

Abstract
Production of ribosomes relies on more than 200 accessory factors to ensure the proper sequence of steps and faultless assembly of ribonucleoprotein machinery. Among trans-acting factors are numerous enzymes, including ribonucleases responsible for processing the large rRNA precursor synthesized by RNA polymerase I that encompasses sequences corresponding to mature 18S, 5.8S, and 25/28S rRNA. In humans, the identity of most enzymes responsible for individual processing steps, including endoribonucleases that cleave pre-rRNA at specific sites within regions flanking and separating mature rRNA, remains largely unknown. Here, we investigated the role of hUTP24 in rRNA maturation in human cells. hUTP24 is a human homolog of the Saccharomyces cerevisiae putative PIN domain-containing endoribonuclease Utp24 (yUtp24), which was suggested to participate in the U3 snoRNA-dependent processing of yeast pre-rRNA at sites A0, A1, and A2. We demonstrate that hUTP24 interacts to some extent with proteins homologous to the components of the yeast small subunit (SSU) processome. Moreover, mutation in the putative catalytic site of hUTP24 results in slowed growth of cells and reduced metabolic activity. These effects are associated with a defect in biogenesis of the 40S ribosomal subunit, which results from decreased amounts of 18S rRNA as a consequence of inaccurate pre-rRNA processing at the 5′-end of the 18S rRNA segment (site A1). Interestingly, and in contrast to yeast, site A0 located upstream of A1 is efficiently processed upon UTP24 dysfunction. Finally, hUTP24 inactivation leads to aberrant processing of 18S rRNA 2 nucleotides downstream of the normal A1 cleavage site.
Keywords: Endoribonuclease, rRNA, rRNA processing, SSU processome, UTP24, U3 snoRNA
Introduction
Ribosome biogenesis is one of the most metabolically-expensive multi-step processes occurring in eukaryotic cells,1 which begins in the nucleolus – a specific sub-nuclear compartment formed around tandemly repeated rDNA units. During the initial step of ribosome biogenesis, RNA polymerase I transcribes rDNA into a large polycistronic precursor RNA molecule: 47S or 35S in human and yeast cells, respectively.2-4 The pre-rRNA subsequently undergoes a series of co-transcriptional and posttranscriptional processing events involving cleavage and maturation through a concerted action of endo- and exoribonucleases as well as chemical modifications.5 In addition, numerous trans-acting proteins and small nucleolar ribonucleoprotein (snoRNP) particles are critical for these modifications.4,6 They also ensure proper rRNA folding, assembly of ribosomal proteins and individual pre-ribosome subunits, and export of the latter across the nuclear membrane.
Ribonucleolytic processing of the pre-rRNA gives rise to the mature 18S, 5.8S, and 28S (25S in yeast) rRNA. The 5S rRNA, which is incorporated into the 60S ribosomal subunit, is synthesized separately by RNA polymerase III. rRNA maturation occurs through elimination of external transcribed sequences (5′-ETS; 3′-ETS) and internal transcribed spacers (ITS1; ITS2), that flank and separate the mature rRNA species, respectively. The order of endoribonucleolytic cleavage events and exoribonucleolytic trimming steps has been studied most thoroughly in S. cerevisiae.2,4 Much less is currently known about rRNA maturation in human cells. Furthermore, several recent studies indicate that pre-rRNA processing steps in yeast cannot always be extrapolated to processes in higher eukaryotes. This is mainly due to the fact that the ETS and ITS regions, which are where the majority of processing events occur, are significantly extended when compared to yeast.7,8 Thus, there are additional processing sites within human 47S pre-rRNA.9-11 Moreover, in some cases the functions of homologous proteins involved in ribosome synthesis in both species are not equivalent.12-14 Furthermore, alternative pre-rRNA processing pathways are utilized in parallel in both model organisms.7,8,15 In humans, the usage of these pathways may be highly dependent on the cell type and physiological state. Finally, there are processing events in humans, but not yeast, that occur either through endonucleolytic cleavage or by exoribonucleolytic maturation.11,16
Processing of human 47S pre-rRNA begins with primary cleavage at site A′ (also known as 01) in the 5′-ETS, which encompasses endoribonucleolytic cleavage sites at 2 adjacent regions between nucleotides 414 and 422 (Fig. 1A).9 This U3 snoRNA-dependent processing site is conserved among higher eukaryotes, but not in yeast.9,17,18 The second cleavage event is thought to take place at site 02 in the vicinity of the 28S/3′-ETS boundary (Fig. 1A). However, unlike in yeast, where pre-rRNA is known to be processed downstream of the mature 25S rRNA 3′-end by the Rnt1 endonuclease,19,20 the exact location of human site 02 has not been determined and it is not known whether processing at sites A′ and 02 occurs simultaneously or sequentially.7 These 2 events, carried out by as yet unknown enzymes, lead to the formation of the 45S pre-rRNA (Fig. 1A), that is further processed through at least 2 alternative pathways.
Figure 1.

Schematic representation of the human pre-rRNA processing pathways. RNA polymerase I transcribes rDNA unit into a large 47S pre-rRNA that encompasses segments corresponding to mature 18S, 5.8S, and 28S rRNA, separated by internal transcribed spacers (ITS1, ITS2) and surrounded by external transcribed sequences (5′-ETS, 3′-ETS). Positions of the processing sites within the 47S precursor are indicated with vertical thin lines together with the names. Nucleotide numbering is according to GenBank (accession number: U13369.1). Arrows indicate known or predicted sites of endoribonucleolytic processing. (A) In the major pathway utilized in HeLa cells, the 45S rRNA arising after primary cleavages at site A' in the 5′-ETS and at site 02 in the vicinity of mature 28S rRNA 3′-end is processed at site 2 within ITS1, giving rise to 30S and 32S rRNA. The 30S is further processed to 18S through a series of endonucleolytic cleavages at sites A0 and A1 at the 5′-end, followed by maturation of the 3′-end involving both 3′–5′ exoribonucleolytic trimming and endoribonucleolytic cleavage. In turn, the 32S is endonucleolytically split into 12S rRNA, which undergoes mainly endonucleolytic processing that generates mature 5.8S rRNA and a 28S' species, which is converted into mature 28S by 5′–3′ exoribonucleolytic trimming. (B) In a parallel pathway, the 45S is first processed at sites A0 and A1, thus generating 43S and 41S, respectively; only then is the 41S rRNA processed at site 2, which produces 21S and 32S precursors. (C) Another branch is based on the endonucleolytic processing of 41S at site E within ITS1 – this event generates an 18S-E intermediate and 36S rRNA. The latter is trimmed by 5′–3′ exoribonuclease to 36S-C, which is eventually cleaved at site 2 to produce the 32S precursor.
In the major processing pathway in human HeLa cell line, the 45S is first cleaved at site 2, which is considered by some to be equivalent to the yeast site A2 cleavage. This event separates RNA destined for the 40S and 60S ribosomal subunits and generates 30S pre-rRNA (containing the 18S rRNA module) and 32S pre-RNA (encompassing the 5.8S and 28S rRNA segments) (Fig. 1A).7,21 Others have argued that processing at site 2 in human cells is independent of 5′-ETS processing, which makes it more similar to the yeast site A3.12 On the other hand, processing at site 2 in human pre-rRNA is not affected by depletion of RNase MRP subunits, while RNase MRP is responsible for A3 cleavage in yeast.16,22,23
The 30S pre-rRNA undergoes further endoribonucleolytic processing at 2 sites within the 5′-ETS, which are roughly equivalent to those in the yeast 35S precursor: A0 (at nucleotide G1643) and A1 (also called site 1), located immediately upstream of the mature 18S 5′-end, which generates 26S and 21S' pre-rRNA, respectively (Fig. 1A).10,13,24,25 Based on in vitro studies, it has been suggested that the 21S pre-rRNA is then trimmed by a 5′–3′ exoribonuclease to yield the 21S′ precursor, the 5′-end of which corresponds to the mature 18S 5′ terminus (nucleotide 3657 with respect to 47S numbering) (Fig. 1A).25,26 The 21S is then progressively shortened to a heterogeneous 21S-C fraction by an as yet unknown 3′–5′ exoribonuclease (Fig. 1A).12 This processing step appears to be unique for higher eukaryotes. The ultimate nuclear processing event involves trimming of 21S-C to the 18S-E pre-rRNA, which can be achieved by 3′–5′ digestion carried out by RNA exosome-associated exoribonuclease RRP6 and/or through direct endoribonucleolytic cleavage at site E, located 78–81 nucleotides downstream of the mature 18S 3′-end (Fig. 1A).11,16 Eventually, 18S-E (an equivalent of the yeast 20S) is exported to the cytoplasm, where it undergoes exoribonucleolytic trimming and ultimate endoribonucleolytic processing at site 3 (Fig. 1A), which is likely catalyzed by an ortholog of the yeast Nob1, cleaving S. cerevisiae 20S pre-rRNA at site D.10,11,27-31 It should be noted that some perceive human site E, rather than site 2, as an equivalent of the yeast site A2. This is due to the fact that the former is more tightly coupled to cleavages at sites A0 and A1 within the 5′-ETS and is dependent on U3 snoRNA and proteins homologous to components of the yeast small subunit (SSU) processome, similar to the relationships between sites A0, A1, and A2 in the yeast pre-rRNA.12,32-34
The 32S rRNA – a second product of 45S processing at site 2 – undergoes trimming by a 5′–3′ exoribonuclease and cleavage at site 3′ within the ITS2. This generates 12S rRNA (the longest known precursor of the mature 5.8S) and 28S' rRNA (Fig. 1A). The latter is most likely processed to mature 28S rRNA by 5′–3′ exoribonucleolytic digestion, similar to yeast, where the ultimate step of 25S rRNA biogenesis is dependent on Rat1 (also known as Xrn2) (Fig. 1A).35,36 The 12S rRNA is further cleaved at site 4a to give rise to 7S rRNA (Fig. 1A). It cannot be excluded that other processing sites (located between 4a and 3′) exist within ITS2 and that the products of cleavage at these sites are matured by 3′–5′ exoribonucleolytic digestion, thus offering an alternative pathway of 7S rRNA generation.37 Eventually, 7S rRNA undergoes either exo- and/or endoribonucleolytic processing, leading to the generation of the mature 5.8S rRNA 3′-end (Fig. 1A).37
In the less frequently utilized processing pathway, which resembles the major pathway in S. cerevisiae where cleavage at site A1 must precede processing at site A2,38 the 45S pre-rRNA that arises after primary cleavage at site A' may be sequentially cleaved at sites A0 and A1 before processing at site 2. These events generate 43S and 41S' pre-rRNA, respectively (Fig. 1B).10 The latter is trimmed by 5′–3′ exoribonuclease to 41S, which in turn is further cleaved at site 2 (Fig. 1B). Eventually, 21S and 32S pre-rRNA are processed to mature 18S, 5.8S, and 28S rRNA in a process similar to the major pathway described above. In yet another concurrent path, 41S is subjected to endoribonucleolytic cleavage at site E to form 18S-E and 36S pre-rRNA (Fig. 1C). The 36S is subsequently progressively shortened by a 5′–3′ exoribonuclease to a heterogeneous 36S-C fraction (Fig. 1C), the 3′-ends of which overlap with the 5′-ends of the 21S-C precursors generated in the major processing pathway.11,16 Finally, cleavage of 36S-C at site 2 gives rise to 32S pre-rRNA (Fig. 1C), which is subsequently processed as outlined above.
Although numerous proteins participating in eukaryotic ribosome biogenesis have been already well-characterized, there are still many factors that remain to be identified. It was previously suggested that a protein with a PIN nuclease domain, namely yUtp24 (also called Fcf1), might be involved in the endoribonucleolytic cleavage of yeast pre-rRNA at sites A0 in the 5′-ETS and A1, thus forming a mature 18S 5′-end, and at site A2 (which was later shown to be processed by a controversial endoribonucleolytic activity of Rcl1 protein, similar to RNA 3′ cyclases).39-42 Nevertheless, yUtp24 has not been shown to have enzymatic activity in vitro, and in vivo analyses, such as primer extension-based mapping of the processing sites affected by yUtp24 mutation, have not been performed to date.
In this study, we analyzed the role of yUtp24 ortholog – hUTP24 – in pre-rRNA processing in human cells. We demonstrate that hUTP24 is involved mainly in processing at site A1, which defines the mature 18S rRNA 5′-end, but – in contrast to yeast – its dysfunction induces aberrant cleavage at a site located 2 nucleotides downstream, which suggests the existence of an alternative, albeit imperfect, processing mechanism. Moreover, since a defect in the putative hUTP24 endoribonucleolytic activity does not impact processing at site A0 in human pre-rRNA, our data show that UTP24 orthologs from 2 evolutionarily distant model eukaryotic organisms play closely related, but not entirely identical, roles in pre-rRNA processing.
Results
Exogenously expressed hUTP24 cannot functionally replace endogenous Utp24 in S. cerevisiae
The 198 amino acid hUTP24 protein contains a C-terminal PIN domain that is potentially associated with ribonucleolytic activity, similar to its yeast counterpart (Fig. 2A). Amino acid sequences of UTP24 proteins from both species are 61% identical and 75% similar to each other. In particular, a conserved tetrad of negatively charged putative catalytic residues (D-E-D-D) is present in the corresponding positions of both proteins (residues 68, 105, 138 and 157 as well as 72, 109, 142 and 161 in yUtp24 and hUTP24, respectively) (Fig. 2A, B), indicating that they may coordinate a divalent cation required for the ribonucleolytic activity.29,43-47
Figure 2.
