this connection can be provided by a mathematical model. Earlier research concerning DNA replication rates in subsequent parts of the S phase showed that replication was slower early in the S phase and gained speed in later sub-phases. Recent data have confirmed these results.37
Regulation of Pre-RC Complex Assembly and Dynamic Evolution of Re-replication Control Mechanisms
To prevent re-replication (the replication of regions already replicated in a nucleus; i.e. >1 < 2 S phases per cell division),13 a cell must ensure that no fork is activated on an already replicated part of the DNA strand.1,38 Also, progression into the S phase shall be delayed if there is a reduced number of licensed origins. Therefore a precise checkpoint mechanism that regulates this transition is of high importance for a cell. Normally, progression into the S phase depends on CDK-based phosphorylation which promotes transcription of the S-phase genes. Low CDK4/6-Cyclin D, CDK2-Cyclin E activities and/or hypophosphorylation of Rb proteins may arrest the cell cycle in the G1 phase.39-43
The first level of licensing regulation involves control of ORC activity. Yeasts have well-defined ORC-binding sites, where ORCs bind and remain until firing. More complex dependence appears in higher eukaryotes where ORC binding to DNA may be facilitated or inhibited by other factors, but – more importantly – one or several ORC subunits may be modified in a cell-cycle-specific manner which changes its stability and chromatin relation.44 These changes are mainly achieved by the regulation of the Orc1 subunit, essential for pre-RC assembly (reviewed by DePamphilis et al.2).
There are many new factors that were reported to have an impact on DNA replication. A factor that collaborates with ORC – ORC associated/leucine-rich repeats and WD repeat domain containing 1 (ORCA/LRWD1) – was shown to co-localize with ORC and its level changes throughout the cell cycle were similar to those of human ORCs. ORCA was shown to associate with ORC core proteins which seemed to facilitate ORC binding to DNA and with geminin in human cells, in a cell-cycle-dependent manner. Moreover, ORCA also localizes to centromeres and telomeres and interacts with recombination proteins (reviewed in Sen and Prasanth,45).
Factors that regulate licensing are also the targets of a cell stress response system mediated by ATR-mediated process. ATR influences the replication locally (at replication forks) or globally by arresting cell cycle and repressing late origins.15 The activation of ATR pathway proceeds gradually – first, it acts at replication forks to stabilize them and then the Chk1 kinase is activated to delay mitosis. Thus, ATR and Chk1 were found to play a crucial role in S-phase DNA damage checkpoint.15,46
MCM, Cdt1 and Cdc6
Factors that regulate cell cycle are also responsible for regulation of origin licensing. Loading of MCM complex requires Cdt1, which is constitutively expressed in yeast, but is the first target of down-regulation in metazoa. In those organisms, Cdt1 levels increase during M and G1 phases while in S and G2 they are significantly low. There are several mechanisms that regulate Cdt1 activity – the most common one is ubiquitination by Skp1-Cullin-F-Box (SCF) ligase, facilitated by CDK-dependent phosphorylation. The cells of higher eukaryota, however, still exhibit Cdt1 degradation even if phosphorylation by CDKs has been shut down. In mammalian and Drosophila cells alternative pathways exist that rely on PCNA and Ddb1-Cul4a-Roc1 (DCR) ubiquitination. In Xenopus there is an APC-dependent mechanism.47 Another pathway of Cdt1 activity regulation involves a metazoa-specific factor called geminin. It is active during S and early M phases where it acts as an inhibitor of licensing. From M to early G1 geminin activity is suppressed by either APC-mediated ubiquitination or inhibition in Xenopus or Drosophila early embryos.48 An inactive geminin may be re-imported into a nucleus just before the S phase and then reactivated.47 Metazoan cells do not get rid of Cdt1 completely throughout the cell cycle. Instead, they use geminin in G2 and M phases to bind to and stabilize Cdt1 in an inactive form. This solution provides quick access to Cdt1 after geminin inactivation at the very end of M phase. Cdt1 has been also reported to be a transcriptional E2F regulation target in A. thaliana.49 Similar down regulation also occurs in Drosophila for its Cdt1 homolog – DUP gene.50
CDKs in general prevent re-replication inhibiting pre-RC components.39,40,42,51 An active Cdc6 protein is present in cells only during early G1 phase and is rapidly degraded from late G1 to S phase.52 Its actions are regulated also by various CDK-dependent inhibition mechanisms, with the most direct one being phosphorylation by CDK which promotes Cdc6 for ubiquitin-mediated proteolysis. This process generates 2 active binding sites for Cdc4 in Cdc6, one at the N-terminal regulatory domain (NDT) and the other on the C-terminus (CDT). Cdc6 is one of the prime factors regulating pre-RC assembly in yeast and it is also inhibited directly by mitotic cyclin Clb2. Binding with Clb2 requires phosphorylation of NDT, prevents Cdc6 from being recruited to ORC and from interacting with Cdc4. This process causes partial stabilization of Cdc6 during mitosis and secures it from rapid proteolysis. Analogously to Clb2 in yeast, geminin binds to and regulates the activity of Cdt1 in metazoans.53 Moreover, Clb2-regulation is shown to be varied among yeast species. Schwanniomyces castellii exhibits an absence of slower mitotic degradation suggesting that Cdc6 may not bind to Clb2. All of the Saccharomyces have the NTD Cl4-binding site, yet only those that bind Clb2 have another one at CTD. This suggests that just one Cdc4 binding site (in this case NTD) is not sufficient for effective Cdc4 binding.14 The role of Cdc6 phosphorylation in regulating pre-RC assembly is still unclear in mammalian cells. Phosphorylation of this factor occurs at the onset of the S phase, but human mutant cells lacking this event are reported to either block initiation of DNA replication or do not (reviewed in DePamphilis et al.2). Cdc6 regulation plays a vital role in the regulation of a cell cycle itself. Its phosphorylation delays mitosis if the replication of DNA is incomplete.54
The most mysterious factor of a pre-RC complex is MCM2–7. Its activity is indeed regulated in budding yeast, where its location within a cell is dictated by the state of phosphorylation, but no similar mechanism has been reported in other organisms. MCM can be phosphorylated either by CDKs or Dbf4-Cdc7 in Xenopus, Drosophila, S. pombe or mammals, but its cellular localization does not change as in S. cerevisiae. A limited number of MCM hexamers is a more important factor for licensing than its activity. There needs to be a certain number of licensed active origins to prevent chromosome instability and there is evidence that in metazoan cells a specific ‘licensing checkpoint’ exists that monitors the process of pre-RC assembly. The precise mechanism of this checkpoint is still unclear, but it is most probably based on downregulation of Cdk2 activity and involves p53 protein.40 In A. thaliana an MCM binding factor has been identified – E2F target gene 1 (ETG1) which appears to be evolutionarily conserved. ETG1 binding to MCM is crucial for successful replication – its depletion triggers replication checkpoints and inhibits a cell cycle.55
At this point one may consider factors that might advance the evolution of control mechanisms. Mutant cells that express Cdc6 lacking N-terminal domain still have normal DNA replication, the same goes for strains with deregulated MCM2–7 or ORC. Also cells with deregulated Cdc6 and MCM2–7 are viable, but lack of both ORC and Cdc6 is lethal.56-58 Some level of redundancy is always present within more complex mechanisms (so that cells may adapt better) and it may be considered as an element providing rapid evolution, as seen in Saccharomyces. Mechanisms that can be verified as interchangeable may also contribute here. Interchangeability is the possibility of preventing re-replication by any of the pre-RC regulatory CDK-dependent systems in a single organism. Many of the pre-RC components have developed some additional functions that are not related to DNA replication. To some extent, this may constrain individual regulation of them in particular organisms.
