

Abstract
Malignant melanoma is the deadliest form of skin cancer; the treatment of advanced and recurrent forms remains a challenge. It has recently been reported that growth hormone-releasing hormone (GHRH) receptor is involved in the pathogenesis of melanoma. Therefore, we investigated the effects of our new GHRH antagonists on a human melanoma cancer cell line. Antiproliferative effects of GHRH antagonists, MIA-602, MIA-606 and MIA-690, on the human melanoma cell line, A-375, were studied in vitro using the MTS assay. The effect of MIA-690 (5 μg/day 28 d) was further evaluated in vivo in nude mice bearing xenografts of A-375. Subcellular localization of p27 was detected with Western blot and immunofluorescent staining. MIA-690 inhibited the proliferation of A-375 cells in a dose-dependent manner (33% at 10 μM, and 19.2% at 5 μM, P < 0 .05 vs. control), and suppressed the growth of xenografted tumors by 70.45% (P < 0.05). Flow cytometric analysis of cell cycle effects following the administration of MIA-690 revealed a decrease in the number of cells in G2/M phase (from 19.7% to 12.9%, P < 0.001). Additionally, Western blot and immunofluorescent studies showed that exposure of A-375 cells to MIA-690 triggered the nuclear accumulation of p27. MIA-690 inhibited tumor growth in vitro and in vivo, and increased the translocation of p27 into the nucleus thus inhibiting progression of the cell cycle. Our findings indicate that patients with malignant melanoma could benefit from treatment regimens, which combine existing chemotherapy agents and novel GHRH-antagonists.
Keywords: growth hormone-releasing hormone antagonist, melanoma, p27, targeted therapy, xenografted mouse model
Abbreviations
- Abu
a-aminobutyric acid
- Ac
acetyl
- Ada
12-aminododecanoyl
- Agm
agmatine
- Amc
8-aminocaprylyl
- ANOVA
one-way analysis of variance
- Cpa
parachlorophenylalanine
- FBS
fetal bovine serum
- Fpa5
pentafluoro-phenylalanine
- GH
growth hormone
- GHRH
growth hormone-releasing hormone
- GHRH-R
growth hormone-releasing hormone receptor
- Har
homoarginine
- hGHRH
human growth hormone-releasing hormone
- IGF-I
insulin-like growth factor I
- mTOR
mammalian target of rapamycin
- MTS
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfonphenyl)-2H-tetrazolium
- Nle
norleucine
- Orn
ornithine
- Ph
phenyl
- PhAc
phenylacetyl
- pGHRH-R
pituitary type GHRH-receptor
- SVs
splice variants
- TBS
tris-buffered saline
- Tyr(Me)
O-methyltyrosine
Introduction
Cutaneous melanomas originate from pigment producing melanocytes or their precursors in the skin and are considered the deadliest form of skin cancer, causing 50000 deaths annually worldwide.1 The increasing incidence exacts a disproportionate toll among the young in the population.2 If the tumor is detected early, surgical excision provides a cure in almost all patients. However, the 5-year-survival rate falls to 15% with a median survival of 1 y for those with advanced, disseminated disease.3 The complexity and aggressiveness of melanoma require multidisciplinary management to achieve optimal care. In recent years the identification of the main genetic aberrations and signaling pathways involved in melanocyte transformation and disease progression has resulted in the development of novel, more effective therapeutic approaches.4
The discovery of certain specific molecular characteristics of cancer cells has led to the development of a new class of drugs known as targeted therapeutics. Targeted therapy can be based on peptide hormones for which receptors are expressed on cancerous cells. Growth hormone-releasing hormone (GHRH) is a hypothalamic neuropeptide that stimulates the secretion of growth hormone (GH) from the anterior pituitary gland upon binding to its receptor (GHRH-R).5 In turn, GH stimulates the production of hepatic insulin-like growth factor I (IGF-I), which is a potent mitogen for many cancers.6 Additionally, GHRH and GHRH-R are not confined to the hypothalamic–pituitary axis, but are also produced by various extrahypothalamic sites.7 GHRH and GHRH-R modulate cell proliferation and apoptosis in many tissues.8-16 Antagonistic analogs of GHRH, synthesized in our laboratory, have been shown to inhibit the growth of various human cancers,5 in part, by suppressing circulating GH/IGF-1 levels.17 Direct mechanisms involved in the dominant antitumor effects of GHRH antagonists appear to be based on blockage of the autocrine action of GHRH on tumors and on inhibition of the local production of IGF-I/II.6,18 Our group has demonstrated the presence of the pituitary type GHRH-receptor (pGHRH-R) and 4 truncated splice variants (SVs) of the GHRH-R in human prostate and breast cancer specimens as well as different cancer cell lines.19-21 SV1 has the greatest structural similarity to the pGHRH-R, differing only in a short segment of the extracellular ligand-binding domain. It has recently been reported that the SV1 receptor is involved in the pathogenesis of melanomas demonstrating that the progression from a state of dysplasia into malignancy is accompanied by expression of SV1 receptor.22 We have shown previously that GHRH antagonists inhibit the growth of diverse human tumors xenografted into nude mice.23-30 Many of these published studies reported the effects of early-stage GHRH antagonists that were later considered unsuitable for clinical development due to limited stability or low potency. In our initial studies with melanomas, for example, early GHRH antagonists MZ-5–156, MZ-7–138 and JMR-132 were shown to inhibit growth of MRI-H187, MRI-H255 and A375 human melanoma cell lines xenografted into nude mice. More potent GHRH antagonists of the MIA series subsequently have become available.
In an effort to identify new drugs with different mechanisms of action than those currently available for malignant melanoma, we have evaluated the in vitro and in vivo effects of 3 highly potent GHRH antagonists of the latest series, MIA-602, MIA-606 and MIA-690, with improved design and resistance to biodegradation on A375 human malignant melanoma.
Results
Presence of GHRH receptors in A-375 human malignant melanoma cell line
In the protein fraction obtained from A-375 cells and from human pituitary, the polyclonal antibody against a common segment in the pGHRH-R and SV1 detected immunoreactive bands at 60 kDa and 39.5 kDa (Fig. 1a). The band at 60 kDa corresponds to pGHRH-R,31 and the 39.5 kDa band is consistent with the receptor protein encoded by SV1.19 Human pituitary was used as a positive control. Protein levels in A-375 cell lysates were quantified by densitometry and were normalized to β-actin levels. The level of SV1 was 4.35-fold higher than that of pGHRH-R (P < 0.05) (Fig. 1b).
Figure 1.