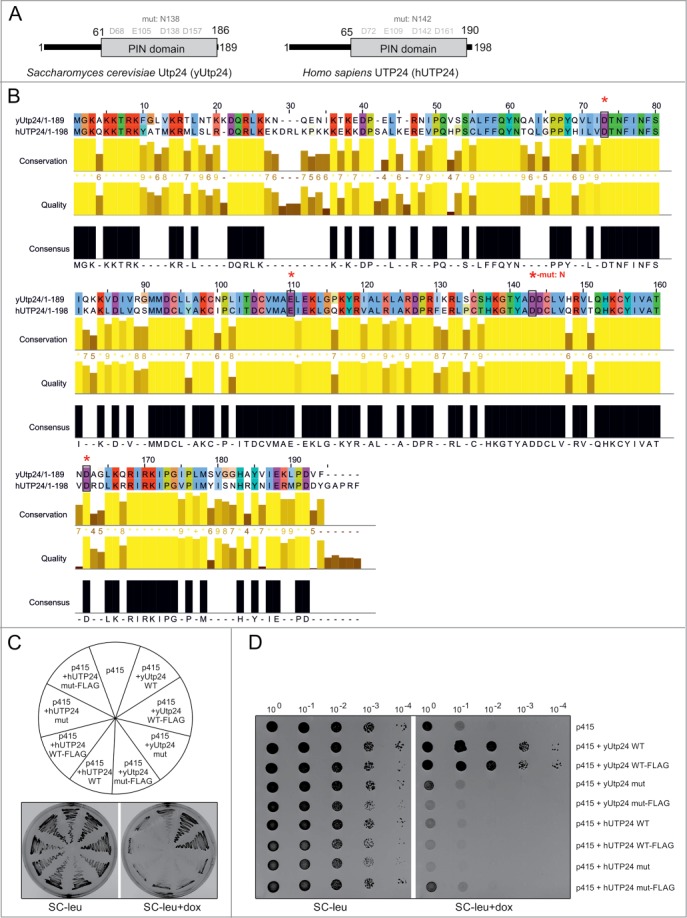
hUTP24 does not complement yUtp24 depletion in S. cerevisiae. (A) Schematic view of yeast and human UTP24 proteins. Both proteins are less than 200 amino acids in length, with an unstructured region at the N-terminus. This is followed by the PIN domain, which is potentially associated with ribonuclease activity. Conserved D-E-D-D tetrads of acidic residues in the putative active site of the PIN domain are highlighted in gray based on the detailed alignment in (B). Positions of D to N substitutions, introduced into yUtp24 (amino acid 138) and hUTP24 (amino acid 142), to generate mutant variants of the proteins are indicated above. (B) Detailed amino acid alignment of yUtp24 and hUTP24. Evolutionarily conserved, negatively charged residues are marked with black rectangles and red asterisks. The position of a D to N amino acid change in the mutant versions of either protein is indicated. (C and D) hUTP24 is not able to replace yUtp24 function in yeast. An S. cerevisiae strain in which endogenous yUTP24 had been placed under the control of a doxycycline-repressible promoter was transformed with an empty p415 vector or its derivatives encoding WT and mut variants of yUtp24 or hUTP24 that possess or lack a FLAG-tag at the C-terminus. In (C), growth of the transformants was analyzed by streaking approximately equal amounts onto media with or without doxycycline. In (D), 10-fold serial dilutions of the liquid cultures of the respective transformed strains were analyzed for growth in the absence or presence of doxycycline. In both cases, plates were incubated for 48 hours at 30°C.
To verify whether human UTP24 can fulfill functions of its homolog in S. cerevisiae, we performed complementation assays in the context of yUtp24 depletion. A yeast strain in which endogenous UTP24 was under the control of a tetracycline-repressible promoter was transformed with a centromeric vector encoding either yUtp24 or hUTP24. Due to the lack of hUTP24-specific antibodies for the initial phase of the study, we prepared plasmid constructs encoding untagged or C-terminal FLAG-tagged UTP24 proteins from both species to confirm expression from the exogenously introduced vectors. In addition, we performed transformations with 2 variants of each construct: WT and mut, which contained an aspartate-to-asparagine substitution at positions 138/142 (D138N/D142N) of yUtp24/hUTP24, to examine the functional role of the putative catalytic site. The analysis revealed that WT yUtp24 was able to complement depletion of endogenous yUtp24 (Fig. 2C, D). In agreement with previous studies, the yUtp24 mut did not sustain growth, indicating that the intact PIN domain is indispensable for protein function (Fig. 2C, D).39 Most importantly, no complementation was observed upon exogenous expression of hUTP24 (Fig. 2C, D), although the cloned human ORFs were efficiently expressed in all relevant yeast strains (Fig. S1A) and the proteins were produced (Fig. S1B). This indicated that the human protein cannot fully replace the molecular function(s) of yUtp24 in S. cerevisiae.
Mutation in the putative catalytic site of yUtp24 results in decreased levels of 40S ribosomal subunit and mature 18S rRNA due to inefficient processing at sites A0, A1, and A2
Previous studies indicated that yUtp24 is required for early cleavage events taking place during 35S pre-rRNA processing.39,40 Northern blot analyses and pulse-chase RNA labeling experiments revealed that its presence in the cell seemed to be indispensable for cleavage at site A0 in the 5′-ETS, while its putative ribonucleolytic activity appeared to be necessary for processing at sites A1 and A2 (see Fig. 3A for positioning of the processing sites in yeast pre-rRNA).
Figure 3.
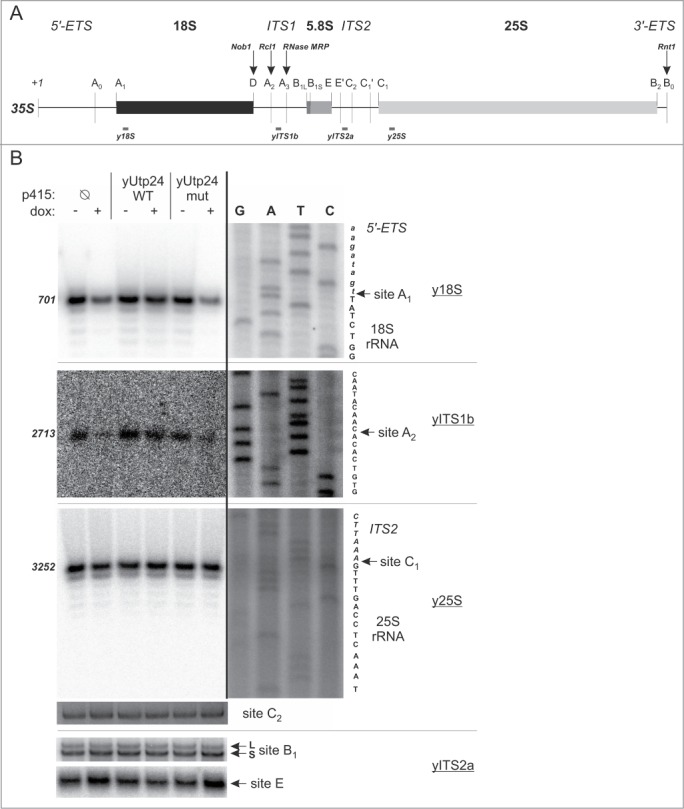
Mutation in the putative active site of yUtp24 leads to inefficient processing of pre-rRNA at sites A1 and A2. (A) A scheme of the S. cerevisiae 35S pre-rRNA. Positions of the processing sites within the 35S precursor are indicated with vertical thin lines together with the names. Nucleotide numbering is based on the GenBank 35S pre-rRNA sequence (accession number: BK006945.2). Arrows indicate sites of endoribonucleolytic processing carried out by known enzymes – their names are provided above with bolded italics. Gray bars below the primary 35S transcript show positions of oligonucleotides used for primer extension in this study. (B) Identification of processing defects by primer extension. The S. cerevisiae strain with endogenous yUTP24 under the control of a doxycycline-regulated promoter was transformed with an empty p415 vector (Ø) or its derivatives encoding WT and mut variants of yUtp24 or hUTP24. Total RNA was isolated from transformants grown either in the absence (“dox: −”) or in the presence (“dox: +”) of doxycycline and subjected to primer extension analysis using oligonucleotides (marked by underline) hybridizing upstream selected processing sites in 35S pre-rRNA (indicated by arrows). Numbers on the left indicate nucleotide positions in 35S corresponding to reverse transcription stops. The same primers were used in parallel with DNA templates comprising appropriate rDNA fragments to generate dideoxynucleotide sequencing ladders, which were co-electrophoresed with primer extension products. Retrieved sequences (in reverse complement) are presented next to the sequencing results. Note that various exposures of gel fragments are presented to optimally visualize the data obtained.
Corroborating observation made by others,40 yUtp24 dysfunction resulted in reduction of the mature 40S subunit and a marked decrease in the amounts of polysomes, with a concomitant increase in the pool of free 60S subunit and monosomes (Fig. S2).
In order to verify whether the growth inhibition and 40S subunit biogenesis defect associated with yUTP24 mutation in our strains was due to the misprocessing of 18S rRNA, we conducted northern blot analyses with probes targeting various regions of the 35S pre-rRNA (see Fig. S3A for location of the probes). In line with previous reports,39,40 we noted that yUtp24 dysfunction (irrespective of whether yUtp24 levels were low or the putative enzymatic activity of the protein was abolished) resulted in a significant decrease of mature 18S rRNA. This was accompanied by a marked reduction of 20S pre-rRNA (Fig. S3B, C), a stable 18S rRNA precursor extending from site A1 to A2. Interestingly, unlike in one of the previous studies,39 both yUtp24 depletion and exogenous expression of the mutant protein variant led to the accumulation of 23S pre-rRNA (Fig. S3B, C), indicating that the yUtp24 activity may be important for U3 snoRNA-dependent processing at sites A0, A1, and A2.
To unequivocally verify whether processing at U3 snoRNA-dependent sites in the yeast pre-rRNA is affected by mutation in the putative catalytic center of yUtp24, we examined these sites by primer extension. We found that in the case of sites A1 and A2 signals corresponding to reverse transcription stops at the expected positions were significantly less intense when endogenous yUtp24 was depleted, unless yUtp24 WT was exogenously produced (Fig. 3B). We attempted to perform similar experiments to directly demonstrate the inefficient processing at site A0, but primer extensions with 2 different primers used previously by other authors19,32 did not produce sufficiently strong signal at the positions predicted for this cleavage site (Fig. S4A, B) to enable drawing unequivocal conclusions. Interestingly, however, we noted an increase of the intensity of the band corresponding to the nucleotide +1 of the primary 35S transcript in the strain producing yUtp24 mut with both primers (Fig. S4A, B). This, together with accumulation of a 23S and 35S species (instead of 22S and 33S, respectively), observed in our RNA gel blot analyses (Fig. S3B, C) clearly indicated that cleavage at this site was also compromised upon yUtp24 dysfunction, irrespective of whether due to the decreased level of endogenous protein or when the mutant yUtp24 variant was exogenously produced. In contrast, processing events at sites C1, C2, E, and B1, which are required for maturation of 5.8S and 25S rRNA, did not appear to be affected (Fig. 3B).
These results collectively demonstrate that although processing at U3 snoRNA-dependent sites upon yUtp24 depletion or when yUtp24 contains a mutation in the putative catalytic center takes place at the previously mapped sites in yeast pre-rRNA, it is significantly less efficient than when in the presence of wild-type yUtp24.
UTP24 interacts with the components of the SSU processome in both yeast and humans
yUtp24 was reported to associate with Faf1 – a 40S ribosomal subunit maturation factor, which, based on 2-hybrid assays, had been postulated to interact with some components of SSU processome.48 A direct identification of Faf1-interacting proteins by tandem affinity purification followed by mass spectrometry revealed that yUtp24 is among numerous yeast SSU processome constituents that co-purify with the bait protein.40
To verify whether yUtp24 is a genuine component of the SSU processome in S. cerevisiae, we performed reciprocal purifications of TAP-tagged yUtp24 WT and mut (to ensure that the catalytic mutation does not affect incorporation of the protein of interest into the assembly) from yeast strains producing fusion proteins under the control of the endogenous promoter, and then analyzed proteins present in the eluates using high resolution mass spectrometry and MaxQuant software for quantification. The results were compared to those obtained from the non-transformed parental yeast strain, which was subjected to the same purification procedure in parallel.
We identified more than one thousand proteins in all experiments. Of these, we focused on 54 known or predicted SSU processome components,49,50 42 proteins classified as pre-rRNA processing or ribosome biogenesis factors and 35 detected ribosomal proteins of the 40S subunit, some of which were previously demonstrated to play an important role in rRNA maturation and synthesis of ribosomes.33,51 We detected 32 SSU processome components co-purifying with yUtp24, 19 of which appeared to interact specifically with both wild-type and mutant bait proteins (Fig. 4). These proteins represent various classes of SSU processome subunits, including those classified by Lim et al.49 and Phipps et al.50 as members of different subcomplexes (mainly UtpA and UtpB, but also U3 snoRNP, Mpp10, and UtpC). In addition, 21 pre-rRNA processing factors and 30 small ribosomal subunit proteins co-purified with yUtp24 (9 and 3 of them, respectively, with high specificity for both wild-type and mutant bait) (Fig. 4). We obtained similar results when the protein extracts were treated with RNase prior to purification, suggesting that the observed interactions were direct (data not shown). These results demonstrate that yUtp24 stably interacts with components of the SSU processome as well as other proteins involved in ribosome biogenesis in yeast.
Figure 4.
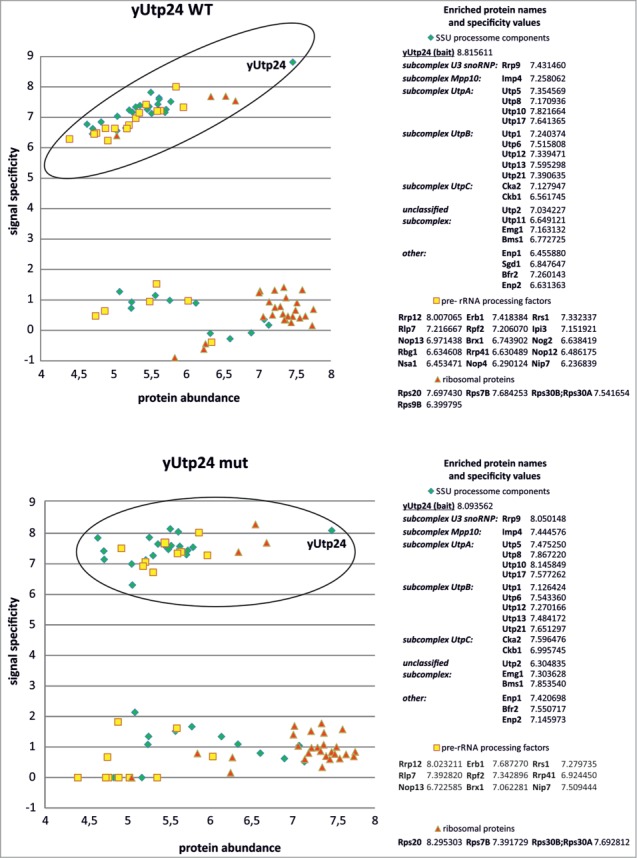
yUtp24 protein interacts with SSU processome components as well as with other factors involved in yeast ribosome biogenesis. Wild-type (top) and mutant yUtp24 (bottom) were purified from the yeast strains as C-terminal fusions with a TAP-tag. Proteins co-purifying with the bait were analyzed by mass spectrometry and the results were compared to the parallel purification, carried out using the unmodified parental strain. To determine whether a given protein specifically co-purified with yUtp24, we compared its intensity to the intensity of that protein in the control sample. Signal specificity (y-axis) was defined as the log10 of the ratio of protein signal intensity measured in the bait purification to background level (which is the protein signal intensity in the negative control purification; background level was arbitrarily set to 1 for proteins not detected in the negative control). Protein abundance (x-axis) was defined as the log10 of the ratio of protein signal intensity divided by its molecular weight in kDa. This parameter was implemented to eliminate differences due to the size of proteins. Points located within the ellipses correspond to high values of both protein abundance and specificity, indicating proteins enriched in bait purification (compared to the control sample) and thus suggest interaction. These hits (subdivided into 3 categories: SSU processome components, pre-rRNA processing factors, and ribosomal proteins) are listed next to the graphs and the calculated specificity values are indicated. In the case of SSU processome subunits, identified proteins belonging to specific subcomplexes within the assembly are indicated, according to ref. 50. The remaining points represent proteins that are present in similar amounts in both bait purification and the control sample.
