

Abstract
Isoforms derived from alternative splicing, mRNA translation initiation or promoter usage extend the functional repertoire of the p53, p63 and p73 genes family and of their regulators MDM2 and MDMX. Here we show cap-independent translation of an N-terminal truncated isoform of hMDMX, hMDMXp60, which is initiated at the 7th AUG codon downstream of the initiation site for full length hMDMXFL at position +384. hMDMXp60 lacks the p53 binding motif but retains the RING domain and interacts with hMDM2 and hMDMXFL. hMDMXp60 shows higher affinity for hMDM2, as compared to hMDMXFL. In vitro data reveal a positive cooperative interaction between hMDMXp60 and hMDM2 and in cellulo data show that low levels of hMDMXp60 promote degradation of hMDM2 whereas higher levels stabilize hMDM2 and prevent hMDM2-mediated degradation of hMDMXFL. These results describe a novel alternatively translated hMDMX isoform that exhibits unique regulatory activity toward hMDM2 autoubiquitination. The data illustrate how the N-terminus of hMDMX regulates its C-terminal RING domain and the hMDM2 activity.
Keywords: alternative translation, isoforms, MDMX, MDM2, protein stability
Introduction
Expression of isoforms extends and differentiates the cell biological activity of genes within the p53 family. The role of different products of p63 and p73 was early on recognized as key cell biological determinants.1,2 The full length p63 and p73 carry transactivation domains and share approximately 60% identity with the p53 core DNA binding domain and behave similar to p53 in some respects such as overlapping promoter binding specificity and gene activation.3 Whereas p53 plays a key role in tumor progression, p63 and p73 are indispensable for squamous epithelia and neural development, respectively.4-6 p53 isoforms are implicated in different stress responses pathways and with specific cell biological activity. While the alternative initiated p53 mRNA translation product, p53/47, specifically induces G2 arrest following stress to the endoplasmic reticulum and the unfolded protein response, the p53β and the Δ133p53 isoforms play a role in controlling cellular senescence.7,8 The MDM2 (mouse double minute 2) is a key regulator of p53 activity and has the capacity to suppress p53 activity either via promoting its degradation and by interfering with its transcriptional activity, or it can stimulate p53s rate of synthesis following genotoxic stress by binding to the p53 mRNA.9-12 However, MDM2 interacts with over 100 different cellular factors, including proteins and nucleotides and exhibits cell biological effects that reach outside the p53 pathway.13-16 While over 40 splice variants of mdm2 have been reported, the main isoforms are the MDM2p90 and the N-terminally truncated MDM2p76.17-19 The latter lacks the p53 binding pocket and is expressed by alternative translation initiation or splicing. The expression of the two MDM2 isoforms is regulated differently and transcripts initiated by the constitutive P1 promoter induce expression of MDM2p76 and MDM2p90 at different levels depending on tissue and stress context and high levels of MDM2p76 have been reported in mice following radiation.20 As MDM2p76 lacks the p53-binding domain, it does not promote p53 ubiquitination but retains the capacity to stimulate p53 synthesis.21 MDM2 and its homolog MDMX are both essential but non-redundant regulators of p53 activity during development but MDMX does not exhibit E3 ligase activity toward p53.11,22 Instead, it has been proposed that the main activity of full length MDMX toward p53 is mediated via an interference with p53's transactivation activity. MDM2 and MDMX form hetero- and homo-dimers via their respective C-terminal RING domains but how these respective formations are regulated and their physiological roles is relatively unknown. Both MDM2 and MDMX are over expressed in certain types of cancers and are implicated in tumor development by the suppression of p53 activity. While amplification of MDM2 is best known to occur in different types of sarcomas it has more recently been shown that approximately 70% of melanomas overexpress MDMX.23,24 The understanding of how MDM2 and MDMX are regulated and how they control p53 activity is the focus of intense research both from the academia and the pharmaceutical industry in search for new therapeutic strategies to activate p53.25,26 In this study we describe the MDMXp60 isoform that lacks the N-terminal p53 binding domain which adds to a previously reported splice variant of mdmx that includes 114 residues of MDMX N-terminus plus 13 unique C-terminal residues, and a caspase cleaved product which lacks the C-terminus.27,28 We show that alternative translation initiation generates MDMXp60 and that different concentration of MDMXp60 either promote, or prevent, MDM2 autoubiquitination activity. These results also reveal that the N-terminus of MDMX controls the interaction between MDM2 and MDMX RING domains.
Results
The translation of hMDMXp60 is initiated at the 7th in frame AUG codon downstream of +1
Expression of a cDNA encoding full length hmdmx mRNA including its 5’UTR (hmdmxwt) in H1299 human lung adenoma cells resulted in the expression of full length hMDMX (hMDMXFL) of approximately 76 kDa as well as a band with the approximate size of 60 kDa (hMDMXp60) as determined using a polyclonal antibody that reacts against an epitope located between residues 125 and 175. To understand the origin of this product we deleted in frame AUG codons downstream of the first +1 AUG, one by one, until we reached the 7th AUG at position +384 (codon 128) (hmdmxΔ2–7 AUG) whereby the expression of hMDMXp60 vanished. If we instead replaced the first AUG alone (data not shown), or the first 6 in frame AUGs with alanines (CGC) (hmdmxΔ1–6 AUG), or deleted the entire sequence upstream of codon 7 (hmdmxp60), we could only detect the hMDMXp60 isoform (Fig. 1A and B). Hence, hMDMXp60 is initiated at the 7th in frame AUG codon and lacks the N-terminus of full length hMDMX, including the p53-binding domain. We next tested expression of endogenous hMDMXp60 in HeLa, MDA-MB-231 and Saos-2 cells and we could identify a band of an approximate similar size as hMDMXp60 using the same polyclonal antibody (Fig. 1C).
Figure 1.

The hMDMXp60 isoform is initiated at the 7th in frame AUG codon. (A) Cartoon illustrating the hmdmx mRNA constructs and the mutated AUG sites (left). The 6 in frame AUG codons after the +1 AUG of the hmdmx coding sequence were substituted with alanine (GCG) (hmdmxΔ2–7AUG) to express only full length hMDMX (hMDMXFL). The hmdmxΔ1–6AUG mRNA lacks the first 6 in frame AUG codons and only expresses hMDMXp60 which is initiated from the 7th in frame AUG (+384). The hMDMXp60 lacks the first 127 amino acids, including the p53 binding domain (right). (B) Western blot showing the expression of the two hMDMX isoforms from indicated constructs in H1299 cells. The hmdmxwt mRNA expresses both isoforms. Actin is used as loading control. (C) The relative expression of the two hMDMX isoforms in different cell lines. H1299 and Saos-2 are p53 negative cell lines whereas HeLa expresses wild type p53 and MDA-MB-231 expresses a mutant p53(R280K). Western blots show one representative experiment out of 3.
