

Abstract
Background & Aims
Liver inflammatory diseases associated with cancer promoting somatic oncogene mutations are increasing in frequency. Preclinical cancer models that allow for the study of early tumor progression are often protracted, which limits the experimental study parameters due to time and expense. Here we report a robust inexpensive approach using Sleeping Beauty Transposition (SBT) delivery of oncogenes along with Gaussia Luciferase expression vector G.luc, to assess de novo liver tumor progression, as well as the detection of immune responses or responses induced by therapeutic intervention.
Methods
Tracking de novo liver tumor progression with G.luc was demonstrated in models of hepatocellular carcinoma (HCC) or adenoma (HCA) initiated by hydrodynamic delivery of SBT oncogenes.
Results
Rising serum luciferase levels correlated directly with increasing liver tumor burden and eventual morbidity. Early detection of hepatocyte apoptosis from mice with MET+CAT transfected hepatocytes was associated with a transient delay in HCC growth mediated by a CD8+ T-cell response against transformed hepatocytes. Furthermore, mice that lack B cells or macrophages had an increase in TUNEL+ hepatocytes following liver MET transfection demonstrating these cells provided protection from MET-induced hepatocyte apoptosis. Treatment of mice bearing established HCC with IL-18+IL-12 decreased tumor burden that was associated with decreased levels of serum luciferase.
Conclusions
Hydrodynamic delivery of the SBT vector G.luc to hepatocytes serves as a simple blood-based approach for real-time tracking of pathologically distinct types of liver cancer, which revealed tumor-induced immunologic responses and was beneficial in monitoring the efficacy of therapeutic interventions.
Keywords: Sleeping Beauty Transposition, HCC, HCA, Gaussia Luciferase, Real-time tracking, Treatment
Introduction
Up to 70%–90% of all liver cancers are detected in patients suffering from chronic inflammatory liver diseases due to obesity, chronic hepatitis C and liver fluke infections that increase the frequency of deleterious somatic mutations in oncogenes and tumor suppressor genes (1–9). Because liver inflammatory diseases due to obesity (10) and hepatitis C infections (11) are observed with increased frequency globally, it is important to understand which somatic mutations in oncogenes drive liver cancer development and/or progression and how inflammation facilitates this process.
The advent of Sleeping Beauty transposition (SBT) technology has provided experimental tools to investigate how mutations of oncogenes lead to the development of liver cancer (12, 13). This non-viral-based gene transfection system can be used to induce liver tumors by stably integrating oncogenes into hepatocytes. The induction of tumors using the SBT system allows the analysis of how inflammation and tumor immunity regulate the entire spectrum of tumor growth including the early stages of neoplasia, which cannot be addressed using transplantable tumors. Although carcinogens such as DEN also allow us to study liver tumor formation, they often induce a myriad of undefined mutations and off target events while the SBT system can be best used to study how specific oncogenes collaborate in the formation of tumors.
Mouse cancer models using SBT, carcinogens and transgenic mice are amenable for the study of tumor formation at the early stages of neoplasia but are limited by the variable and often protracted time for development/progression of tumors, depending on the oncogene combinations used for initiation. In vivo imaging could be used to follow tumor progression but this approach can be prohibitively expensive, limited by the number of mice that can be scanned and requires dedicated staff to run complex instrumentation. Thus, a tumor-tracking method that could be used sequentially in individual mice over a long period of time would allow an assessment of tumor status in real-time. Such an approach may reveal therapeutic windows, be used to extrapolate tumor burden and survival times and potentially elucidate immunoregulatory events.
In this study, we investigated the use of the SBT G.luc vector for tracking nonlinear oncogene-induced liver tumor progression. Using two distinct de novo liver tumor models, we demonstrated that the level of serum luciferase released by G.luc vector integrated in hepatocytes (G.luc+ hepatocytes) significantly correlated with the volume of liver tumors, thereby providing a minimally-invasive method to track in living mice the growth of de novo-generated liver tumors. Using this minimally-invasive method, parameters can be measured that would otherwise require euthanasia of mouse cohorts with an associated higher experimental cost. The evaluation of immunogenic de novo liver tumors using G.luc+ hepatocytes also allowed us to identify inflection points that revealed CD8+ T-cell mediated transient tumor inhibitory effects during tumor progression. Concordantly, we show for the first time that stable expression of luciferase by hepatocytes can also be used to efficiently track, in real-time, the outcome of therapies against oncogene-driven de novo liver tumors.
Materials and Methods
Plasmid constructs
Plasmids encoding the sleeping beauty transposase (HSB2) and transposons containing human c-Met (MET) and empty vector control (PT3), were a generous gift from Dr. Xin Chen (UCSF, San Francisco, Ca.). Transposon constructs containing Δ90Nβ-catenin (CAT) and Myr-AKT (AKT) were previously reported (14). The gene for Gaussia Luciferase was PCR-amplified and cloned into pENTR (Invitrogen) and subsequently Gateway-cloned into the PT3 destination vector to produce a Gaussia luciferase transposon (G.luc) vector.
Experimental model
C57/BL6J (WT), B6.129S2-Cd8a (CD8−/−), B6.129S2-Cd4/J (CD4−/−), B6.129S2-lgh-6 (Bcell−/−) and CD1d−/− (NKT−/−) mice were purchased from The Jackson Laboratory and were bred and maintained at the Frederick National Laboratory in a specific pathogen free facility in accordance with an approved Animal Care and Use Protocol. Oncogene constructs were delivered to hepatocytes using the hydrodynamic transfection technique. Briefly, constructs containing either MET+CAT or AKT+CAT along with HSB and G.luc vector were mixed in saline at a volume of 10% vol/mouse weight and injected into aged and sex matched 8–12 week old C57/BL6J or B6.129S2-Cd8a mice via tail vein over 5–7 seconds as previously described (12). MET+CAT induces liver tumors with a pathology consistent with hepatocellular carcinoma while AKT+CAT induces liver tumors histopathologically characterized as hepatocellular adenoma (14).
