

Abstract
MicroRNA (miRNA) acts as a critical regulator of gene expression at post-transcriptional and occasionally transcriptional levels in plants. Identification of reliable miRNA genes, monitoring the procedures of transcription, processing and maturation of the miRNAs, quantification of the accumulation levels of the miRNAs in specific biological samples, and validation of miRNA–target interactions become the basis for thoroughly understanding of the miRNA-mediated regulatory networks and the underlying mechanisms. Great progresses have been achieved for sequencing technology. Based on the high degree of sequencing depth and coverage, the high-throughput sequencing (HTS, also called next-generation sequencing) technology provides unprecedentedly efficient way for genome-wide or transcriptome-wide studies. In this review, we will introduce several HTS platform-based methods useful for plant miRNA research, including RNA-seq (RNA sequencing), RNA-PET-seq (paired end tag sequencing of RNAs), sRNA-seq (small RNA sequencing), dsRNA-seq (double-stranded RNA sequencing), ssRNA-seq (single-stranded RNA sequencing) and degradome-seq (degradome sequencing). In particular, we will provide some special cases to illustrate the novel use of HTS methods for investigation of the processing modes of the miRNA precursors, identification of the RNA editing sites on miRNA precursors, mature miRNAs and target transcripts, re-examination of the current miRNA registries, and discovery of novel miRNA species and novel miRNA–target interactions. Summarily, we opinioned that integrative use of the above mentioned HTS methods could make the studies on miRNAs more efficient.
Keywords: degradome-seq, dsRNA-seq, plant microRNA, RNA-PET-seq, sRNA-seq, ssRNA-seq
MicroRNA Genes in Plants
Similar to the protein-coding genes,1 most of the plant microRNA (miRNA) genes are transcribed by RNA polymerase II,2,3 resulting in poly(A)-tailed, single-stranded primary transcripts called pri-miRNAs (primary microRNAs). Specific sequence region of a pri-miRNA could form an internal hairpin-like structure constituted by a highly complementary double-stranded “stem” and a single-stranded “loop.” Facilitated by HYPONASTIC LEAVES 1 (HYL1), the pri-miRNA is recognized by Dicer-like 1 (DCL1), an RNase III-like enzyme. After two-step cropping by DCL1, the pri-miRNA is processed into the pre-miRNA (precursor microRNA), and then into the miRNA:miRNA* short duplex (Fig. 1). To protect the duplex from being digested by SMALL RNA DEGRADING NUCLEASE (SDN, an exonuclease), the 3′ ends of the miRNA and the miRNA* are methylated by HUA ENHANCER 1 (HEN1). To exert the regulatory impact on specific target transcript(s), the miRNA strand (also named as the “guide” strand) separated from the methylated short duplex is incorporated into a silencing complex called miRISC (miRNA-induced silencing complex), while the miRNA* (“passenger” or “degraded” strand) is generally thought to be degraded rapidly. An Argonaute (AGO) protein containing a PAZ small RNA (sRNA)-binding domain and a Piwi RNase H-like domain is included in the miRISC.4 According to the previous study on the sequence features of the sRNAs associated with different AGO protein complexes in Arabidopsis (Arabidopsis thaliana), the AGO1-associated sRNAs are predominantly 5′ U (uridine)-started and 21 nt in length, the AGO2-associated sRNAs are predominantly 5′ A (adenine)-started and 21 nt in length, the AGO4-associated ones are predominantly 5′ A-started and 24 nt in length, and the AGO5-associated ones are predominantly 5′ C (cytosine)-started.5 According to the current registries in miRBase,6 a large portion of the plant miRNAs are 5′ U-started and 21 nt in length. Thus, AGO1 has been proved to be the major performer in miRNA-guided target cleavages.4,7. A target transcript is specifically recognized by a miRNA based on sequence complementarity. Subsequently, the AGO1-mediated target slicing occurs between 10th and 11th nucleotide positions of the miRNA. To see more detailed introduction of the origin, biogenesis and action model of the plant miRNAs, please refer to the excellent reviews by Jones-Rhoades et al.8 and Voinnet9. Notably, a more complicated case was recently discovered for the “non-coding” pri-miRNAs in plant. Lauressergues and his colleagues10 showed that several plant pri-miRNAs (such as pri-miR171b of Medicago truncatula and pri-miR165a of Arabidopsis thaliana) contained short open reading frame sequences encoding regulatory peptides. By increasing the transcription of the pri-miRNAs, the miRNA-encoded peptides (miPEPs) enhance the accumulation of the corresponding mature miRNAs, resulting in the down-regulation of the target genes.10 Thus, the above discovery refreshed our traditional view that the “junk” regions of the miRNA precursors could play a coding role.11
Figure 1.