Cap-independent translation of hMDMXp60and hMDMXFL
The fact that hMDMXp60 is initiated at the 7th in frame AUG codon downstream of the +1 AUG makes leaky scanning or canonical cap-dependent translation initiation unlikely mechanisms to explain its expression.29 We therefore tested if synthesis of hMDMXp60 could be mediated via cap-independent mechanism of translation initiation that allows the ribosomal pre-initiation complex to enter direct on the hmdmx mRNA. We created a bicistronic construct by placing an open reading frame (GFP) followed by a hairpin structure upstream of the full length hmdmx mRNA including its 5′UTR. This construct prevents cap-dependent initiation of translation from the hmdmx mRNA (Fig. 2A). After expression of the bicistronic hmdmx in H1299 cells we could observe the expression of GFP and approximately equal amounts of hMDMXFL and hMDMXp60 (Fig. 2B). The relative levels of hMDMXFL as compared to hMDMXp60 are approximately 4 fold higher when expressed from the hmdmxwt construct, as compared to the bicistronic setting, indicating that even though both isoforms can be synthesized by cap-independent translation, this mechanism of initiation is more important for the synthesis of hMDMXp60 (Fig. 2B). In order to ensure that the bicistronic mRNA remains intact in cells and that we did not create a cryptic promoter between the 5’UTR of the hmdmx mRNA and the initiation site for hMDMXp60 by inserting the hmdmx mRNA downstream of the GFP-hairpin sequences, we carried out a series of RT-PCR and RT-qPCR using different sets of primers giving similar size products (Fig. 2A). Using a forward primer starting upstream of the hairpin structure (P1) together with a reverse primer spanning the +1 AUG site (P3) resulted in a similar amount of RT-PCR products as compared with a forward primer starting from within the 5’UTR (P2) plus the P3. Using a primer pair from the coding sequence (P4 and P5) also revealed a similar amount of RT-PCR products (Fig. 2C). Using these primer pairs we could verify that the relative amount of RT-PCR products derived from the primers covering the 5’ UTR (P2 and P3) and the primers spanning the initiation site for hMDMXp60(P4 and P5) is similar. To verify the specificity of the RT-PCR reactions and to ensure that transfected cDNA or cellular DNA did not interfere with the RT-PCR reactions we also carried out the control reactions in which we did not add reverse transcriptase (Fig. 2D). Finally, by adding siRNA toward GFP, the expression levels of GFP and hMDMX isoforms were reduced similarly from cells expressing the bicistronic construct but had, as expected, no effect on hMDMX isoform levels in cells expressing the hmdmxwt construct (Fig. 2E; Fig. S1). To test if cap-independent translation from the hmdmx mRNA is responsive to regulatory pathways we subjected cells expressing the bicistronic construct to different levels of UV irradiation and analyzed the effect hMDMX expression after 4 hours (the suppression is similar after 2 hours, data not shown). The reduction of hMDMX expression was similarly suppressed by increasing the levels of UV in the presence, or not, of proteasome inhibitor, indicating that cap-independent translation of the hmdmx mRNA was affected by this treatment. The effect of UV radiation on cap-dependent GFP expression from the same construct was less affected (Fig. 2F; Fig. S2). These results support the notion that both hMDMX isoforms can be expressed via cap-independent mechanisms of translation and that, at least under these conditions, cap-independent translation is relatively more important for controlling the synthesis of hMDMXp60. The data also suggest that the cap-independent translation of the hmdmx mRNA is under regulation of UV stress pathways.
Figure 2.

(See previous page) hMDMXp60 and hMDMXFL are derived from cap-independent mRNA translation initiation. (A) Cartoon illustrating the bicistronic hmdmx mRNA inserted downstream of the GFP open reading frame and downstream of a hairpin structure preventing ribosomal read through from cap-dependent translation initiation of the GFP open reading frame. The location of primers (P1 to P5) is indicated. These were used for RT-PCR and RT-qPCR to ensure that the bicistronic mRNA remains intact in cells and that no cryptic promoter activity causes alternative RNA species. (B) The expression of the two hMDMX isoforms from the wild type mRNA and from the bicistronic construct in H1299 cells. The relative level of expression of the two hMDMX isoforms from wild type vs. bicistronic constructs is estimated (upper graph). (C) RT-PCR using indicated primer pairs repeated 4 times each for the bicistroninc hmdmx mRNA. (D) The relative amount of RT-PCR products from the bicistronic and the wild type hmdmx mRNAs as estimated using quantitative RT-PCR from indicated primers. The ratio of RT-PCR products derived from the primers covering the 5’UTR and the initiation site for hmdmxFL (P2 and P3) and from a sequence covering the initiation site of hMDMXp60 (P4 and P5) are similar between the wild type and the bicistronic hmdmx mRNAs. This shows that no cryptic promoter or alternative splicing was created by fusing the hmdmx mRNA to the bicistronic construct. No added RT enzyme (-RT) shows that the quantified PCR products are derived from mRNA. Actin mRNA serves as control. The graph shows data from 3 independent experiments plus SD. (E) The use of siRNA against the GFP results in a similar reduction in expression of GFP and hMDMX from the bicistronic hmdmx mRNA. There is no effect on hMDMX expression form the wild type mRNA (Fig. S1). (F) Western blot showing the effect of indicated UV doses 4 hours after treatment on cap-independent translation of the hmdmx mRNA as compared to cap-dependent translation of gfp (Fig. S2). Western blots show one representative experiment out of 3.