There is no single mechanism that is completely effective, thus plurality is a huge advantage for efficient re-replication inhibition. On the other hand, when the number of mechanisms increases, relative importance of a single one is decreased. These regulatory mechanisms may be lost or gained during evolution. Geminin in metazoa is an additional system which might be gained in response to the increasing genome size. Re-replication is blocked better when a cell has the ability to swap between regulation mechanisms, and this may be the cause of their rapid evolution.
Dynamics of Origin Initiation
When the S phase begins, a licensed locus activates and 2 replication forks that are created at the origin start moving in opposite directions with approximately constant speed, duplicating DNA along the way. As explained earlier, slight fluctuations in the formation of replication origins and their subsequent activation lead to different replication times. Origins also differ in their initiation timing and efficiency which results from frequency of initiation. In fact, efficiency of adjacent origins is interrelated with replication timing – more efficient origins passively replicate their later (or less efficient) neighbors.
One of the major factors that control origin firing is the level of dNTPs. It determines both the number of activated origins and the speed of replication forks. Lower levels of dNTPs cause activation of more origins and slow down the replication forks. Below the critical level of dNTPs, replication checkpoint becames activated and stops the replication.15 Some parts of genome such as chromosome fragile sites (CFSs, they are origin-poor regions overlapping mostly large genes) are especially endangered under such conditions - their replication fully depends on whether slowed forks manage to end the process in time. If not, the cell may enter mitosis with some of the chromosomes still being unreplicated.59 A wide range of chromosomal aberrations: translocations, complex duplications, deletions, double-minute chromosomes, homogenously stainined regions, multicentric chromosomes, lagging chromosomes, sticky chromosomes, small supernumerary marker chromosomes and multiradial chromosomes can induce genetic instability. Aberrant mitotic structures, chromosome fragmentation (C-Frag) and premature chromosome condensation (PCC) are defined as early events in genome chaos leading to chromosome instability (CIN) which can be numerical or structural.18,59-61 CIN is also one of the mechanisms crucial for cancer progression and evolution.17
Chromatin Structure and RO Positions on the Chromosome
The chromatin structure and context appear to be major determinants of origin timing. Origins located near centromeres initiate replication earlier than ROs from subtelomeric regions regardless of their efficiency. The most comprehensive data regarding this subject are provided by experiments with autonomously replicating sequences (ARS) in S. cerevisiae. If ARS1 (which is an early-firing origin) is relocated from its centromeric locus to the telomeric residue of ARS501, its initiation time is delayed, suggesting that early-firing may be the normal primal state and that late-firing is a consequence of chromatin repressive features.62 Thus the chromosomal environment of the origin locus has an impact on its timing. In some circumstances this delay may be extended to the point of dormancy.63 One could presume that this may be caused by interference in origin licensing, but studies of dormant ARS301 reveal that it is indeed capable of initiating if given additional time;64 however, this RO is fired very late which may suggest that passive replication is a favored mechanism.
Different cells of a single population use different sets of origins according to need.65-66 Nonetheless, populational profiles still reflect the timings and efficiencies of single ROs. Comprehensive studies of chromosomes III and VI of S. cerevisiae, confirmed by whole-genome analysis, reveal that near each CEN at least one RO is initiated early and some ROs are initiated even earlier than those of the CEN region (Fig. 5).25,67,68 The thesis of origins being controlled by chromosome structure or subnuclear localization appears to be validated by experiments that analyze the effects of deletion of chromatin modifiers such as SIR3 (required for subtelomeric chromatin assembly) which results in earlier replication of normally late telomeric parts.69 Deletions of other corresponding factors such as YKU70, RPD3 or GCN5 result in a similar hastened initiation of late ROs.70-73 However, increased histone acetylation due to GCN5 tethering or RPD3 deletion, does not cause late ROs to initiate as early as the earliest origins. It has been suggested that there may be other elusive determinants of early-firing. Current data suggest that a functional CEN establishes and promotes early origin initiation.74 In the yeast Candida albicans, excision of the CEN sequence resulted in formation of a new functional CEN region (neo-CEN) and recruitment of a new ORC to this site. As one may expect, the new origin fired early. This phenomenon was repeated subsequently at several loci in distant strains, but this might have been determined by other determinants or cryptic ROs, not by the neo-CEN. The best evidence for CEN defining early replication has been provided by Pohl et al.75 Their research on S. cerevisiae strains with CENs relocated from original positions shows that this treatment advanced the initiation time of normally late origins if the relocated CEN was near and delayed the early ROs present in the proper CEN locus. The same reliance exists in the fission yeast S. pombe, although its mechanism seems to differ from S. cerevisiae.76 In S. pombe an equivalent of DDK-activating subunit Dbf4 – Dfp1 – is directly bound by Swi6.77 Tethering of this binding slows down origin activation in centromeric regions of S. pombe but does not affect euchromatin fraction of the genome.
Figure 5.
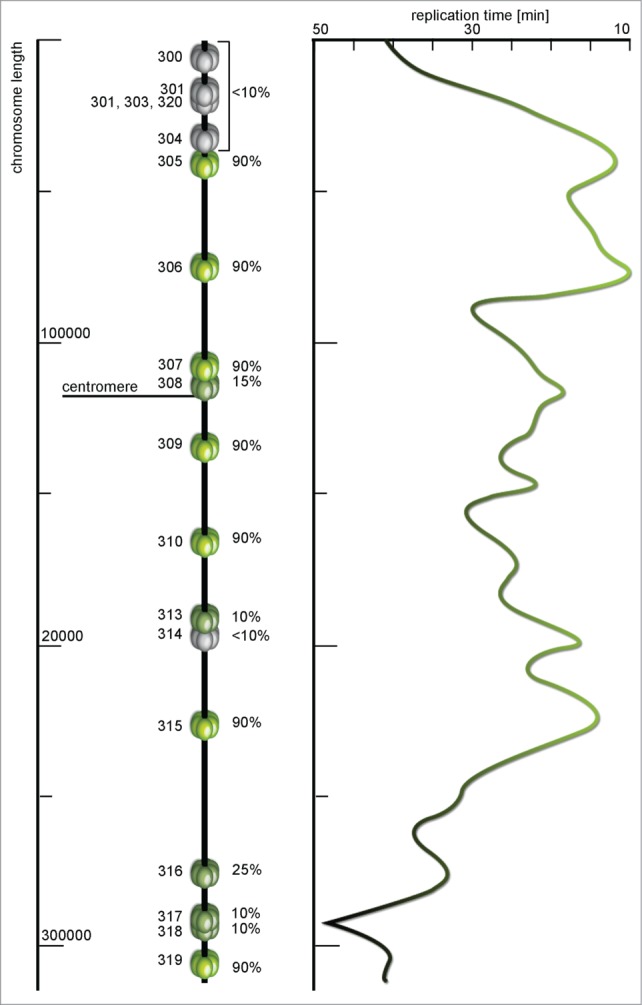
A map of ARS sequences and replication time profile of Saccharomyces cerevisiae chromosome III. Each miniature MCM helicase depicts a replication origin site with the name on the left and efficiency (%) on the right side of the model. The efficiency here is the percentage of cell cycles in which specific origin were initiated. Origins with less than 10% of efficiency are marked dormant (gray), these are origins for which initiation has not been reported, and highly efficient origins are colored green. The timing profile shows distribution of activation time along the chromosome – from green parts replicated in about 10 min to gray regions that are replicated in late subperiods of the S phase. The timing profile seems to almost perfectly reflect the activity and efficiency of particular origins (according to Raghuraman et al.67 and Poloumienko et al.113, modified).
In eukaryotic cells the rates of replication were proved to respond to chromatin structure, with high gene density regions tending to replicate earlier (Fig. 5). The mammalian genome is characterized by its large size and occurrence of large GC regions (isochores). For mouse and human cells replication time has been determined and gene-rich isochores tend to replicate earlier, but similar isochoric structures were not shown for Drosophila or Arabidopsis. This may explain why gene content, expression profile and epigenetic modifications have a more subtle influence on replication dynamics in plants,78 still the replication profiles remain very similar (if not identical). In A. thaliana chromosome 4 the majority of euchromatin is replicated early while the heterochromatin replication begins in the late S phase. This observation is consistent with observations in mouse, human and Drosophila cells.
