

Abstract
To maintain genome stability, mammalian cells have developed a delicate, yet efficient, system to sense and repair damaged DNA, including two evolutionarily conserved DNA damage repair (DDR) pathways: homologous recombination (HR) and non-homologous-end-joining (NHEJ). Deregulation in these repair pathways may lead to genomic instability and subsequent human diseases, including cancer. On the other hand, hyper-activation of the oncogenic Akt signaling pathway has been observed in almost all solid tumors. Emerging evidence has begun to reveal a possible role of active Akt in regulating DDR, possibly through suppression of HR. However, whether and how Akt regulates NHEJ remains largely undefined. To this end, we recently reported that Akt impairs NHEJ by phosphorylating XLF at T181, to trigger its dissociation from the functional DNA ligase IV (LIG4)/XRCC4 complex. Here, we provide an additional perspective discussing how Akt is activated upon DNA damage to regulate DNA repair pathways as well as the cellular apoptotic responses.
Keywords: Akt, cell cycle, cell signaling, DNA repair, HR, NHEJ, Oncogene, phophorylation, signaling, tumorigenesis, XLF
DDR Pathways in Eukaryotic Cells
In normal somatic cells, many DNA-damaging lesions occur daily. These DNA lesions may result from either endogenous cellular processes such as DNA replication and transcription, or various exogenous challenges, including ionizing radiation (IR) and ultraviolet (UV) light. The damaged DNA may lead to genomic instability by forming mismatched DNA base pairs, single-strand breaks (SSBs), or double-strand breaks (DSBs) that are the most lethal type of DNA lesions. To antagonize these threats triggered by various types of DNA damage, cells have evolved multiple DDR repair mechanisms specific for different types of DNA lesions. To date, at least five major DDR pathways have been identified in eukaryotic cells, including base excision repair (BER), nucleotide excision repair (NER), mismatch repair (MMR), HR and NHEJ (Fig. 1).1
Figure 1.
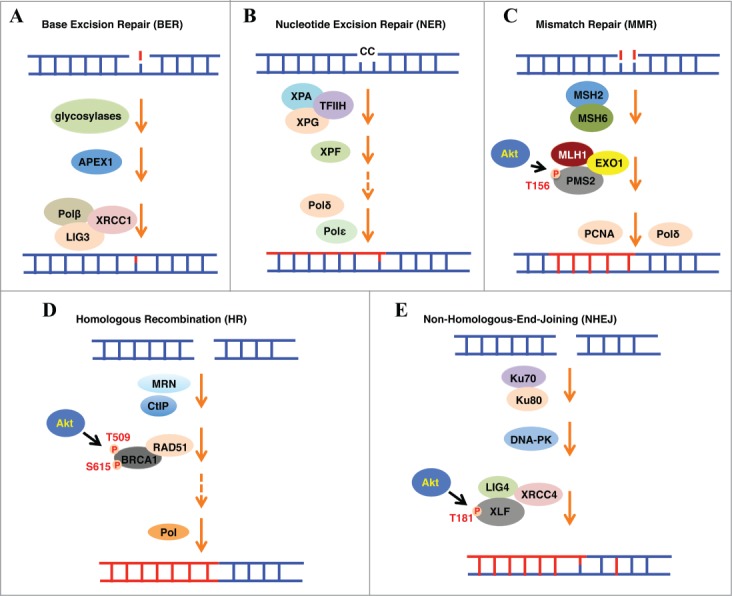
Five major DNA repair mechanisms have been identified in eukaryotic cells, including BER (A), NER (B), MMR (C), HR (D) and NHEJ (E). These pathways are comprised of multi-subunit complexes, which deal with different types of DNA lesions. On the other hand, oncogenic Akt regulates these pathways in part by phosphorylating DNA repair factors, such as phosphorylating hPMS2, BRCA1 and XLF to impair MMR, HR and NHEJ, respectively.