hMDMXp60 binds hMDM2 with higher affinity as compared with hMDMXFL
As hMDMXp60 does not retain the N-terminal p53 binding domain we first tested its putative function by comparing the binding of hMDMXp60 to hMDMXFL and hMDM2, which is mediated by respective C-terminal RING domains. An equal amount of hmdmxp60 and hmdmxfl were expressed in H1299 cells together with hmdm2. When hMDM2 was immunoprecipitated (IPed) using the 4B2 antibody against an N-terminal epitope we observed the co-IP of both hMDMXFL and hMDMXp60 (Fig. 3A). If we instead used anti-HA antibodies against an hMDMXp60 fused to an HA-tag (hmdmxp60HA) from cell lysates expressing hmdmxfl, we observed a relative small amount of co-IPed hMDMXFL, indicating that the two hMDMX isoforms are found in complexes with each other and/or with hMDM2. Similarly, hMDM2 was also found to coIP with hMDMXp60HA (Fig. 3B, left and right panels). In order to study the interaction between hMDM2 and the hMDMX isoforms in more detail, we carried out ELISA assays using a fixed amount of recombinant hMDMXFL or hMDMXp60 (500 ng) and increasing levels of recombinant hMDM2. This revealed a higher affinity between hMDMXp60 and hMDM2, as compared to hMDMXFL and hMDM2 (Fig. 3C). Interestingly, the ELISA data also indicated a positive cooperative binding between hMDM2-hMDMXp60 and hMDM2-hMDMXFL. Taken together, these data show that hMDMXp60 forms a stronger interaction with hMDM2 as compared to hMDMXFL.
Figure 3.
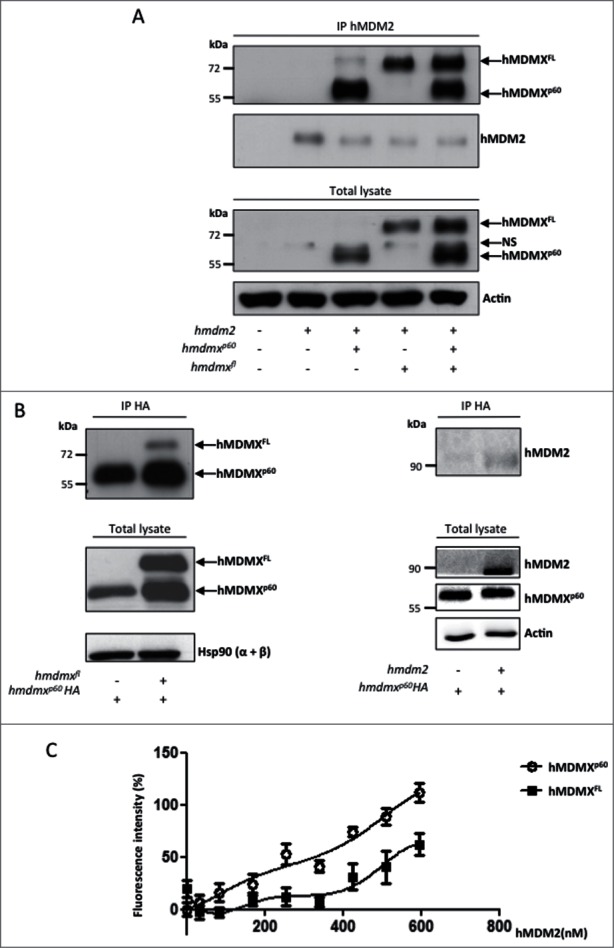
hMDMXp60 shows higher affinity toward hMDM2 as compared to hMDMXFL. (A) Immunoprecipitation (IP) against hMDM2 shows that hMDM2 interacts with both hMDMX isoforms (upper panel). The protein levels in whole lysates are shown below. (NS stands for non-specific). (B) Co-immunoprecipitation of hMDMX isoforms (left) or hMDM2 (right) using an HA-tagged hMDMXp60. Hsp90 and actin serves as loading controls of total lysates. (C) ELISA using a fixed (500 ng) amount of recombinant purified hMDMXp60 or hMDMXFL and increasing amounts of hMDM2. The affinity of hMDMXp60 to hMDM2 is higher as compared to hMDMXFL. Both proteins bind hMDM2 in a biphasic fashion. The data shows the average of 7 independent experiments and SD. Western blots (A and B) show one representative experiment out of 3.
hMDMXp60 stabilizes hMDMXFL from hMDM2-mediated degradation
The differences in affinities between hMDM2-hMDMXFL and hMDM2-hMDMXp60 were surprising as the interaction between hMDMX and hMDM2 takes place between the C-terminal RING-RING domains. This prompted us to test if the capacity of hMDM2 to control the stability of each isoform is also affected by the hMDMX N-terminus. The expression of a fixed amount of either hmdmxfl or hmdmxp60 (200ng cDNA) in the presence of increasing levels of hmdm2 (10 to 200 ng cDNA) showed an approximately 90% reduction in hMDMXFL levels at 50 ng transfected hmdm2 cDNA. The corresponding reduction in hMDMXp60 expression was approximately 20% at the same concentration of hmdm2. At the maximal 200 ng of hmdm2 the levels of hMDMXp60 were reduced by 80% (Fig. 4A, left and right panels). When we instead expressed a fixed amount (200 ng) of both hmdmx isoforms in the presence of increasing levels of hmdm2 we observed that the presence of hMDMXp60 stabilized hMDMXFL (Fig. 4B). This stabilizing effect of hMDMXp60 on hMDMXFL was further demonstrated by increasing the levels of hmdmxp60 in the presence of a fixed amount of hmdmxfl, indicating that hMDMXp60 also affects endogenous hMDM2-mediated regulation of hMDMXFL stability (Fig. 4C). These results could be explained by hMDMXp60 being a poor substrate for hMDM2 and hMDMXp60 could, thus, stabilize hMDMXFL by competing for binding to hMDM2. An easy explanation for hMDMXp60 being a poor substrate could be that hMDM2 promotes ubiquitination of hMDMX N-terminal lysine residues. To test this hypothesis we mutated each, or all together, of the 6 in frame lysines residues upstream of the 7th AUG initiation codon of hMDMX to arginines. This did, however, not affect the capacity of hMDM2 to promote degradation, indicating that hMDM2 mediated ubiquitination of hMDMX takes place on lysines residues further downstream of lysine 104 (Fig. 4D). The idea that the N-terminus of hMDMX is not the target for hMDM2 E3 ubiquitin ligase activity was further supported by in vitro ubiquitination assays using recombinant purified proteins showing that hMDM2 promotes equally well polyubiquitination of hMDMXFL as of hMDMXp60 (Fig. S3A). Furthermore, in vivo ubiquitination assays showed that mHMDMXp60 is a substrate for hMDM2 but that the degradation of the full length form is more efficient (Fig. S3B). Hence, while these results show that hMDMXp60 has a higher affinity for hMDM2 as compared to hMDMXFL and that hMDMXp60 protects hMDMXFL from hMDM2-mediated degradation, they offer no support to the idea that hMDMXp60 being a poor ubiquitin substrate for hMDM2 and instead indicates that the N-terminus of hMDMX regulates hMDM2 E3 ligase activity.