Immunohistochemistry (IHC) staining and analysis
Liver tissues were fixed with 10% normal buffered formalin overnight and then transferred to 70% ethanol. Paraffin blocks were made from the fixed tissues. Slides cut from the blocks were analyzed for apoptosis using TUNEL staining, proliferation using BrdU at 1:50 (Invitrogen Life Technologies), β-catenin at 1:200 (Abcam) and N-Ras (F155 Santa Cruz). For IHC slide analysis, five non-overlapping pictures were taken from at least two separate lobes at and images were analyzed using CellProfiler (http://www.cellprofiler.org/) with modified Ki67 pipeline.
Immunotherapeutic regimen
Recombinant murine IL-12 and IL-18 was purchased from PeproTech Inc. (Rocky Hill, NJ). Stock aliquots of cytokines were diluted with HBSS and mice were injected intraperitoneally (i.p.) with vehicle control (VC) or IL-12 (0.3μg) + IL-18 (0.3μg) on days 20–24 and 28–31 post initiation of liver tumors with MET+CAT oncogenes.
qPCR quantitation of oncogenes in the liver
Livers were harvested and snap frozen on a dry ice and 2-Methylbutane mixture for later manipulations. Frozen livers were mechanically shattered into small fragments and genomic DNA was isolated using a Gentra Puregene Tissue Kit (Qiagen) according to manufactures instructions. Oncogene copy number per 100 ng of liver DNA was determined using qPCR analysis with TaqMan probes MET (Hs 01564484_m1), CTNNB1 (CAT) (Hs 00355049_m1) and AKT (mm 01331626_m1) (ABI) and comparing it to a standard curve made using the oncogene plasmids.
Tumor assessment by MRI
Animal imaging was performed by Frederick National Laboratory Small Animal Imaging Program. Magnetic resonance imaging (MRI) was performed with a 3.0T clinical scanner (Philips Intera Achieva, Philips Medical Systems, Einthoven, NL) to detect the onset of tumors, monitor their progression, and track the changes in the liver volume biweekly. Data was acquired using a 40-mm diameter solenoid receiver coil (Philips Research, Hamburg, Germany). The mice were anesthetized in an induction chamber with 3% isoflurane in O2 at a 1 L/min flow rate and then placed in a custom made mouse holder within the imaging coil. During an imaging session, the anesthesia level was modified between 1.5–2.0% isoflurane to maintain a 40 bpm pulmonary rate, and their physiology monitored (Biopac System Inc., Goleta, CA). Body temperature was maintained at 37°C by supplying warm air (Small Animal Instruments, Inc., Stony Brook, NY) around the mouse holder. Multi slice T2 weighted turbo spin echo (T2w-TSE) sequence was applied in coronal view with respiratory triggering to minimize the motion artifacts. An imaging volume of 36×27×18 mm was chosen to cover the mouse abdominal cavity. The images were acquired with a repetition time (TR) 4500ms, echo time (TE) 65ms, with an in plane resolution of 0.190×0.190mm, and slice thickness 0.5mm. Serial images obtained in 8 bi-weekly consecutive imaging sessions were used to calculate the changes in tumor and liver volumes.
Gaussia Luciferase assay
Mouse blood was collected using retro orbital bleeds in a serum separator tube (Becton Dickinson co.) and then serum was recovered by centrifuging tubes for 3 minutes at 20,000 Xg and then 5 μl of serum was added to 100μl of PBS and plated on a 96 well flat bottom white polystyrene assay plate (Costar 3362). Serum luciferase levels were determined using the BioLux Gaussia Luciferase Assay kit (New England BioLabs) according to manufacturer’s direction and luminescence measurements were acquired using FLUOstar Omega microplate reader (BMG LABTECH) after controlled injection of 50 μl of substrate mixture into plates containing the serum PBS mixture.
Liver leukocyte isolation and flow cytometric analysis
Liver leukocytes were isolated as previously described (15). Isolated leukocytes were counted and 1×106 cells were Fc-blocked with monoclonal 2.4G2 prior to staining with appropriately titered monoclonal antibodies (CD8a PerCP and PD-1 PE; eBioscience) used in conjunction with Live/Dead reagent; Invitrogen). Data was collected using a LSRII Special Order System equipped with solid state blue (488nm), red (640nm) and violet (405nm) lasers and then analyzed using FloJo Vx0.6.
Transfection of mouse hepatoma Hepa1–6 cells
Hepa1–6 cells were maintained in DMEM containing 1.45 g of glucose/liter (Sigma), supplemented with 2 mM glutamine, 50 μg/ml gentamycin sulfate, and 10% (v/v) heat-inactivated fetal calf serum. Targefect-Hepatocyte reagent (Targeting Systems, CA) was used for PT3-GFP or PT3-MET plasmid transfection into Hepa1-6 cells according to the manufacturer’s protocol. Cells were harvested 24h or 72h post transfection for apoptosis detection. Cell apoptosis was detected using PE Annexin V Apoptosis Detection Kit I according to manufacturer’s protocol (BD Bioscience).
Statistical analysis
Data in this study were analyzed with linear mixed effects hierarchical models, analysis of variance and covariance, Kaplan-Meier and Cox Proportional Hazards regression methods, correlation and (weighted) regression analysis, post hoc tests (e.g., Tukey’s test for multiple comparisons) and non-parametric methods (e.g., Wilcoxon’s test). Repeated measures analyses (in which ‘repeated’ measurements are recorded for the same animal on more than one occasion) and mixed effects models take into consideration the correlation/covariation of responses within the same animal over time (16, 17). Homogeneity of variance and covariance assumptions were routinely tested in model fits. Probability values less than 0.05 were considered significant.