The BER and NER pathways are mechanistically similar. BER predominantly corrects a single damaged base, while NER fixes more complex lesions, such as pyrimidine dimers and intra-strand crosslinks. In the BER pathway, the mismatched bases could be recognized and excised by DNA glycosylases such as uracil-DNA glycosylase (UNG), resulting in an apurinic/apyrimidinic (AP) site, which can be further processed by the AP endonuclease, APEX1. The DNA polymerase β (Polβ) then fills the nucleotide gaps followed by the rejoining of the DNA ends by an enzyme complex comprising of DNA ligase III (LIG3) and XRCC1.2 The NER pathway is more complicated and comprised of more than 30 proteins, including XPA, XPG, XRCC1, XPC-Rad23B-CEN2, DDB1-XPE, and the DNA polymerase δ or ε (Polδ or Polε).3 On the other hand, the MMR pathway is responsible for correcting mismatched bases generated during DNA replication, thereby preventing the accumulation of mutations in newly duplicated genes. The major components of the MMR pathway include the recognition complex MSH2-MSH6 and MutL, the excision proteins ExoI and the DNA synthesis enzymes Polδ and PCNA.4
Importantly, HR and NHEJ pathways are mainly responsible for the repair of DSBs, the most abundant and lethal types of DNA damage in cells. HR-mediated repair is considered as an error-free mechanism and requires similar or identical parental DNA strands as repair templates, thereby is relatively restricted to the S and G2 phases of the cell cycle. On the other hand, NHEJ-directed DSBs repair is considered error-prone and actually occurs in all phases of the cell cycle. Mechanistically, HR is in large mediated by BRCA1, Mre11-Rad50-Nbs1 (MRN), CtIP, Rad51, Exo1 and BRCA2, whereas NHEJ is carried out sequentially by a set of core complexes including 53BP1, Ku70/Ku80, DNA-PKcs and LIG4/XRCC4/XLF. More recently, PAXX, homologous to XLF and XRCC4, was identified to be essential for NHEJ by directly binding Ku70/Ku80,5,6 further expanding our understanding of the signaling cascades mediating DNA damage responses. Importantly, these pathways are temporally and spatially regulated to direct the cells to make correct choices to repair different types of DNA lesions.7
Akt Is Activated in Response to DNA Damage
Akt, also known as protein kinase B (PKB), is a serine/threonine kinase that plays a critical role in promoting cell proliferation, survival and metabolism.8 In response to stimulation by various growth factors, such as insulin or IGF-1, Akt is activated through PDK1 and mTORC2-mediated phosphorylation at Thr308 and Ser473, respectively.8 More recently, accumulating evidence indicates that Akt can also be activated upon DNA damage in a PIKK family member ATM, ATR or DNA-PKcs-dependent manner (Fig. 2). Viniegra et al. demonstrated that ATM is a major upstream activator of Akt in response to insulin or γ-radiation.9 As such, cells derived from ataxia telangiectasia (AT) patients or ATM-KO mice with low ATM activity are defective in Akt-Ser473 phosphorylation upon insulin or γ-radiation treatment.9 On the other hand, Caporali et al. demonstrated that TMZ, a methylation agent activating the MMR pathway, depends on ATR, but not ATM, to trigger Akt activation.10 Furthermore, Bozulic et al. found that DNA-PKcs is required for the activation of Akt and co-localizes with Akt-pS473 at DNA damage sites in response to γ-radiation or doxorubicin.11 Although these studies suggest that ATM, ATR and DNA-PK are involved in activating Akt in response to various DNA damaging agents, it is still unclear whether these kinases directly phosphorylate Akt, or act through other adaptor proteins to indirectly promote Akt activation. Interestingly, Fraser et al. demonstrated that in prostate and lung cancer cells, DSB-induced Akt phosphorylation is dependent on the MRE11-ATM-RNF168 signaling pathway, but not DNA-PKcs, PI3K, or ATR.12 Therefore, the activation of Akt in response to DNA damage may be DNA lesion and cell context-dependent, which warrants additional in-depth investigation.