We next investigated whether hUTP24 associates with other factors involved in 40S subunit maturation in human cells. To this end, we established stable, inducible HEK293 Flp-In T-REx cell lines producing either wild-type or mutant hUTP24 fused to eGFP at the C-terminus. We first confirmed that both variants of the protein display nucleolar localization (Fig. S5A). In addition, we demonstrated that the intracellular localization of hUTP24 was independent of the positioning of the epitope (Fig. S5B), not restricted to the eGFP-tag (Fig. S5C), and not unique for HEK293 cells (Fig. S5D). Following those preliminary experiments, we carried out purification of the eGFP-tagged WT and mut variants from HEK293 Flp-In T-REx derived cell lines in the presence of RNase A and subjected the eluates to mass spectrometry analysis. Parallel purification performed using protein extract from non-transfected HEK293 Flp-In T-REx cells served as a negative control. Using this approach, we identified more than 1500 proteins in our experimental samples and selected 29 homologs of the yeast (SSU) processome components, 13 known or putative pre-rRNA processing proteins and 5 proteins of the small ribosomal subunit for further analysis. We found that 21 SSU processome components co-purified with hUTP24 from both cell lines, of which 8 and 7 were specifically enriched compared to the background in WT and mut samples, respectively (Fig. 5). Four of these proteins (CIRH1A, PWP2, PDCD11, and NAT10) were common for hUTP24 WT and mut (Fig. 5). We did not identify any homologs of proteins belonging to U3 snoRNP and Mpp10 subcomplexes in either purification. Moreover, we identified 8 and 6 pre-rRNA processing factors co-purifying with hUTP24 WT and mut, respectively, of which 5 and 3 were significantly enriched compared to the control sample, respectively (Fig. 5). The 5 ribosomal proteins (RPS2, RPS3, RPS12, RPS13, and RPS15) were not particularly enriched (Fig. 5), most likely due to the high abundance of these proteins in the background.
Figure 5.
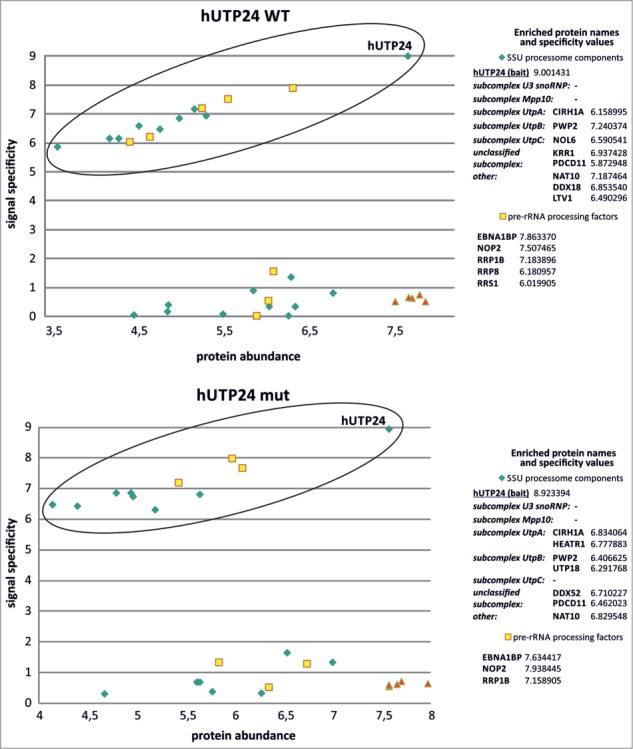
hUTP24 protein is associated with homologs of the yeast SSU processome subunits and other pre-rRNA processing factors in human cells. Wild-type (top) and mutant hUTP24 (bottom) were purified as C-terminal fusions with an eGFP-tag. Proteins co-purifying with the bait were analyzed by mass spectrometry and the results were compared to the parallel purification, carried out using the unmodified parental cell line. For a more detailed description of the graphs and data analysis refer to Figure 4.
Taken together, these findings indicate that hUTP24 appears to interact with other proteins participating in the biogenesis of the small ribosomal subunit, similar to its yeast counterpart. Nevertheless, the association might be somewhat weaker than in the case of S. cerevisiae or the network of interactions between proteins involved in pre-rRNA processing in human cells may differ from that in yeast.
Mutation in the putative hUTP24 catalytic site impairs cell growth and decreases cell metabolic activity
Defects in rRNA processing were previously reported upon siRNA-mediated depletion of hUTP24 or its homolog in mouse cells.16,37,52,53 However, this approach does not allow for differentiation between the effects resulting from the lower protein levels and the deficit of its potential ribonucleolytic activity. Therefore, we constructed a human cellular model to study the outcome of the mutation in the hUTP24 putative catalytic site, wherein endogenous hUTP24 was downregulated with sh-miRNA, concomitantly with exogenous expression of RNAi-insensitive FLAG-tagged hUTP24 WT or mut variants (Fig. 6A). A similar approach was previously used successfully to investigate mutations in the hDIS3 gene, which encodes the major catalytic subunit of the human nuclear RNA exosome.54 The detailed characterization of constructed cell lines revealed that the model was working properly (Fig. S6).
Figure 6.
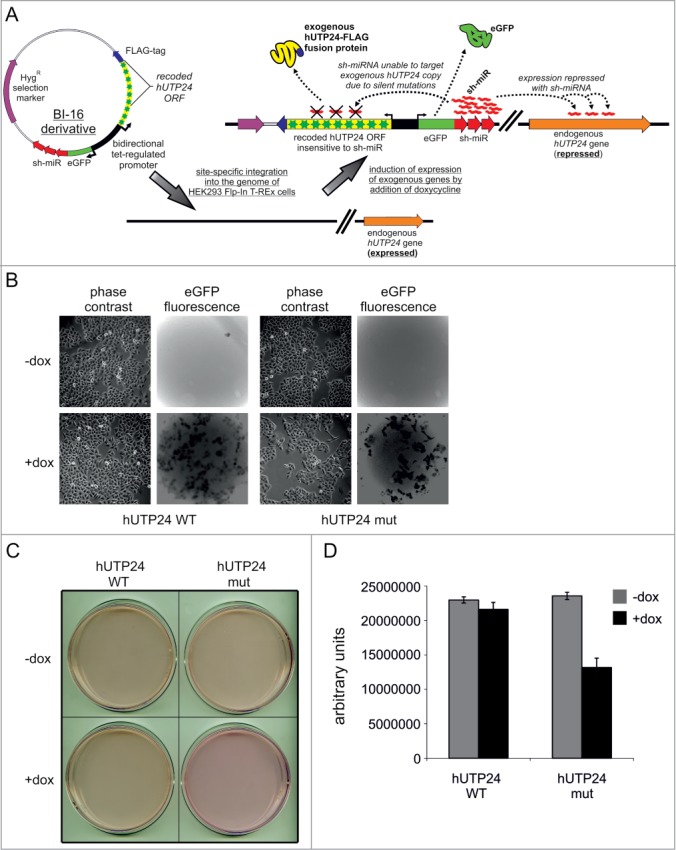
Human cellular model for analysis of hUTP24 function reveals that it is important for cell physiology. (A) General principle of the constructed cellular model. Derivatives of BI-16 vector (compatible with Flp-In™ T-Rex™ system from Invitrogen) containing wild-type or mutated version of FLAG-tagged hUTP24 and an eGFP-sh-miRNA fusion, which were both under the control of a bidirectional tetracycline-regulated promoter, were integrated into the HeLa Flp-In T-REx cell line genome. The FLAG-tagged hUTP24 ORF was recoded in a way rendering it insusceptible to sh-miRNA silencing. Upon induction with doxycycline, stable cell lines produced either wild-type or mutated hUTP24-FLAG fusion and sh-miRNA, thereby silencing expression of only the endogenous hUTP24. Production of sh-miRNA was monitored by eGFP co-expression. (B) Cell-growth analysis. Equal amounts of cells from each model line were seeded in culture dishes, subjected to doxycycline-mediated induction (48 h + 48 h), and analyzed by microscopy. An appropriate filter was used to visualize eGFP fluorescence in situ. The cells harboring mutant hUTP24 grew worse than the cell line with WT protein. (C) Acidification of culture medium was significantly less pronounced for cells producing hUTP24 mut than the wild-type counterpart. The cells were seeded as in (B) and the culture dishes were scanned using an Epson Perfection V750 Pro Scanner. (D) Metabolic activity assay. Approximately equal numbers of cells from each stable model cell line were grown in triplicate in 96-well plates, either untreated (“−dox”) or treated with doxycycline (“+dox”). AlamarBlue® reagent was added after 72 h, and the metabolic status was assessed by fluorescence measurements. Cells producing hUTP24 mut displayed lower metabolic activity than cells expressing the wild-type protein.
We immediately noticed that cells producing hUTP24 mut grew significantly slower than those synthesising hUTP24 or any line maintained in media lacking the inducer (Fig. 6B). In concordance, acidification of the growth medium was much less pronounced for the cell line exogenously expressing the mutant hUTP24 variant than its wild-type counterpart (Fig. 6C). In addition, the metabolic activity of the mutant cell line was approximately 1.8-fold lower than that of hUTP24 WT cells (Fig. 6D), further indicating the proliferation defect and/or decreased viability. These data indicate that the putative catalytic activity of hUTP24 is essential for cell growth.
Expression of the mutant form of hUTP24 has a negative impact on the production of 18S rRNA and biogenesis of the small ribosome subunit
To investigate the impact of the hUTP24 mutation on ribosome biogenesis, we analyzed ribosome profiles using cytoplasmic extracts from model cell lines grown in the absence or presence of doxycycline after cycloheximide treatment. Expression of the mutant hUTP24 variant resulted in a marked decrease of the levels of the 40S subunit, elevated levels of the free 60S subunit, and a reduced amount of monosomes (Fig. 7, upper panel). Intriguingly, in contrast to the situation in yeast cells, there was no significant effect on the formation of polysomes (Fig. 7, upper panel). Electrophoretic analysis of RNA isolated from the gradient fractions corresponding to small and large ribosomal subunits as well as to the assembled monosomes revealed that the small peak corresponding to the 40S subunit contained severely diminished amounts of 18S rRNA, whereas the fractions under the peak representing the 60S subunit contained a majority of the mature 28S rRNA molecules present in the cytoplasmic extract (Fig. 7, upper panel). When the extracts were prepared from cells not treated with cycloheximide either in the absence of magnesium (Fig. 7, middle panel) or using a high-salt buffer (Fig. 7, bottom panel), reduction of the 40S subunit levels was the most apparent phenotype, while the levels of the 60S subunit were relatively similar between the 2 cell lines irrespective of the absence or presence of doxycycline. These results indicate that the increased levels of the free large ribosomal subunit observed in the presence of cycloheximide (Fig. 7, upper panel) were not due to their enhanced biogenesis, but rather resulted from their excess over the 40S subunit, which prevented incorporation of overabundant 60S into 80S monosomes. This is reflected by the joint reduction of 18S and 28S rRNA levels in the monosome fraction (Fig. 7, upper panel).
Figure 7.
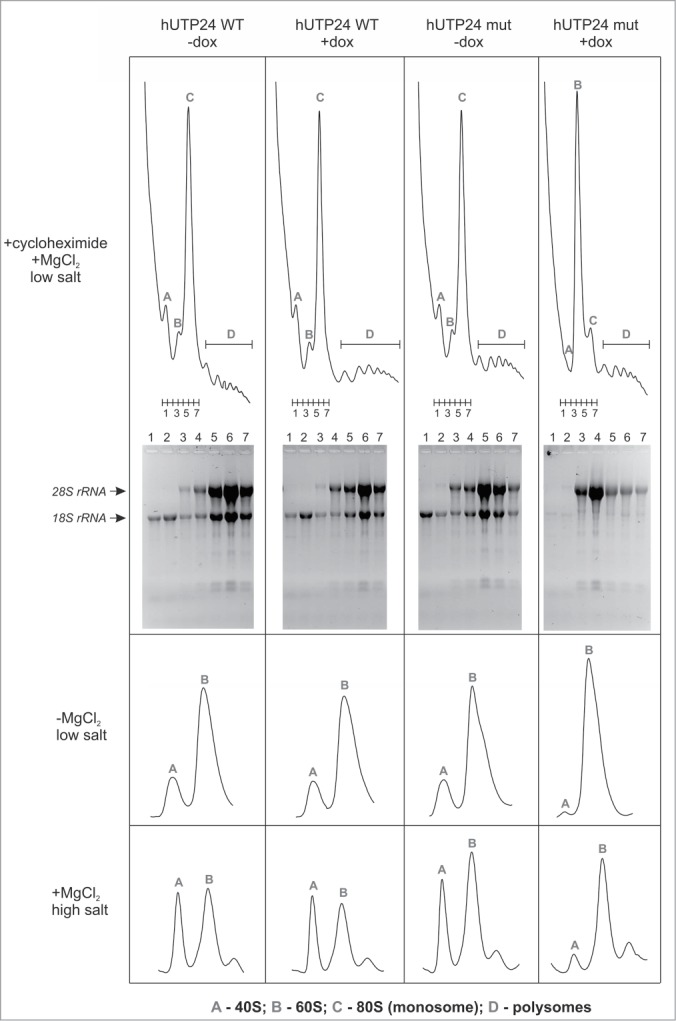
Mutation in the hUTP24 putative catalytic site leads to a ribosome biogenesis defect. Analysis of polysome and ribosome subunits profiles was conducted by preparing native cytoplasmic extracts from model cell lines, grown in the absence (“−dox”) or presence (“+dox”) of doxycycline, using buffers with cycloheximide (top panel), lacking magnesium (middle panel), or containing salt at high concentration (bottom panel), and separated by centrifugation in linear sucrose gradients. Graphs show distribution of absorbance at 254 nm from the top (left) to the bottom (right). Peaks corresponding to individual subunits (40S and 60S), monosomes (80S) and polysomes are indicated. In the experiment performed using cycloheximide, 7 fractions were collected from each gradient (numbered 1–7), as indicated below the graphs. RNA was then isolated from these fractions and separated in denaturing agarose-formaldehyde gels. The bottom part of the panel demonstrates results of the electrophoretic analysis. Expression of hUTP24 mut leads to diminished levels of the 40S subunit and monosomes, which contain reduced amounts of both 18S and 28S rRNA.