Ongoing replication fork may encounter several obstacles during replication process, such as actively transcribed genes, DNA lesions (damaged bases, crosslinks) or protein complexes with DNA which can make it slack or even stall. Some parts of a chromosome, such as centromeres or telomeres, tend to slow down replication forks probably to avoid collisions.15,79
Genome-wide Determinants of Origin Initiation
Most of the early and late S. cerevisiae origins are affected by forkhead box (Fox) transcription factors Fkh1 and Fkh2 (Fkh1/2), that are thought to regulate replication timing (compare with Fig. 6.73,80 Evidence demonstrates that cells lacking Fkh1 and Fkh2 exhibit different replication profiles where 106 of normally early ROs were delayed (entitled Fkh-activated), and 82 of late origins (Fkh-repressed) initiated earlier. Interestingly, CEN-proximal origins up to 25 kb apart are not affected. CENs, therefore, determine early firing locally without Fkh1/2 dependence. Fkh1/2-binding sites tend to be denser near Fkh-activated ROs, and the removal of 2 binding sites adjacent to (early) origin ARS305 significantly delayed its firing compared to a wild type. On the contrary, Fkh1/2 consensus binding sites were found to be depleted near Fkh-repressed ROs.
Figure 6.
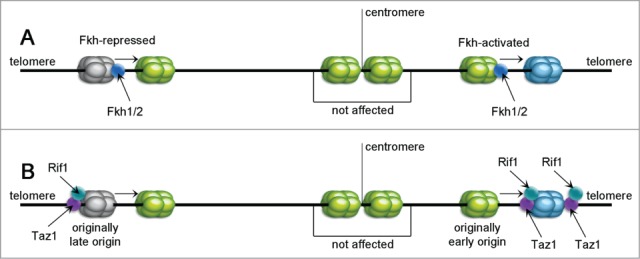
Sequence summarizing the effects of Fkh1/2, Rif1 and Taz1 proteins. Fkh1/2 are reported to control replication timing by acting as activators or inhibitors of origin activity (A). Some origins are reported to be Fkh-activated (shown on the right) and demonstrate lower efficiency upon Fkh depletion. Others act conversely. Taz1 and Rif1 (B) are shown to bind mostly to the telomeric regions with replication timing control (RTC) sequences, but they can also affect some internal loci. If the RTC sequence near an internal late origin is deleted, its firing time accelerates (left) and also an active origin can be delayed if placed between 2 RTCs (right).
Other regulators of replication time appear to be telomere-binding proteins Rif1 and Taz1, examined in S. pombe (Fig. 6).81-82 Taz1 binds directly to telomeric sequences and afterwards recruits Rif1.83 Telomeric chromatin was discussed previously in regard to replication timing of subtelomeric sites, and Taz1 and Rif1 were shown to affect replication dynamics in some internal loci as well.82 Deletion of the replication timing control (RTC) sequence neighboring an internal late origin, advances its firing. Additionally, insertion of 2 copies of RTC next to an early origin extends its initiation. RTCs have a Taz1-binding sequence (tandem-repeated telomere sequence), and Taz1 binding to chromatin was shown in 13 of the internal ROs with neighboring RTCs. Yet, Taz1 aggregation is greatest near telomeres and it does not seem to bind some of the more internal origins that may be affected by its deletion. This may reflect some distant regulation by telomeric Taz1 (compare with Fig. 6).
Rif1 may play an another role in timing regulation. It was shown that it acts contrary to DDK,81 and in some loci, Rif1 binding is independent of Taz1. The sequence binding Rif1 near LE origins resembles the late consensus sequence of ars727.84 Deletion of rif1+ caused later replication of 134 normally early origins (EL – early to late firing transition) while the LE group (late to early transisted ROs) consisted of all Taz1-regulated ROs together with 47 internal late or even dormant ROs. Rif1 also may bind internally with increased density near LE origins, which suggests that it affects them directly by delaying their firing and that EL actions are distal. Exceptionally, pericentic origins have many EL groups and bind Rif1, suggesting that it may have a direct influence on timing these ROs. It seems probably that in order to regulate the pericentric region, Rif1 may bind to Swi6, but this is still unclear. All the data point to the conclusion that Rif1 acts by direct promotion of pericentric ROs and delay of subtelomeric ROs.
The evolutionary role of Rif1 seems to be well preserved. Despite Taz1 not being present in S. cerevisiae, Rap1 protein fulfills that role of recruitment of Rif1.85 Moreover, replication timing of subtelomeric origins in budding yeast is connected with the loss of Rif1.86 This may result from loss of a Rif1-dependent telomere length-sensing mechanism which promotes early replication of normal-length telomeres instead of critically short ones. This mechanism can be also halted by deletion of YKU70. A simpler mechanism of advancing replication time may also arise from reduced Taz1 and Rif1 binding at shorter telomeres, which promotes earlier initiation of ROs in subtelomeric regions. Out of the handful of analyzed origins, in one particular case CEN-proximal RO ARS1 replicated later in rif1Δ cells.86 This phenomenon may suggest that budding yeast CENs may be able to bind Rif1, but this needs elucidation. The latest data show that Rif1 also plays a significant role in replication timing in mammals as observed in HeLa and mouse cells.87-88 Both cases show a significant change in a standard replication pattern corresponding to the deletion of Rif1, with increased early initiation events. These data evidence once more that Rif1 acts as an inhibitor for origin initiation.
Regulation of origin timing by Fkh1/2, Taz1 and Rif1 seems to be distinct from the Abf1-dependent mechanism. Various studies imply that the role of Abf1 is to provide a nucleosome-free region that is essential for MCM loading.89 It is unclear whether Abf1 affects replication timing or not – some experiments reveal that a nucleosome-free region is essential for establishing origin competence but does not determine its timing.90-91 Accordingly, deletion or different levels of Fkh1/2, Taz1 and/or Rif1 do not affect ORC or MCM2–7 levels;81-82 hence these factors do not determine licensing (compare with Fig. 6).
Transcription directly through RO disrupts the origin by interference with ORC and MCM binding,92-93 so some changes in transcription can also cause alterations in origin competence (either positive or negative). Nevertheless, no changes in ORC and/or MCM binding (level- and time-dependent) were observed in cells with deregulated timing mechanisms in fkh1Δ, fkh2Δ, rif1Δ, taz1Δ yeast or Rif1-depleted HeLa cells.80-82,87,94 Moreover, most of the transcriptional deregulations can be rescued by shortening of the C-terminus of Fkh2 in fkh1Δfkh2Δ cells, but this treatment does not affect origin deregulation.80 In addition, transcriptome analysis revealed no correlation between residual changes in transcription and origin deregulation.88 This evidence strongly implies that origin placement, licensing and timing control are independent of transcription.
Among more interesting factors regulating the timing and speed of replication there is checkpoint kinase 1 (Chk1) which inhibits origin firing when a cell is under replication stress. Current data reveal that at low levels of Chk1 activity (low replication stress), this factor suppresses firing of new replication factories more preferentially than it inhibits dormant origin activation within already active factories. This phenomenon occurs probably because of an ability of ATR/Chk1 to slightly reduce levels of Cdks, which further reduces the level of active replication factories. In such a case the probability of double fork stalls is increased and this may trigger dormant origins to become active.95
Cdc45-Sld3 Loading
The timing of the DDK-dependent recruitment of Cdc45 and its loading factor – Sld3 – is a temporally regulated process that reflects different timing of RO initiation.96 Experiments by Aparicio et al.97 and Kamimura et al.96 showed that Cdc45 was bound to DNA at early origins in the G1 phase, which might suggest its limited level throughout the cell cycle. Early origin timing is established in the early G1 phase,98 and therefore association of Cdc45 with G1 is essential. Analyses of Fkh1/2 dependence of Cdc45 revealed that Fkh-activated ROs have a high affinity for Cdc45 in G1 phase, but CEN-proximal regions act oppositely – their Cdc45 binding ability increases in the absence of Fkh1/2, which confirms the mechanisms of earlier initiation of centromeric regions.