Figure 2.
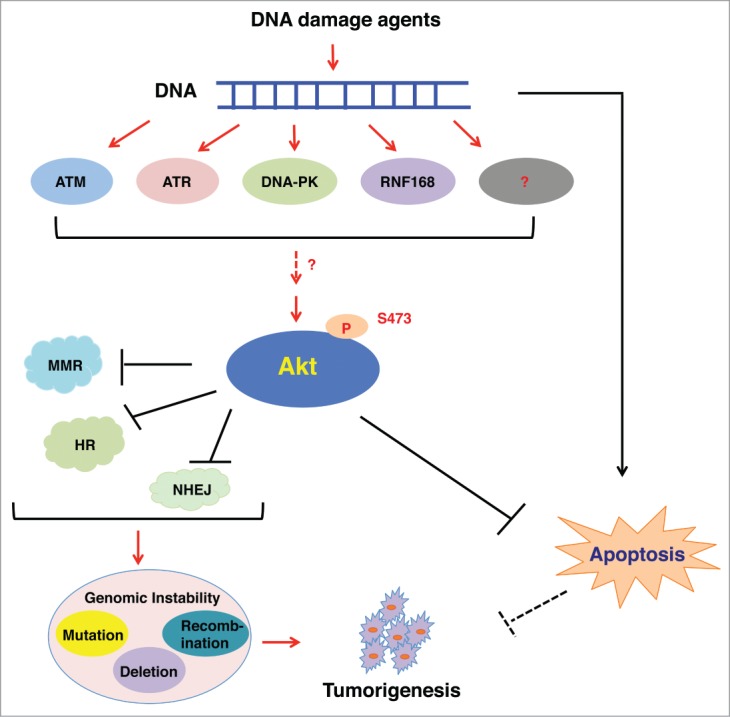
In response to DNA damage agents, Akt is activated through different mediators, including ATM, ATR, DNA-PK and RNF168. In turn, activated Akt inhibits DNA repair pathways, resulting in genomic instability, such as gene mutation, deletion and recombination. At the same time, Akt blocks the DNA damage stress-induced apoptosis to promote cell survival. As a result, these 2 events allow cells to select for favorable genetic alterations to facilitate tumorigenesis.
Akt Modulates DNA Repair Pathways
Cumulative studies have demonstrated that Akt plays a critical role in regulating DDR through directly phosphorylating multiple DNA damage repair factors in response to genotoxic agents.13 Specifically, Akt negatively regulates MMR through directly interacting with, and phosphorylating hPMS2 at Thr156, to promote hPMS2 cytoplasmic translocation and degradation (Fig. 1C). In keeping with this conclusion, inactivation of Akt by siRNA or an Akt inhibitor leads to increased nuclear accumulation of hPMS2.14 Similarly, hyper-activated Akt was also observed to impair HR. Overexpression of the constitutively active Akt inhibited BRCA1 foci formation at DNA damage sites through inducing the cytoplasmic retention of BRCA1 and Rad51 (Fig. 1D).15,16 However, Akt-dependent phosphorylation of the two canonical Akt sites, Thr509 and Ser615 within the nuclear localization sequence of BRCA1, didn't significantly affect Akt-induced BRCA1 cytoplasmic translocation. These results thus argue for a yet undefined molecular mechanism underlying Akt-mediated cytoplasmic translocation and inactivation of BRCA1 and Rad51,15,17 which warrants further investigation.