Figure 4.
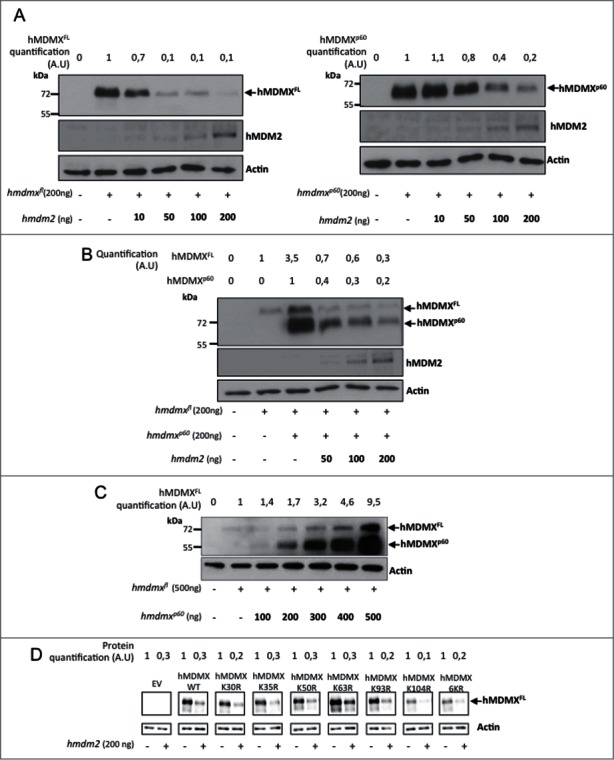
hMDMXp60 stabilizes hMDMXFL in the presence of hMDM2. (A) The levels of expression of hMDMXFL in H1299 cells following increasing amounts of hMDM2 (left). A similar experiment but using a fixed amount of hMDMXp60 and increasing levels of hMDM2 (right). Compare differences in the expression of hMDMX isoform in cells transfected with 50 ng hmdm2 cDNA. (B) hMDMXFL is protected from hMDM2-mediated degradation in the presence of hMDMXp60. (C) Increasing levels of hMDMXp60 stabilizes hMDMXFL in cells expressing endogenous hMDM2. (D) Mutation of each, or all 6 together, of the lysine residues in the N-terminus of hMDMX upstream of the initiation site for hMDMXp60 do not affect hMDM2-mediated degradation of hMDMX. The data are representative from 3 independent experiments (Fig. S3A and B).
hMDMX isoforms induce oscillations in hMDM2 expression
Having observed that hMDMXp60 affects the stability of hMDMXFL in the presence of hMDM2, we next asked the question if hMDMXp60 might affect the stability of hMDM2. E3 RING ligases generally require dimerization to promote E3 ubiquitin ligase activity30 and we tested the effect of hMDMXp60 on hMDM2's autoubiquitination activity. Increasing levels of hmdmxp60 resulted in an increase in hMDM2 levels at the lowest concentrations of transfected cDNA (10ng). But as the levels of hmdmxp60 increased to 50 ng the expression of hMDM2 dropped. Further increase in hmdmxp60 resulted in a subsequent increase in hMDM2 expression and at 200 ng of hmdmxp60 there was a significant increase in hMDM2 expression (Fig. 5A, panel a). This was not observed using an hMDM2 protein carrying a mutation in residue cysteine 464 which prevents its E3 ligase activity or reduced when proteasome inhibitors were added (Figs. 5A, panel d; Fig. S4A). This oscillation was reproducible even though the fluctuation pattern of hMDM2 levels varies from one experiment to the next and with different combinations of hMDMX isoforms (Figs. 5A; Fig. S4B). When we carried out the same increase in hmdmxp60 but in the presence of a fixed amount of hmdmxfl (200 ng) we observed less fluctuation in hMDM2 expression levels (Fig. 5A, panel b). However, when we instead increased the levels of both hmdmxfl and hmdmxp60, the oscillation of hMDM2 was restored, or even enhanced (Fig. 5A, panel c). To further test the effect of hMDMXp60 on hMDM2 stability, we carried out in vitro autoubiquitination of hMDM2. This showed that the amount of polyubiquitinated hMDM2 increases in the presence of increasing amounts of hMDMXp60 (Fig. 5B; Fig. S5A). The corresponding experiment using similar amounts of hMDMXFL resulted in less polyubiquitinated hMDM2 as compared to hMDMXp60 (Fig. S5B). These results indicate that hMDMXp60 has a more profound effect on hMDM2 autoubiquitination, as compared to hMDMXFL. To some extent, this difference might be attributed to the higher affinity of hMDMXp60 for hMDM2 but it also indicates that the N-terminus of hMDMX not only regulates the affinity to hMDM2 but also influences its E3 ubiquitin ligase activity.
Figure 5.