Taz1 and Rif1 can also determine Cdc45 binding to chromatin – in rif1Δ and taz1Δ fission yeast cells one can observe an increased rate of Cdc45 binding to earlier origins at mutated strands, according to repressive nature of Taz1 and Rif1.81-82 Rif1 may act prior to initiation in order to inhibit Cdc45 access to ROs which is consistent with other data showing that Rif1 binds to chromatin in G1.81,87,88 Several studies confirmed that Cdc45-Sld3 indeed limited origin firing in vivo. In X. laevis egg extracts, a decrease in Cdc45 level reduced the efficiency of replication,99 in S. pombe, its overexpression increased the early S phase replication.100-101 The same correlation exists in mammalian cells, where Cdc45 is present at a relatively low level compared to the total number of licensed origins, and its increase resulted in a higher number of replisomes.12
It was shown that in S. cerevisiae, late-firing ROs were directly dependent not only on Cdc45 levels, but also on Sld3, Sld7 or DDK subunits of Cdc7 and Dbf4.102 Another analysis showed that overexpression of Sld3, Dbf4, Sld2 and Dbp11 advanced late origin firing.103 In addition, overexpression of Cdc45 and Sld7 together with the previously listed factors increases the general number of active origins. All these data imply that abundance of initiation factors, especially DDK-dependent ones, limits initiation of DNA replication.
Cdc45 is a component of replisomes and because of its limited concentration in G1, only a small number of origins can fire at the onset of S phase.104-105 Origins described as relatively late (compared to the very early ROs) have to wait until early replicons terminate and Cdc45s can be recycled to another licensed origin – this is the major role of temporal firing distribution.106 Cdc45-Sld3 loading and recycling onto origins may be also regulated by the checkpoint systems modulating Sld3 and DDK activities but despite this the timing program is established nonetheless.107-109
Consequences of Replication Stress
According to Zeman and Cimprich,110 the most common human disease associated with defects in DNA replication process is cancer. On the other hand, a significant heterogeneity in more than 20 phenotypes associated with defects in replication stress response-ralated proteins was shown.110 Current research indicates that cancer cells overexpress many of the factors involved in replication control.15 In fact, almost half of the deletions observed in cancer cells evolved from CFSs.15,111
The excess of factors promoting initiation cause almost all origins to fire in early S phase in S. cerevisiae which deplets the level of dNTPs and causes destabilization of replication forks. Insufficient DNA replication leads to failure in chromosome duplication and impacts their segregation during mitosis.18,59-61 As mentioned earlier, CFSs sites lack the excess origin mechanism that can compensate for slowed or stalled forks. As a result, incompletely duplicated chromosomes may form anaphase bridges. Further, these bridges are the source of kinetic tension, which together with additional centromeres formed also as a result of chronic replication stress, may lead to chromosome breaks and their uneven segregation (the majority of extra centromeres are functional). The fact that these events occur indicates that some of the low-level replication stress events are not detected by cells.15 The anaphase bridges, however, activate a mitotic checkpoint that inhibits mitosis progression until the problem is resolved or induce aneuploidy and chromosomal instability, as well as cell death.61 On the other hand, in yeast, a process called adaptation has been described, which allows yeast cells to overcome mitotic arrest and continue cell division.15
The phenomenon of genome chaos itself may be triggered by a single event or by a drug treatment (e.g., C-Frag or PCC). In both cases, one can observe various types of shattered chromosomes at different time points. Due to a huge amount of DSBs occurring during C-Frag, it is supposed that non-homologous end joining (NHEJ) may play a vital role in the creation of chaotic genomes. The fact that cells lacking Ku70 and Ku80 proteins still undergo C-Frag but are not experiencing genome chaos holds this theory strong. In addition, recent research using sequencing has revealed that chaotic genome reorganization occurs mainly through NHEJ).18,112 Interestingly, despite a high amount of cells dying during and after induction of genome chaos, there is a small fraction of surviving cells. This means that genome chaos can also act as an adaptive mechanism because the cells that manage to survive are further selected and mostly those with the least chaotic new karyotype live on and proliferate. This event may be one of the prerequisites of tumor surviving.18 This theory is confirmed by the fact that majority of tumors have altered genomes that are the key to cancer evolution even though most of the chaotic genomes are evolutionally eliminated in later phases of cancer.
Perspectives and Conclusions
Origin placement is evolutionarily dictated by some essential factors such as replication timing or stall event prevention, and is the case with evolution, none of the determining mechanisms act alone. Recent experiments and theoretical analyses have shed new light on the subject of origin placement which had been thought to be more random. In fact, although it is not possible to determine a precise locus for origin placement in higher eukaryotes, RO distribution is defined by much more complex correlations and mechanisms. Stochasticity is a fundamental feature of DNA replication and mathematical models are indispensable tools allowing understanding of the dynamics and robustness.
Theoretical analyses show that only by proper distribution of ROs the chance of replication failure can be minimized to almost none. Moreover, biological factors that affect timing mostly do not determine licensing. In accordance with this, we conclude that: (i) empirical analyses of replication in different groups of organisms confirm that mathematical models are accurate for all eukaryotes (plants, animals and even humans,36) and (ii) even the simplest of the mathematical models can be used to determine the probability that a given locus is the site of replication, provided that analysis takes a limited scope of factors affecting replication. Actually, simultaneous analysis of in silico-created data with in vivo-obtained results is also possible to achieve full description of the behavior of replication origins during DNA replication process. All the above mentioned analyses enabled us to present a comprehensive review of current knowledge about the spatio-temporal dynamics of activation and licensing of replication origins, supported by the valid mathematical models.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
The work was funded by “POMOST” fellowship from the Foundation for Polish Science (contract no. POMOST/2011–4/8).