In contrast to the reported suppressive role of Akt in the MMR and HR repair processes, the function of Akt in the regulation of NHEJ is largely unknown. Notably, it has been reported that Akt could interact with and activate DNA-PK in the initiation step of NHEJ, as well as discharge DNA-PK from the DNA damage site at the late repair stage, suggesting both an activation and a suppression function for Akt in regulating NHEJ.18,19 However, the detailed mechanistic role of Akt in regulating NHEJ remains elusive. To this end, we have recently identified that Akt1 is the physiological upstream kinase for XLF phosphorylation at Thr181, but not other components of NHEJ we examined, including XRCC4 and LIG4.20 More importantly, phosphorylation of XLF at Thr181 dissociated it from the LIG4/XRCC4 complex, and triggered its interaction with 14-3-3 for XLF cytoplasmic retention (Fig. 1E). Furthermore, the SCFβ-TRCP E3 ligase could target the cytoplasmic, Thr181-phosphorylated-XLF for ubiquitination and subsequent degradation in a CKI-dependent manner. As a result, Akt-mediated XLF phosphorylation largely impairs NHEJ, leading to DSB accumulation and subsequent genomic instability.20
Notably, previous studies have demonstrated that DNA repair pathways are functionally redundant and deficiency in one pathway may be compensated by others.21 Therefore, Akt might be only one node within a regulator network that dictates the choice of DNA repair pathways by regulating the activity of various repair factors. To this end, we have recently discovered that the activity of Akt is cell cycle-dependent, with peaked activity in late S and G2/M phases,22 thus Akt may govern NHEJ and HR efficiencies in a cell cycle-dependent manner. Specifically, in the G1-phase with low Akt activity, NHEJ is the predominant repair mechanism for DSBs, whereas higher activity of Akt in the S/G2 phase may suppress NHEJ by phosphorylating XLF, thereby HR is primarily responsible for the DSB repair. However, additional in-depth studies are warranted to further examine this hypothesis.
Interestingly, a recent report also identified a Cdk1-dependent phosphorylation and inactivation mechanism on Xlf1, the fission yeast homolog of XLF/Cernunnos.23 Similar to the cellular effects of our identified Akt-mediated XLF phosphorylation event in mammalian cells, Hentges et al. demonstrated that phosphorylation of Xlf1 in fission yeast by Cdk1 inhibits NHEJ.23 Hence, NHEJ can override HR when Cdk1-mediated Xlf1 phosphorylation is inhibited, arguing for a critical role of phosphorylation of XLF in suppression of NHEJ. Intrigued by the similar consequences of XLF phosphorylation events, we have aligned human XLF and fission yeast Xlf1 sequences and interestingly observed that neither the mammalian Akt site (Thr181), nor the yeast Cdk1 sites (Thr180 and Ser192) are conserved between these two species (Fig. 3). Moreover, the Cyclin A/Cdk2-mediated Akt phosphorylation sites (Ser477/Thr479)22 are also not present in fission yeast Gad8 (homologous to mammalian Akt), indicating the cell-cycle dependent activation mechanism for Akt may not be present in yeast as well. Together, these results suggest that the Akt-mediated regulatory mechanism by governing XLF phosphorylation and suppression of NHEJ may be an event acquired during evolution.
Figure 3.
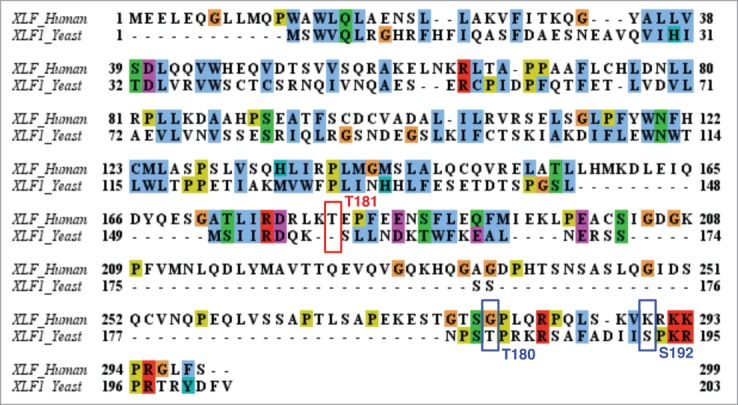
Alignment of the XLF protein sequence in human and fission yeast. The canonical Akt site (T181) in human and the Cdk1 sites (T180 and S192) in yeast are not conserved between these two species.