Results
Serum G.luc levels correlate with MET+CAT oncogene-induced liver tumor burden
Ectopic expression of foreign proteins in mice can lead to an immune response against the protein and the tissues expressing the protein (18, 19). To determine whether secretable G.luc could be stably expressed in the liver and not immunologically rejected, we delivered Gaussia Luciferase expression vector (G.luc) hydrodynamically to the liver via Sleeping Beauty translocation (SBT), with or without transposase (HSB2) (12). Transposase HSB2 causes genomic insertion of the transposon randomly at AT-dinucleotide sites. In the absence of HSB2, the G.luc transposon does not integrate into the hepatocyte genome. Serum expression kinetics of luciferase was analyzed in C57/Bl6 mice injected with G.luc (non-integrated) or G.luc+HSB2 vectors (integrated) (Supplementary Fig. 1). Stable constitutive expression of luciferase was observed by day 3 in mice given G.luc+HSB2. The levels of serum Gaussia Luciferase were significantly higher in mice given G.luc+HSB2 vectors compared to mice given G.luc vector alone (post hoc Tukey’s test; p < 0.0001). Conversely, there was no significant difference in luciferase levels after day 3 between the serum of mice given G.luc alone and control serum from naïve mice. These data demonstrate that hepatocytes with integrated G.luc vector (G.luc+ hepatocytes) stably express constitutive levels of luciferase while luciferase levels of non-integrated G.luc are rapidly lost, returning to baseline level by day 3.
Because hepatocytes with integrated G.luc stably express luciferase under homeostatic conditions, we next tested whether this technique could be used to track hepatocyte proliferation. We hypothesized that the constitutively expressed levels of luciferase should rise when G.luc+ hepatocytes are induced by oncogenes to proliferate. De novo HCC formation was initiated by hydrodynamic transfection of 4ug of G.luc vector and escalating concentrations of MET+CAT oncogenes in combination with HSB2 (14). At days 30 through 56 mice that have received MET+CAT doses of 25, 12.5 and 5ug showed significantly higher serum luciferase levels compared to mice injected with PT3 controls or lower concentration of MET+CAT (Supplementary Fig. 2A). To confirm that the significant rise in luciferase levels with the higher MET+CAT oncogene groups was due to tumor burden, mice were euthanized at day 55 and tumor-bearing livers were weighed (Supplementary Fig. 2B). A proportional relationship was observed between the amount of MET+CAT oncogenes used to initiate tumors and their respective tumor-bearing liver weights (Supplementary Fig. 2C). Linear regression analysis revealed a significant correlation between serum luciferase levels and the weights of the tumor-bearing livers. To specifically determine if serum luciferase levels correlated with liver tumor burden, we quantitated the amount of integrated oncogenes in liver tissue using qPCR and compared it with the levels of serum luciferase. The copy number of MET and CAT oncogenes in 100ng of genomic DNA was then plotted against serum luciferase levels from these same mice at day 55 (Supplementary Fig. 2D, E). Linear regression analysis showed a significant correlation between the liver levels of either oncogene and serum luciferase levels.
Progression of AKT+CAT-initiated liver tumors originated in G.luc+ hepatocytes is accurately tracked by serum luciferase levels
To validate that hepatocytes with integrated G.luc vector could also be used to track alternative models of de novo-initiated liver tumors, we used AKT+CAT oncogenes that induce liver tumors (hepatocellular adenomas) that are histopathologically distinct from those induced by MET+CAT (hepatocellular carcinomas) (14). Furthermore, we wanted to determine whether G.luc+ hepatocytes could be used to track liver tumor progression throughout the oncogenenic process. This is critical because tumors can grow in complex three-dimensional nonlinear tracks (20, 21). Mice were hydrodynamically-transfected with G.luc vector along with AKT+CAT oncogenes and HSB2. Tumor progression was analyzed in real time by measuring liver size using MRI analysis on days 27, 41, 59, 66, 95 and 103. At day 27, normal liver morphology was evident. However, as AKT+CAT-initiated tumors progress to day 103, tumor nodules develop causing the liver to distend (Supplementary Fig. 3A). Serum luciferase levels were quantitated on the same days of MRI examination and a comparative analysis was done between these two parameters (Supplementary Fig. 3B). Both MRI liver analysis and serum luciferase levels revealed that AKT+CAT-initiated liver tumors progress in a nonlinear manner. Pearson correlations between serum luciferase levels and tumor sizes for all three animals are extremely high: r4401 = 0.99 (p < 0.0001), r4403 = 0.98 (p = 0.0005), and r4404 = 0.99 (p < 0.0001). In addition, analysis of covariance (ANCOVA) showed that for each animal, the proportionate RLU and Liver Size measures were statistically parallel, demonstrating the association between induced size changes in liver tumors and levels of serum luciferase. Taken together, these data demonstrate a high degree of correlation exists between liver tumor growth kinetics and levels of serum luciferase. Moreover, G.luc+ hepatocytes can be used to track the nonlinear progression of de novo-initiated liver tumors.
Serum luciferase levels detect growth rate changes in oncogene-induced liver tumors
The changes in kinetics of tumor progression relative to morbidity can provide insights into the diversity of basic biological mechanisms and/or immune recognition of tumors initiated by different oncogenic events. We tested whether integration of G.luc into hepatocytes provides a useful tool for detecting subtle variations in tumor progression of pathologically distinct hepatocellular adenoma (AKT+CAT) or carcinoma (MET+CAT) (14). By day 340, 90% and 70% of mice transfected with AKT+CAT and MET+CAT, respectively, had succumbed to the tumors (Fig. 1A) while all the mice given the PT3 control vector survived. Although the mortality rate of AKT+CAT and MET+CA- transfected mice was similar (Fig 1A), serum luciferase levels measured at various time points indicated intriguing differences in the kinetics and therefore in the biology of AKT+CAT and MET+CAT-initiated tumor growth (Fig. 1B). During the early phase (days 10–42) of tumor growth, luciferase levels in the sera from AKT+CAT-induced tumors increased, while the levels in mice that received MET+CAT fell slightly below the PT3 control (Fig. 1C). These data suggest the AKT+CAT oncogene combination initially causes sustained proliferation of transformed hepatocytes more effectively than the MET+CAT oncogenes. However, starting at day 59, luciferase levels from both groups rise approximately co-linearly (Fig. 1B). It remained to be determined whether this difference in kinetics was due to intrinsic characteristics in the induction and maintenance of proliferation in the transformed hepatocytes, or rather to some other biological factors such as differences in the ability of the immune system to recognize and respond to these tumors. We speculated that the human MET protein might be immunogenic in mice, and when expressed on hepatocytes might elicit an immune response resulting in detectable changes in tumor progression. To determine whether hepatocytes expressing the MET oncogene undergo apoptosis we injected MET or CAT oncogenes into mice and collected livers on day 7. Apoptotic hepatocytes were detected by TUNEL staining of liver tissue sections (Fig. 1D). TUNEL staining was more prevalent in the livers of mice that received the MET oncogene compared to those that received CAT.