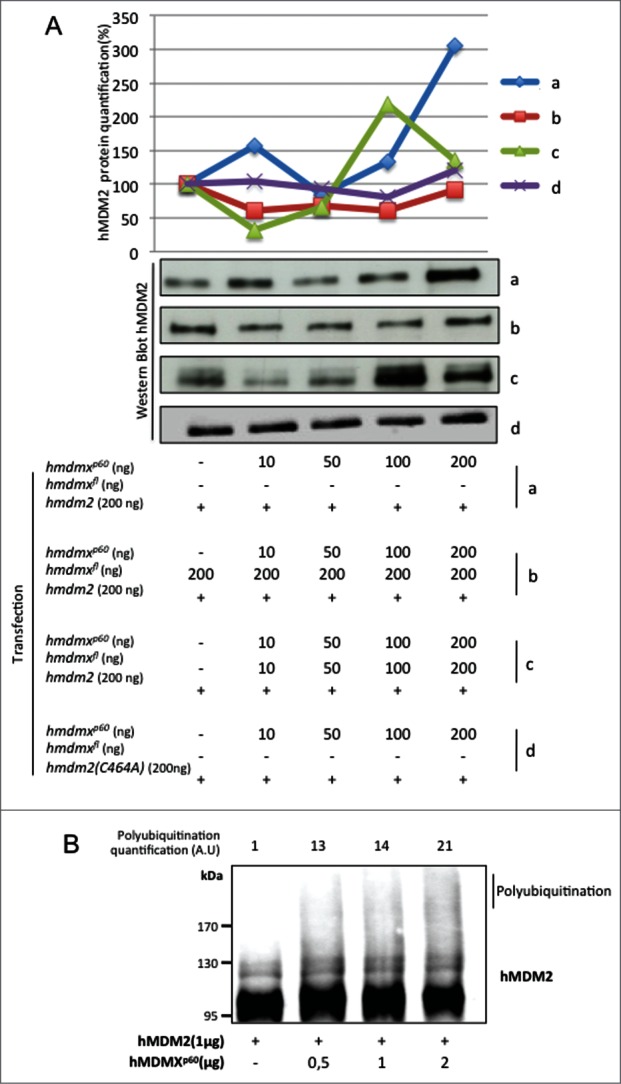
Increasing levels of hMDMXp60 induces oscillation in hMDM2 expression levels in H1299 cells. (A) Expression of a fixed amount of hmdm2 and increasing levels of either (a) hMDMXp60 alone or (b) together with a fixed amount of hmdmxfl, or (c) increasing levels of both isoforms. The hmdm2(C464A) mutant (d) is E3 ligase dead and is not affected by hMDMXp60 (Fig. S4A and B). (B) In vitro autoubiquitination of recombinant hMDM2 in the presence of increasing levels of hMDMXp60. Data shows one representative experiment out of 3 (Fig. S5A and B).
Discussion
Isoforms within the p53 pathway and its extended family of p63 and p73 as well as of hMDM2, have been shown to play important roles in expanding the functional repertoire of these genes. Each of these gene products forms dimers or multimers either as hetero- or homo oligomers and these different complexes determine the activity of respective protein. Most of the factors in these pathways can interact with more than one partner and, for example, h-h-p53 can be co-immunoprecipitated together and it has been suggested that all isoforms of p53, p63 and p73 form hetero-oligomers. However, certain complexes seem to prevail under certain conditions, indicating that the formations of these complexes are regulated.31 Hence, the regulation of affinity between different isoforms is a common theme in how these gene products exert their activity. And together with the fact that certain complexes show a dominant, or specific, phenotype can help to explain how a certain cellular biological effect of one isoform can be achieved even in the presence of higher levels of another. Hence, the deletion of a functional domain that at the same time changes the affinity for a partner can thus render an isoform to become dominant. p53, for example, is transcriptionally active as a tetramer but in vitro studies have indicated that the concentration required to form tetramers is very high, suggesting this can be a regulated step in the cells. The N-terminally truncated alternative translation product p53/47 lacks the first of p53s 2 transactivation domains (TAs) and shows a 10-fold higher affinity for its homo-oligomers during the unfolded protein response, which can help to explain how relatively low levels of p53/47 can impose a G2 cell cycle arrest following ER stress even in the presence of higher levels of the full length p53 product which instead drives toward a G1 arrest.7
The regulation of hMDMX and its cellular biological functions are less clear as compared to the more intensively studied hMDM2. However, both proteins are critical for normal embryonic development. But whereas the role of hMDM2 is linked to the control of p53 stability, the activity of hMDMX has been proposed to mainly relate to interference with p53's gene regulatory capacity. It has been shown that increase of hMDMX expression is frequent (app. 70%) in melanomas and it is interesting to notice that cap-independent translation of hmdmx is sensitive to UV irradiation at relative low doses (5 j/m2) whereas it is not possible to estimate the effect of UV irradiation on cap-dependent translation of hmdmx under these conditions (Fig. 2F and Fig. S2). This is, thus, an interesting observation and the identification of the cellular pathway governing synthesis of hMDMX might shed light on a pathway controlling p53 activity during UV exposure.
There are conflicting reports regarding the regulation of hMDMX expression by hMDM2 and both a stabilizing and destabilizing effect have been reported.32-34 The results presented here indicate that both scenarios could in fact be correct and the net outcome in terms of expression levels might depend on the ratio of hMDM2 to the hMDMXFL and the hMDMXp60 isoforms. The surprising observation that deletion of the N-terminal p53 binding domain of hMDMX results in an increase in the affinity to hMDM2 offers an explanation to hMDMXp60-mediated stabilization of hMDMX based on competition between the 2 isoforms for hMDM2. What is likewise surprising is that increasing levels of hMDMXp60 at lower concentrations results in a decrease in hMDM2 levels and at higher concentrations in a subsequent increase in expression. The effect is dependent on hMDM2 E3 ligase activity as the expression levels of an E3 ligase inactive hMDM2 protein is not affected under similar conditions. This biphasic expression curve is independent of p53 and suggests that the ratio of hMDM2 to hMDMXp60 determines hMDM2 stability. It is, however, difficult to see how this can take place, unless the hMDM2-hMDMXp60 complex includes more than one copy of hMDMXp60. A possible scenario could be that as long as the ratio hMDM2:hMDMXp60 is in favor of hMDM2, hMDMXp60 has a destabilizing effect but when the ratio is in favor of MDMXp60 it switches to instead stabilize MDM2 expression. This suggests that N-terminus of hMDMX regulates the interaction between the RING domains of hMDM2 and hMDMX and that the increase in affinity between hMDM2 and hMDMXp60 alterspu hMDM2 E3 ligase activity toward itself. It is interesting to compare this with the model whereby the N-termini interaction between the Box-1 domain of p53 and the hydrophobic pocket of hMDM2 leads to allosteric changes in the core and RING domains of hMDM2.35 A more detailed study on how differences in ligand binding to hMDM2 affect its E3 ligase activity is currently under way. In addition to a direct effect of MDM2 on the ubiquitination of the two hMDMX isoforms, the observed differences in protein stability in cells (Fig. 4A and S3B) vs. a similar MDM2-mediated ubiquitination of the two substrates in vitro (Fig. S3A), indicate that post-ubiquitination events might play an additional role in controlling the turnover rates. For example, it has been shown that hMDM2 interacts directly with the proteasome and it cannot be ruled out that something similar does not take place for hMDMX.36
MDM2 and MDMX are both key regulators of p53 activity and it can be assumed that the effects of MDMXp60 will also affect p53 activity and expression. This can be envisioned to take place on several levels. In light of the role of the N-terminus of MDMX in controlling the hetero-dimerization of the RING domains it is possible that the p53-MDMX interaction could affect the MDMX-MDM2 interface. Secondly, it is also possible that certain stoichiometric conditions of the p53-MDM2-MDMXFL/MDMXp60 interactome will affect p53 stability and/or p53 activity in different ways. However, as MDMX and MDM2 are both involved in positive and negative regulation of p53, the addition of p53 into this model adds a substantial increase in complexity that will require a separate study.