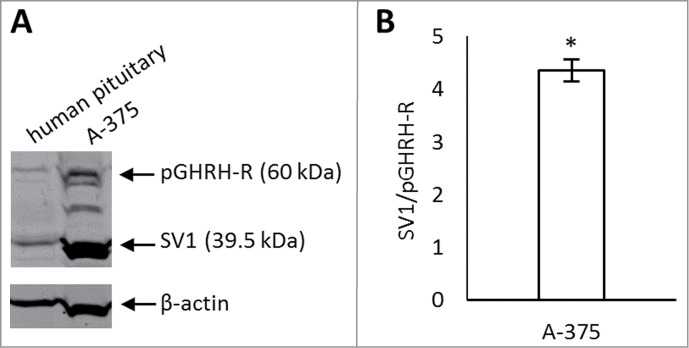
(A) Expression of pituitary GHRH receptor (pGHRH-R) and its predominant splice variant (SV1) in A-375 human malignant melanoma cell line by Western blot. A polyclonal antibody generated against GHRH-R detected its 2 variants, pGHRH-R at 60 kDa and SV1 at 39.5 kDa. Human pituitary was used as a positive control and β-actin as a loading control. (B) Densitometric analysis of SV1 compared to pGHRH-R levels. The level of SV1 was significantly higher than that of pGHRH-R in A-375 cells. Values were calculated from 3 different experiments, normalized to β-actin levels and expressed as SV1/GHRH-R ratio. Error bars represent SEM, *: P < 0.05.
Antiproliferative effects of GHRH antagonists on A-375 cells in vitro
GHRH antagonists MIA-602, MIA-606, and MIA-690 significantly inhibited the proliferation of A-375 cells at 10 μM concentration (33.8%, 34.8% and 33% inhibition, respectively, P < 0.05, Fig. 2a) in vitro. At 5 μM concentration, only the antiproliferative effect of MIA-602 and MIA-690 was significant (13.2% and 19.2% inhibition, P < 0.05). There was no significant reduction in proliferation rate when the compounds were added at 1 μM concentration. Of the 3 analogs tested, the inhibitory effect of MIA-690 was greatest and therefore this compound was subjected to further studies.
Figure 2.
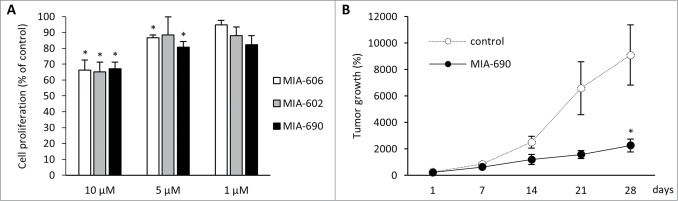
(A) In vitro antiproliferative effects of GHRH antagonists MIA-602, MIA-606, and MIA-690 on the proliferation of A-375 human malignant melanoma cells. Compounds were used at 10 μM, 5 μM and 1 μM concentrations for 72 hours. The results were calculated from 3 independent experiments and were expressed as % of control. Error bars represent SEM, *: P < 0.05. (B) Effect of MIA-690 on the growth of A375 malignant melanoma tumors xenografted into nude mice. MIA-690 was administered at 5 μg/day dose subcutaneously for 28 d. Control animals received vehicle. Error bars represent SEM, *: P < 0.05.
Inhibition of growth of A-375 xenografts by GHRH antagonist in vivo
The antitumor effect of the GHRH antagonist, MIA-690, was investigated in A-375 human malignant melanoma xenografted into athymic nude mice. Animals received daily subcutaneous injections of MIA-690 (5 μg/animal) or vehicle (control group) for 28 d (Fig. 2b). In GHRH antagonist treated animals the rate of tumor growth was reduced throughout the experiment although the difference between the groups was only significant at the last time point. Four weeks after study initiation we found a 70.45% reduction in mean tumor volume in response to GHRH antagonist treatment compared to the control group (P < 0.05).
Effect of MIA-690 on the expression of genes involved in signal transduction pathway activation or inhibition
We used real-time PCR arrays to measure changes in the expression of genes that occur in response to the stimulation of different signal transduction pathways in order to identify the key elements of GHRH antagonist-induced signaling. For this, we used RNA from excised tumors of control and MIA-690-treated animals and reverse transcribed into cDNA which was then subjected to analysis with the Human Signal Transduction PathwayFinder RT2 Profiler PCR array. We found important functional molecules affected by treatment with MIA-690 and also selected genes potentially related to tumor growth inhibition (Table 1.). Genes altered significantly after treatment with GHRH antagonist, were the following: Interleukin 8 (5.98-fold decrease; P < 0.05), Jun proto-oncogene (3.68-fold decrease; P < 0.05), and Lymphotoxin α (TNF superfamily, member 1) (3.08-fold increase; P < 0.05).
Table 1.
Relative expression of genes involved in distinct signal transduction pathways
| Gene | Fold change after therapy with GHRH antagonist |
|---|---|
| Interleukin 8 |
−5.98* |
| Jun proto-oncogene |
−3.68* |
| Lymphotoxin α | 3.08* |
In vivo melanoma specimens were evaluated by Human Signal Transduction PathwayFinder RT2 Profiler PCR Array system. Only genes with statistically different changes are presented (*: P < 0.05 vs. control).
Effect of GHRH antagonist on cell cycle progression of A-375 cells
Analysis of the A-375 cells by flow cytometry revealed that their treatment with MIA-690 at 5 μM concentration produced major changes in the distribution of the cells across the different phases of cell cycle (Fig. 3). In the control group 52.8% ± 0.42% of the cells were in G0/G1 phase, 27.5% ± 0.73% in S phase, and 19.69% ± 0.4% in G2/M phase. After a 1-hour-treatment with the GHRH antagonist, the distribution was the following: 55.13% ± 1.3%, 32% ± 1.38%, and 12.86% ± 1.34% for G0/G1, S and G2/M phases, respectively. The GHRH-antagonist significantly inhibited the proportion of cells in the G2/M phase (P < 0.001). In addition, there was a significant increase in the percentage of apoptotic cells following the incubation with the GHRH antagonist (8.26% ± 1.64% in treated vs. 1.27% ± 0.63 in control cells, P < 0.05).
Figure 3.