References
- 1.Blow JJ, Dutta A. Preventing re-replication of chromosomal DNA. Nat Rev Mol Cell Biol 2005; 6:476-86; PMID:15928711; http://dx.doi.org/ 10.1038/nrm1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DePamphilis ML, Blow JJ, Ghosh S, Saha T, Noguchi K, Vassilev A. Regulating the licensing of DNA replication origins in metazoa. Curr Opin Cell Biol 2006; 18:231-9; PMID:16650748; http://dx.doi.org/ 10.1016/j.ceb.2006.04.001 [DOI] [PubMed] [Google Scholar]
- 3.Evrin C, Clarke P, Zech J, Lurz R, Sun J, Uhle S, Li H, Stillman B, Speck C. A double-hexameric MCM2-7 complex is loaded onto origin DNA during licensing of eukaryotic DNA replication. Proc Natl Acad Sci USA 2009; 106:20240-5; PMID:19910535; http://dx.doi.org/ 10.1073/pnas.0911500106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Remus D, Beuron F, Tolun G, Griffith JD, Morris EP, Diffley JF. Concerted loading of Mcm2-7 double hexamers around DNA during DNA replication origin licensing. Cell 2009; 139:719-30; PMID:19896182; http://dx.doi.org/ 10.1016/j.cell.2009.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moyer SE, Lewis PW, Botchan MR. Isolation of the Cdc45/Mcm2-7/GINS (CMG) complex, a candidate for the eukaryotic DNA replication fork helicase. Proc Natl Acad Sci USA 2006; 103:10236-41; PMID:16798881; http://dx.doi.org/ 10.1073/pnas.0602400103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ilves I, Petojevic T, Pesavento JJ, Botchan MR. Activation of the MCM2-7 helicase by association with Cdc45 and GINS proteins. Mol Cell 2010; 37:247-58; PMID:20122406; http://dx.doi.org/ 10.1016/j.molcel.2009.12.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kelly TJ, Brown GW. Regulation of chromosome replication. Annu Rev Biochem 2000; 69:829-80; PMID:10966477; http://dx.doi.org/ 10.1146/annurev.biochem.69.1.829 [DOI] [PubMed] [Google Scholar]
- 8.Burkhart R, Schulte D, Hu D, Musahl C, Göhring F, Knippers R. Interactions of human nuclear proteins P1Mcm3 and P1Cdc46. Eur J Biochem 1995; 228:431-8; PMID:7705359; http://dx.doi.org/ 10.1111/j.1432-1033.1995.tb20281.x [DOI] [PubMed] [Google Scholar]
- 9.Donovan S, Harwood J, Drury LS, Diffley JF. Cdc6p-dependent loading of Mcm proteins onto pre-replicative chromatin in budding yeast. Proc Natl Acad Sci 1997; 94:5611-6; PMID:9159120; http://dx.doi.org/ 10.1073/pnas.94.11.5611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mahbubani HM, Chong JP, Chevalier S, Thömmes P, Blow JJ. Cell cycle regulation of the replication licensing system: involvement of a Cdk-dependent inhibitor. J Cell Biol 1997; 136:125-35; PMID:9008708; http://dx.doi.org/ 10.1083/jcb.136.1.125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blow JJ, Ge XQ, Jackson DA. How dormant origins promote complete genome replication. Trends Biochem Sci 2011; 36:405-14; PMID:21641805; http://dx.doi.org/ 10.1016/j.tibs.2011.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wong PG, Winter SL, Zaika E, Cao TV, Oguz U, Koomen JM, Hamlin JL, Alexandrow MG. Cdc45 limits replicon usage from a low density of preRCs in mammalian cells. PLoS One 2011; 6:e17533; PMID:21390258; http://dx.doi.org/ 10.1371/journal.pone.0017533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mazurczyk M, Rybaczek D. Replication and re-replication: different implications of the same mechanism. Biochimie 2015; 108:25-32; PMID:25446651; http://dx.doi.org/ 10.1016/j.biochi.2014.10.026 [DOI] [PubMed] [Google Scholar]
- 14.Drury LS, Diffley JF. Factors affecting the diversity of DNA replication licensing control in eukaryotes. Curr Biol 2009; 19:530-5; PMID:19285403; http://dx.doi.org/ 10.1016/j.cub.2009.02.034 [DOI] [PubMed] [Google Scholar]
- 15.Magdalou I, Lopez BS, Pasero P, Lambert SAE. The causes of replication stress and their consequences on genome stability and cell fate. Semin Cell Dev Biol 2014; 30:154-64; PMID:24818779; http://dx.doi.org/ 10.1016/j.semcdb.2014.04.035 [DOI] [PubMed] [Google Scholar]
- 16.Truong LN, Wu X. Prevention of DNA re-replication in eukaryotic cells. J Mol Cell Biol 2011; 3:13-22; PMID:21278447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heng HH, Bremer SW, Stevens JB, Horne SD, Liu G, Abdallah BY, Ye KJ, Ye CJ. Chromosomal instability (CIN): what it is and why it is crucial to cancer evolution. Cancer Metastasis Rev 2013; 32:325-340; PMID:23605440 [DOI] [PubMed] [Google Scholar]
- 18.Liu G, Stevens JB, Horne SD, Abdallah BY, Ye KJ, Bremer SW, Ye CJ, Chen DJ, Heng HH. Genome chaos: survival strategy during crisis. Cell Cycle 2014; 13:528-37; PMID:24299711; http://dx.doi.org/ 10.4161/cc.27378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Horne SD, Chowdhury SK, Heng HHQ. Stress, genomic adaptation, and the evolutionary trade-off. Front Genet 2014; 5:1-6; PMID:24567736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stevens JB, Abdallah SA, Liu G, Ye CJ, Horne SD, Wang G, Savasan S, Shekhar M, Krawetz SA, Hüttemann M, Tainsky MA, Wu GS, Xie Y, Zhang K, Heng HHQ. Diverse system stresses: common mechanisms of chromosome fragmentation. Cell Death Dis 2011; 2:e178; PMID:21716293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sirbu BM, Couch FB, Feigerle JT, Bhaskara S, Hiebert SW, Cortez D. Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev 2011; 25:1320-7; PMID:21685366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Woodward AM, Göhler T, Luciani MG, Oehlmann M, Ge X, Gartner A, Jackson DA, Blow JJ. Excess Mcm2-7 license dormant origins of replication that can be used under conditions of replicative stress. J Cell Biol 2006; 173:673-83; PMID:16754955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ge XQ, Jackson DA, Blow JJ. Dormant origins licensed by excess Mcm2-7 are required for human cells to survive replicative stress. Genes Dev 2007; 21:3331-41; PMID:18079179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ibarra A, Schwob E, Mendez J. Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proc Natl Acad Sci USA 2008; 105:8956-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamashita M, Hori Y, Shinomiya T, Obuse C, Tsurimoto T, Yoshikawa H, Shirahige K. The efficiency and timing of initiation of replication of multiple replicons of Saccharomyces cerevisiae chromosome VI. Genes Cells 1997; 2:655-65; PMID:9491800 [DOI] [PubMed] [Google Scholar]
- 26.Yang SC, Rhind N, Bechhoefer J. Modeling genome-wide replication kinetics reveals a mechanism for regulation of replication timing. Mol Syst Biol 2010; 6:404-16; PMID:20739926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rhind N. DNA replication timing: random thoughts about origin firing. Nat Cell Biol 2006; 8:1313-6; PMID:17139278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guilbaud G, Rappailles A, Baker A, Chen CL, Arneodo A, Goldar A, d'Aubenton-Carafa Y, Thermes C, Audit B, Hyrien O. Evidence for sequential and increasing activation of replication origins along replication timing gradients in the human genome. PLoS Comput Biol 2011; 7:e1002322; PMID:22219720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saner N, Karschau J, Natsume T, Gierlinski M, Retkute R, Hawkins M, Nieduszynski CA, Blow JJ, de Moura AP, Tanaka TU. Stochastic association of neighboring replicons creates replication factories in budding yeast. J Cell Biol 2013; 202:1001-12; PMID:24062338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karschau J, Blow JJ, de Moura APS. Optimal placement of origins for DNA replication. Phys Rev Lett 2012; 108:058101; PMID:22400964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blow JJ, Gillespie PJ, Jackson DA. Replication origins in Xenopus egg extract are 5–15 kgbases apart and are activated in clusters that fire at different times. J Cell Biol 2001; 152:15-26; PMID:11149917; http://dx.doi.org/ 10.1083/jcb.152.1.