Fig. 1.

Morbidity analysis of mice with AKT+CAT and MET+CAT- initiated liver tumors using G.luc integrated in the liver. G.luc (4ug), AKT (20ug)+CAT (20ug) and MET (20ug)+CAT (20ug) oncogenes along with HSB2 (4.4ug) were delivered to C57/Bl6 mice using hydrodynamic tail vein injection. (A) Kaplan-Meier (KM) survival plot. (B) Serum luciferase levels days 0–218. (C) Serum luciferase levels days 0–42. PT3 n=5, AKT+CAT n=10, MET+CAT n=10, mean ± SEM. (D) Liver sections were ApopTag Peroxidase stained using the TUNEL method on day 7 following initiation with MET or CAT oncogenes. (E,F) MET was hydrodynamically injected into WT, CD1d−/−, CD8−/−, CD4 −/− or lgh-6 mice. For NK cell and macrophage depletion (F) mice were treated with asGM1 or Clodronate respectively on days −3 and −1 prior to MET delivery. On day 7 liver tissue sections from (E,F) were stained for TUNEL counted on 5 non-overlapping 100X fields from 3 individual mice per group using CellProfiler. n=3, ***p<0.0001 mean ± SEM. Mann-Whitney U test was used to determine significance.
To determine if the increased MET-induced hepatocyte apoptosis was immune mediated, we first examined the level of nonspecific TUNEL+ hepatocytes by immunohistochemistry in naïve WT, NKT deficient (CD1d−/−), CD8−/−, CD4−/− and B cell−/− (lgh-6) mice. We also examined the level of nonspecific TUNEL+ hepatocytes in naïve mice depleted of NK cells and macrophages using asGM1 and clodronate respectively. No change in the frequency of TUNEL+ hepatocytes was observed in CD1d−/−, CD8−/−, CD4−/−, B cell−/− or in mice treated with clodronate or asGM1 compared to WT mice (Supplementary Fig. 4A). Next to determine if the increased MET-induced hepatocyte apoptosis was immune mediated, we injected MET into CD1d−/−, CD8−/−, CD4−/− or B cell−/− mice. We also injected MET into mice depleted of NK cells and macrophages using asGM1 and clodronate respectively. Livers were harvested on day 7 and TUNEL+ hepatocytes were quantitated using immunohistochemistry. A significant decrease in TUNEL+ hepatocytes was observed in the CD8−/− mice compared to WT mice (Fig 1E), suggesting CD8 cells are at least partially required for MET-induced hepatocyte apoptosis. In contrast, the frequency of TUNEL+ hepatocytes was increased in mice depleted of macrophages and B cell deficient mice (Fig. 1F), suggesting these cells may offer a partial protection from MET-induced hepatocyte apoptosis.
Recently it was shown that oncogenic N-ras can induce senescence in pre-malignant cells that are eliminated by CD4+ T-cells (22). However, mice with MET transduced livers did not express hepatocyte N-ras (Fig. 2A) or senescence marker beta-galactosidase at days 10 and 30 (Fig. 2B), suggesting MET does not induce the senescence pathway in hepatocytes.
Fig. 2.

MET does not induce N-ras-mediated hepatocyte senescence or oncogene-induced apoptosis. (A) MET was delivered to WT (n=5) or CD8−/− (n=5) mice or N-ras (control) was delivered to WT mice and on days 10 and 30 liver tissue sections were stained for N-ras. (B) MET was delivered to WT (n=5) and CD8−/− mice (n=5) and on days 10 and 30 liver tissue sections from these mice and B6.129S4-Gt(ROSA)26Sortm1Sor/J (control) mice were stained for beta-galactosidase. (C) Hepa1–6 cells were transfected with SBT vectors MET or GFP (control) using Targefect-Hepatocyte. At 24 and 72 hrs cell apoptosis was determined by staining for Annexin V and 7-AAD and analyzed by flow cytometry. For cell apoptosis control Hepa1-6 cells were treated for 24hrs with 100nM Sorafenib. Two independent experiments were performed with similar results.
The ability of MET oncogene to induce apoptosis directly was also examined. Mouse hepatoma Hepa1–6 cells were transfected with MET or GFP SBT vectors with greater than 20% transfection efficiency determined by the number of GFP+ Hepa1–6 cells (Supplementary Fig. 4B). For a cell apoptosis control, Hepa1-6 cells were treated for 24hrs with 100nM Sorafenib. At 24 and 72hrs. apoptotic Hepa1–6 cells were detected by Annexin V and 7-AAD staining. No increase in the proportion of apoptotic Hepa1–6 cells was observed compared to GFP-transfected controls (Fig. 2C), suggesting at least in this setting MET does not cause oncogene-induced apoptosis.