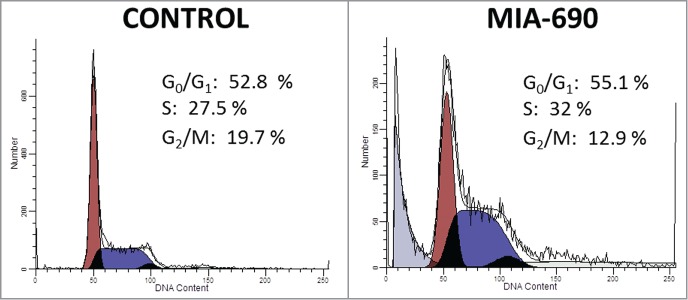
Effect of GHRH antagonist on cell cycle progression of A-375 human malignant melanoma cells. Cells were incubated with 5 μM MIA-690 for 1 h. Cell cycle analysis was measured by laser flow cytometry with a Coulter XL flow cytometer using 488 nm excitation. Numbers indicate the percentage of cells in G0/G1, S and G2/M cell phases as determined by the ModFit software. Data is representative of 2 independent experiments with triplicates in each group.
Induction of nuclear translocation of p27 by GHRH antagonist in A-375 cells
To assess the cellular redistribution of the cell cycle regulator, p27, in A-375 human malignant melanoma cells following GHRH treatment we used 2 approaches. Firstly, cells incubated with 10 μM MIA-690 for 5, 10, 20, 30 and 60 min were lysed and separated into cytosolic and nuclear fractions. Samples were processed for Western blot analysis of p27 protein levels (Fig. 4a). GAPDH was used for loading control. We found an increase in nuclear p27 levels following stimulation with the GHRH antagonist that became more pronounced with time and was paired with a simultaneous decrease in cytosolic p27 levels.
Figure 4.
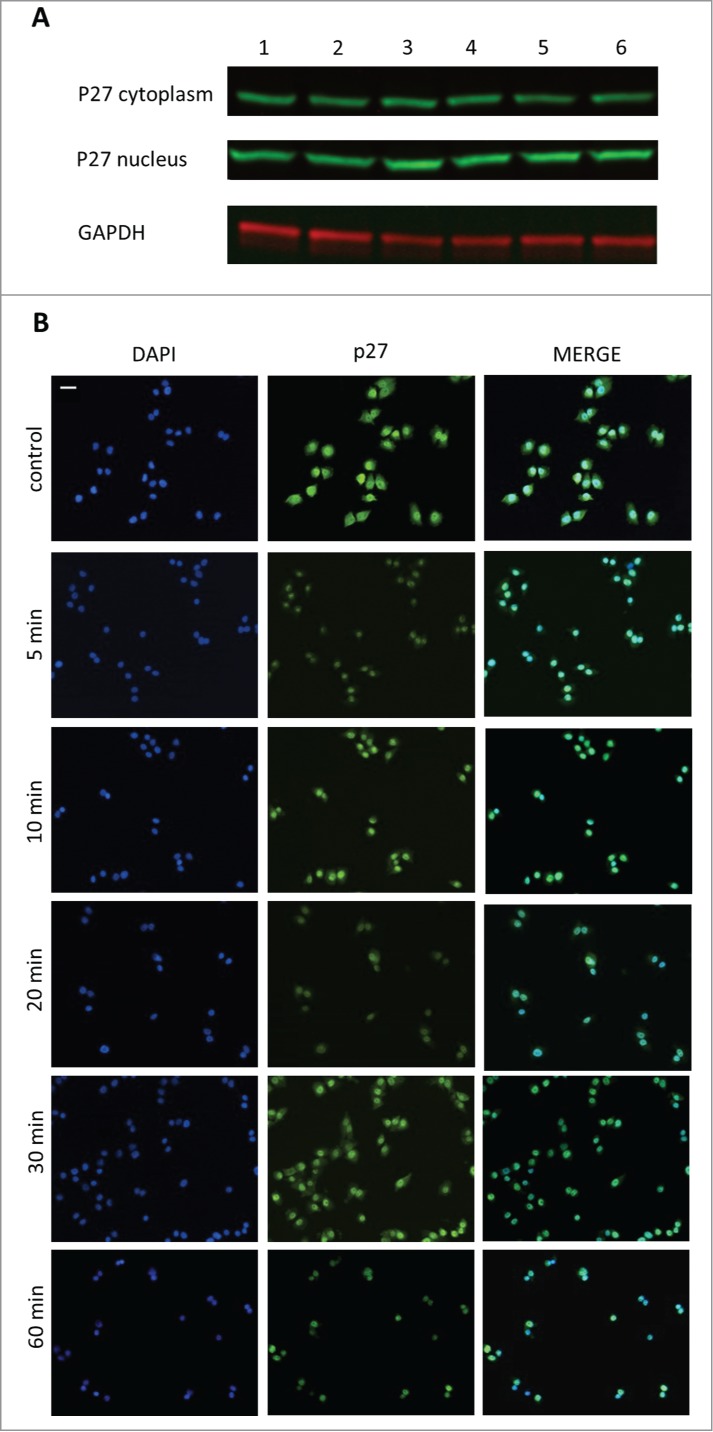
The effect of GHRH antagonist on the localization of p27 in A-375 human malignant melanoma cells determined by Western blot (A) and immunocytochemistry (B). The level of p27 was detected in cytoplasmic and nuclear fractions of A-375 cells with Western blot following incubation with 10 μM MIA-690 for 5, 10, 20, 30 and 60 min (A). Internal standard for the cytoplasmic fraction was GAPDH. Changes in the intracellular localization of p27 following incubation with 10 μM MIA-690 was also detected by immunocytochemistry. In this experiment GHRH antagonist was again added for 5, 10, 20, 30 and 60 min. Immunostained coverslip samples were imaged with a Nikon Eclipse Ti Inverted Microscope fitted with epifluorescence optics and images were recorded using a Nikon DS-Qi1Mc camera. Nuclei are labeled with DAPI (blue). Bars correspond to 100 μM.
In a different approach, cells were incubated with 10 μM MIA-690 for 5, 10, 20, 30 and 60 min and were fixed for immunocytochemistry. In control cells, p27 was localized both in the nucleus and in the cytoplasm. Just 5 min after the addition of GHRH antagonist to the medium, a great proportion of p27 was translocated into the nucleus and it was practically undetectable in the cytoplasm after 60 min.
Discussion
The present study demonstrates that 3 GHRH-antagonists of the new series developed in our laboratory can powerfully inhibit the growth of the human malignant melanoma cell line A375, both in vitro and in vivo. We used proliferation assays to determine the most effective analog among these newly synthesized peptides. Our results showed that all 3 compounds tested, MIA-602, MIA-606, and MIA-690, inhibited the growth of the A375 cell line in vitro. The effects of these GHRH antagonists were similar and the seemingly most effective peptide, MIA-690, was used in further studies. MIA-690 tested at a dose of 5 μg daily inhibited the growth of A375 xenografts in nude mice. Through the treatment, its antitumor effect was not accompanied by weight loss or macroscopic changes of the heart, liver, spleen, ovaries, or kidneys of the tested animals, suggesting that this new GHRH antagonist may not possess untoward adverse effects.