To confirm that the defect in 40S subunit biogenesis is a consequence of decreased 18S rRNA synthesis de novo, a pulse-chase metabolic RNA labeling experiment was performed in the model cell lines with an exogenous copy of hUTP24 WT or mut, either treated or not treated with doxycycline. The increase in the amounts of 28S rRNA during the chase phase was similar for hUTP24 WT and mut cell lines, irrespective of the presence of inducer in the medium (Fig. 8). Similarly, synthesis of 5.8S and 5S rRNA did not seem to be significantly affected by the hUTP24 mutation (Fig. S7). In contrast, a much slower increase of 18S rRNA levels was observed in the case of cells producing hUTP24 mut compared to their non-induced counterparts or to the cell line expressing the hUTP24 WT variant (Fig. 8). As a consequence, the steady-state level of 18S RNA in these cells was also significantly diminished (see methylene blue staining of the blot in Fig. 8).
Figure 8.
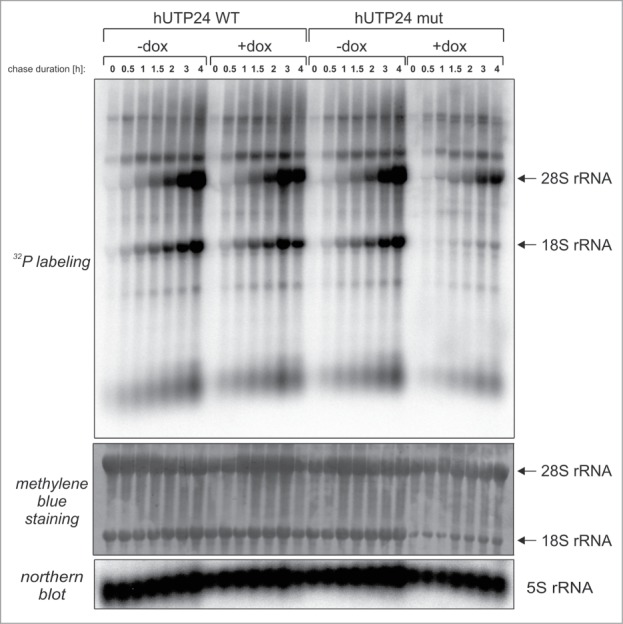
hUTP24 mutation results in decreased production of 18S rRNA, but not 28S rRNA. Model cell lines cultured in a medium lacking doxycycline (“−dox”) or in the presence of inducer (“+dox”) were pulse labeled with 32P orthophosphoric acid, followed by chase in normal media for varying times (indicated above each lane). RNA was then isolated from the cells, separated in a denaturing agarose-formaldehyde gel and transferred onto nylon membrane. The blot was first stained with methylene blue (upper panel) and then subjected to phosphorimaging (middle panel). Positions of 28S and 18S rRNA are indicated with arrows on the right. Hybridization with a probe complementary to 5S rRNA (bottom panel) served as a loading control.
hUTP24 dysfunction results in the inhibition of cleavage at site A1, which leads to aberrant processing, generating 18S rRNA shortened by 2 nucleotides at the 5′ end, while processing at site A0 is unaffected
In order to further investigate rRNA biogenesis defect in hUTP24 mutant cells, we analyzed pre-rRNA processing intermediates and examined several cleavage sites within the rRNA precursor molecule.
We first conducted northern blot analyses using probes targeting mature rRNA species (see Fig. 9A for location of the probes), which demonstrated that the production of the mutant hUTP24 variant led to decreased steady-state levels of 18S rRNA, but not 28S, 5.8S, or 5S rRNA (Fig. 9B). Further analyses with probes hybridizing to different parts of 5′-ETS, ITS1, and ITS2 regions (Fig. 9A) revealed that there was a slight increase of unprocessed 47S precursor (Fig. 9C, probe h5ETS) and an apparent decrease in the amounts of RNA species migrating slightly slower than 18S rRNA (Fig. 9C, probe hITS1a). Based on the location of hITS1a probe (between 3′-end of 18S rRNA and processing site E), this species most likely corresponds to the 18S-E intermediate that is normally processed to a mature 18S by endonucleolytic cleavage at site 3 or by 3′–5′ exoribonucleolytic trimming. This finding suggests that when mutant hUTP24 is produced, maturation of 18S rRNA is impaired at a step preceding 18S-E formation. Accordingly, the other most pronounced phenotypes were accumulation of 26S (Fig. 9C, probes hA0, hITS1a, and hITS1b) and 43S (Fig. 9C, probes hA0, hITS1a, hITS1b, hITS2a, and hITS2b) processing intermediates extending from site A0 to sites 2 or 02, respectively.10 These observations strongly indicate that the production of hUTP24 mut resulted in an inefficient cleavage at site A1. Accumulation of 26S pre-rRNA may also suggest impairment of the cleavage at site E. This was supported by the drop in the levels of 18S-E pre-rRNA and E-2 fragment (Fig. 9C, probes hITS1a and hITS1b) and corroborated previous observation that processing at site E is tightly coupled to upstream cleavage events in the human pre-rRNA.12 The levels of 30S pre-rRNA were significantly lower in the cells producing hUTP24 mut compared to hUTP24 WT (Fig. 9C, probes hA', hA0, hITS1a, and hITS1b). This could suggest that the cleavage at site 2 may also be impaired. However, the levels of 32S pre-rRNA, which is the second direct product of processing at this site, were normal (Fig. 9C, probes hITS2a and hITS2b). Moreover, accumulation of 45S, an intermediate directly preceding 30S and 32S formation was not visible. Furthermore, if cleavage at site 2 was impaired, one would not expect accumulation of 26S pre-rRNA, which was clearly the most prominent phenotype resulting from hUTP24 dysfunction. Thus, cleavage at site 2 seems to be unaffected, which was also confirmed by the primer extension analysis (see below). Decreased levels of 30S pre-rRNA may in turn result partly from less efficient cleavage at site A', which is suggested by slight downregulation of 45S intermediate. Intriguingly, levels of 12S and 7S precursors of 5.8S appeared to be slightly increased in the hUTP24 mutant cell line (Fig. 9C, probes hITS2a and hITS2b). In addition, we noticed accumulation of some pre-rRNA degradation intermediates, depending on the production of the hUTP24 mut protein variant (such as those indicated with question marks in the Fig. 9C for probes hA' and hITS1a). Finally there was a modest accumulation of 41S rRNA in the cells producing hUTP24 mut protein, which is quite difficult to explain based on the available data. It should be emphasized that the overall picture of pre-rRNA processing aberrations that we observed in our RNA gel blot analyses is not easy to interpret, since it may result not only from the possible misprocessing at site A1 (due to the impairment of hUTP24 activity), but probably also from some secondary, compensatory and degradative effects, reflected by the unexpected phenotypes listed above.
Figure 9.
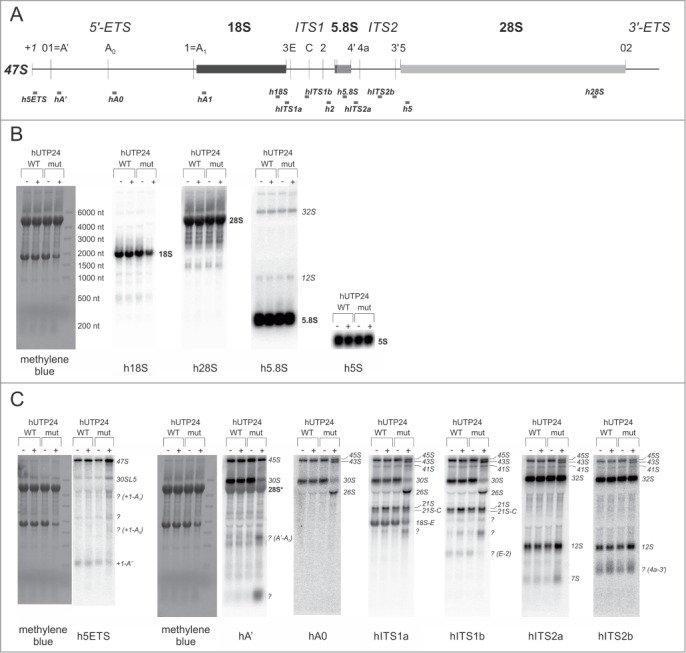
hUTP24 dysfunction results in multiple changes in the pattern of pre-rRNA processing intermediates. (A) A scheme of the human 47S pre-rRNA. Positions of the processing sites within the 47S precursor are indicated with vertical thin lines together with the names. Nucleotide numbering is according to GenBank (accession number: U13369.1). Gray bars below the primary transcript show positions of northern blot probes and oligonucleotides used for primer extension in this study. (B) Northern blot analysis of the mature rRNA levels. Total RNA was isolated from model cell lines grown either in the absence (“−dox”) or presence (“+dox”) of doxycycline, separated in a denaturing agarose-formaldehyde gel, and transferred onto nylon membrane, which was then stained with methylene blue and sequentially hybridized with probes complementary to mature rRNA, as indicated at the bottom. Positions of different RNA species are indicated on the right. hUTP24 mutation leads to decreased levels of 18S rRNA, but not 28S, 5.8S, and 5S rRNA. (C) Northern blot analysis of pre-rRNA processing intermediates. Experiment was performed as in (B), but using probes targeting various regions of 5′-ETS, ITS1, or ITS2, as indicated at the bottom. Positions of different RNA species are indicated on the right. The major visible phenotypes resulting from hUTP24 mutation are accumulation of 26S (probes hA0, hITS1a, hITS1b) and 43S (probes hA0, hITS1a, hITS1b, hITS2a, hITS2b) intermediates as well as decreased levels of 18S-E (probe hITS1a) and 30S (probes hA', hA0, hITS1a, and hITS1b). All of these differences with regard to the cell line producing hUTP24 WT suggest that processing at site A1 at the boundary of 5′-ETS and the 18S rRNA segment is compromised.
Therefore, to gain more direct insight into the possible defects of processing at sites surrounding the 18S rRNA module, primer extension analyses were performed. The cleavages at site A0 in the 5′-ETS, at site 2 (a major processing site within ITS1), which separates 18S from 5.8S/28S, site 5 (at the 5′-end of 28S rRNA) and processing at the 5′-end of 5.8S rRNA seemed to be unaffected in the cells producing hUTP24 mut (Fig. 10; Fig. S8). We observed a slight decrease in the efficiency of processing at the primary site A' (Fig. 10; Fig. S8) and noted clear differences in the patterns of products obtained for the wild-type and mutant variants of hUTP24, using a primer located immediately downstream of the 5′-end of 18S rRNA (Fig. 10). The boundary between 5′-ETS and 18S rRNA was previously mapped between nucleotides 3656 and 3657 in HEK293 cells.13 We confirmed that this was also a major cleavage site in our model HeLa cell line producing hUTP24 WT (Fig. 10) and in other human cells from different sources (Fig. S9A). In contrast, reverse transcription stops detected for hUTP24 mut were more heterogeneous, and the most prominent one was shifted 2 nucleotides downstream of the normal cleavage site (i.e. located between nucleotides 3658 and 3659) (Fig. 10). This effect was not restricted to HeLa cells, since we observed the exact same differences for HEK293 stable cell lines producing hUTP24 WT and mut (Fig. S9B). This is in stark contrast to the situation in yeast, where impairment of yUtp24 function resulted in a less efficient cleavage at the equivalent site A1 with no qualitative changes (see Fig. 3B for comparison).
Figure 10.
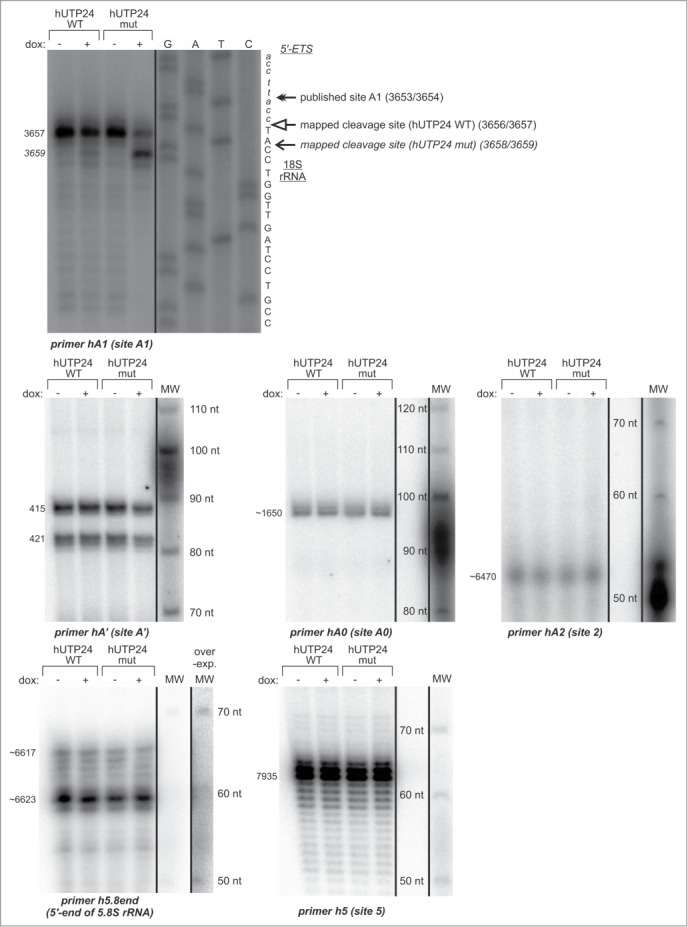
Pre-rRNA processing defects resulting from hUTP24 mutation are mainly due to aberrant cleavage at site A1. Primer extension analysis of pre-rRNA processing at selected sites within the 47S pre-rRNA. Total RNA was isolated from model cell lines grown either in the absence (“−dox”) or presence (“+dox”) of doxycycline and subjected to primer extension using oligonucleotides hybridizing upstream of the sites A', A0, A1, 2, 5, as well as of the 5′-end of 5.8S rRNA. Numbers on the left indicate nucleotide positions in 47S corresponding to reverse transcription stops. In the case of site A1, the same primer was used in parallel with a DNA template comprising an appropriate rDNA fragment to generate dideoxynucleotide sequencing ladders, which were co-electrophoresed with primer extension products. The retrieved sequence (in reverse complement) is presented next to the sequencing results. Production of hUTP24 mut leads to a shift of the major reverse transcription stop 2 nucleotides downstream with respect to the controls. For the remaining pre-rRNA processing sites, a 5′-labeled DNA molecular weight marker (MW) was run in parallel to estimate sizes of the primer extension products. The lengths of the MW fragments are indicated next to the bands. Quantification of the results is presented in the Supplemental Figure 8.