CD8+ T-cells regulate early growth of MET+CAT oncogene-induced liver cancer
For these studies we used a lower dose of oncogenes (10ug) to allow more time for nuanced immune reactions to occur and to increase sensitivity for G.luc tracking. Cytotoxic CD8+ T-cells that recognize tumor antigen can cause tumor cell apoptosis. However, chronic exposure to excessive antigen can lead to CD8+ T-cell exhaustion (23), characterized in part by the up-regulation of inhibitory receptors such as PD-1 (24, 25). We hypothesized that early CD8-mediated immune responses against MET occur and result in apoptosis of transformed hepatocytes that was detected by the early decrease in luciferase following delivery of MET+CAT oncogenes to the liver. If this theory is correct, we reasoned that as CD8+ T-cells become exhausted, remaining transformed G.luc+ hepatocytes would begin to proliferate resulting in rising serum luciferase levels, in association with accelerated tumor growth and eventual mortality. To test this hypothesis, tumor infiltrating CD8+ T-cells from mice injected with MET+CAT or AKT+CAT oncogenes were analyzed by flow cytometry for the expression of PD-1 antigen as a marker of antigen-experienced exhausted T cells (Fig. 3A). At day 14, PD-1 expression was induced on 33% of the liver CD8+ T-cells from mice where tumors were initiated with the MET+CAT oncogenes. In contrast, no PD-1 was detected on CD8+ T-cells from mice where tumors were initiated by the AKT+CAT oncogene combination, or in non-tumor bearing mice injected with control PT3 vector. These data suggest that CD8+ T-cells mediate immune responses against hepatocytes expressing MET+CAT, but not AKT+CAT oncogenes.
Fig. 3.

CD8+ T-cells regulate early MET+CAT-induced liver oncogenesis. (A) C57/Bl6 mice were hydrodynamically- transfected with MET+CAT (10ug), AKT+CAT (10ug) or PT3 (control) and on day 14 liver leukocytes were isolated and CD8+ T-cells were analyzed for PD-1 expression by flow cytometry. n=4, mean ± SEM (B) Serum luciferase levels from WT or CD8−/− mice hydrodynamically transfected with G.luc and MET+CAT (10ug) or PT3 (control). WT (MET+CAT n=5, CD8−/− (MET+CAT) n=4, WT (PT3) n=5, CD8−/− n=5, mean ± SEM. (C) Kaplan-Meier (KM) survival plot of WT and CD8−/− mice hydrodynamically transfected with MET+CAT oncogenes. WT n=25, CD8−/− n=10, mean ± SEM. (D) MET+CAT (10ug) was hydrodynamically injected into WT and CD8−/− mice and on day 42 liver tissue sections were stained for β-catenin and (E) 5 non-overlapping 100X fields were quantified for tumor area, mean tumor size and number of tumor nodules using CellProfiler. n=5, ****p<0.00001 mean ± SEM. Mann-Whitney U test was used to determine significance.
To determine whether the early differences in serum levels of luciferase were reflecting tumor control by a CD8-mediated response, luciferase levels after injection of MET+CAT oncogenes were compared in CD8-deficient and WT mice. CD8 deficiency did not affect the ability of hepatocytes to take up and express SBT vectors. When CD8−/− and WT mice were hydrodynamically transfected with G.luc+HSB2 and MET-CAT, serum luciferase levels at day 10 were indistinguishable but by days 15 and 22 they increased in CD8−/− mice and not in WT mice (Fig. 3B) confirming that serum luciferase levels accurately captured the early CD8-mediated immune response against MET+CAT transformed hepatocytes. Since the data presented above confirms that MET+CAT transduced hepatocytes are immunogenic, we next wanted to determine whether the transient CD8-mediated response confers a survival advantage. When CD8−/− and WT mice were hydrodynamically transfected with MET+CAT oncogenes CD8−/− mice had a significantly shorter survival time than WT mice (Fig. 3C). The shorter survival from MET+CAT-induced liver tumors in CD8−/− mice signified tumor load developed more quickly in these mice. However, gross morphology at time of morbidity did not reveal if the shortened survival time was due to quicker tumor development and growth or if more tumor nodules were formed. To determine if CD8+ T-cells regulated tumor size (growth) and/or the number of tumor nodules that develop, we analyzed mice with MET+CAT-initiated livers at day 42, before frank tumor is observed, by staining for the surrogate oncogene β-catenin (CAT) (Fig. 3D). Tumor nodules were quantitated for percent field (100X magnification), mean tumor nodule size in microns and the number of tumor nodules. CD8−/− mice exhibited significantly more tumor area and mean tumor nodule size compared to WT mice (Fig 3E). However, no difference was observed in the number of tumor nodules between the two stains of mice (Fig 3E), suggesting CD8+ T-cells regulate tumor growth rather than the number of malignant foci.