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Newman TJ, Mamun MA, Nieduszynski CA, Blow JJ. Replisome stall events have shaped the distribution of replication origins in the genomes of yeasts. Nucleic Acids Res 2013; 41:9705-18; PMID:23963700; http://dx.doi.org/ 10.1093/nar/gkt728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cotobal C, Segurado M, Antequera F. Structural diversity and dynamics of genomic replication origins in Schizosaccharomyces pombe. EMBO J 2010; 29:934-42; PMID:20094030; http://dx.doi.org/ 10.1038/emboj.2009.411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu J, Yanagisawa Y, Tsankov A, Hart C, Aoki K, Kommajosyula N, Steinmann KE, Bochicchio J, Russ C, Regev A, et al.. Genome-wide identification and characterization of replication origins by deep sequencing. Genome Biol 2012; 13:R27; PMID:22531001; http://dx.doi.org/ 10.1186/gb-2012-13-4-r27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Theis JF, Irene C, Dershowitz A, Brost RL, Tobin ML, di Sanzo FM, Wang JY, Boone C, Newlon CS. The DNA damage response pathway contributes to the stability of chromosome III derivatives lacking efficient replicators. PLoS Genet 2010; 6:e1001227; PMID:21151954; http://dx.doi.org/ 10.1371/journal.pgen.1001227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gindin Y, Valenzuela MS, Aladjem MI, Meltzer PS, Bilke S. A chromatin structure-based model accurately predicts DNA replication timing in human cells. Mol Syst Biol 2014; 10:722; PMID:24682507; http://dx.doi.org/ 10.1002/msb.134859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li B, Zhao H, Rybak P, Dobrucki JW, Darzynkiewicz Z, Kimmel M. Different rates of DNA replication at early versus late S-phase sections: multiscale modeling of stochastic events related to DNA content/EdU (5-ethynyl-2'deoxyuridine) incorporation distributions. Cytometry Part A 2014; 85:785-97; http://dx.doi.org/ 10.1002/cyto.a.22484 [DOI] [PubMed] [Google Scholar]
- 38.Arias EE, Walter JC. Strength in numbers: preventing rereplication via multiple mechanisms in eukaryotic cells. Genes Dev 2007; 21:497-518; PMID:17344412; http://dx.doi.org/ 10.1101/gad.1508907 [DOI] [PubMed] [Google Scholar]
- 39.Shreeram S, Sparks A, Lane DP, Blow JJ. Cell type-specific responses of human cells to inhibition of replication licensing. Oncogene 2002; 21:6624-32; PMID:12242660; http://dx.doi.org/ 10.1038/sj.onc.1205910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Machida YJ, Teer JK, Dutta A. Acute reduction of an origin recognition complex (ORC) subunit in human cells reveals a requirement of ORC for Cdk2 activation. J Biol Chem 2005; 280:27624-30; PMID:15944161; http://dx.doi.org/ 10.1074/jbc.M502615200 [DOI] [PubMed] [Google Scholar]
- 41.Teer JK, Machida YJ, Labit H, Novac O, Hyrien O, Marheineke K, Zannis-Hadjopoulos M, Dutta A. Proliferating human cells hypomorphic for origin recognition complex 2 and pre-replicative complex formation have a defect in p53 activation and Cdk2 kinase activation. J Biol Chem 2006; 281:6253-60; PMID:16407251; http://dx.doi.org/ 10.1074/jbc.M507150200 [DOI] [PubMed] [Google Scholar]
- 42.Liu P, Slater DM, Lenburg M, Nevis K, Cook JG, Vaziri C. Replication licensing promotes cyclin D1 expression and G1 progression in untransformed human cells. Cell Cycle 2009; 8:125-36; PMID:19106611; http://dx.doi.org/ 10.4161/cc.8.1.7528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nevis KR, Cordeiro-Stone M, Cook JG. Origin licensing and p53 status regulate Cdk2 activity during G(1). Cell Cycle 2009; 8:1952-63; PMID:19440053; http://dx.doi.org/ 10.4161/cc.8.12.8811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.DePamphilis ML. Cell cycle dependent regulation of the origin recognition complex. Cell Cycle 2005; 4:70-9; PMID:15611627; http://dx.doi.org/ 10.4161/cc.4.1.1333 [DOI] [PubMed] [Google Scholar]
- 45.Shen Z, Prasanth SG. Emerging players in the initiation of eukaryotic DNA replication. Cell Div 2012; 7:22; PMID:23075259; http://dx.doi.org/ 10.1186/1747-1028-7-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Iyer DR, Rhind N. Checkpoint regulation of replication forks: global or local. Biochem Soc Trans 2013; 41:1701-5; PMID:24256278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li A, Blow JJ. Cdt1 downregulation by proteolysis and geminin inhibition prevents DNA re-replication in Xenopus. EMBO J 2005; 24:395-404; PMID:15616577; http://dx.doi.org/ 10.1038/sj.emboj.7600520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li A, Blow JJ. Non-proteolytic inactivation of geminin requires CDK-dependent ubiquitination. Nat Cell Biol 2004; 6:260-7; PMID:14767479; http://dx.doi.org/ 10.1038/ncb1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Castellano MM, Boniotti MB, Caro E, Schnittger A, Gutierrez C. DNA replication licensing affects cell proliferation or endoreplication in a cell type-specific manner. Plant Cell 2004; 16:2380-93; PMID:15316110; http://dx.doi.org/ 10.1105/tpc.104.022400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sloan RS, Swanson CI, Gavilano L, Smith KN, Malek PY, Snow-Smith M, Duronio RJ, Key SC. Characterization of null and hypomorphic alleles of the Drosophila I(2)dtl/cdt2 gene: larval lethality and male fertility. Fly 2012; 6:173-83; PMID:22722696; http://dx.doi.org/ 10.4161/fly.20247 [DOI] [PubMed] [Google Scholar]
- 51.Feng D, Tu Z, Wu W, Liang C. Inhibiting the expression of DNA replication-initiation proteins induces apoptosis in human cancer cells. Cancer Res 2003; 63:7356-64; PMID:14612534 [PubMed] [Google Scholar]
- 52.Piatti S, Lengauer C, Nasmyth K. Cdc6 is an unstable protein whose de novo synthesis in G1 is important for the onset of S phase and for preventing a ‘reductional’ anaphase in the budding yeast Saccharomyces cerevisiae. EMBO J 1995; 14:3788-99; PMID:7641697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mailand N, Diffley JF. CDKs promote DNA replication origin licensing in human cells by protecting Cdc6 from APC/C-dependent proteolysis. Cell 2005; 122:915-26; PMID:16153703; http://dx.doi.org/ 10.1016/j.cell.2005.08.013 [DOI] [PubMed] [Google Scholar]
- 54.Oehlmann M, Score AJ, Blow JJ. The role of Cdc6 in ensuring complete genome licensing and S phase checkpoint activation. J Cell Biol 2004; 168:181-90; http://dx.doi.org/ 10.1083/jcb.200311044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Takahashi N, Lammens T, Boudolf V, Maes S, Takeshi Y, De Jaeger G, Witters E, Inzé D, De Veylder L. The DNA replication checkpoint aids survival of plants deficient in the novel replisome factor ETG1. EMBO 2008; 27:1840-51; http://dx.doi.org/ 10.1038/emboj.2008.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nguyen VQ, Co C, Li JJ. Cyclin-dependent kinases prevent DNA re-replication through multiple mechanisms. Nature 2001; 411:1068-73; PMID:11429609; http://dx.doi.org/ 10.1038/35082600 [DOI] [PubMed] [Google Scholar]
- 57.Mimura S, Seki T, Tanaka S, Diffley JFX. Phosphorylation-dependent binding of mitotic cyclins to Cdc6 contributes to DNA replication control. Nature 2004; 431:1118-23; PMID:15496876; http://dx.doi.org/ 10.1038/nature03024 [DOI] [PubMed] [Google Scholar]