Based on the nature of the processing defect, we postulated that the altered mature 5′-end of 18S rRNA observed in absence of hUTP24 activity might be due to the action of a 5′–3′ exoribonuclease, with the hXRN2 protein being the most likely candidate. To verify this hypothesis, we subjected our model HeLa cell lines, either untreated or treated with doxycycline, to RNA interference using siRNA against hXRN2 mRNA or unrelated control siRNA. Western blot analyses confirmed that hXRN2 was efficiently silenced and that the hUTP24-FLAG fusions as well as eGFP-sh-miRNA were properly synthesized (Fig. S10A). Nevertheless, the pattern of pre-rRNA misprocessing at the boundary of 5′-ETS and 18S in the cell line producing hUTP24 mut remained unchanged upon hXRN2 silencing (Fig. S10B). Northern blot analyses revealed that while the levels of mature rRNA did not differ between siRNA-treated and untreated cells (Fig. S10C), downregulation of hXRN2 expression resulted in the characteristic phenotypes previously reported, such as accumulation of unprocessed 47S pre-rRNA (Fig. S10D, probe h5ETS), 30SL5 species (Fig. S10D, probes hA' and hA0) (extending from the transcription start site to the processing site 2 – see Fig. 1), 36S pre-rRNA (Fig. S10D, probe hITS1b), as well as +1-A' (Fig. S10D, probe h5ETS) and E-2 (Fig. S10D, probe hITS1b) processing intermediates, which are normally eliminated from the cell by the 5′–3′ exoribonucleolytic activity of hXRN2.11,36 However, in agreement with the primer extension results, hXRN2 silencing did not significantly influence any of the phenotypes associated with hUTP24 mutation.
Moreover, we performed similar experiments using siRNAs that downregulated the expression of genes encoding 2 other nuclear proteins with 5′–3′ exoribonuclease activity, namely NOL12 (a homolog of the yeast Rrp17) and DOM3Z (also known as DXO1; an enzyme homologous to S. cerevisiae Rai1, shown to have pyrophosphohydrolase, decapping, and 5′–3′ exoribonuclease activities in mammalian cells).55-57 Although the efficiency of silencing was very high for both proteins (Fig. S11A, D), similarly to hXRN2 downregulation we did not observe any changes in the pattern of processing at site A1 upon hUTP24 dysfunction (Fig. S11B, E). It should be pointed out that NOL12 knockdown led to the accumulation of 47S and 26S pre-rRNA, while downregulation of DOM3Z expression resulted in the increased levels of CamKI mature mRNA and its inefficiently spliced pre-mRNAs (Fig. S11C, F). Since these phenotypes were observed previously,16,57 we conclude that both NOL12 and DOM3Z silencing was functional in our hands.
Taken together, these data raise an interesting possibility that an as yet unidentified 5′–3′ exoribonuclease or endoribonuclease might be involved in the maturation of 18S rRNA 5′ terminus. Our results indicate that mutation in the putative active site of hUTP24 results in aberrant 18S rRNA maturation at the 5′-end.
Discussion
The role of UTP24 proteins in rRNA processing in eukaryotic cells
In this study we have comprehensively characterized the role of hUTP24 protein in pre-rRNA processing in human cells and compared it to the function of the homologous yUtp24 in S. cerevisiae. Both yeast and human UTP24 proteins are involved in early cleavages of the long pre-rRNA precursor at sites dependent on U3 snoRNA action. Their dysfunction results in diminished production of mature 18S rRNA and accumulation of its misprocessed precursors. Importantly, we identified biologically significant differences in the molecular phenotypes caused by mutation in the active site of UTP24 proteins from both species, which are likely to be the reason why hUTP24 cannot functionally replace yUtp24 in yeast. For instance, while the putative endoribonuclease activity of the PIN domain is essential for cleavage at processing site A1 located at the 5′ end of 18S rRNA in both organisms, processing at site A0 of the human rRNA precursor does not appear to be UTP24-dependent and occurred efficiently in cells expressing the mutant hUTP24. Interestingly, a recent study suggested that depletion of hUTP24 leads to accumulation of 30S pre-rRNA in addition to increased levels of 47S.37 Similar results were reported by other research group also utilizing an RNAi strategy.16,52 Such a phenotype would indicate that cleavage at site A0 is less efficient when the expression of hUTP24 is downregulated by siRNA. However, in our experiments we did not observe accumulation of 30S; instead, levels of this processing intermediate were lower when the mutant form of hUTP24 was produced in cells, suggesting that the upstream processing event – that is, cleavage at site A' – might be impaired in such conditions. This was supported by a decreased level of 45S pre-rRNA and – more directly – by the results of primer extension analysis. More intriguingly, accumulation of 26S pre-rRNA was not observed previously upon siRNA-mediated hUTP24 silencing, while it was the major phenotype in our case, implying that the putative hUTP24 enzymatic activity is involved in processing at site A1. Accumulation of 26S rRNA may indicate some impairment of cleavage at site E, as suggested also by the downregulation of 18S-E pre-rRNA and E-2 fragment. These discrepancies may be related to different experimental conditions, and therefore it is possible that the sole presence (but not the nucleolytic activity) of hUTP24 is indispensable for cleavage at site A0, and that the SSU processome does not assemble/function properly in the absence of exogenously produced protein, thus leading to aberrant processing at this site. However, we should note that our own attempts to detect phenotypes related to rRNA misprocessing upon simple siRNA-mediated depletion of hUTP24 were unsuccessful (data not shown). The lack of dependence of processing at site A0 in the human pre-rRNA on hUTP24 activity is contrary to the situation in yeast, where both A1 and A0 cleavages are dependent on U3 snoRNA and believed to be mediated by UTP24.
U3 snoRNA is a central component of the SSU processome complex, which assembles from sub-complexes on the nascent pre-rRNA molecules and includes UTP24 (see below). U3 snoRNA is essential for processing of the yeast primary transcript at sites A0, A1, and A2 or their equivalents in metazoa, partially by acting as a chaperone for 18S rRNA folding.32,58-61 U3 snoRNA is involved in elaborate interactions with pre-rRNA in all model organisms analyzed to date (depicted schematically in Fig. S12), which guide U3 and associated protein components of the SSU processome to the target cleavage sites on the pre-rRNA. Conformational switches occurring in both U3 snoRNA and pre-rRNA during establishment of intermolecular interactions are believed to avert premature or incorrect processing at sites A0, A1, and A2. In yeast, A0 and A1 processing sites are located in close proximity to each other, and there is a significant potential for base-pairing between sequences surrounding these sites (Fig. S12A). A similar situation is observed in the case of Trypanosoma and Xenopus pre-rRNA (Fig. S12B, C).61,62 This interaction positions both sites more or less opposite to one another at the base of a stem-loop structure (Fig. S12A–C). Such a structural organization most likely facilitates coordinated processing at both sites. Contrary to species mentioned above, the relative spatial positioning of sites A0 and A1 in human pre-rRNA remains to be determined, since there are apparently no such mutually complementary sequences adjacent to these sites (Fig. S12D). It is also worth emphasizing that while the distance separating A0 and A1 sites in S. cerevisiae, Trypanosoma, and Xenopus is between 90 and 220 nucleotides (Fig. S12A–C), it exceeds 2000 nt in the human rRNA precursor (Fig. S12D). Therefore, it is possible that the detailed mechanism of A1 site recognition in human cells diverged during evolution from the one observed in yeast in terms of losing coordination with cleavage at site A0, which appears not to be UTP24-dependent.
The observation discussed above may partially explain another difference between effects of yUtp24 and hUTP24 mutations. While in yeast it resulted in less efficient processing at A1 (but the site of processing was preserved), in human cells it led to the shift of the cleavage site by 2 nucleotides downstream of the normal mature 18S rRNA 5′-end. In yeast, 2 signals define the position of the mature 18S rRNA 5′-end: the first one is the 5′-ETS sequence immediately upstream of the A1 cleavage site, while the second one is a stem of a central pseudoknot structure within 18S rRNA with 3 nucleotides separating the A1 site from this stem.38,63 The conservation of 5′-ETS sequence preceding A1 is observed in Ascomycete fungi and among plants, but does not exist in vertebrates, suggesting that only the second mechanism of A1 site positioning mentioned above may operate in the latter, including humans. This may also influence the difference in phenotypes observed in yeast and human cells upon inactivation of yUtp24 and hUTP24, respectively.
Furthermore, differences in U3 snoRNA-pre-rRNA interactions in various organisms (see Fig. S12A–D and refer to the Supplemental Discussion for a more detailed description), raise an intriguing possibility that there may exist some interplay between processing at sites A' and A1 in human cells, which is functionally equivalent to coordinated processing at spatially juxtaposed sites A0 and A1 in yeast. These distinct relationships would help to explain differences in molecular phenotypes resulting from UTP24 inactivation in both species. There is actually some parallel between the yeast A0 site and the human A' site, namely these are the 5′-most processing sites within 5′-ETS of the pre-rRNA molecule in both organisms. Indeed, results of northern blot analyses performed herein for both organisms demonstrate that the mutation in the active site of UTP24 protein results in the accumulation of the full-length 35S/47S precursor, which is not cleaved at site A0/A' in yeast/humans. However, this phenotype is less pronounced than the molecular effects documenting misprocessing at site A1. We therefore cannot definitively conclude that hUTP24 is directly involved in cleavage at site A' in the human pre-rRNA, since such accumulation of the full-length precursors might be a general secondary consequence of rRNA misprocessing. Furthermore, unlike the A0 processing site in yeast, the human A' cleavage site is heterogeneous and we were not able to propose any RNA-RNA interactions that could mimic those juxtaposing sites A0 and A1 in S. cerevisiae. All discrepancies described are fully supported by our notion that hUTP24 produced exogenously in S. cerevisiae is not able to complement decreased levels of endogenous yUTP24 gene expression. Despite significant conservation of the PIN domain sequences of both UTP24 paralogs, hUTP24 most likely has some requirements for its activity that are not fulfilled in the yeast cell.
Reconstitution of U3 snoRNA-pre-rRNA interactions and the presence of RNA chaperones may be required to demonstrate endoribonucleolytic activity of UTP24 proteins in vitro
A fundamental question concerning UTP24 proteins that still remains unanswered is whether they actually display endoribonucleolytic activity, since it was not directly demonstrated in vitro for either yeast or human homolog. Our intensive attempts to detect this activity using recombinant, N-terminal MBP-tagged (proteins devoid of the MBP entirely lost their solubility following protease-mediated cleavage) yUtp24/hUTP24 or native hUTP24 purified from human cells by GFP-trap chromatography have also failed. Regardless of whether employing oligoribonucleotides of random sequence or short RNA substrates encompassing respective A1 processing sites, we could barely detect any activity above the background level (data not shown).
The lack of observable UTP24 endoribonucleolytic activity in vitro on linear RNA substrates, even if their sequences cover A1 processing sites, may not only be due to the absence of appropriate secondary structure, which in vivo is ensured by complex pre-rRNA-U3 snoRNA interactions described above and in the Supplemental Discussion, but also because of the lack of Imp3 and Imp4 proteins. These proteins were shown to act as RNA chaperones, enabling breakage of the intramolecular base-pairing within U3 snoRNA and pre-rRNA and establishment of the intermolecular interaction between these 2 RNA molecules, which is a prerequisite for processing at site A1.64-66 In yeast, both Imp3 and Imp4, which bind to the U3 snoRNA 5′-domain and the hinge region, significantly increase stability of the duplex formed by U3 snoRNA 5′-hinge and 5′-ETS site I,64,65 while Imp3 alone is indispensable for liberating U3 snoRNA box A segment from a stem-loop structure and unwinding helix 1 at the 5′-end of the 18S rRNA segment to allow for their reciprocal hybridization.66 Indeed, formation of the U3 snoRNA box A-18S duplex does not occur in vitro unless Imp3 and Imp4 are present. Interactions between Imp3, Imp4, and the U3 snoRNA-pre-rRNA hybrid are also believed to assist in docking the SSU processome onto the pre-rRNA, thereby facilitating subsequent cleavage events at sites A0, A1, and A2 by UTP24 and other, as yet unidentified endoribonuclease(s) recruited together with other components of the SSU processome. The proposed role of these interactions in vivo is to provide mechanisms that enable temporal control of the initiation of the 40S subunit biogenesis. In particular, cleavage sites A0 and A1 may become accessible for respective endoribonucleases exclusively due to the unfolding properties of Imp3. This ensures that U3 snoRNA does not act prematurely on nascent pre-rRNA.
Aberrant pathway of 18S rRNA processing in cells producing hUTP24 mutant
Results from in vitro studies suggest that the human pre-rRNA is cleaved at site A1 located a few nucleotides upstream of the mature 18S rRNA 5′-terminus and that this maturation step is completed by trimming the resulting extension by an unknown 5′–3′ exoribonuclease.25,26 Since this observation has not been confirmed in vivo and – besides that – in all other species, including the closely related mouse, the A1 site and the 5′-end of 18S rRNA apparently coincide, it should be treated with caution. Nevertheless, our results demonstrating that hUTP24 dysfunction leads to the appearance of an alternative processing site located 2 nucleotides downstream of the mature 18S 5′-end suggest an involvement of some exoribonuclease working in the 5′–3′ direction, while such phenotype was not observed in yeast. Such an alternative aberrant processing pathway may be responsible for residual translation in cells with hUTP24 dysfunction, which was not the case in yeast. Moreover, the impaired processing must also be ineffective, since we found that 18S rRNA maturation was strongly inhibited. Interestingly, this phenotype was retained after siRNA-mediated downregulation of the expression of genes encoding the 3 known nuclear 5′–3′ exoribonucleases (XRN2, NOL12, and DXO1). Therefore, the enzyme responsible for aberrant processing at site A1 when hUTP24 is mutated remains to be identified. Nevertheless, we cannot exclude the possibility that some other endoribonuclease executes this cleavage when the enzymatic function of hUTP24 is impaired.
hUTP24 may not be a stable component of the putative human U3 processome
Our proteomic analyses suggest that mutations in the putative catalytic centers of yeast and human UTP24 do not impair their interactions with remaining components of the SSU processome. Although we are aware that the results of purifications carried out for both organisms cannot be compared directly, the lower number of protein partners detected in the case of hUTP24 suggest that its interactions with other SSU processome subunits are weaker than those mediated by yUTP24 in S. cerevisiae. Moreover, even in yeast, yUtp24 has not been assigned to any known stable subcomplex of the SSU processome and it is possible that the networks of interactions between the UTP24 proteins in yeast and human cells are different. This may expand a list of reasons explaining dissimilarities in pre-rRNA processing defects observed upon UTP24 mutation in both organisms. The UTP24 region responsible for interactions with other proteins remains to be identified; however it is likely to be located outside the conserved PIN domain associated with enzymatic activity (i.e., in the N-terminal regions where the degree of evolutionary conservation between yUtp24 and hUTP24 is lower), which may account for the different number of identified UTP24 protein partners in yeast and humans.