Western blot results revealed a high expression of SV1 and a lower expression of pGHRH-R. This finding can further support the theories that, on certain cancer cells and tumors, SV1 functions as the main therapeutic target,32 and in melanomas has a crucial role in the progression of the disease.22
The exposure of A375 cells to MIA-690 altered the subcellular localization of p27, an event that is, at least in part, responsible for the observed changes in cell cycle progression. In many cancers the level of p27 is reduced in the nucleus, and cytoplasmic p27 appears to acquire a cell cycle-independent oncogenic function to promote cancer cell invasion.33 Reduced levels or mislocalized p27 is associated with poor clinical outcome in a variety of human malignancies.34 Chen et al. recently demonstrated that nuclear p27 expression was remarkably reduced in primary melanomas and further reduced in metastatic melanoma compared with dysplastic nevi; cytoplasmic p27 expression, on the other hand, was significantly increased from dysplastic nevi to primary melanomas and further increased in melanoma metastases, and that gain of cytoplasmic p27 was associated with a poor 5-year survival of metastatic melanoma patients.35 In the present study, both Western blot analysis and immunocytochemistry provided evidence that GHRH triggers a translocation of p27 from the cytoplasm into the nucleus thus enabling the restoration of its local antiproliferative effect.
The mechanism behind this action is still unclear. A serine threonine protein kinase, the mammalian target of rapamycin (mTOR) integrates nutrient sensing and mitogenic signals to regulate protein synthesis, cell growth, and proliferation.36 Activation of Akt mediated by mTOR is known to promote p27 phosphorylation, which impairs p27 nuclear import 37. It has been recently discovered that inhibition of tumor growth by GHRH antagonists involves inactivation of Akt.6,38 Consequently, the GHRH antagonist may have induced p27 translocation indirectly by interfering with the PI3K/Akt/mTOR pathway.
Impaired ability to undergo apoptosis is one of the main reasons for the resistance to therapy seen in most advanced melanoma cases. The c-Jun protein, encoded by Jun proto-oncogene, in combination with c-Fos, forms the AP-1 early response transcription factor. Members of the AP-1 family have a significant role in controlling the balance between cell death and survival.39 Early-phase-melanoma-derived cells have low basal levels of AP-1, and this activity increases during tumor progression.40 Treatment with GHRH-antagonist, MIA-690, significantly decreased the level of Jun proto-oncogene compared to the control samples which may greatly contribute to its antitumor effect.
Serum concentrations of angiogenic factors, such as IL-8 and VEGF, have been reported to correlate with tumor burden and prognosis in metastatic melanoma. Previous studies have demonstrated that elevated IL-8 levels were associated with metastatic melanoma, and a decrease in serum levels was a promising sign of positive response to chemotherapy and immunotherapy.41,42 In the present study, after treatment with MIA-690, the mRNA level of IL-8 markedly dropped. Our laboratory has earlier shown that treatment with GHRH-antagonists inhibits growth and vascularization of experimental ovarian cancer via decreasing VEGF secretion,32 suggesting that one of the cornerstones of the antitumor effect of GHRH-antagonists might be derived from its angiogenesis inhibiting potential.
Many antitumor therapies aim at enhancing T-cell based immune reactions, such as IL-2, which has a response rate of 10–20% in advanced and recurrent melanoma.43 Lymphotoxin-α is another potent mediator of proinflammatory and tumoricidal activities, as well as lymphoid genesis, and it has a critical role in tumor necrosis factor receptor 1 signaling.44,45 A study from Scharma et al.46 has shown that a recombinant antibody-lymphotoxin-α fusion protein induced an adaptive immune response protecting mice from melanoma, and elicited the formation of lymphoid-like tissue in the tumor microenvironment. The mRNA level of Lymphotoxin-α in our study was significantly elevated in the tumors of MIA-690-treated mice compared to the control group suggesting that an elevated immune response may also have contributed to the beneficial effects of this GHRH antagonist.
Our results demonstrate that novel antagonists of GHRH suppress the growth of human experimental malignant melanoma both in vitro and in vivo. This effect may be explained in part by restoring the nuclear p27 function, which is a promising prognostic marker for advanced and recurrent melanoma. Our work and previous results elucidate the importance of pGHRH-R and SV1 in the pathogenesis of melanoma. Preclinical studies suggest that GHRH antagonists lack significant adverse effects, which makes them, in combination with conventional chemotherapy drugs, a great potential asset in the treatment of melanoma, which is considered as one of the most therapeutically challenging malignancies.
Material and Methods
Peptides and reagents
GHRH antagonists MIA-602, MIA-606, and MIA-690 were synthesized in our laboratory by the solid phase method and purified by reversed-phase high-performance liquid chromatography as described previously.47 The chemical structures are the following:
MIA-602 [(PhAc-Ada)0-Tyr1, D-Arg2, Fpa56, Ala8, Har9, Tyr(Me)10, His11, Orn12, Abu15, His20, Orn21, Nle27, D-Arg28, Har29]hGH-RH(1–29)NH2;
MIA-606 [(PhAc-Ada)0-Tyr1, D-Arg2, Fpa56, N-MeAla8, Har9, Tyr(Me)10, His11, Orn12, Abu15, His20, Orn21, Nle27, D-Arg28, Har29,Agm30]hGH-RH(1–29)NH2;
MIA-690 [(PhAc-Ada)0-Tyr1, D-Arg2, Cpa6, Ala8, Har9, Fpa510, His11, Orn12, Abu15, His20, Orn21, Nle27, D-Arg28, Har29]hGH-RH(1–29)NH2.
Non-coded amino acids and acyl groups are abbreviated as follows: Abu, α-aminobutyric acid; Ac, acetyl; Ada, 12-aminododecanoyl; Agm, agmatine; Amc, 8-aminocaprylyl; Cpa, para-chlorophenylalanine; Fpa5, pentafluoro-phenylalanine; Har, homoarginine; hGHRH, human GHRH; Nle, norleucine; Orn, ornithine; Ph, phenyl; PhAc, phenylacetyl; Tyr(Me), O-methyl-tyrosine. The peptides were lyophilized; stability and purity were verified by HPLC before experimental use. For in vitro studies, the peptides were dissolved in dimethyl sulfoxide (DMSO) to final concentrations that did not exceed 0.1%. For in vivo studies, the peptides were dissolved in 0.1% DMSO in sterile aqueous 10% propylene glycol (vehicle).