- 58.Wilmes GM, Archambault V, Austin RJ, Jacobson MD, Bell SP, Cross FR. Interaction of the S-phase cyclin Clb5 with an ‘RXL’ docking sequence in the initiator protein Orc6 provides an origin-localized replication control switch. Genes Dev 2004; 18:981-91; PMID:15105375; http://dx.doi.org/ 10.1101/gad.1202304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mankouri HW, Huttner D, Hickson ID. How unfinished business from S-phase affects mitosis and beyond. EMBO J 2013; 32:2661-71; PMID:24065128; http://dx.doi.org/ 10.1038/emboj.2013.211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rybaczek D. Ultrastructural changes associated with the induction of premature chromosome condensation in Vicia faba root meristem cells. Plant Cell Rep 2014; 33:1547-64; PMID:24898011; http://dx.doi.org/ 10.1007/s00299-014-1637-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Potapova TA, Zhu J, Li R. Aneuploidy and chromosomal instability: a vicious cycle driving cellular evolution and cancer genome chaos. Cancer Metastasis Rev 2013; 32:377-89; PMID:23709119; http://dx.doi.org/ 10.1007/s10555-013-9436-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ferguson BM, Fangman WL. A position effect on the time of replication origin activation in yeast. Cell 1992; 68:333-9; PMID:1733502; http://dx.doi.org/ 10.1016/0092-8674(92)90474-Q [DOI] [PubMed] [Google Scholar]
- 63.Dubey DD, Davis LR, Greenfeder SA, Ong LY, Zhu JG, Broach JR, Newlon CS, Huberman JA. Evidence suggesting that the ARS elements associated with silencers of the yeast mating-type locus HML do not function as chromosomal DNA replication origins. Mol Cell Biol 1991; 11:5346-55; PMID:1922050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vujcic M, Miller CA, Kowalski D. Activation of silent replication origins at autonomously replicating sequence elements near the HML locus in budding yeast. Mol Cell Biol 1999; 19:6098-109; PMID:10454557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Patel PK, Arcangioli B, Baker SP, Bensimon A, Rhind N. DNA replication origins fire stochastically in fission yeast. Mol Biol Cell 2006; 17:308-16; PMID:16251353; http://dx.doi.org/ 10.1091/mbc.E05-07-0657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Czajkowsky DM, Liu J, Hamlin JL, Shao Z. DNA combing reveals intrinsic temporal disorder in the replication of yeast chromosome VI. J Mol Biol 2008; 375:12-9; PMID:17999930; http://dx.doi.org/ 10.1016/j.jmb.2007.10.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Raghuraman MK, Winzeler EA, Collingwood D, Hunt S, Wodicka L, Conway A, Lockhart DJ, Davis RW, Brewer BJ, Fangman WL. Replication dynamics of the yeast genome. Science 2001; 294:115-21; PMID:11588253; http://dx.doi.org/ 10.1126/science.294.5540.115 [DOI] [PubMed] [Google Scholar]
- 68.Yabuki N, Terashima H, Kitada K. Mapping of early firing origins on a replication profile of budding yeast. Genes Cells 2002; 7:781-9.; PMID:12167157; http://dx.doi.org/ 10.1046/j.1365-2443.2002.00559.x [DOI] [PubMed] [Google Scholar]
- 69.Stevenson JB, Gottschling DE. Telomeric chromatin modulates replication timing near chromosome ends. Genes Dev 1999; 13:146-51; PMID:9925638; http://dx.doi.org/ 10.1101/gad.13.2.146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cosgrove AJ, Nieduszynski CA, Donaldson AD. Ku complex controls the replication time of DNA in telomere regions. Genes Dev 2002; 16:2485-90; PMID:12368259; http://dx.doi.org/ 10.1101/gad.231602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vogelauer M, Rubbi L, Lucas I, Brewer BJ, Grunstein M. Histone acetylation regulates the time of replication origin firing. Mol Cell 2002; 10:1223-33; PMID:12453428; http://dx.doi.org/ 10.1016/S1097-2765(02)00702-5 [DOI] [PubMed] [Google Scholar]
- 72.Aparicio JG, Viggiani CJ, Gibson DG, Aparicio OM. The Rpd3-Sin3 histone deacetylase regulates replication timing and enables intra-S origin control in Saccharomyces cerevisiae. Mol Cell Biol 2004; 24:4769-80; PMID:15143171; http://dx.doi.org/ 10.1128/MCB.24.11.4769-4780.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Knott SR, Viggiani CJ, Tavaré S, Aparicio OM. Genome-wide replication profiles indicate an expansive role for Rpd3L in regulating replication initiation timing or efficiency, and reveal genomic loci of Rpd3 function in Saccharomyces cerevisiae. Genes Dev 2009; 23:1077-90; PMID:19417103; http://dx.doi.org/ 10.1101/gad.1784309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Koren A, Tsai HJ, Tirosh I, Burrack LS, Barkai N, Berman J. Epigenetically-inherited centromere and neocentromere DNA replicates earliest in S-phase. PLoS Genet 2010; 6:e1001068; PMID:20808889; http://dx.doi.org/ 10.1371/journal.pgen.1001068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Pohl TJ, Brewer BJ, Raghuraman MK. Functional centromeres determine the activation time of pericentric origins of DNA replication in Saccharomyces cerevisiae. PLoS Genet 2012; 8:e1002677; PMID:22589733; http://dx.doi.org/ 10.1371/journal.pgen.1002677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 2001; 410:120-4; PMID:11242054; http://dx.doi.org/ 10.1038/35065138 [DOI] [PubMed] [Google Scholar]
- 77.Hayashi MT, Takahashi TS, Nakagawa T, Nakayama J, Masukata H. The heterochromatin protein Swi6/HP1 activates replication origins at the pericentromeric region and silent mating-type locus. Nat Cell Biol 2009; 11:357-62; PMID:19182789; http://dx.doi.org/ 10.1038/ncb1845 [DOI] [PubMed] [Google Scholar]
- 78.Lee TJ, Pascuzzi PE, Settlage SB, Shultz RW, Tanurdzic M, Rabinowicz PD, Menges M, Zheng P, Main D, Murray JA, et al.. Arabidopsis thaliana chromosome 4 replicates in two phases that correlate with chromatin state. PLoS Genet 2010; 6:e1000982; PMID:20548960; http://dx.doi.org/ 10.1371/journal.pgen.1000982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lambert S, Carr AM. Impediments to replication fork movement: stabilisation, reactivation and genome instability. Chromosoma 2013; 122:33-45; PMID:23446515; http://dx.doi.org/ 10.1007/s00412-013-0398-9 [DOI] [PubMed] [Google Scholar]
- 80.Knott SR, Peace JM, Ostrow AZ, Gan Y, Rex AE, Viggiani CJ, Tavare S, Aparicio OM. Forkhead transcription factors establish origin timing and long-range clustering in S. cerevisiae. Cell 2012; 148:99-111; PMID:22265405; http://dx.doi.org/ 10.1016/j.cell.2011.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hayano M, Kanoh Y, Matsumoto S, Renard-Guillet C, Shirahige K, Masai H. Rif1 is a global regulator of timing of replication origin firing in fission yeast. Genes Dev 2012; 26:137-50; PMID:22279046; http://dx.doi.org/ 10.1101/gad.178491.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tazumi A, Fukuura M, Nakato R, Kishimoto A, Takenaka T, Ogawa S, Song JH, Takahashi TS, Nakagawa T, Shirahige K, et al.. Telomere-binding protein Taz1 controls global replication timing through its localization near late replication origins in fission yeast. Genes Dev 2012; 26:2050-62; PMID:22987637; http://dx.doi.org/ 10.1101/gad.194282.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kanoh J, Ishikawa F. spRap1 and spRif1, recruited to telomeres by Taz1, are essential for telomere function in fission yeast. Curr Biol 2001; 11:1624-30; PMID:11676925; http://dx.doi.org/ 10.1016/S0960-9822(01)00503-6 [DOI] [PubMed] [Google Scholar]
- 84.Yompakdee C, Huberman JA. Enforcement of late replication origin firing by clusters of short G-rich DNA sequences. J Biol Chem 2004; 279:42337-44; PMID:15294892; http://dx.doi.org/ 10.1074/jbc.M407552200 [DOI] [PubMed] [Google Scholar]
- 85.Marcand S, Wotton D, Gilson E, Shore D. Rap1p and telomere length regulation in yeast. Ciba Found Symp 1997; 211:76-93; PMID:9524752 [DOI] [PubMed] [Google Scholar]