Conclusion
In summary, our study clearly shows that hUTP24 plays a crucial role in human rRNA processing and is essential for accurate endonucleolytic cleavage at the 5′-end of 18S rRNA. Moreover, our results indicate that an alternative aberrant processing pathway exists in human cells, which generates 18S rRNA molecules shortened by 2 nucleotides at the 5′ terminus.
Materials and Methods
Human cell cultures and generation of stable cell lines
Human HeLa, HEK293 Flp-In T-REx (Invitrogen) and HeLa Flp-In T-REx (kindly provided by Dr. Matthias W. Hentze67) cells were cultured as monolayers in Dulbecco's modified Eagle's medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (Gibco) or tetracycline-free FBS (TET System Approved FBS; Clontech) (in the case of T-REx cell lines) and antibiotics (penicillin-streptomycin; Sigma-Aldrich) at 37°C in a 5% CO2 humidified atmosphere.
The stable inducible HEK293 Flp-In T-REx cell lines were obtained in this study using highly pure DNA midipreps of pU24–17-pU24–23 plasmid constructs and the Flp-In™ T-REx™ system (Invitrogen), according to the protocol of the manufacturer. Established cell lines were grown in the same medium as above supplemented with hygromycin B (100 μg/ml) and blasticidin (10–15 μg/ml) (both from Invitrogen). Transfections were performed with Lipofectamine2000 (Invitrogen). Expression of exogenous genes was induced by addition of doxycycline to the culture medium at a final concentration of 100 ng/ml. The stable inducible HeLa Flp-In T-REx model cell lines bearing sh-miRNA-insensitive FLAG-tagged hUTP24 (WT or mut) and sh-miRNA-eGFP insert for silencing of the endogenous hUTP24 were constructed likewise, using pU24–22 or pU24–23 construct, respectively.
Purification of UTP24 proteins from yeast and human cell cultures for proteomic analyses
The yeast strains ADZY799 and ADZY800 producing TAP-tagged yUtp24 WT or mut variant, respectively, as well as the parental BMA64 strain (negative control), were grown in 8 l of YPD to OD600 = 2. Following centrifugation at 3200×g for 5 minutes at 4°C, the cell pellet was resuspended in 30 ml of lysis buffer (40 mM Hepes-KOH pH = 8.0; 250 mM NaCl; 1 mM DTT), frozen in liquid nitrogen and stored at −80°C. Cells were broken in a laboratory blender chilled with dry ice. The homogenate was melt in the presence of protease inhibitors and centrifuged in 35Ti rotor (Beckman) in a Beckman ultracentrifuge at 20000 rpm for 20 minutes at 4°C. Supernatant was spun again at 32000 rpm for 75 min at 4°C and afterwards the supernatant was dialyzed for 3 hours against 3 Ls of dialysis buffer (40 mM Hepes-KOH, pH = 8.0; 150 mM NaCl; 1 mM DTT; 1 mM PMSF; 20 mM benzamidine-HCl; 20% glycerol). The dialyzed extract was incubated on a rotating wheel overnight with 1 ml of IgG Sepharose 6 Fast Flow (GE Healthcare) equilibrated with lysis buffer, in the presence of 0.1% reduced Triton X-100 (rTX-100) at 4°C. The beads were transferred onto the column compatible with ÄKTA Purifier system (GE Healthcare) and washed with 30 ml of IPP150 (10 mM Tris-HCl, pH = 8.0; 150 mM NaCl; 0.1% rTX-100), followed by wash with 30 ml of TEV protease cleavage buffer (10 mM Tris-HCl, pH = 8.0; 150 mM NaCl; 0.5 mM EDTA; 1 mM DTT). The on-column TEV protease cleavage with home-made enzyme was performed for 4 hours at 18°C. The eluate from IgG beads was collected and the eluted proteins were precipitated with PRM reagent (0.05 mM pyrogallol red; 0.16 mM sodium molybdate; 1 mM sodium oxalate; 50 mM succinic acid, pH = 2.5).
For purification of C-terminally eGFP-tagged hUTP24 WT and mut, HEK293 Flp-In T-REx cell lines obtained following transfection with constructs pU24–20 or pU24–21, respectively, were grown in the presence of doxycycline on 2 150 mm plates until the confluence reached approximately 90%. Non-transfected HEK293 Flp-In T-REx cells were cultured in parallel as a control. Following aspiration of the medium, cells were washed with PBS, detached with trypsin-EDTA, suspended in PBS and centrifuged for 5 minutes at 500×g, 4°C. The cell pellet was frozen in liquid nitrogen and stored at −80°C. After thawing the cells on ice, the pellet was resuspended in 1.7 ml of the lysis buffer [20 mM Hepes-KOH, pH = 7.1; 100 mM NaCl; 3 mM MgCl2; 10% glycerol; 0.5% Igepal-CA630; 1 mM PMSF; 1x Protease Inhibitor Cocktail (2 μM pepstatin A; 0.6 μM leupeptin; 2 mM benzamidine; 2 μg/ml chymostatin)] and incubated for 30 minutes on a rotating wheel at 4°C in the presence of 0.1 mg/ml RNase A. The cells were then disrupted by sonication in a Bioruptor® XL sonicator (Diagenode) at high-power setting by applying 30 cycles of 30 seconds pulse: 30 seconds break. Cell debris was removed by centrifugation at 16000×g, 4°C for 15 minutes and the supernatant (protein extract) was incubated for 90 minutes with 80 µl of GFP-Trap® magnetic resin (Chromotek; pre-washed 2x 20 minutes with the lysis buffer) on a rotating wheel at 4°C. Following aspiration of the solution containing unbound proteins, the beads were washed 3 times for 5 minutes with 1 ml of the lysis buffer on a rotating wheel at 4°C. Eventually, bound proteins were stripped off the beads with 100 µl of the elution buffer (50 mM-Tris-HCl, pH = 8.0; 10% glycerol; 3% SDS; 50 mM DTT; 1 mM PMSF; 1x Protease Inhibitor Cocktail) in a thermomixer set at 100°C for 7 minutes. Fifteen µl aliquot of the eluate was analyzed by a standard SDS-PAGE electrophoresis and silver-staining of the gel. Proteins from the remainder of the eluate were precipitated by chloroform-methanol method.68
Mass spectrometry analysis
Precipitated proteins were dissolved in 100 μl of 100 mM ammonium bicarbonate buffer and digested overnight with 10 ng/ml trypsin (Promega) at 37°C. The peptides were reduced in 10 mM DTT for 30 min at RT and alkylated in 55 mM iodoacetamide for 20 min at RT. Finally, trifluoroacetic acid was added at a final concentration of 0.1%. MS analysis was performed by LC-MS in the Laboratory of Mass Spectrometry (IBB PAS, Warsaw) using a nanoAcquity UPLC system (Waters) coupled to an LTQ-Orbitrap Velos mass spectrometer (Thermo Scientific). Peptides were separated by a 160-min linear gradient of 95% solution A (0.1% formic acid in water) to 35% solution B (acetonitrile and 0.1% formic acid). The mass spectrometer was operated in the data-dependent MS-MS2 mode, acquiring data in the m/z range of 300–2000. Data were analyzed with MaxQuant (Version 1.5.0.30) platform. We used reference proteome database from Uniprot, for both yeast and human samples. Identified proteins were analyzed in the following way: protein abundance was defined as the signal intensity calculated by MaxQuant software for a protein (sum of intensities of identified peptides of given protein) divided by its molecular weight. Specificity was defined as the ratio of protein signal intensity measured in the bait purification to background level (which is the protein signal intensity in the negative control purification; background level was arbitrarily set to 1 for proteins not detected in the negative control). High values of both protein abundance and specificity indicate a proteins that are enriched in purification, and can suggest interaction.
RNA isolation and RNA gel blot analysis
RNA was isolated from 50 ml of fresh yeast cultures (OD600≈0.5) and human HeLa Flp-In-derived cell lines (5 × 106 cells) using the standard hot acidic phenol procedure or TRI Reagent (Sigma-Aldrich), respectively. Total RNA (5–10 μg) was fractionated by electrophoresis in a 1% formaldehyde-agarose gel [prepared using either NBC buffer (50 mM boric acid; 1 mM sodium acetate; 5 mM NaOH) or, to achieve good separation of the large human pre-rRNA precursors, using TT buffer (30 mM tricine; 30 mM triethanolamine, pH= 8.0), according to ref. 69], followed by RNA immobilization on the Hybond N+ membrane (Amersham) by overnight capillary transfer in 20xSSC (3 M NaCl; 0.3 M sodium citrate). RNA was fixed on membranes by UV-crosslinking. Hybridizations were performed in PerfectHyb Plus hybridization buffer (Sigma-Aldrich). The blots were handled according to standard procedures and probed at 42°C (5′-labeled oligonucleotide probes) or 63°C (DNA fragments labeled by random-priming). Between successive hybridizations, probes were stripped off the membranes at 65°C using boiling 0.1% SDS.
For detection of hUTP24 transcript, 10 ng of gel-purified insert excised from pU24–9 plasmid with XbaI/SalI enzymes was labeled by random-priming with α-32P[dATP] and DecaLabel DNA Labeling Kit (Thermo Scientific) according to manufacturer's instructions and utilized as a probe. For all other transcripts, 32P-labeled oligonucleotides (listed in Table S2) were used as probes. After hybridization, membranes were washed with 2xSSC, 0.1% SDS in the appropriate temperature and eventually exposed to a PhosphorImager screen (FujiFilm), which was scanned following exposure using a FLA 7000 scanner (FujiFilm).
Primer extension analysis
DNA oligonucleotides for primer extension were labeled with T4 PNK (NEB) and 2.5 µl [γ-32P]ATP (Hartmann Analytic; 3000 Ci/mmol) in a total volume of 10 µl, according to the instructions of the enzyme's manufacturer. Reaction was terminated at 90°C for 5 minutes. Labeled DNA was diluted to 50 µl with sterile deionised water and extracted once with an equal volume of phenol:chloroform (1:1). Upper phase was transferred to a new tube and DNA was precipitated with 500 µl of isopropanol in the presence of 5 µl of 3M sodium acetate (pH = 5.2) and 1 µl of RNase-free glycogen (20 mg/ml). Following wash with 80% ethanol and air-drying, DNA pellet was dissolved in 20 µl of RNase-free water.
Four µg of total RNA was mixed with 2 µl of 5′-labeled DNA oligo and 2 µl of 5× ssHyb buffer (50 mM Tris-HCl, pH = 7.5; 1.5 M NaCl; 10 mM EDTA, pH = 8.0) in a total volume of 10 µl. The mixture was incubated at 85°C for 7 minutes and then the thermoblock was set to 42°C to allow annealing of the primer to RNA. In the meantime, a master mix containing 26.75 µl of RNase-free water, 6.25 µl of 10× AMV RT buffer (NEB), 5 µl of 10 mM dNTP (dATP, dGTP, dCTP, dTTP) Mix, 1 µl of RiboLock™ RNase Inhibitor (ThermoScientific) and 1 µl of AMV Reverse Transcriptase (NEB) per sample, was prepared and pre-warmed at 42°C. After annealing, 40 µl of the master mix was added to the sample and the incubation was carried out at 42°C for 90 min. Reaction was stopped at 95°C for 5 minutes and the products was subjected to single extraction with an equal volume of phenol:chloroform (1:1). DNA was precipitated, washed, dried and dissolved as described above. Subsequently 20 µl of formamide loading dye (90% formamide in 1x TBE; 0.03% xylene cyanol; 0.03% bromophenol blue).
DNA templates for generation of sequencing ladders were prepared by linearization of pU24–13, pU24–14, pU24–15 and pU24–16 plasmids (for sequencing with primers y18S, yITS1b, y25S and hA1 respectively) with BamHI or PstI restriction enzyme, depending on the orientation of the insert. Following standard phenol:chloroform:isoamyl alcohol and chloroform extractions, DNA was precipitated and washed with ethanol and resuspended in a sterile water. Five µg of linearized plasmid were subjected to alkaline denaturation by adding 1/10th volumes of solution containing 2 M NaOH and 2 mM EDTA and incubating for 30 min at 37°C, followed by neutralization with 1/10th volumes of 3 M sodium acetate, pH = 5.2 and DNA precipitation with 2.5 volumes of 96% ethanol for 15 min at −80°C. Single-stranded DNA templates prepared this way were eventually resuspended in 7 µl of sterile water. Sequencing reaction were carried out using Sequenase™ Version 2.0 DNA Sequencing Kit (USB/Affymetrix), according to the manufacturer's instructions. Three µg of the appropriate ssDNA template and 1 – pmol of DNA oligonucleotide (the same as present in the respective primer extension reaction) were used for sequencing. We utilized dGTP termination mixes and [α-32P]dATP for labeling. 10× diluted labeling mix and Mn buffer were used to enable more facile reading of the sequences located close to the sequencing primer.
In cases when sequencing with a given primer did not work, a molecular weight marker was prepared instead by 5′-labeling of 10 bp DNA Ladder (Invitrogen) with T4 PNK and [γ-32P]ATP to enable estimation of the reverse transcription products' size.
Eventually, products of primer extension and sequencing reactions (or a molecular weight marker) were analyzed in parallel by electrophoresis in 6% denaturing polyacrylamide-urea gels (0.4 mm thick). Following electrophoretic separation, the gel was transferred onto Whatman 3MM filter paper, covered with saran wrap and dried for 90 minutes at 70°C under vacuum. The gel was exposed to a PhosphorImager screen, which was subsequently scanned using a FLA 7000 scanner.