Cell line
Human malignant melanoma cancer cell line A-375 was obtained from American Type Culture Collection (ATCC, Manassas, VA) and maintained in culture using Dulbecco's Modified Eagle Medium (Invitrogen, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS) and antibiotics (100 U/ml penicillin, 100 mg/ml streptomycin). The cells were grown at 37°C in a humidified 95% air/5% CO2 atmosphere.
Cell proliferation assay (MTS assay)
Cells (104 per well) were seeded onto 96-well plates with 100 μl of culture medium containing 10% FBS, cultured overnight, starved for 24 hours in medium without FBS, then exposed to GHRH antagonists MIA-602, MIA-606, or MIA-690 at concentrations of 1–10 μM for 72 h. Cell viability was evaluated by using a 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfonphenyl)-2H-tetrazolium (MTS) assay (CellTiter 96 Aqueous 1 Solution Cell Proliferation Assay; Promega, Madison, WI) according to the manufacturer's instructions. Absorbance was measured at 490 nm in a Victor 3 Multilabel Counter (Perkin-Elmer, Waltham, MD). All experiments were done in quadruplicate and were repeated 3 times. The inhibition of cell proliferation was expressed as the percentage of vehicle control (0.1% DMSO in the culture medium).
Animals and experimental protocol
Six-week-old female athymic nude mice (Ncr nu/nu) were obtained from the National Cancer Institute (Bethesda, MD). The animals were housed in sterile cages under laminar flow hoods in a temperature-controlled room with a 12-h light/12-h dark schedule and were fed autoclaved chow and water ad libitum. Xenografts were initiated by subcutaneous injection of A-375 human melanoma cancer cells into both flanks of nude mice. The developed tumors were dissected, minced and approximately 1 mm3 pieces of tumor tissue were transplanted subcutaneously into both flank areas of the animals. When tumors reached an appropriate size (approximately 50 mm3) mice were randomly divided into 2 groups (n = 7) and received the following daily treatment as s.c. injections: group 1, vehicle solution; group 2, 5 μg of GHRH antagonist MIA-690. Tumor volume (length × width × height × 0.5236) was measured weekly for 28 d. Mice were then sacrificed with cervical dislocation under isoflurane anesthesia (Baxter, Deerfield, IL), tumors were excised and weighed, and necropsy was performed. Tumor specimens were snap-frozen and stored at − 70°C. All experiments were approved by the Veterans Affairs Animal Care and Use Committee and were performed in accordance with institutional guidelines for the use of experimental animals.
Total RNA isolation and RT2 profiler PCRTM arrays
Total RNA was isolated from 30 mg samples of tumor tissue using NucleoSpin kit (Macherey-Nagel Inc., Bethlehem, PA). Three samples from each group were analyzed. The yield and quality of total RNA was determined spectrophotometrically using 260 nm and 260/280 nm ratio, respectively. Two μg of RNA with a final volume of 40 μl were reverse transcribed into cDNA with the QuantiTect® Reverse transcription Kit (Qiagen, Valencia, CA) using a VeritiTM 96-well thermal cycler (Applied Biosystems, Foster City, CA). Human Signal Transduction PathwayFinder RT2 Profiler PCR array (SABiosciences, Frederick, MD) was used to examine the mRNA levels of 84 key genes responsible for signal transduction pathway activation or inhibition. Fold-changes in gene expression were calculated using the ΔΔCt method. Normalization was performed using 5 housekeeping genes on the arrays.
Cell cycle analysis
Cell cycle analysis was measured by laser flow cytometry. A-375 cells were seeded into 25 ml flasks at a density of 2 × 105/ml cells, cultured overnight, starved for 24 hours in medium not containing FBS, and then exposed to MIA-690 at a concentration of 5 μM for 1 hour. Cells were washed then with PBS and covered with 1 ml of hypotonic propidium-iodide/sodium-citrate staining solution containing 0.3 μl/ml of Nonidet-40 detergent. A rubber policeman was used to scrape the monolayer and stained nuclei were isolated by vigorous pipetting. The nuclear suspension was transferred into 2 ml tubes for analysis on a Coulter XL flow cytometer using 488 nm laser excitation. The forward and side scatter profile gated out debris and dead cells, and DNA content of 10000 cells was used for generation of DNA histograms. Cell cycle distribution analysis was performed by ModFit Program (Verity Software House, Topsham, ME).
Cell fractionation and Western blot analysis
Cells (106 per well) were seeded onto 6-well plates with 2 ml of culture medium containing 10% FBS, cultured overnight, starved for 24 hours in medium without FBS, then exposed to GHRH antagonist MIA-690 at a concentration of 10 μM for 5, 10, 20, 30 and 60 min. Cells were then washed 2 times in PBS and were scraped in lysis buffer containing 50 mM Tris-HCl pH 7.4, 150 mM NaCl, 10% glycerol, 1 mM sodium-orthovanadate, 1 mM sodium fluoride, 1% Non-iodet and protease inhibitor cocktail on ice. Lysates were spun at 8000 rpm for 2.5 min. Half of the supernatant was saved as cytosolic fraction and the pellet was resuspended with the remaining lysis buffer. Lysates were centrifuged at 14000 rpm for 15 min at 4°C and the supernatant was collected as nuclear fraction.
When whole cell lysates were generated, cells were scraped in lysis buffer. Lysates were spun at 10000 rpm for 10 min at 4°C and the supernatant was collected. Protein concentrations were determined by NanoDrop (NanoDrop Technologies Inc., Wilmington, DE).
Equal amounts of protein were resuspended in 4X sample loading buffer (0.25 M Trizma Base, 8% SDS, 40% glycerol, 0.004% bromophenol blue, 4% β-mercaptoethanol; pH 6.8), boiled for 3 min and separated by 12% SDS-polyacrylamide gel electrophoresis. Proteins from the gel were then transferred onto nitrocellulose membrane, which was blocked with 50–50% Tris-buffered saline (TBS) (20 mM Tris-HCl pH:7.5, 150 mM NaCl): Odyssey blocking buffer for 1 h at room temperature, followed by an overnight incubation at 4°C with primary antibodies for β-actin (1:5000; A-5441, Sigma-Aldrich, St. Louis, MO), GHRH-R (1:3000; ab-28692, AbCam Inc., Cambridge, MA), and p27 (1:200; sc-528, Santa Cruz Biotechnology Inc., Santa Cruz, CA). Signals were developed by incubating the membrane with the appropriate Infrared IRDye®-labeled secondary antibodies (1:10000; LI-COR) for 1 h at room temperature and were then visualized with the Odyssey Infrared Imaging System (LI-COR). Densitometry was performed using ImageJ software (NIH, Bethesda, MD).