MET+CAT oncogene-induced liver tumor response to IL-18+IL-12 therapies can be accurately tracked by luciferase expression
MET is a broadly expressed oncogene in patients with numerous malignancies (26) and is recognized by cytotoxic T-cells (27). We reported previously that IL-18+IL-12 therapy was effective against transplantable tumors seeded in the liver (28). Since MET+CAT induced a CD8+ T-cell immune response we hypothesized that IL-18+IL-12 therapy would augment naturally occurring responses against MET+CAT induced tumors. Furthermore, we postulate that serum luciferase could be used to track in real-time the effectiveness of IL-18+IL-12 therapy against MET+CAT- induced liver tumors originated from G.luc+ hepatocytes. Mice were hydrodynamically transfected with MET+CAT, HSB2 and G.luc initiating de novo liver tumor formation in G.luc+ hepatocytes. Liver tumors were allowed to form and on days 20–24 and 28–31 mice were treated with VC or with IL-18+IL-12. Serum luciferase levels were not significantly different between the two groups of mice for the first 19 days (Fig. 4A). However, during days 31–61 post therapy, serum luciferase levels for the VC-treated group increased while the IL-18+IL-12 treatment group significantly decreased to near undetectable levels (p<0.0001). This result suggests that therapy induced a loss of MET+CAT- transformed G.luc+ hepatocytes. To confirm whether the decreased serum luciferase levels following IL-18+IL-12 therapy were due to reduced MET+CAT tumor burden, mice were euthanized at day 61. Liver weights, as well as genomic copy number of MET and CAT oncogenes measured by qPCR, were quantitated as parameters of tumor size (Fig. 4B,C). VC-treated mice showed a significantly higher liver weight and MET and CAT copy number than IL-18+IL-12-treated mice. (Fig. 4B,C). Gross morphological examination of livers reveals a broad spectrum of tumor burden from nothing observable to substantial burden in the VC group. On the other hand, no visible tumor nodules were observed on the livers from mice treated with IL-18+IL-12 (Fig 5A). Histological analysis reveals livers with frank tumor in the VC group, which had extensive regions that stained positively for β-catenin, while none of the livers in IL-18+IL-12 group stained positive for β-catenin (Fig. 5B). H&E analysis of the IL-18+IL-12 group shows normal liver structure (Fig. 5C). H&E analysis of the VC group shows disrupted liver structure with a thick trabecular pattern containing large eosin+ tumor cells displaying prominent nucleoli. Serum luciferase levels were significantly higher in mice with morphologically-distinguishable gross tumors (5 out 8 mice in VC group), which reflected liver weight (Fig. 5D). Serum luciferase levels in IL-18+IL-12-treated mice were at background levels and liver weights were normal (1g or less). Linear regression analysis comparing serum luciferase levels and liver weights for all mice were highly significant (Fig. 5E). Collectively we demonstrate for the first time that SBT-delivered, oncogene-induced liver tumors are amenable for studying the efficacy of immunotherapeutic approaches and that G.luc vector can be used to assess the effectiveness of these therapies.
Fig. 4.
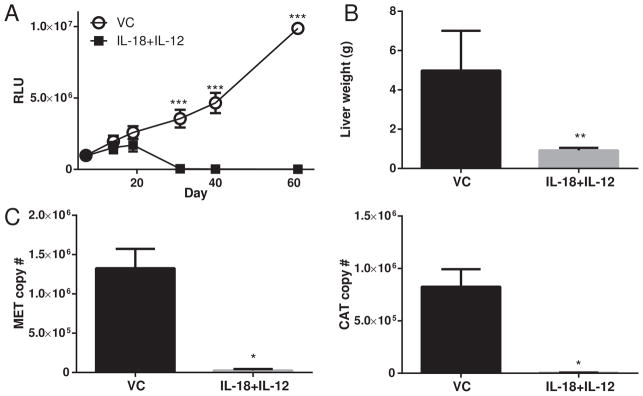
IL-18+IL-12 therapy against MET+CAT oncogene-induced liver tumors. MET (10ug)+CAT (10ug) oncogenes along with G.luc (4ug) and HSB2 (2.4ug) were hydrodynamically-transfected into C57/Bl6 mice. On days 20–24 and 28–31 mice were treated with vehicle control (VC) or IL-18+IL-12. (A) Serum Gaussia Luciferase levels vs. Day. (B) Livers were weighed on day 61. (C) DNA was isolated from livers on day 61 and MET and CAT oncogene copy number was qPCR quantitated from 100 ng of DNA. VC n=5, IL-18+12 n=10, mean ± SEM. For B and C Wilcoxon Rank Sum test was used to determine significance.
Fig. 5.

Liver morphology analysis from mice with MET+CAT initiated liver tumors treated with IL-18+IL-12. MET+CAT was hydrodynamically transfected and on days 22–24, 28–31 mice were treated with VC or IL-18+IL-12. (A) Livers were harvested on day 55 (B) and tissue sections were stained for β-catenin surrogate marker or (C) H&E. (D) Serum luciferase levels (left y-axis) and liver weight (right y-axis) was compared per mouse. (E) Linear regression analysis was performed on serum luciferase levels vs tumor weight dotted live represents 95% confidence band.
Discussion
Hydrodynamic transfection of Sleeping Beauty transposition (SBT) vectors provides an efficient and rapid system to functionally evaluate oncogenic collaboration in the formation of de novo liver tumors. Furthermore, this technique permits the assessment of inflammation- induced somatic mutations, delineating cancer “drivers” or “passengers” (7, 29). SBT delivery of oncogenes initiates tumor development and subsequent progression, which permits the study of inflammatory events driving liver tumor formation, progression and immune escape. However, the time to morbidity for tumors initiated by the SBT system can vary significantly. Thus, a tracking approach that quantifies tumor growth patterns and therapeutic interventions in living mice, without the need for costly and time-consuming imaging, or euthanasia and analysis of cohorts, would allow for efficient delineation of molecular and immune regulatory events that regulate tumor initiation and growth. We found co-delivery of a Gaussia luciferase expression vector (G.lu) in combination with oncogenes accurately tracked liver tumor growth initiated using the SBT system. Decreasing the amount of G.luc decreases the background serum luciferase levels. Reducing the ratio of oncogene to G.luc allows a higher frequency of hepatocytes to take up both oncogenes and G.luc allowing for a higher frequency of luciferase expressing transformed hepatocytes and increased sensitivity for tracking tumor growth with G.luc.
Solid tumors often grow in a complex nonlinear fashion (20, 21). Tumor proliferation and apoptosis can be influenced in various ways by different factors, such as immune recognition (30, 31) and the presence of angiogenic factors needed to form tumor vasculature (32). Determining these tumor growth inflection points could offer valuable biological insights into tumor growth as well as informing the timing and outcome of possible therapeutic interventions. We determined that serum luciferase levels closely tracked tumor-induced changes in liver size as detected and quantitated by MRI analysis. Thus, we show here for the first time that the nonlinear growth patterns of oncogene-induced liver tumors can be tracked in real-time by serum luciferase levels from hepatocytes using an integrated G.luc expression vector. In addition to tracking SBT-induced tumor growth, the stable expression of integrated G.luc should also be useful in tracking tumors initiated by carcinogens/mutagen such as DEN. Furthermore, the changes in stable luciferase expression from G.luc could be useful in monitoring liver toxicity or studies employing partial hepatectomy.