RNA metabolic labeling
HeLa Flp-In T-REx-derived cell lines were grown on 60 mm plates in a standard DMEM medium, either lacking doxycycline or containing the inducer, until reaching ∼80% confluence. The cells were then pre-incubated for 60 minutes in DMEM without phosphates (Gibco), supplemented with 10% FBS and antibiotics. Subsequently, [32P]orthophosphoric acid (15 µCi/ml) was added to the medium and the plates were incubated with isotope for 60 minutes before changing the medium for a standard DMEM. Cells were harvested in PBS following trypsin digestion and collected by centrifugation at the following time-points after final medium change: 0.5, 1, 2, 3, 4 and 5 h. Total RNA was extracted using TRI reagent and resolved in 1% agarose-formaldehyde gel or in 6% polyacrylamide-urea gel. RNA was then transferred onto Hybond N+ membrane and the results were analyzed by phosphorimaging.
Preparation of cytoplasmic extracts from yeast and human cells and their separation in sucrose gradients
In the case of yeast, cytoplasmic extracts for polysome analysis in the presence of low salt were prepared as follows. Yeast were grown in 200 ml of appropriate medium (lacking or containing doxycycline) at 30°C to OD600≈0.5–0.75. Next, cycloheximide was added to the final concentration of 100 µg/ml and the cultures were immediately chilled on ice. Cells were centrifuged at 3000×g for 5 minutes at 4°C and washed 3 times in ice-cold buffer A (20 mM Tris-HCl, pH = 7.5; 100 mM NaCl; 30 mM MgCl2; 100 µg/ml cycloheximide; 200 µg/ml heparin). After final wash, cell pellet was resuspended in 0.5–1 ml of buffer A and cells were lysed by 10 cycles of vortexing (45 seconds pulse/45 seconds break on ice) in the presence of 0.5 mm glass beads. Cell lysate was cleared by centrifugation at 21000xg for 15 minutes at 4°C and RNA concentration in collected supernatant was measured using Nanodrop 2000c device (Thermo Scientific). Twelve OD260 units of cytoplasmic extract in 500 μl of buffer A were layered onto 7–47% (w/v) sucrose gradient (prepared using filtered sucrose solutions in buffer B: 50 mM Tris-HCl, pH = 7.5; 50 mM KCl; 12 mM MgCl2; 1 mM DTT) and ultracentrifuged for 3 hours at 39000 rpm, 4°C in SW-41Ti rotor (Beckman Coulter). Subsequently, 0.5 ml fractions were collected from each gradient by pumping 60% sucrose solution (prepared as above) to the bottom of tubes and OD260 was monitored on ÄKTA Purifier.
Yeast cytoplasmic extracts for analysis of ribosomal subunit stoichiometry were prepared as above (omitting cycloheximide addition), either in the absence of Mg2+, using low-salt buffer C (50 mM Tris-HCl, pH = 7.5; 50 mM NaCl; 1 mM DTT) or in the presence of Mg2+, but employing high-salt buffer D (50 mM Tris-HCl, pH = 7.5; 800 mM KCl; 10 mM MgCl2; 1 mM DTT). In either case, 5 OD260 units of cytoplasmic extract were separated by ultracentrifugation in 10–30% (w/v) sucrose gradient, made with buffer E (50 mM Tris-HCl; 50 mM KCl; 1 mM DTT) or with buffer D, respectively, for 6 hours at 39000 rpm, 4°C in SW-41Ti rotor. Gradient fractions were collected (using high-density sucrose solutions in respective buffer) and analyzed as described above.
For human cells, cytoplasmic extracts for polysome analysis were prepared as follows. Stable cell lines were grown in a medium lacking or containing doxycycline on one ø145 mm plate until reaching ∼95% confluence. Cells were treated with cycloheximide (200 μg/ml) at 37°C for 15 minutes, harvested by trypsinization, spun down for 1 minute at 500×g; 4°C and then washed 3 times with ice-cold PBS containing 100 μg/ml cycloheximide. After final wash and complete removal of PBS, the cells were suspended in 0.5 ml of lysis buffer Ah [10 mM Hepes-KOH, pH = 7.5; 100 mM KCl; 2.5 mM MgCl2; 1 mM DTT; 100 μg/ml cycloheximide; 1 mg/ml heparin (Sigma-Aldrich); 1% reduced Igepal-CA630 (Sigma-Aldrich); 80 u/ml RiboLock™ RNase Inhibitor (Thermo Scientific); 1 × Protease Inhibitor Cocktail, Complete EDTA-free (Roche)], lysed by thorough pipetting and incubation for 15 minutes at 4°C on a rotating wheel. Lysates were then centrifuged for 10 minutes at 10000×g; 4°C and RNA concentration in collected supernatants was measured using Nanodrop. Twelve OD260 units of cytoplasmic extracts in 500 μl of lysis buffer were ultracentrifuged in 7–47% sucrose gradients (prepared using filtered sucrose solutions in lysis buffer, lacking detergent and ribonuclease inhibitor), followed by collection of the fractions and analysis as described above. RNA was extracted from gradient fractions with TRI reagent and resolved in denaturing agarose-formaldehyde gel.
Cytoplasmic extracts from human cells for analysis of ribosomal subunit stoichiometry were prepared as above (omitting cycloheximide addition), either in the absence of Mg2+, with low-salt buffer Ch (50 mM Hepes-KOH, pH = 7.5; 50 mM KCl; 1 mM DTT) or in the presence of Mg2+, but with high-salt buffer Dh (50 mM Hepes-KOH, pH = 7.5; 800 mM KCl; 2.5 mM MgCl2; 1 mM DTT). In either case, reduced Igepal-CA630 was added to cell suspension (final concentration 0.4%) prior to centrifugation; 5 OD260 units of cytoplasmic extract were separated by ultracentrifugation in 10–30% (w/v) sucrose gradient, made with buffer Ch or with buffer Dh, respectively, for 6 hours at 39000 rpm, 4°C in SW-41Ti rotor. Gradient fractions were collected (using high-density sucrose solutions in respective buffer) and analyzed as described above.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
Authors would like to thank Prof. Joanna Kufel for critical reading of the manuscript and Dr Pawel Sikorski for insightful discussions. Katarzyna Kowalska is acknowledged for excellent technical help with construction of the plasmids. We are indebted to Krystian Stodus and Kamil Kobylecki for their attempts to purify UTP24 proteins and demonstrate their enzymatic activity in vitro.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Authors' Contributions
RT conceived the study, designed the experiments, analyzed the data and wrote the manuscript. AL established stable HeLa Flp-In T-Rex-derived cell lines for the analysis of hUTP24 function and performed the experiments shown in Figure 6D and Supplemental Figures 6A, B, and D, as well as contributed to primer extension analyses. KD generated ADZY799 and ADZY800 strains, purified yUtp24 variants from yeast and participated in RNA metabolic labeling experiments and in northern-blot analyses. DC analyzed mass-spectrometry data. All remaining experiments were carried out by RT. AD participated in data interpretation and writing the manuscript.
Funding
This work was supported by the National Science Center within the grant allocated to RT on the basis of the decision number DEC-2011/01/D/NZ1/03510. Experiments were carried out with the use of CePT infrastructure that was financed by the European Union via the European Regional Development Fund (Innovative economy 2007–13, Agreement POIG.02.02.00–14–024/08–00). KD is the recipient of the “START” Stipend for Young Researchers from the Foundation for Polish Science. AD is a recipient of fellowships form Foundation for Polish Science (“Master” and “Ideas for Poland”).
References
- 1.Warner JR. The economics of ribosome biosynthesis in yeast. Trends Biochem Sci 1999; 24:437-40; PMID:10542411; http://dx.doi.org/ 10.1016/S0968-0004(99)01460-7 [DOI] [PubMed] [Google Scholar]
- 2.Venema J, Tollervey D. Processing of pre-ribosomal RNA in Saccharomyces cerevisiae. Yeast 1995; 11:1629-50; PMID:8720068; http://dx.doi.org/ 10.1002/yea.320111607 [DOI] [PubMed] [Google Scholar]
- 3.Tschochner H, Hurt E. Pre-ribosomes on the road from the nucleolus to the cytoplasm. Trends Cell Biol 2003; 13:255-63; PMID:12742169; http://dx.doi.org/ 10.1016/S0962-8924(03)00054-0 [DOI] [PubMed] [Google Scholar]
- 4.Woolford JL Jr., Baserga SJ. Ribosome biogenesis in the yeast Saccharomyces cerevisiae. Genetics 2013; 195:643-81; PMID:24190922; http://dx.doi.org/ 10.1534/genetics.113.153197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fernandez-Pevida A, Kressler D, de la Cruz J. Processing of preribosomal RNA in Saccharomyces cerevisiae. Wiley Interdiscip Rev RNA 2015; 6:191-209; PMID:25327757; http://dx.doi.org/ 10.1002/wrna.1267 [DOI] [PubMed] [Google Scholar]
- 6.Kressler D, Hurt E, Bassler J. Driving ribosome assembly. Biochim Biophys Acta 2010; 1803:673-83. PMID:19879902; http://dx.doi.org/ 10.1016/j.bbamcr.2009.10.009 [DOI] [PubMed] [Google Scholar]
- 7.Mullineux ST, Lafontaine DL. Mapping the cleavage sites on mammalian pre-rRNAs: where do we stand? Biochimie 2012; 94:1521-32; PMID:22342225; http://dx.doi.org/ 10.1016/j.biochi.2012.02.001 [DOI] [PubMed] [Google Scholar]
- 8.Henras AK, Plisson-Chastang C, O'Donohue MF, Chakraborty A, Gleizes PE. An overview of pre-ribosomal RNA processing in eukaryotes. Wiley Interdiscip Rev RNA 2015; 6:225-42; PMID:25346433; http://dx.doi.org/ 10.1002/wrna.1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kass S, Craig N, Sollner-Webb B. Primary processing of mammalian rRNA involves two adjacent cleavages and is not species specific. Mol Cell Biol 1987; 7:2891-8; PMID:3670298; http://dx.doi.org/ 10.1128/MCB.7.8.2891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rouquette J, Choesmel V, Gleizes PE. Nuclear export and cytoplasmic processing of precursors to the 40S ribosomal subunits in mammalian cells. EMBO J 2005; 24:2862-72; PMID:16037817; http://dx.doi.org/ 10.1038/sj.emboj.7600752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Preti M, O'Donohue MF, Montel-Lehry N, Bortolin-Cavaille ML, Choesmel V, Gleizes PE. Gradual processing of the ITS1 from the nucleolus to the cytoplasm during synthesis of the human 18S rRNA. Nucleic Acids Res 2013; 41:4709-23; PMID:23482395; http://dx.doi.org/ 10.1093/nar/gkt160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carron C, O'Donohue MF, Choesmel V, Faubladier M, Gleizes PE. Analysis of two human pre-ribosomal factors, bystin and hTsr1, highlights differences in evolution of ribosome biogenesis between yeast and mammals. Nucleic Acids Res 2011; 39:280-91; PMID:20805244; http://dx.doi.org/ 10.1093/nar/gkq734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morello LG, Hesling C, Coltri PP, Castilho BA, Rimokh R, Zanchin NI. The NIP7 protein is required for accurate pre-rRNA processing in human cells. Nucleic Acids Res 2011; 39:648-65; PMID:20798176; http://dx.doi.org/ 10.1093/nar/gkq758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morello LG, Coltri PP, Quaresma AJ, Simabuco FM, Silva TC, Singh G, Nickerson JA, Oliveira CC, Moore MJ, Zanchin NI. The human nucleolar protein FTSJ3 associates with NIP7 and functions in pre-rRNA processing. PloS One 2011; 6:e29174; PMID:22195017; http://dx.doi.org/ 10.1371/journal.pone.0029174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hadjiolova KV, Nicoloso M, Mazan S, Hadjiolov AA, Bachellerie JP. Alternative pre-rRNA processing pathways in human cells and their alteration by cycloheximide inhibition of protein synthesis. Eur J Biochem 1993; 212:211-5; PMID:8444156; http://dx.doi.org/ 10.1111/j.1432-1033.1993.tb17652.x [DOI] [PubMed] [Google Scholar]
- 16.Sloan KE, Mattijssen S, Lebaron S, Tollervey D, Pruijn GJ, Watkins NJ. Both endonucleolytic and exonucleolytic cleavage mediate ITS1 removal during human ribosomal RNA processing. J Cell Biol 2013; 200:577-88; PMID:23439679; http://dx.doi.org/ 10.1083/jcb.201207131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kass S, Tyc K, Steitz JA, Sollner-Webb B. The U3 small nucleolar ribonucleoprotein functions in the first step of preribosomal RNA processing. Cell 1990; 60:897-908; PMID:2156625; http://dx.doi.org/ 10.1016/0092-8674(90)90338-F [DOI] [PubMed] [Google Scholar]
- 18.Venema J, Tollervey D. Ribosome synthesis in Saccharomyces cerevisiae. Annual Rev Genet 1999; 33:261-311; PMID:10690410; http://dx.doi.org/ 10.1146/annurev.genet.33.1.261 [DOI] [PubMed] [Google Scholar]
- 19.Kufel J, Dichtl B, Tollervey D. Yeast Rnt1p is required for cleavage of the pre-ribosomal RNA in the 3′ ETS but not the 5′ ETS. RNA 1999; 5:909-17; PMID:10411134; http://dx.doi.org/ 10.1017/S135583829999026X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elela SA, Igel H, Ares M Jr.. RNase III cleaves eukaryotic preribosomal RNA at a U3 snoRNP-dependent site. Cell 1996; 85:115-24; PMID:8620530; http://dx.doi.org/ 10.1016/S0092-8674(00)81087-9 [DOI] [PubMed] [Google Scholar]
- 21.Idol RA, Robledo S, Du HY, Crimmins DL, Wilson DB, Ladenson JH, Bessler M, Mason PJ. Cells depleted for RPS19, a protein associated with Diamond Blackfan Anemia, show defects in 18S ribosomal RNA synthesis and small ribosomal subunit production. Blood Cells Mol Dis 2007; 39:35-43; PMID:17376718; http://dx.doi.org/ 10.1016/j.bcmd.2007.02.001 [DOI] [PubMed] [Google Scholar]
- 22.Lygerou Z, Allmang C, Tollervey D, Seraphin B. Accurate processing of a eukaryotic precursor ribosomal RNA by ribonuclease MRP in vitro. Science 1996; 272:268-70; PMID:8602511; http://dx.doi.org/ 10.1126/science.272.5259.268 [DOI] [PubMed] [Google Scholar]