- 86.Lian HY, Robertson ED, Hiraga S, Alvino GM, Collingwood D, McCune HJ, Sridhar A, Brewer BJ, Raghuraman MK, Donaldson AD. The effect of Ku on telomere replication time is mediated by telomere length but is independent of histone tail acetylation. Mol Biol Cell 2011; 22:1753-65; PMID:21441303; http://dx.doi.org/ 10.1091/mbc.E10-06-0549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yamazaki S, Ishii A, Kanoh Y, Oda M, Nishito Y, Masai H. Rif1 regulates the replication timing domains on the human genome. EMBO J 2012; 31:3667-77; PMID:22850674; http://dx.doi.org/ 10.1038/emboj.2012.180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cornacchia D, Dileep V, Quivy JP, Foti R, Tili F, Santarella-Mellwig R, Antony C, Almouzni G, Gilbert DM, Buonomo SB. Mouse Rif1 is a key regulator of the replication-timing programme in mammalian cells. EMBO J 2012; 31:3678-90; PMID:22850673; http://dx.doi.org/ 10.1038/emboj.2012.214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lipford JR, Bell SP. Nucleosomes positioned by ORC facilitate the initiation of DNA replication. Mol Cell 2001; 7:21-30; PMID:11172708; http://dx.doi.org/ 10.1016/S1097-2765(01)00151-4 [DOI] [PubMed] [Google Scholar]
- 90.Berbenetz NM, Nislow C, Brown GW. Diversity of eukaryotic DNA replication origins revealed by genome- wide analysis of chromatin structure. PLoS Genet 2010; 6:e1001092; PMID:20824081; http://dx.doi.org/ 10.1371/journal.pgen.1001092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Eaton ML, Galani K, Kang S, Bell SP, MacAlpine DM. Conserved nucleosome positioning defines replication origins. Genes Dev 2010; 24:748-53; PMID:20351051; http://dx.doi.org/ 10.1101/gad.1913210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Snyder M, Sapolsky RJ, Davis RW. Transcription interferes with elements important for chromosome maintenance in Saccharomyces cerevisiae. Mol Cell Biol 1988; 8:2184-94; PMID:3290652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Blitzblau HG, Chan CS, Hochwagen A, Bell SP. Separation of DNA replication from the assembly of break-competent meiotic chromosomes. PLoS Genet 2012; 8:e1002643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhu G, Spellman PT, Volpe T, Brown PO, Botstein D, Davis TN, Futcher B. Two yeast forkhead genes regulate the cell cycle and pseudohyphal growth. Nature 2000; 406:90-4; PMID:10894548; http://dx.doi.org/ 10.1038/35021046 [DOI] [PubMed] [Google Scholar]
- 95.Ge XQ, Blow JJ. Chk1 inhibits replication factory activation but allows dormant origin firing in existing factories. J Cell Biol 2010; 191:1285-97; PMID:21173116; http://dx.doi.org/ 10.1083/jcb.201007074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kamimura Y, Tak YS, Sugino A, Araki H. Sld3, which interacts with Cdc45 (Sld4), functions for chromosomal DNA replication in Saccharomyces cerevisiae. EMBO J 2001; 20:2097-107; PMID:11296242; http://dx.doi.org/ 10.1093/emboj/20.8.2097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Aparicio OM, Stout AM, Bell SP. Differential assembly of Cdc45p and DNA polymerases at early and late origins of DNA replication. Proc Natl Acad Sci USA 1999; 96:9130-5; PMID:10430907; http://dx.doi.org/ 10.1073/pnas.96.16.9130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Dimitrova DS, Gilbert DM. The spatial position and replication timing of chromosomal domains are both established in early G1 phase. Mol Cell 1999; 4:983-93; PMID:10635323; http://dx.doi.org/ 10.1016/S1097-2765(00)80227-0 [DOI] [PubMed] [Google Scholar]
- 99.Edwards MC, Tutter AV, Cvetic C, Gilbert CH, Prokhorova TA, Walter JC. MCM2–7 complexes bind chromatin in a distributed pattern surrounding the origin recognition complex in Xenopus egg extracts. J Biol Chem 2002; 277:33049-57; PMID:12087101; http://dx.doi.org/ 10.1074/jbc.M204438200 [DOI] [PubMed] [Google Scholar]
- 100.Patel PK, Kommajosyula N, Rosebrock A, Bensimon A, Leatherwood J, Bechhoefer J, Rhind N. The Hsk1(Cdc7) replication kinase regulates origin efficiency. Mol Biol Cell 2008; 19:5550-8; PMID:18799612; http://dx.doi.org/ 10.1091/mbc.E08-06-0645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wu PY, Nurse P. Establishing the program of origin firing during S phase in fission yeast. Cell 2009; 136:852-64; PMID:19269364; http://dx.doi.org/ 10.1016/j.cell.2009.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tanaka S, Nakato R, Katou Y, Shirahige K, Araki H. Origin association of Sld3, Sld7, and Cdc45 proteins is a key step for determination of origin-firing timing. Curr Biol 2011; 21:2055-63; PMID:22169533; http://dx.doi.org/ 10.1016/j.cub.2011.11.038 [DOI] [PubMed] [Google Scholar]
- 103.Mantiero D, Mackenzie A, Donaldson A, Zegerman P. Limiting replication initiation factors execute the temporal programme of origin firing in budding yeast. EMBO J 2011; 30:4805-14; PMID:22081107; http://dx.doi.org/ 10.1038/emboj.2011.404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Aparicio OM, Weinstein DM, Bell SP. Components and dynamics of DNA replication complexes in S. cerevisiae: redistribution of MCM proteins and Cdc45p during S phase. Cell 1997; 91:59-69; PMID:9335335; http://dx.doi.org/ 10.1016/S0092-8674(01)80009-X [DOI] [PubMed] [Google Scholar]
- 105.Tercero JA, Labib K, Diffley JF. DNA synthesis at individual replication forks requires the essential initiation factor Cdc45p. EMBO J 2000; 19:2082-93; PMID:10790374; http://dx.doi.org/ 10.1093/emboj/19.9.2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ma E, Hyrien O, Goldar A. Do replication forks control late origin firing in Saccharomyces cerevisiae? Nucleic Acids Res 2012; 40:2010-9; PMID:22086957; http://dx.doi.org/ 10.1093/nar/gkr982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zegerman P, Diffley JF. Checkpoint-dependent inhibition of DNA replication initiation by Sld3 and Dbf4 phosphorylation. Nature 2010; 467:474-8; PMID:20835227; http://dx.doi.org/ 10.1038/nature09373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Santocanale C, Diffley JF. A Mec1- and Rad53-dependent checkpoint controls late-firing origins of DNA replication. Nature 1998; 395:615-8; PMID:9783589; http://dx.doi.org/ 10.1038/27001 [DOI] [PubMed] [Google Scholar]
- 109.Shirahige K, Hori Y, Shiraishi K, Yamashita M, Takahashi K, Obuse C, Tsurimoto T, Yoshikawa H. Regulation of DNA-replication origins during cell-cycle progression. Nature 1998; 395:618-21; PMID:9783590; http://dx.doi.org/ 10.1038/27007 [DOI] [PubMed] [Google Scholar]
- 110.Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol 2014; 16:2-9; PMID:24366029; http://dx.doi.org/ 10.1038/ncb2897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Le Tallec B, Millot GA, Blin ME, Brison O, Dutrillaux B, Debatisse M. Common fragile site profiling in epithelial and erythroid cells reveals that most recurrent cancer deletions lie in fragile sites hosting large genes. Cell Rep 2013; 4:420-8; PMID:23911288; http://dx.doi.org/ 10.1016/j.celrep.2013.07.003 [DOI] [PubMed] [Google Scholar]
- 112.Malhotra A, Lindberg M, Faust GG, Leibowitz ML, Clark RA, Layer RM, Quinlan AR, Hall IM. Breakpoint profiling of 64 cancer genomes reveals numerous complex rearrangements spawned by homology-independent mechanisms. Genome Res 2013; 23:762-76; PMID:23410887; http://dx.doi.org/ 10.1101/gr.143677.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Polumienko A, Dershowitz A, De J, Newlon CS. Completion of replication map of Saccharomyces cerevisiae chromosome III. Mol Biol Cell 2001; 15:3317-27; http://dx.doi.org/ 10.1091/mbc.12.11.3317 [DOI] [PMC free article] [PubMed] [Google Scholar]