MET, which is widely expressed in numerous malignancies (26), was also demonstrated to be a tumor antigen recognized by cytotoxic T-cells (CTLs) (27). In our model we found human MET, but not human β-catenin (CAT), induced hepatocyte apoptosis that was at least partially mediated by CD8+ T-cells. This result suggested human MET in a mouse model may also induce cytotoxic T-cells. Some MET-induced TUNEL staining was still present in CD8−/− mice suggesting MET may also induce some oncogene-driven apoptosis. Interestingly delivering MET to lgh-6 (B cell deficient) mice or depleting macrophages with Clodronate increased the number of TUNEL+ hepatocytes suggesting these cells offer partial protection from MET-induced apoptosis. Examination of differences in early growth patterns revealed that CD8+ T-cells regulated the persistence of MET+CAT-transduced hepatocytes during the time span shortly after transformation. Furthermore, CD8-deficient mice succumbed to MET+CAT-induced liver tumors more rapidly than did their WT counterpart. Histological examination of MET+CAT-initiated tumors shows CD8−/− mice had larger tumor nodules, as well as a similar number of tumor nodules. Taken together, this suggests CD8 T-cells regulate the growth of MET+CAT-induced liver tumors. However, the larger tumor nodules could also be masking the total number of nodules in CD8 deficient mice as they may be coalescing in some cases.
To our knowledge this is the first report to use immunotherapy to treat SBT-induced liver tumors. We previously reported that IL-18+IL-12 was a potent immunotherapeutic regimen against implanted tumors (28). Because human CTLs have been shown to respond to MET (27) and we found mouse CD8+ T-cells responded to human MET, we examined the immunotherapeutic potential of treating mice bearing MET+CAT tumors with IL-18+IL-12. We found treating mice with IL-18+IL-12 ablated MET+CAT-initiated liver tumors and restored liver morphology that was accurately tracked by serum luciferase quantitation, suggesting that this technology can be used to monitor preclinical immunotherapeutic approaches against liver cancer. Immunohistochemical analysis of β-catenin staining demonstrated that IL-18+IL-12 also cleared all detectable microscopic tumors, in addition to the absence of grossly detectable visible tumors as described above. This data suggests IL-18+IL-12 therapy may elicit an anti-tumor response in humans mediated by MET responding CTLs.
In summary, we have shown that hydrodynamic delivery of G.luc vector can be used as a simple cost-effective, blood-based approach for tracking the progression of pathologically-distinct liver tumors, evaluation of immunologic responses and efficacy of therapeutic interventions in individual mice. This approach can be used to track nonlinear tumor progression in real-time and identify potentially important biological regulatory points at which therapeutic intervention may hold greater potential for controlling liver tumor burden. Furthermore, IL-18+IL-12 is an effective therapy against antigenic MET-induced liver tumors.
Supplementary Material
Acknowledgments
Financial support: This work was supported by the Intramural Research Program of the National Cancer Institute, National Institutes of Health (NCI/NIH).
We thank Dr. Giorgio Trinchieri for his critical editing of the manuscript and Megan Karwan for her assistance with animal experiments. This work was supported by the Intramural Research Program of the National Cancer Institute, National Institutes of Health (NCI/NIH).
Abbreviations
- SBT
Sleeping Beauty Transposition
- G.luc
Gaussia Luciferase expression SBT vector
- HCC
hepatocellular carcinoma
- HCA
hepatocellular adenoma
- G.luc+ hepatocytes
G.luc vector integrated in hepatocytes
- HSB2
Sleeping Beauty transposase
- MET
human c-Met
- PT3
empty SBT vector
- CAT
Δ90Nβ-catenin
- AKT
Myr-AKT
- ConA
concanavalin A
- WT
wild type
Footnotes
Author’s contributions
Study conception: Subleski and Stauffer
Acquisition of data: Subleski, Scarzello, Kronfili, Saleh, Back, Jiang
Analysis and interpretation of data: Subleski, Scarzello, Alvord, Kronfili, Saleh Weiss and Wiltrout
Drafting of manuscript: Subleski, Weiss and Wiltrout
Critical revision: Subleski, Scarzello, Weiss and Wiltrout
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organization imply endorsement by the U.S. government.