- 23.Lygerou Z, Mitchell P, Petfalski E, Seraphin B, Tollervey D. The POP1 gene encodes a protein component common to the RNase MRP and RNase P ribonucleoproteins. Genes Dev 1994; 8:1423-33; PMID:7926742; http://dx.doi.org/ 10.1101/gad.8.12.1423 [DOI] [PubMed] [Google Scholar]
- 24.Kent T, Lapik YR, Pestov DG. The 5′ external transcribed spacer in mouse ribosomal RNA contains two cleavage sites. RNA 2009; 15:14-20; PMID:19029311; http://dx.doi.org/ 10.1261/rna.1384709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hannon GJ, Maroney PA, Branch A, Benenfield BJ, Robertson HD, Nilsen TW. Accurate processing of human pre-rRNA in vitro. Mol Cell Biol 1989; 9:4422-31; PMID:2586517; http://dx.doi.org/ 10.1128/MCB.9.10.4422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu YT, Nilsen TW. Sequence requirements for maturation of the 5′ terminus of human 18 S rRNA in vitro. J Biol Chem 1992; 267:9264-8; PMID:1577760 [PubMed] [Google Scholar]
- 27.Fatica A, Oeffinger M, Dlakic M, Tollervey D. Nob1p is required for cleavage of the 3′ end of 18S rRNA. Mol Cell Biol 2003; 23:1798-807; PMID:12588997; http://dx.doi.org/ 10.1128/MCB.23.5.1798-1807.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fatica A, Tollervey D, Dlakic M. PIN domain of Nob1p is required for D-site cleavage in 20S pre-rRNA. RNA 2004; 10:1698-701; PMID:15388878; http://dx.doi.org/ 10.1261/rna.7123504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pertschy B, Schneider C, Gnadig M, Schafer T, Tollervey D, Hurt E. RNA helicase Prp43 and its co-factor Pfa1 promote 20 to 18 S rRNA processing catalyzed by the endonuclease Nob1. J Biol Chem 2009; 284:35079-91; PMID:19801658; http://dx.doi.org/ 10.1074/jbc.M109.040774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lamanna AC, Karbstein K. Nob1 binds the single-stranded cleavage site D at the 3′-end of 18S rRNA with its PIN domain. Proc Natl Acad Sci USA 2009; 106:14259-64; PMID:19706509; http://dx.doi.org/ 10.1073/pnas.0905403106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lamanna AC, Karbstein K. An RNA conformational switch regulates pre-18S rRNA cleavage. J Mol Biol 2011; 405:3-17; PMID:20934433; http://dx.doi.org/ 10.1016/j.jmb.2010.09.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hughes JM, Ares M Jr.. Depletion of U3 small nucleolar RNA inhibits cleavage in the 5′ external transcribed spacer of yeast pre-ribosomal RNA and impairs formation of 18S ribosomal RNA. EMBO J 1991; 10:4231-9. PMID:1756730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O'Donohue MF, Choesmel V, Faubladier M, Fichant G, Gleizes PE. Functional dichotomy of ribosomal proteins during the synthesis of mammalian 40S ribosomal subunits. J Cell Biol 2010; 190:853-66; PMID:20819938; http://dx.doi.org/ 10.1083/jcb.201005117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Choesmel V, Fribourg S, Aguissa-Toure AH, Pinaud N, Legrand P, Gazda HT, Gleizes PE. Mutation of ribosomal protein RPS24 in Diamond-Blackfan anemia results in a ribosome biogenesis disorder. Hum Mol Genet 2008; 17:1253-63; PMID:18230666; http://dx.doi.org/ 10.1093/hmg/ddn015 [DOI] [PubMed] [Google Scholar]
- 35.Geerlings TH, Vos JC, Raue HA. The final step in the formation of 25S rRNA in Saccharomyces cerevisiae is performed by 5′–3′ exonucleases. RNA 2000; 6:1698-703; PMID:11142370; http://dx.doi.org/ 10.1017/S1355838200001540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang M, Pestov DG. 5′-end surveillance by Xrn2 acts as a shared mechanism for mammalian pre-rRNA maturation and decay. Nucleic Acids Res 2011; 39:1811-22; PMID:21036871; http://dx.doi.org/ 10.1093/nar/gkq1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tafforeau L, Zorbas C, Langhendries JL, Mullineux ST, Stamatopoulou V, Mullier R, Wacheul L, Lafontaine DL. The complexity of human ribosome biogenesis revealed by systematic nucleolar screening of Pre-rRNA processing factors. Mol Cell 2013; 51:539-51; PMID:23973377; http://dx.doi.org/ 10.1016/j.molcel.2013.08.011 [DOI] [PubMed] [Google Scholar]
- 38.Venema J, Henry Y, Tollervey D. Two distinct recognition signals define the site of endonucleolytic cleavage at the 5′-end of yeast 18S rRNA. EMBO J 1995; 14:4883-92; PMID:7588617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bleichert F, Granneman S, Osheim YN, Beyer AL, Baserga SJ. The PINc domain protein Utp24, a putative nuclease, is required for the early cleavage steps in 18S rRNA maturation. Proc Natl Acad Sci USA 2006; 103:9464-9; PMID:16769905; http://dx.doi.org/ 10.1073/pnas.0603673103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rempola B, Karkusiewicz I, Piekarska I, Rytka J. Fcf1p and Fcf2p are novel nucleolar Saccharomyces cerevisiae proteins involved in pre-rRNA processing. Biochem Biophys Res Commun 2006; 346:546-54; PMID:16762320; http://dx.doi.org/ 10.1016/j.bbrc.2006.05.140 [DOI] [PubMed] [Google Scholar]
- 41.Horn DM, Mason SL, Karbstein K. Rcl1 protein, a novel nuclease for 18 S ribosomal RNA production. J Biol Chem 2011; 286:34082-7; PMID:21849504; http://dx.doi.org/ 10.1074/jbc.M111.268649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tanaka N, Smith P, Shuman S. Crystal structure of Rcl1, an essential component of the eukaryal pre-rRNA processosome implicated in 18S rRNA biogenesis. RNA 2011; 17:595-602; PMID:21367972; http://dx.doi.org/ 10.1261/rna.2571811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lebreton A, Tomecki R, Dziembowski A, Seraphin B. Endonucleolytic RNA cleavage by a eukaryotic exosome. Nature 2008; 456:993-6; PMID:19060886; http://dx.doi.org/ 10.1038/nature07480 [DOI] [PubMed] [Google Scholar]
- 44.Schaeffer D, Tsanova B, Barbas A, Reis FP, Dastidar EG, Sanchez-Rotunno M, Arraiano CM, van Hoof A. The exosome contains domains with specific endoribonuclease, exoribonuclease and cytoplasmic mRNA decay activities. Nat Struct Mol Biol 2009; 16:56-62; PMID:19060898; http://dx.doi.org/ 10.1038/nsmb.1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eberle AB, Lykke-Andersen S, Muhlemann O, Jensen TH. SMG6 promotes endonucleolytic cleavage of nonsense mRNA in human cells. Nat Struct Mol Biol 2009; 16:49-55; PMID:19060897; http://dx.doi.org/ 10.1038/nsmb.1530 [DOI] [PubMed] [Google Scholar]
- 46.Huntzinger E, Kashima I, Fauser M, Sauliere J, Izaurralde E. SMG6 is the catalytic endonuclease that cleaves mRNAs containing nonsense codons in metazoan. RNA 2008; 14:2609-17; PMID:18974281; http://dx.doi.org/ 10.1261/rna.1386208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Skruzny M, Schneider C, Racz A, Weng J, Tollervey D, Hurt E. An endoribonuclease functionally linked to perinuclear mRNP quality control associates with the nuclear pore complexes. PLoS Biol 2009; 7:e8; PMID:19127978; http://dx.doi.org/ 10.1371/journal.pbio.1000008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Karkusiewicz I, Rempola B, Gromadka R, Grynberg M, Rytka J. Functional and physical interactions of Faf1p, a Saccharomyces cerevisiae nucleolar protein. Biochem Biophys Res Commun 2004; 319:349-57; PMID:15178413; http://dx.doi.org/ 10.1016/j.bbrc.2004.05.012 [DOI] [PubMed] [Google Scholar]
- 49.Lim YH, Charette JM, Baserga SJ. Assembling a protein-protein interaction map of the SSU processome from existing datasets. PloS One 2011; 6:e17701; PMID:21423703; http://dx.doi.org/ 10.1371/journal.pone.0017701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Phipps KR, Charette J, Baserga SJ. The small subunit processome in ribosome biogenesis-progress and prospects. Wiley Interdiscip Rev RNA 2011; 2:1-21; PMID:21318072; http://dx.doi.org/ 10.1002/wrna.57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Robledo S, Idol RA, Crimmins DL, Ladenson JH, Mason PJ, Bessler M. The role of human ribosomal proteins in the maturation of rRNA and ribosome production. RNA 2008; 14:1918-29; PMID:18697920; http://dx.doi.org/ 10.1261/rna.1132008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sloan KE, Bohnsack MT, Schneider C, Watkins NJ. The roles of SSU processome components and surveillance factors in the initial processing of human ribosomal RNA. RNA 2014; 20:540-50; PMID:24550520; http://dx.doi.org/ 10.1261/rna.043471.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang M, Anikin L, Pestov DG. Two orthogonal cleavages separate subunit RNAs in mouse ribosome biogenesis. Nucleic Acids Res 2014; 42:11180-91; PMID:25190460; http://dx.doi.org/ 10.1093/nar/gku787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tomecki R, Drazkowska K, Kucinski I, Stodus K, Szczesny RJ, Gruchota J, Owczarek EP, Kalisiak K, Dziembowski A. Multiple myeloma-associated hDIS3 mutations cause perturbations in cellular RNA metabolism and suggest hDIS3 PIN domain as a potential drug target. Nucleic Acids Res 2014; 42:1270-90; PMID:24150935; http://dx.doi.org/ 10.1093/nar/gkt930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Oeffinger M, Zenklusen D, Ferguson A, Wei KE, El Hage A, Tollervey D, Chait BT, Singer RH, Rout MP. Rrp17p is a eukaryotic exonuclease required for 5′ end processing of Pre-60S ribosomal RNA. Mol Cell 2009; 36:768-81; PMID:20005841; http://dx.doi.org/ 10.1016/j.molcel.2009.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chang JH, Jiao X, Chiba K, Oh C, Martin CE, Kiledjian M, Tong L. Dxo1 is a new type of eukaryotic enzyme with both decapping and 5′–3′ exoribonuclease activity. Nat Struct Mol Biol 2012; 19:1011-7; PMID:22961381; http://dx.doi.org/ 10.1038/nsmb.2381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jiao X, Chang JH, Kilic T, Tong L, Kiledjian M. A mammalian pre-mRNA 5′ end capping quality control mechanism and an unexpected link of capping to pre-mRNA processing. Mol Cell 2013; 50:104-15; PMID:23523372; http://dx.doi.org/ 10.1016/j.molcel.2013.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hughes JM. Functional base-pairing interaction between highly conserved elements of U3 small nucleolar RNA and the small ribosomal subunit RNA. J Mol Biol 1996; 259:645-54; PMID:8683571; http://dx.doi.org/ 10.1006/jmbi.1996.0346 [DOI] [PubMed] [Google Scholar]
- 59.Dutca LM, Gallagher JE, Baserga SJ. The initial U3 snoRNA:pre-rRNA base pairing interaction required for pre-18S rRNA folding revealed by in vivo chemical probing. Nucleic Acids Res 2011; 39:5164-80; PMID:21349877; http://dx.doi.org/ 10.1093/nar/gkr044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Borovjagin AV, Gerbi SA. U3 small nucleolar RNA is essential for cleavage at sites 1, 2 and 3 in pre-rRNA and determines which rRNA processing pathway is taken in Xenopus oocytes. J Mol Biol 1999; 286:1347-63; PMID:10064702; http://dx.doi.org/ 10.1006/jmbi.1999.2527 [DOI] [PubMed] [Google Scholar]
- 61.Borovjagin AV, Gerbi SA. Xenopus U3 snoRNA GAC-Box A' and Box A sequences play distinct functional roles in rRNA processing. Mol Cell Biol 2001; 21:6210-21; PMID:1150966; http://dx.doi.org/ 10.1128/MCB.21.18.6210-6221.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hartshorne T, Toyofuku W. Two 5′-ETS regions implicated in interactions with U3 snoRNA are required for small subunit rRNA maturation in Trypanosoma brucei. Nucleic Acids Res 1999; 27:3300-9; PMID:10454637; http://dx.doi.org/ 10.1093/nar/27.16.3300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sharma K, Venema J, Tollervey D. The 5′ end of the 18S rRNA can be positioned from within the mature rRNA. RNA 1999; 5:678-86; PMID:10334338; http://dx.doi.org/ 10.1017/S1355838299990052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gerczei T, Correll CC. Imp3p and Imp4p mediate formation of essential U3-precursor rRNA (pre-rRNA) duplexes, possibly to recruit the small subunit processome to the pre-rRNA. Proc Natl Acad Sci USA 2004; 101:15301-6; PMID:15489263; http://dx.doi.org/ 10.1073/pnas.0406819101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gerczei T, Shah BN, Manzo AJ, Walter NG, Correll CC. RNA chaperones stimulate formation and yield of the U3 snoRNA-Pre-rRNA duplexes needed for eukaryotic ribosome biogenesis. J Mol Biol 2009; 390:991-1006; PMID:19482034; http://dx.doi.org/ 10.1016/j.jmb.2009.05.072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shah BN, Liu X, Correll CC. Imp3 unfolds stem structures in pre-rRNA and U3 snoRNA to form a duplex essential for small subunit processing. RNA 2013; 19:1372-83; PMID:23980203; http://dx.doi.org/ 10.1261/rna.039511.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BM, Strein C, Davey NE, Humphreys DT, Preiss T, Steinmetz LM, et al.. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 2012; 149:1393-406; PMID:22658674; http://dx.doi.org/ 10.1016/j.cell.2012.04.031 [DOI] [PubMed] [Google Scholar]
- 68.Wessel D, Flugge UI. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal Biochem 1984; 138:141-3; PMID:6731838; http://dx.doi.org/ 10.1016/0003-2697(84)90782-6 [DOI] [PubMed] [Google Scholar]
- 69.Mansour FH, Pestov DG. Separation of long RNA by agarose-formaldehyde gel electrophoresis. Anal Biochem 2013; 441:18-20; PMID:23800830; http://dx.doi.org/ 10.1016/j.ab.2013.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.