We observed that translation of both hMDMXFL and MDMXp60 can be initiated by cap-independent mechanisms. It is likely that the initiation of hMDMXp60 at the 7th in frame AUG reflects an internal initiation mechanism, suggesting that the 5’UTR of the encoded sequence of hmdmx is highly structured, presumably together with the 5’ sequence. We do not yet know under what cellular conditions the initiation of hMDMXp60 is regulated or, for that matter, if there is a mechanisms in place that allows the cell to express one, or the other, of these isoforms. But as the relative levels of expression of the two isoforms differs between cell lines, this is at least a possibility. The notion that translation of the hmdmx mRNA is regulated is further supported by the observation that UV-irradiation suppresses cap-independent expression of both isoforms when expressed from a bi-cistronic hmdmx construct. We also observe a similar suppression of expression using a mono-cistrionic construct (Fig. S2) but it cannot be concluded if UV irradiation also affects cap-dependent synthesis of the two isoforms as this construct does not allow us to distinguish between these two forms of translation initiation. The fact that UV irradiation can suppress hMDMX synthesis might play a role in physiological conditions to activate the p53 pathway should be considered in light of the fact that down regulation of hMDMX is observed at a high frequency in malignant melanomas.
The hMDMXp60 isoform shows a similarity with hMDM2p76, which also lacks the N-terminal p53 binding domain. This isoform is induced under conditions of genotoxic stress and since it does not have the p53 interactive domain it does not have a direct negative effect on p53 and instead exerts a positive effect. On the contrary, it retains its capacity to bind the p53 mRNA and stimulate p53 mRNA translation. Similarly, hMDMXp60 is also initiated just after the p53 binding domain and can thus not suppress p53 activity, but it has been shown that the N-terminus of hMDMX is not required for its capacity to fold the nascent p53 mRNA and promote hMDM2-dependent synthesis of p53 following activation by the ATM kinase. Further studies will tell if this isoform can also play a positive role in the activation of p53.
Material and methods
Cell culture and transfections
H1299 cells were cultured in RPMI-1640 (GIBCO), HeLa and MDA cells were cultured in DMEM (GIBCO), in a humidified 5% CO2 atmosphere at 37°C. All medias were supplemented with 10% SVF, 1% L-glutamine (GIBCO) and 1% penicillin/streptomycin (GIBCO).
Cells were transfected using Genejuice (Novagen) with plasmids indicated in the figures according to the manufacturer's protocol and the total amount of DNA was kept constant by adding empty pcDNA3 vector. Cells were also transfected with siRNA (Qiagen) as indicated on the figure, using Ribojuice (Novagen) according to the manufacturer's protocol. Cells expressing indicated hmdmx constructs were treated with UV irradiation and harvested 4 h after. MG132 was used at a concentration of 25 μM.
Western blotting and immunoprecipitation (IP)
For western blots, cells were lysed in buffer containing: HEPES KOH pH7,5; 50 mM Beta-glycero-phosphate; 1 mM EDTA pH8; 1 mM EGTA pH8; 0,5 mM Na3VO4; 100 mM KCl; 10% Glycerol; 1% Triton X-100. For IP, cells were lysed in buffer containing 1% NP40; 150 mM NaCl; 20 mM Tris-HCl. Both buffers were complemented with Complete Protease Inhibitor Cocktail Tablets (Roche). The following antibodies were used: MDMX (Bethyl), Actin (Sigma) and 4B2, GFP, HA (gift from B. Vojtesek, Masaryk Memorial Cancer Institute, Brno, Czech Republic). IPs were performed using protein G–Sepharose beads (Sigma).
Protein expression quantifications were performed using Bio1d® software.
RT-qPCR
RNA extraction from cells was performed using RNeasy® Mini kit (Qiagen) according to the manufacturer's procedure. Reverse transcription was performed using the Moloney Murine Leukemia Virus M-MLV (Invitrogen) reverse transcriptase and oligo dT (Invitrogen). The quantitative PCR were performed on a StepOne RealTime PCR system (Applied Bioystem) using the PerfeCTa SYBR Green mix (Quanta BioSciences) according to the manufacturer's procedure. The following primers were used, P1: CGGCATGGACGAGCTGTACAAG; P2: GGGAGGCCGGAAGTTGCGGCTTCA; P3: GAAAATGATGTCATTTTGGTAGTG; P4: ACTTGGAATATCCATACTGTG; and P5: CAGCAGGTGCGCAAGGTGAA.
Purification of recombinant protein
Histidine tagged recombinant proteins were produced in BL21 (DE3) Escherichia coli and purified on HiTrap Nickel column (GE healthcare) on ÄKTA purifier system. Lysis and binding buffer contain 25 mM HEPES, 100 mM NaCl, 15 mM Imidazol, 10% glycerol, 1 mM Tris, 10 μM ZnSO4 at pH 8 and were complemented with Complete Protease Inhibitor Cocktail EDTA-free Tablets (Roche). Recombinant proteins were eluted from Nickel column with buffer containing 25 mM HEPES, 100 mM NaCl, 300 mM Imidazol, 10μM ZnSO4, 5 mM β-Mercaptoethanol, 1 mM Tris at pH 8 and dialyzed upon gel filtration step on Superdex 200 in pH8 buffer containing 25 mM HEPES, 100 mM NaCl, 1 mM Tris, 10 μM ZnSO4.