Conflict of interest: The authors who have taken part in this study declared that they do not have anything to disclose regarding funding or conflict of interest with respect to this manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Totoki Y, Tatsuno K, Yamamoto S, Arai Y, Hosoda F, Ishikawa S, et al. High-resolution characterization of a hepatocellular carcinoma genome. Nat Genet. 2011 May;43(5):464–469. doi: 10.1038/ng.804. [DOI] [PubMed] [Google Scholar]
- 2.Thorgeirsson SS, Grisham JW. Molecular pathogenesis of human hepatocellular carcinoma. Nat Genet. 2002 Aug;31(4):339–346. doi: 10.1038/ng0802-339. [DOI] [PubMed] [Google Scholar]
- 3.Thamavit W, Bhamarapravati N, Sahaphong S, Vajrasthira S, Angsubhakorn S. Effects of dimethylnitrosamine on induction of cholangiocarcinoma in Opisthorchis viverrini-infected Syrian golden hamsters. Cancer Res. 1978 Dec;38(12):4634–4639. [PubMed] [Google Scholar]
- 4.Levrero M. Viral hepatitis and liver cancer: the case of hepatitis C. Oncogene. 2006 Jun 26;25(27):3834–3847. doi: 10.1038/sj.onc.1209562. [DOI] [PubMed] [Google Scholar]
- 5.Khandekar MJ, Cohen P, Spiegelman BM. Molecular mechanisms of cancer development in obesity. Nat Rev Cancer. 2011 Dec;11(12):886–895. doi: 10.1038/nrc3174. [DOI] [PubMed] [Google Scholar]
- 6.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010 Sep;60(5):277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 7.Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012 Jun;44(6):694–698. doi: 10.1038/ng.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007 Jun;132(7):2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 9.Bhamarapravati N, Virranuvatti V. Liver diseases in Thailand. An analysis of liver biopsies. Am J Gastroenterol. 1966 Apr;45(4):267–275. [PubMed] [Google Scholar]
- 10.Ng M, Fleming T, Robinson M, Thomson B, Graetz N, Margono C, et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2014 May 28; doi: 10.1016/S0140-6736(14)60460-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mohd HK, Groeger J, Flaxman AD, Wiersma ST. Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology. 2013 Apr;57(4):1333–1342. doi: 10.1002/hep.26141. [DOI] [PubMed] [Google Scholar]
- 12.Bell JB, Podetz-Pedersen KM, Aronovich EL, Belur LR, McIvor RS, Hackett PB. Preferential delivery of the Sleeping Beauty transposon system to livers of mice by hydrodynamic injection. Nat Protoc. 2007;2(12):3153–3165. doi: 10.1038/nprot.2007.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen X, Calvisi DF. Hydrodynamic transfection for generation of novel mouse models for liver cancer research. Am J Pathol. 2014 Apr;184(4):912–923. doi: 10.1016/j.ajpath.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stauffer JK, Scarzello AJ, Andersen JB, De Kluyver RL, Back TC, Weiss JM, et al. Coactivation of AKT and beta-catenin in mice rapidly induces formation of lipogenic liver tumors. Cancer Res. 2011 Apr 1;71(7):2718–2727. doi: 10.1158/0008-5472.CAN-10-2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Subleski JJ, Hall VL, Wolfe TB, Scarzello AJ, Weiss JM, Chan T, et al. TCR-dependent and -independent activation underlie liver-specific regulation of NKT cells. J Immunol. 2011 Jan 15;186(2):838–847. doi: 10.4049/jimmunol.1001735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crowder MJ, Hand DJ. Analysis of repeated measures. 1 1990. [Google Scholar]
- 17.Pinheiro JC, Bates DM. Mixed-effects models in S and S-PLUS. 2000. [Google Scholar]
- 18.Bonini C, Ferrari G, Verzeletti S, Servida P, Zappone E, Ruggieri L, et al. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science. 1997 Jun 13;276(5319):1719–1724. doi: 10.1126/science.276.5319.1719. [DOI] [PubMed] [Google Scholar]
- 19.Riddell SR, Elliott M, Lewinsohn DA, Gilbert MJ, Wilson L, Manley SA, et al. T-cell mediated rejection of gene-modified HIV-specific cytotoxic T lymphocytes in HIV-infected patients. Nat Med. 1996 Feb;2(2):216–223. doi: 10.1038/nm0296-216. [DOI] [PubMed] [Google Scholar]
- 20.Shimizu S, Shirato H, Xo B, Kagei K, Nishioka T, Hashimoto S, et al. Three-dimensional movement of a liver tumor detected by high-speed magnetic resonance imaging. Radiother Oncol. 1999 Mar;50(3):367–370. doi: 10.1016/s0167-8140(98)00140-6. [DOI] [PubMed] [Google Scholar]
- 21.Wise SM, Lowengrub JS, Frieboes HB, Cristini V. Three-dimensional multispecies nonlinear tumor growth--I Model and numerical method. J Theor Biol. 2008 Aug 7;253(3):524–543. doi: 10.1016/j.jtbi.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011 Nov 24;479(7374):547–551. doi: 10.1038/nature10599. [DOI] [PubMed] [Google Scholar]
- 23.Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJ, Suresh M, Altman JD, et al. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med. 1998 Dec 21;188(12):2205–2213. doi: 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013 Dec;3(12):1355–1363. doi: 10.1158/2159-8290.CD-13-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fourcade J, Sun Z, Pagliano O, Guillaume P, Luescher IF, Sander C, et al. CD8(+) T cells specific for tumor antigens can be rendered dysfunctional by the tumor microenvironment through upregulation of the inhibitory receptors BTLA and PD-1. Cancer Res. 2012 Feb 15;72(4):887–896. doi: 10.1158/0008-5472.CAN-11-2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sierra JR, Tsao MS. c-MET as a potential therapeutic target and biomarker in cancer. Ther Adv Med Oncol. 2011 Nov;3(1 Suppl):S21–S35. doi: 10.1177/1758834011422557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schag K, Schmidt SM, Muller MR, Weinschenk T, Appel S, Weck MM, et al. Identification of C-met oncogene as a broadly expressed tumor-associated antigen recognized by cytotoxic T-lymphocytes. Clin Cancer Res. 2004 Jun 1;10(11):3658–3666. doi: 10.1158/1078-0432.CCR-03-0640. [DOI] [PubMed] [Google Scholar]
- 28.Subleski JJ, Hall VL, Back TC, Ortaldo JR, Wiltrout RH. Enhanced antitumor response by divergent modulation of natural killer and natural killer T cells in the liver. Cancer Res. 2006 Nov 15;66(22):11005–11012. doi: 10.1158/0008-5472.CAN-06-0811. [DOI] [PubMed] [Google Scholar]
- 29.Reimand J, Bader GD. Systematic analysis of somatic mutations in phosphorylation signaling predicts novel cancer drivers. Mol Syst Biol. 2013;9:637. doi: 10.1038/msb.2012.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004 Aug;21(2):137–148. doi: 10.1016/j.immuni.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 31.Pardoll D. Does the immune system see tumors as foreign or self? Annu Rev Immunol. 2003;21:807–839. doi: 10.1146/annurev.immunol.21.120601.141135. [DOI] [PubMed] [Google Scholar]
- 32.Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature. 2005 Dec 15;438(7070):967–974. doi: 10.1038/nature04483. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.