As in a uni-cellular organism like fission yeast, a single cell carries all cellular processes, while in multi-cellular organisms such as human, cells are subtyped and differentiated for maintaining only a certain subset of cellular functions, we speculate that the additional layer of regulation provided in mammals (the Cdk-Akt-XLF axis in mammals versus the Cdk-XLF axis in fission yeast) would allow each molecule in the cell within a multi-cell organism to be more tightly controlled to afford more diverse cellular functions. In this scenario, the Akt-mediated XLF phosphorylation mechanism may develop during the transition from single cell organism toward multi-cell organism. However, in-depth evolutionary and molecular studies are necessary to rigorously examine this hypothesis.
Akt Prevents Cellular Apoptosis Triggered by DNA Damage Stress
Previous studies have demonstrated that Akt promotes cellular survival in large part by inhibiting apoptosis. To this end, activated Akt could directly phosphorylate and inhibit the function of pro-apoptotic Bcl-2 family members, including Bad, Bax, caspase-9, GSK-3 and FOXO1.24 Non-repaired DNA lesions are reported to either cause genetic mutations, or trigger apoptosis and subsequent cell death.25 Importantly, we found that upon treatments by etoposide or bleocin to induce DSBs, Akt promotes the phosphorylation of XLF and at the same time, Akt also phosphorylates pro-apoptotic proteins including Bad and GSK-3β to exert an anti-apoptosis function. These observations suggest that Akt may suppress NHEJ and simultaneously block DNA damage-induced apoptosis, thereby providing a time window that allows cells to accumulate cancerous mutations without rendering cell death (Fig. 2).20 Interestingly, we also observed that Akt-mediated XLF phosphorylation is not critical for cell survival following treatments with apoptotic agents such as Taxol or ABT-737 that do not trigger DNA damage, indicating the specificity of Akt activation and subsequent phosphorylation of XLF events in preventing apoptosis in response to DNA damage.
Concluding Remarks
Altogether, our results demonstrate that upon DNA damage, activated Akt phosphorylates XLF to suppress NHEJ, which leads to an accumulation of genetic alterations that may facilitate tumorigenesis in part by inactivating tumor suppressor genes and/or activating oncogenes. On the other hand, Akt could also protect cells from apoptosis triggered by DNA damage stress, allowing cells to buy time to accumulate driver mutations to facilitate tumorigenesis. Together, our study provides a possible molecular mechanism for the hyper-activated Akt event in promoting genomic instability and tumorigenesis. Nonetheless, additional investigation is necessary to examine whether Akt also modulates other DNA damage repair mechanisms in addition to HR and NHEJ, as well as to fully understand the role(s) of Akt in the regulation of DNA repair. In addition, given the intensive cross-talk between the Akt and DDR pathways, it will be critical to pinpoint the upstream kinase cascades through which distinct types of DNA damage utilize activation of the Akt oncoprotein. Further understanding of these pathways will provide the rationale and guidance for future application of Akt inhibitors to combat human cancers.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
WW is an ACS scholar. PL is supported by 1K99CA181342. Our work was supported in part by the NIH grants (R01CA177910 and R01GM094777 to WW).