ELISA
96 well plates (Thermo scientific) were coated with 500 ng of either hMDMXFL or hMDMXp60 proteins purified from bacteria in 0.1 M NaHCO3 (200 μl/well) overnight at 4°C. After incubation plates were washed 6x 200 μl with 0.1% PBS-Tween and blocked for 1 h at RT with 0.1% PBS-Tween containing milk 3%. Plates were washed 6x 200 μl with 0.1% PBS-Tween. The second bound protein was hMDM2 and was diluted (from 0 to 40 μg/μl) and incubated 2 h at RT. Plates were washed 6 × 200 μl with 0.1% PBS-Tween and were incubated with the anti-MDM2 4B2 mAb (1:1000) for 1 h at RT. After washing 6 × 200 μl with 0.1% PBS-Tween, secondary mouse antibody was incubated for 1 h at RT. Plates were washed and incubated with 50 μl/well of ECL mix and luminescence was measured.
In vitro ubiquitination assays
In vitro ubiquitination assays were performed 2 h at 37°C, using reaction mix containing: E3 hMDM2 recombinant protein (amount as indicated on the figure) and an equal amount of Ubiquitin-Activating Enzyme E1, (VWR); Ubiquitin Conjugating Enzyme 5 b (VWR) and in presence or not of Ubiquitin His-Tagged (VWR) in a solution containing 1 μM ATP (Invitrogen); 1 μM DTT (Invitrogen). Equal amount of leammli buffer was added and samples were loaded on acrylamide gels. After transfer on nitrocellulose membrane and blocking with milk, hybridation with MDMX antibody (Bethyl) or 4B2 antibody and the corresponding secondary antibody, ubiquitination pattern were revealed with SuperSignal West Dura Chemoluminescent Substrate (Thermo scientific).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Borek Vojtesek for antibodies and to Sebastien Apcher for bicistronic cDNA constructs.
Funding
This work was supported by Inserm and la Ligue Contre le Cancer. A-S T is supported by the ARC. VO-I is funded by CONACyT CB-166233.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1. Blandino G, Dobbelstein M. p73 and p63: why do we still need them? Cell Cycle 2004; 3:886-94; PMID:15254416; http://dx.doi.org/ 10.4161/cc.3.7.996 [DOI] [PubMed] [Google Scholar]
- 2. Levrero M, De Laurenzi V, Costanzo A, Gong J, Wang JY, Melino G. The p53/p63/p73 family of transcription factors: overlapping and distinct functions. J Cell Sci 2000; 113 (Pt 10):1661-70; PMID:10769197 [DOI] [PubMed] [Google Scholar]
- 3. Dotsch V, Bernassola F, Coutandin D, Candi E, Melino G. p63 and p73, the ancestors of p53. Cold Spring Harbor Perspec Biol 2010; 2:a004887; PMID:20484388; http://dx.doi.org/ 10.1101/cshperspect.a004887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Truong AB, Khavari PA. Control of keratinocyte proliferation and differentiation by p63. Cell Cycle 2007; 6:295-9; PMID:17264679; http://dx.doi.org/ 10.4161/cc.6.3.3753 [DOI] [PubMed] [Google Scholar]
- 5. Murray-Zmijewski F, Lane DP, Bourdon JC. p53/p63/p73 isoforms: an orchestra of isoforms to harmonise cell differentiation and response to stress. Cell Death Differ 2006; 13:962-72; PMID:16601753; http://dx.doi.org/ 10.1038/sj.cdd.4401914 [DOI] [PubMed] [Google Scholar]
- 6. Moll UM, Slade N. p63 and p73: roles in development and tumor formation. Mol Cancer Res 2004; 2:371-86; PMID:15280445 [PubMed] [Google Scholar]
- 7. Bourougaa K, Naski N, Boularan C, Mlynarczyk C, Candeias MM, Marullo S, Fahraeus R. Endoplasmic reticulum stress induces G2 cell-cycle arrest via mRNA translation of the p53 isoform p53/47. Mol Cell 2010; 38:78-88; PMID:20385091; http://dx.doi.org/ 10.1016/j.molcel.2010.01.041 [DOI] [PubMed] [Google Scholar]
- 8. Fujita K, Mondal AM, Horikawa I, Nguyen GH, Kumamoto K, Sohn JJ, Bowman ED, Mathe EA, Schetter AJ, Pine SR, et al. p53 isoforms Delta133p53 and p53beta are endogenous regulators of replicative cellular senescence. Nat Cell Biol 2009; 11:1135-42; PMID:19701195; http://dx.doi.org/ 10.1038/ncb1928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Candeias MM, Malbert-Colas L, Powell DJ, Daskalogianni C, Maslon MM, Naski N, Bourougaa K, Calvo F, Fahraeus R. P53 mRNA controls p53 activity by managing Mdm2 functions. Nat Cell Biol 2008; 10:1098-105; PMID:19160491; http://dx.doi.org/ 10.1038/ncb1770 [DOI] [PubMed] [Google Scholar]
- 10. Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature 1997; 387:299-303; PMID:9153396; http://dx.doi.org/ 10.1038/387299a0 [DOI] [PubMed] [Google Scholar]
- 11. Marine JC, Francoz S, Maetens M, Wahl G, Toledo F, Lozano G. Keeping p53 in check: essential and synergistic functions of Mdm2 and Mdm4. Cell Death Differ 2006; 13:927-34; PMID:16543935; http://dx.doi.org/ 10.1038/sj.cdd.4401912 [DOI] [PubMed] [Google Scholar]
- 12. Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett 1997; 420:25-7; PMID:9450543; http://dx.doi.org/ 10.1016/S0014-5793(97)01480-4 [DOI] [PubMed] [Google Scholar]
- 13. Maslon MM, Hupp TR. Drug discovery and mutant p53. Trends Cell Biol 2010; 20:542-55; PMID:20656489; http://dx.doi.org/ 10.1016/j.tcb.2010.06.005 [DOI] [PubMed] [Google Scholar]
- 14. Ganguli G, Wasylyk B. p53-independent functions of MDM2. Mol Cancer Res : MCR 2003; 1:1027-35; PMID:14707286 [PubMed] [Google Scholar]
- 15. Manfredi JJ. The Mdm2-p53 relationship evolves: Mdm2 swings both ways as an oncogene and a tumor suppressor. Genes Dev 2010; 24:1580-9; PMID:20679392; http://dx.doi.org/ 10.1101/gad.1941710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fåhraeus R, Olivares-Illana V. MDM2s social network. Oncogene 2014; 33, 4365-4376; http://dx.doi.org/ 10.1038/onc.2013.410 [DOI] [PubMed] [Google Scholar]