Immunofluorescence staining
Cells (106 per well) were seeded onto 22-mm2 glass coverslips placed in a 6-well plate with 2 ml of culture medium containing 10% FBS, cultured overnight, starved for 24 hours in medium without FBS, then exposed to GHRH antagonist MIA-690 at a concentration of 10 μM for 5, 10, 20, 30 and 60 min. After treatment, A-375 cells were fixed with 4% paraformaldehyde for 7 min, then permeabilized with methanol at 4°C for 5 min. Coverslip samples were blocked with 2% goat serum, followed by a 60-min incubation with primary antibodies for p27 (1:50; sc-528, Santa Cruz Biotechnology). Signals were developed by incubating the coverslip samples for 60 min with the appropriate secondary fluorescent antibodies (1:500; Alexa fluor Goat anti-Rabbit 488 (A11008) and Alexa fluor Goat anti-mouse 555 (A21422, Molecular Probes, Eugene, OR). Fixed cultures were co-stained with Vectashield Mounting Media with DAPI (Vector Laboratories, Inc., Burlingame, CA) to detect nuclei. Immunostained coverslip samples were imaged with a Nikon Eclipse Ti Inverted Microscope fitted with epifluorescence optics (Nikon, Melville NY). Microscopic images were recorded digitally using a Nikon DS-Qi1Mc camera and NIS Elements BR Imaging Software (Nikon).
Statistical analysis
For statistical evaluation, SigmaStat 3.0 software (Sytat Software, San Jose, CA) was used. Results are expressed as mean ± SEM. One-way analysis of variance (ANOVA) followed by Bonferroni t-test or a 2-tailed Student's t-test were used where appropriate, and significance was accepted at P < 0.05.
Funding Statement
Medical Research Service of the Veterans Affairs Department; Departments of Pathology, and Medicine, Division of Hematology/Oncology and Sylvester Comprehensive Cancer Center of the Miller Medical School; University of Miami; South Florida Veterans Affairs Foundation for Research and Education (all to A.V.S); Urology Care Foundation Research Scholars Program and the AUA Southeastern Section (F.G.R.); and L. Austin Weeks Endowment for Urologic Research (N.L.B.).
Disclosure of Potential Conflicts of Interest
A.V.S., M.Z., and R.-Z.C. are listed as co-inventors on patents for GHRH antagonists, which have been assigned to the University of Miami and the Veterans Affairs Department; however, this study was purely experimental. The rest of the authors have nothing to disclose.
References
- 1. Slipicevic A, Herlyn M. Narrowing the knowledge gaps for melanoma. Upsala J Med Sci 2012; 117:237-43; PMID:22339359; http://dx.doi.org/ 10.3109/03009734.2012.658977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Usher-Smith JA, Emery J, Kassianos AP, Walter FM. Risk Prediction Models for Melanoma: A Systematic Review. Cancer Epidemiol Biomarkers Prev 2014; 23:1450-63; PMID:24895414; http://dx.doi.org/ 10.1158/1055-9965.EPI-14-0295 [DOI] [PubMed] [Google Scholar]
- 3. Tsao H, Atkins MB, Sober AJ. Management of cutaneous melanoma. N Engl J Med 2004; 351:998-1012; PMID:15342808; http://dx.doi.org/ 10.1056/NEJMra041245 [DOI] [PubMed] [Google Scholar]
- 4. Lee B, Mukhi N, Liu D. Current management and novel agents for malignant melanoma. J Hematol Oncol 2012; 5:3; PMID:22333219; http://dx.doi.org/ 10.1186/1756-8722-5-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schally AV, Varga JL, Engel JB. Antagonists of growth-hormone-releasing hormone: an emerging new therapy for cancer. Nat Clin Pract Endocrinol Metab 2008; 4:33-43; PMID:18084344; http://dx.doi.org/ 10.1038/ncpendmet0677 [DOI] [PubMed] [Google Scholar]
- 6. Rick FG, Schally AV, Szalontay L, Block NL, Szepeshazi K, Nadji M, Zarandi M, Hohla F, Buchholz S, Seitz S. Antagonists of growth hormone-releasing hormone inhibit growth of androgen-independent prostate cancer through inactivation of ERK and Akt kinases. Proc Natl Acad Sci USA 2012; 109:1655-60; PMID:22307626; http://dx.doi.org/ 10.1073/pnas.1120588109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cai R, Schally AV, Cui T, Szalontay L, Halmos G, Sha W, Kovacs M, Jaszberenyi M, He J, Rick FG, et al. Synthesis of new potent agonistic analogs of growth hormone-releasing hormone (GHRH) and evaluation of their endocrine and cardiac activities. Peptides 2014; 52:104-12; PMID:24373935; http://dx.doi.org/ 10.1016/j.peptides.2013.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fahrenholtz CD, Rick FG, Garcia MI, Zarandi M, Cai RZ, Block NL, Schally AV, Burnstein KL. Preclinical efficacy of growth hormone-releasing hormone antagonists for androgen-dependent and castration-resistant human prostate cancer. Proc Natl Acad Sci USA 2014; 111:1084-9; PMID:24395797; http://dx.doi.org/ 10.1073/pnas.1323102111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kiaris H, Chatzistamou I, Papavassiliou AG, Schally AV. Growth hormone-releasing hormone: not only a neurohormone. Trends Endocrinol Metab 2011; 22:311-7; PMID:21530304; http://dx.doi.org/ 10.1016/j.tem.2011.03.006 [DOI] [PubMed] [Google Scholar]
- 10. Rick FG, Saadat SH, Szalontay L, Block NL, Kazzazi A, Djavan B, Schally AV. Hormonal manipulation of benign prostatic hyperplasia. Curr Opin Urol 2013; 23:17-24; PMID:23202285; http://dx.doi.org/ 10.1097/MOU.0b013e32835abd18 [DOI] [PubMed] [Google Scholar]
- 11. Rick FG, Schally AV, Block NL, Abi-Chaker A, Krishan A, Szalontay L. Mechanisms of synergism between antagonists of growth hormone-releasing hormone and antagonists of luteinizing hormone-releasing hormone in shrinking experimental benign prostatic hyperplasia. Prostate 2013; 73:873-83; PMID:23280565; http://dx.doi.org/ 10.1002/pros.22633 [DOI] [PubMed] [Google Scholar]