References
- 1.Helleday T, Eshtad S, Nik-Zainal S. Mechanisms underlying mutational signatures in human cancers. Nat Rev Genet 2014; 15:585-98; PMID:24981601; http://dx.doi.org/ 10.1038/nrg3729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zharkov DO. Base excision DNA repair. Cell Mol Life Sci 2008; 65:1544-65; PMID:18259689; http://dx.doi.org/ 10.1007/s00018-008-7543-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Costa RM, Chigancas V, Galhardo Rda S, Carvalho H, Menck CF. The eukaryotic nucleotide excision repair pathway. Biochimie 2003; 85:1083-99; PMID:14726015; http://dx.doi.org/ 10.1016/j.biochi.2003.10.017 [DOI] [PubMed] [Google Scholar]
- 4.Li GM. Mechanisms and functions of DNA mismatch repair. Cell Res 2008; 18:85-98; PMID:18157157; http://dx.doi.org/ 10.1038/cr.2007.115 [DOI] [PubMed] [Google Scholar]
- 5.Ochi T, Blackford AN, Coates J, Jhujh S, Mehmood S, Tamura N, Travers J, Wu Q, Draviam VM, Robinson CV, et al.. DNA repair. PAXX, a paralog of XRCC4 and XLF, interacts with Ku to promote DNA double-strand break repair. Science 2015; 347:185-8; PMID:25574025; http://dx.doi.org/ 10.1126/science.1261971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xing M, Yang M, Huo W, Feng F, Wei L, Jiang W, Ning S, Yan Z, Li W, Wang Q, et al.. Interactome analysis identifies a new paralogue of XRCC4 in non-homologous end joining DNA repair pathway. Nat Commun 2015; 6:6233; PMID:25670504; http://dx.doi.org/ 10.1038/ncomms7233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell 2010; 40:179-204; PMID:20965415; http://dx.doi.org/ 10.1016/j.molcel.2010.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell 2007; 129:1261-74; PMID:17604717; http://dx.doi.org/ 10.1016/j.cell.2007.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Viniegra JG, Martinez N, Modirassari P, Hernandez Losa J, Parada Cobo C, Sanchez-Arevalo Lobo VJ, Aceves Luquero CI, Alvarez-Vallina L, Ramon y Cajal S, Rojas JM, et al.. Full activation of PKB/Akt in response to insulin or ionizing radiation is mediated through ATM. J Biol Chem 2005; 280:4029-36; PMID:15546863; http://dx.doi.org/ 10.1074/jbc.M410344200 [DOI] [PubMed] [Google Scholar]
- 10.Caporali S, Levati L, Starace G, Ragone G, Bonmassar E, Alvino E, D'Atri S. AKT is activated in an ataxia-telangiectasia and Rad3-related-dependent manner in response to temozolomide and confers protection against drug-induced cell growth inhibition. Mol Pharmacol 2008; 74:173-83; PMID:18413665; http://dx.doi.org/ 10.1124/mol.107.044743 [DOI] [PubMed] [Google Scholar]
- 11.Bozulic L, Surucu B, Hynx D, Hemmings BA. PKBalpha/Akt1 acts downstream of DNA-PK in the DNA double-strand break response and promotes survival. Mol Cell 2008; 30:203-13; PMID:18439899; http://dx.doi.org/ 10.1016/j.molcel.2008.02.024 [DOI] [PubMed] [Google Scholar]
- 12.Fraser M, Harding SM, Zhao H, Coackley C, Durocher D, Bristow RG. MRE11 promotes AKT phosphorylation in direct response to DNA double-strand breaks. Cell Cycle 2011; 10:2218-32; PMID:21623170; http://dx.doi.org/ 10.4161/cc.10.13.16305 [DOI] [PubMed] [Google Scholar]
- 13.Xu N, Lao Y, Zhang Y, Gillespie DA. Akt: a double-edged sword in cell proliferation and genome stability. J Oncol 2012; 2012:951724; PMID:22481935; http://dx.doi.org/ 10.1155/2012/951724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jia J, Zhang Y, Cai J, Wang J, Ding H, Zhou J, Fang F, Wang Z. A novel function of protein kinase B as an inducer of the mismatch repair gene hPMS2 degradation. Cell Signal 2013; 25:1498-504; PMID:23499907; http://dx.doi.org/ 10.1016/j.cellsig.2013.02.021 [DOI] [PubMed] [Google Scholar]