- 17. Giglio S, Mancini F, Pellegrino M, Di Conza G, Puxeddu E, Sacchi A, Pontecorvi A, Moretti F. Regulation of MDM4 (MDMX) function by p76(MDM2): a new facet in the control of p53 activity. Oncogene 2010; 29:5935-45; PMID:20697359; http://dx.doi.org/ 10.1038/onc.2010.324 [DOI] [PubMed] [Google Scholar]
- 18. Bartel F, Taubert H, Harris LC. Alternative and aberrant splicing of MDM2 mRNA in human cancer. Cancer Cell 2002; 2:9-15; PMID:12150820; http://dx.doi.org/ 10.1016/S1535-6108(02)00091-0 [DOI] [PubMed] [Google Scholar]
- 19. Chandler DS, Singh RK, Caldwell LC, Bitler JL, Lozano G. Genotoxic stress induces coordinately regulated alternative splicing of the p53 modulators MDM2 and MDM4. Cancer Res 2006; 66:9502-8; PMID:17018606; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-4271 [DOI] [PubMed] [Google Scholar]
- 20. Saucedo LJ, Myers CD, Perry ME. Multiple murine double minute gene 2 (MDM2) proteins are induced by ultraviolet light. J Biol Chem 1999; 274:8161-8; PMID:10075719; http://dx.doi.org/ 10.1074/jbc.274.12.8161 [DOI] [PubMed] [Google Scholar]
- 21. Candeias MM, Malbert-Colas L, Powell DJ, Daskalogianni C, Maslon MM, Naski N, Bourougaa K, Calvo F, Fahraeus R. p53 mRNA controls p53 activity by managing Mdm2 functions. Nat Cell Biol 2008; 10(9):1098–105; PMID:19160491; http://dx.doi.org/ 10.1038/ncb1770 [DOI] [PubMed] [Google Scholar]
- 22. Marine JC, Jochemsen AG. Mdmx and Mdm2: brothers in arms? Cell Cycle 2004; 3:900-4; PMID:15254433; http://dx.doi.org/ 10.4161/cc.3.7.998 [DOI] [PubMed] [Google Scholar]
- 23. Danovi D, Meulmeester E, Pasini D, Migliorini D, Capra M, Frenk R, de Graaf P, Francoz S, Gasparini P, Gobbi A, et al. Amplification of Mdmx (or Mdm4) directly contributes to tumor formation by inhibiting p53 tumor suppressor activity. Mol Cell Biol 2004; 24:5835-43; PMID:15199139; http://dx.doi.org/ 10.1128/MCB.24.13.5835-5843.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gembarska A, Luciani F, Fedele C, Russell EA, Dewaele M, Villar S, Zwolinska A, Haupt S, de Lange J, Yip D, et al. MDM4 is a key therapeutic target in cutaneous melanoma. Nat Med 2012; 18:1239-47; PMID:22820643; http://dx.doi.org/ 10.1038/nm.2863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pei D, Zhang Y, Zheng J. Regulation of p53: a collaboration between Mdm2 and Mdmx. Oncotarget 2012; 3:228-35; PMID:22410433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang X. p53 regulation: teamwork between RING domains of Mdm2 and MdmX. Cell Cycle 2011; 10:4225-9; PMID:22134240; http://dx.doi.org/ 10.4161/cc.10.24.18662 [DOI] [PubMed] [Google Scholar]
- 27. Rallapalli R, Strachan G, Cho B, Mercer WE, Hall DJ. A novel MDMX transcript expressed in a variety of transformed cell lines encodes a truncated protein with potent p53 repressive activity. J Biol Chem 1999; 274:8299-308; PMID:10075736; http://dx.doi.org/ 10.1074/jbc.274.12.8299 [DOI] [PubMed] [Google Scholar]
- 28. Gentiletti F, Mancini F, D’Angelo M, Sacchi A, Pontecorvi A, Jochemsen AG, Moretti F. MDMX stability is regulated by p53-induced caspase cleavage in NIH3T3 mouse fibroblasts. Oncogene 2002; 21:867-77; PMID:11840332; http://dx.doi.org/ 10.1038/sj.onc.1205137 [DOI] [PubMed] [Google Scholar]
- 29. Mathews M, Sonenberg N, Hershey J. Translational Control in Biology and Medicine, Mathews M. B., Sonenberg N., Hershey J. W. B., eds. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2007. [Google Scholar]
- 30. Metzger MB, Pruneda JN, Klevit RE, Weissman AM. RING-type E3 ligases: master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Biochimica et Biophy Acta 2014; 1843:47-60; PMID:23747565; http://dx.doi.org/ 10.1016/j.bbamcr.2013.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rocco JW, Ellisen LW. p63 and p73: life and death in squamous cell carcinoma. Cell Cycle 2006; 5:936-40; PMID:16687923; http://dx.doi.org/ 10.4161/cc.5.9.2716 [DOI] [PubMed] [Google Scholar]
- 32. Sharp DA, Kratowicz SA, Sank MJ, George DL. Stabilization of the MDM2 oncoprotein by interaction with the structurally related MDMX protein. J Biol Chem 1999; 274:38189-96; PMID:10608892; http://dx.doi.org/ 10.1074/jbc.274.53.38189 [DOI] [PubMed] [Google Scholar]
- 33. Stad R, Ramos YF, Little N, Grivell S, Attema J, van Der Eb AJ, Jochemsen AG. Hdmx stabilizes Mdm2 and p53. J Biol Chem 2000; 275:28039-44; PMID:10827196 [DOI] [PubMed] [Google Scholar]
- 34. Linares LK, Hengstermann A, Ciechanover A, Muller S, Scheffner M. HdmX stimulates Hdm2-mediated ubiquitination and degradation of p53. Proc Natl Acad Sci U S A 2003; 100:12009-14; PMID:14507994; http://dx.doi.org/ 10.1073/pnas.2030930100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wallace M, Worrall E, Pettersson S, Hupp TR, Ball KL. Dual-site regulation of MDM2 E3-ubiquitin ligase activity. Mol Cell 2006; 23:251-63; PMID:16857591; http://dx.doi.org/ 10.1016/j.molcel.2006.05.029 [DOI] [PubMed] [Google Scholar]
- 36. Kulikov R, Letienne J, Kaur M, Grossman SR, Arts J, Blattner C. Mdm2 facilitates the association of p53 with the proteasome. Proc Natl Acad Sci U S A 2010; 107:10038-43; PMID:20479273; http://dx.doi.org/ 10.1073/pnas.0911716107 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.