- 12. Rick FG, Szalontay L, Schally AV, Block NL, Nadji M, Szepeshazi K, Vidaurre I, Zarandi M, Kovacs M, Rekasi Z. Combining growth hormone-releasing hormone antagonist with luteinizing hormone-releasing hormone antagonist greatly augments benign prostatic hyperplasia shrinkage. J Urol 2012; 187:1498-504; PMID:22341819; http://dx.doi.org/ 10.1016/j.juro.2011.11.081 [DOI] [PubMed] [Google Scholar]
- 13. Jaszberenyi M, Rick FG, Szalontay L, Block NL, Zarandi M, Cai RZ, Schally AV. Beneficial effects of novel antagonists of GHRH in different models of Alzheimer's disease. Aging 2012; 4:755-67; PMID:23211425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kanashiro-Takeuchi RM, Takeuchi LM, Rick FG, Dulce R, Treuer AV, Florea V, Rodrigues CO, Paulino EC, Hatzistergos KE, Selem SM, et al. Activation of growth hormone releasing hormone (GHRH) receptor stimulates cardiac reverse remodeling after myocardial infarction (MI). Proc Natl Acad Sci USA 2012; 109:559-63; PMID:22203988; http://dx.doi.org/ 10.1073/pnas.1119203109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lucas R, Sridhar S, Rick FG, Gorshkov B, Umapathy NS, Yang G, Oseghale A, Verin AD, Chakraborty T, Matthay MA, et al. Agonist of growth hormone-releasing hormone reduces pneumolysin-induced pulmonary permeability edema. Proc Natl Acad Sci U S A 2012; 109:2084-9; PMID:22308467; http://dx.doi.org/ 10.1073/pnas.1121075109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lucas R, Czikora I, Sridhar S, Zemskov E, Gorshkov B, Siddaramappa U, Oseghale A, Lawson J, Verin A, Rick FG, et al. Mini-review: novel therapeutic strategies to blunt actions of pneumolysin in the lungs. Toxins 2013; 5:1244-60; PMID:23860351; http://dx.doi.org/ 10.3390/toxins5071244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. LeRoith D, Roberts CT, Jr. The insulin-like growth factor system and cancer. Cancer Lett 2003; 195:127-37; PMID:12767520; http://dx.doi.org/ 10.1016/S0304-3835(03)00159-9 [DOI] [PubMed] [Google Scholar]
- 18. Schally AV. New approaches to the therapy of various tumors based on peptide analogues. Horm Metab Res 2008; 40:315-22; PMID:18491250; http://dx.doi.org/ 10.1055/s-2008-1073142 [DOI] [PubMed] [Google Scholar]
- 19. Havt A, Schally AV, Halmos G, Varga JL, Toller GL, Horvath JE, Szepeshazi K, Koster F, Kovitz K, Groot K, et al. The expression of the pituitary growth hormone-releasing hormone receptor and its splice variants in normal and neoplastic human tissues. Proc Natl Acad Sci USA 2005; 102:17424-9; PMID:16299104; http://dx.doi.org/ 10.1073/pnas.0506844102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pozsgai E, Schally AV, Hocsak E, Zarandi M, Rick F, Bellyei S. The effect of a novel antagonist of growth hormone releasing hormone on cell proliferation and on the key cell signaling pathways in nine different breast cancer cell lines. Int J Oncol 2011; 39:1025-32; PMID:21701777; http://dx.doi.org/ 10.3892/ijo.2011.1098 [DOI] [PubMed] [Google Scholar]
- 21. Hohla F, Buchholz S, Schally AV, Seitz S, Rick FG, Szalontay L, Varga JL, Zarandi M, Halmos G, Vidaurre I, et al. GHRH antagonist causes DNA damage leading to p21 mediated cell cycle arrest and apoptosis in human colon cancer cells. Cell Cycle 2009; 8:3149-56; PMID:19755849; http://dx.doi.org/ 10.4161/cc.8.19.9698 [DOI] [PubMed] [Google Scholar]
- 22. Chatzistamou I, Volakaki AA, Schally AV, Kiaris H, Kittas C. Expression of growth hormone-releasing hormone receptor splice variant 1 in primary human melanomas. Regul Peptides 2008; 147:33-6; PMID:18255167; http://dx.doi.org/ 10.1016/j.regpep.2007.12.008 [DOI] [PubMed] [Google Scholar]
- 23. Jaszberenyi M, Schally AV, Block NL, Zarandi M, Cai RZ, Vidaurre I, Szalontay L, Jayakumar AR, Rick FG. Suppression of the proliferation of human U-87 MG glioblastoma cells by new antagonists of growth hormone-releasing hormone in vivo and in vitro. Targeted Oncol 2013; 8:281-90; PMID:23371031; http://dx.doi.org/ 10.1007/s11523-013-0264-y [DOI] [PubMed] [Google Scholar]
- 24. Papadia A, Schally AV, Halmos G, Varga JL, Seitz S, Buchholz S, Rick F, Zarandi M, Bellyei S, Treszl A, et al. Growth hormone-releasing hormone antagonists inhibit growth of human ovarian cancer. Horm Metab Res 2011; 43:816-20; PMID:22009378; http://dx.doi.org/ 10.1055/s-0031-1287766 [DOI] [PubMed] [Google Scholar]
- 25. Perez R, Schally AV, Vidaurre I, Rincon R, Block NL, Rick FG. Antagonists of growth hormone-releasing hormone suppress in vivo tumor growth and gene expression in triple negative breast cancers. Oncotarget 2012; 3:988-97; PMID:22941871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rick FG, Seitz S, Schally AV, Szalontay L, Krishan A, Datz C, Stadlmayr A, Buchholz S, Block NL, Hohla F. GHRH antagonist when combined with cytotoxic agents induces S-phase arrest and additive growth inhibition of human colon cancer. Cell Cycle 2012; 11:4203-10; PMID:23095641; http://dx.doi.org/ 10.4161/cc.22498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Seitz S, Rick FG, Schally AV, Treszl A, Hohla F, Szalontay L, Zarandi M, Ortmann O, Engel JB, Buchholz S. Combination of GHRH antagonists and docetaxel shows experimental effectiveness for the treatment of triple-negative breast cancers. Oncol Rep 2013; 30:413-8; PMID:23624870 [DOI] [PubMed] [Google Scholar]
- 28. Stangelberger A, Schally AV, Rick FG, Varga JL, Baker B, Zarandi M, Halmos G. Inhibitory effects of antagonists of growth hormone releasing hormone on experimental prostate cancers are associated with upregulation of wild-type p53 and decrease in p21 and mutant p53 proteins. Prostate 2012; 72:555-65; PMID:21796649; http://dx.doi.org/ 10.1002/pros.21458 [DOI] [PubMed] [Google Scholar]