- 15.Plo I, Laulier C, Gauthier L, Lebrun F, Calvo F, Lopez BS. AKT1 inhibits homologous recombination by inducing cytoplasmic retention of BRCA1 and RAD51. Cancer Res 2008; 68:9404-12; PMID:19010915; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-0861 [DOI] [PubMed] [Google Scholar]
- 16.Tonic I, Yu WN, Park Y, Chen CC, Hay N. Akt activation emulates Chk1 inhibition and Bcl2 overexpression and abrogates G2 cell cycle checkpoint by inhibiting BRCA1 foci. J Biol Chemi 2010; 285:23790-8; PMID:20495005; http://dx.doi.org/ 10.1074/jbc.M110.104372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Altiok S, Batt D, Altiok N, Papautsky A, Downward J, Roberts TM, Avraham H. Heregulin induces phosphorylation of BRCA1 through phosphatidylinositol 3-Kinase/AKT in breast cancer cells. J Biol Chem 1999; 274:32274-8; PMID:10542266; http://dx.doi.org/ 10.1074/jbc.274.45.32274 [DOI] [PubMed] [Google Scholar]
- 18.Toulany M, Kehlbach R, Florczak U, Sak A, Wang S, Chen J, Lobrich M, Rodemann HP. Targeting of AKT1 enhances radiation toxicity of human tumor cells by inhibiting DNA-PKcs-dependent DNA double-strand break repair. Mol Cancer Ther 2008; 7:1772-81; PMID:18644989; http://dx.doi.org/ 10.1158/1535-7163.MCT-07-2200 [DOI] [PubMed] [Google Scholar]
- 19.Toulany M, Lee KJ, Fattah KR, Lin YF, Fehrenbacher B, Schaller M, Chen BP, Chen DJ, Rodemann HP. Akt promotes post-irradiation survival of human tumor cells through initiation, progression, and termination of DNA-PKcs-dependent DNA double-strand break repair. Mol Cancer Res 2012; 10:945-57; PMID:22596249; http://dx.doi.org/ 10.1158/1541-7786.MCR-11-0592 [DOI] [PubMed] [Google Scholar]
- 20.Liu P, Gan W, Guo C, Xie A, Gao D, Guo J, Zhang J, Willis N, Su A, Asara JM, et al.. Akt-mediated phosphorylation of XLF impairs non-homologous end-joining DNA repair. Mol Cell 2015; 57:648-61; PMID:25661488; http://dx.doi.org/ 10.1016/j.molcel.2015.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Q, Turner KM, Alfred Yung WK, Chen K, Zhang W. Role of AKT signaling in DNA repair and clinical response to cancer therapy. Neuro-oncology 2014; 16:1313-23; PMID:24811392; http://dx.doi.org/ 10.1093/neuonc/nou058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu P, Begley M, Michowski W, Inuzuka H, Ginzberg M, Gao D, Tsou P, Gan W, Papa A, Kim BM, et al.. Cell-cycle-regulated activation of Akt kinase by phosphorylation at its carboxyl terminus. Nature 2014; 508:541-5; PMID:24670654; http://dx.doi.org/ 10.1038/nature13079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hentges P, Waller H, Reis CC, Ferreira MG, Doherty AJ. Cdk1 restrains NHEJ through phosphorylation of XRCC4-like factor Xlf1. Cell Rep 2014; 9:2011-7; PMID:25533340; http://dx.doi.org/ 10.1016/j.celrep.2014.11.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C. PI3K/Akt and apoptosis: size matters. Oncogene 2003; 22:8983-98; PMID:14663477; http://dx.doi.org/ 10.1038/sj.onc.1207115 [DOI] [PubMed] [Google Scholar]
- 25.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature 2009; 461:1071-8; PMID:19847258; http://dx.doi.org/ 10.1038/nature08467 [DOI] [PMC free article] [PubMed] [Google Scholar]