- 29. Kovacs M, Schally AV, Hohla F, Rick FG, Pozsgai E, Szalontay L, Varga JL, Zarandi M. A correlation of endocrine and anticancer effects of some antagonists of GHRH. Peptides 2010; 31:1839-46; PMID:20633588; http://dx.doi.org/ 10.1016/j.peptides.2010.07.006 [DOI] [PubMed] [Google Scholar]
- 30. Abdel-Wahab M, Schally AV, Rick FG, Szalontay L, Block NL, Jorda M, Mahmoud O, Markoe A, Shi Y-F, Reiner T, et al. Antagonists of growth hormone releasing hormone (GHRH) given before whole body radiation lead to modulation of radiation response and organ-specific changes in the expression of angiogenesis. J Radiat Oncol 2012; 1:389-96; http://dx.doi.org/ 10.1007/s13566-012-0031-1 [DOI] [Google Scholar]
- 31. Rick FG, Schally AV, Block NL, Nadji M, Szepeshazi K, Zarandi M, Vidaurre I, Perez R, Halmos G, Szalontay L. Antagonists of growth hormone-releasing hormone (GHRH) reduce prostate size in experimental benign prostatic hyperplasia. Proc Natl Acad Sci U S A 2011; 108:3755-60; PMID:21321192; http://dx.doi.org/ 10.1073/pnas.1018086108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Klukovits A, Schally AV, Szalontay L, Vidaurre I, Papadia A, Zarandi M, Varga JL, Block NL, Halmos G. Novel antagonists of growth hormone-releasing hormone inhibit growth and vascularization of human experimental ovarian cancers. Cancer 2012; 118:670-80; PMID:21751186; http://dx.doi.org/ 10.1002/cncr.26291 [DOI] [PubMed] [Google Scholar]
- 33. Slingerland J, Pagano M. Regulation of the cdk inhibitor p27 and its deregulation in cancer. J Cell Physiol 2000; 183:10-7; PMID:10699961; http://dx.doi.org/ 10.1002/(SICI)1097-4652(200004)183:1<10::AID-JCP2>3.0.CO;2-I [DOI] [PubMed] [Google Scholar]
- 34. Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer 2008; 8:253-67; PMID:18354415; http://dx.doi.org/ 10.1038/nrc2347 [DOI] [PubMed] [Google Scholar]
- 35. Chen G, Cheng Y, Zhang Z, Martinka M, Li G. Prognostic significance of cytoplasmic p27 expression in human melanoma. Cancer Epidemiol Biomar Prev: A Pub Am Assoc Cancer Res Cosponsored Am Soc Prev Oncol 2011; 20:2212-21; http://dx.doi.org/ 10.1158/1055-9965.EPI-11-0472 [DOI] [PubMed] [Google Scholar]
- 36. Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer 2006; 6:729-34; PMID:16915295; http://dx.doi.org/ 10.1038/nrc1974 [DOI] [PubMed] [Google Scholar]
- 37. Hong F, Larrea MD, Doughty C, Kwiatkowski DJ, Squillace R, Slingerland JM. mTOR-raptor binds and activates SGK1 to regulate p27 phosphorylation. Mol Cell 2008; 30:701-11; PMID:18570873; http://dx.doi.org/ 10.1016/j.molcel.2008.04.027 [DOI] [PubMed] [Google Scholar]
- 38. Siejka A, Barabutis N, Schally AV. GHRH antagonist MZ-5-156 increases the expression of AMPK in A549 lung cancer cells. Cell Cycle 2011; 10:3714-8; PMID:22041656; http://dx.doi.org/ 10.4161/cc.10.21.17904 [DOI] [PubMed] [Google Scholar]
- 39. Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol 2002; 4:E131-6; PMID:11988758; http://dx.doi.org/ 10.1038/ncb0502-e131 [DOI] [PubMed] [Google Scholar]
- 40. Rutberg SE, Goldstein IM, Yang YM, Stackpole CW, Ronai Z. Expression and transcriptional activity of AP-1, CRE, and URE binding proteins in B16 mouse melanoma subclones. Mol Carcin 1994; 10:82-7; PMID:8031468; http://dx.doi.org/ 10.1002/mc.2940100205 [DOI] [PubMed] [Google Scholar]
- 41. Scheibenbogen C, Mohler T, Haefele J, Hunstein W, Keilholz U. Serum interleukin-8 (IL-8) is elevated in patients with metastatic melanoma and correlates with tumour load. Melanoma Res 1995; 5:179-81; PMID:7640519; http://dx.doi.org/ 10.1097/00008390-199506000-00006 [DOI] [PubMed] [Google Scholar]
- 42. Brennecke S, Deichmann M, Naeher H, Kurzen H. Decline in angiogenic factors, such as interleukin-8, indicates response to chemotherapy of metastatic melanoma. Melanoma Res 2005; 15:515-22; PMID:16314737; http://dx.doi.org/ 10.1097/00008390-200512000-00006 [DOI] [PubMed] [Google Scholar]
- 43. Atkins MB, Kunkel L, Sznol M, Rosenberg SA. High-dose recombinant interleukin-2 therapy in patients with metastatic melanoma: long-term survival update. Cancer J Sci Am 2000; 6(Suppl 1):S11-4; PMID:10685652 [PubMed] [Google Scholar]
- 44. Sacca R, Cuff CA, Lesslauer W, Ruddle NH. Differential activities of secreted lymphotoxin-alpha3 and membrane lymphotoxin-alpha1beta2 in lymphotoxin-induced inflammation: critical role of TNF receptor 1 signaling. J Immunol 1998; 160:485-91 [PubMed] [Google Scholar]
- 45. Cuff CA, Schwartz J, Bergman CM, Russell KS, Bender JR, Ruddle NH. Lymphotoxin alpha3 induces chemokines and adhesion molecules: insight into the role of LT alpha in inflammation and lymphoid organ development. J Immunol 1998; 161:6853-60; PMID:9862717 [PubMed] [Google Scholar]
- 46. Schrama D, thor Straten P, Fischer WH, McLellan AD, Brocker EB, Reisfeld RA, Becker JC. Targeting of lymphotoxin-alpha to the tumor elicits an efficient immune response associated with induction of peripheral lymphoid-like tissue. Immunity 2001; 14:111-21; PMID:11239444; http://dx.doi.org/ 10.1016/S1074-7613(01)00094-2 [DOI] [PubMed] [Google Scholar]
- 47. Zarandi M, Horvath JE, Halmos G, Pinski J, Nagy A, Groot K, Rekasi Z, Schally AV. Synthesis and biological activities of highly potent antagonists of growth hormone-releasing hormone. Proc Natl Acad Sci U S A 1994; 91:12298-302; PMID:7991622; http://dx.doi.org/ 10.1073/pnas.91.25.12298 [DOI] [PMC free article] [PubMed] [Google Scholar]