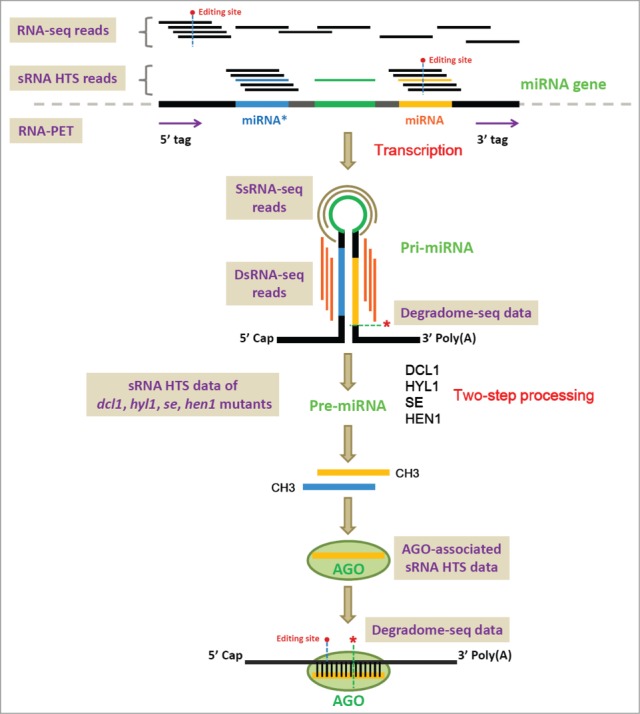
Graphic summarization of the transcription, processing, maturation and action of the plant microRNAs (miRNAs). Different kinds of sequencing methods based on the high-throughput sequencing platform, including RNA-seq (RNA sequencing), RNA-PET-seq (paired end tag sequencing of RNAs), sRNA-seq (small RNA sequencing), dsRNA-seq (double-stranded RNA sequencing), ssRNA-seq (single-stranded RNA sequencing) and degradome-seq (degradome sequencing), are shown on the figure. Please refer to the text of this review for detailed utilities of these sequencing methods for plant miRNA studies.
Considering the sequence features of the miRNAs, and the huge sRNA population existing in plants,12 high-throughput approaches are desired for cloning, characterization and quantification of the miRNA genes. Fortunately, high-throughput sequencing (HTS, also called next generation sequencing) platform was developed, and has been widely used for genome sequencing or resequencing, transcriptome sequencing [also called RNA sequencing (RNA-seq)], small RNA sequencing (sRNA-seq), chromatin immunoprecipitation sequencing (ChIP-Seq) and so on.13-21 Different from the traditional Sanger sequencing, billions of short reads could be generated in parallel by using HTS platform, enabling genome-wide or transcriptome-wide sequencing with unprecedented coverage and depth. In this review, the applications of RNA-seq, RNA-PET-seq (paired end tag sequencing of RNAs), sRNA-seq, dsRNA-seq (double-stranded RNA sequencing), ssRNA-seq (single-stranded RNA sequencing) and degradome-seq (degradome sequencing) in plant miRNA studies will be introduced. To make the brief introduction more sensible, detailed examples of the utilities of the HTS data for plant miRNA research will be provided.
Introduction of the HTS Methods for Plant miRNA Research
RNA-seq
The RNA-seq technology is widely used for whole-transcriptome mRNA (mRNA) analyses. The poly(A)-tailed transcripts are extracted from a biological sample, and (1) are first fragmented and then converted to short double-stranded cDNAs or (2) first converted to full-length double-stranded cDNAs and then fragmented to short duplexes. Finally, the short double-stranded cDNA fragments with ligated adapters are sent for sequencing.19,22 The sequenced reads could be assembled to discover novel transcripts, or mapped to the already known templates for expression level quantification of specific genes.
As introduced in the above section, most of the miRNA genes are transcribed by RNA polymerase II.2,3 In this regard, the poly(A)-tailed pri-miRNAs are included in the extracted mRNA samples. However, once transcribed, the pri-miRNAs will be subjected to DCL1-mediated 2-step cropping, resulting in processed miRNA:miRNA* duplexes without poly(A) tails. Thus, the pri-miRNAs might not be stable enough, and their abundances might be low. Fortunately, relying on the high degree of sequencing depth, RNA-seq enabled us to detect the transcripts encoded by weakly expressed genes or the transcripts with low stability. From this point of view, it is reasonable to propose that the pri-miRNAs could be detected by RNA-seq. In other words, by mapping RNA-seq reads onto the pri-miRNAs, the abundances of the primary transcripts of the miRNA genes could be quantified (Fig. 1). Although the abundances of the pri-miRNAs could not accurately reflect the in vivo levels of the mature miRNAs [because the processing of the pri-miRNAs is influenced by many factors as summarized in our previous work23], the detected spatio-temporal expression patterns of the pri-miRNAs could be directly linked to the features of their host genes including the characteristics of their promoter regions. For the plant miRNA genes, both the pri-miRNAs and the pre-miRNAs are highly variable in sequence length, which becomes an obstacle for their cloning. The traditional 5′ or 3′ RACE (rapid amplification of cDNA ends) was previously used for the identification of miRNA precursors.3 Obviously, this kind of work is laborious and time-consuming. Instead, transcript assembly by using RNA-seq reads will enable us to obtain full-length or partial-length sequences of novel pri-miRNAs.
RNA-PET-seq
As introduced above, RNA-seq reads could be utilized for transcript assembly, which could also be employed for pri-miRNA cloning. However, we should notice that although the high degree of sequencing coverage and depth is the superiority of the HTS platform, the poor stability of the primary transcripts of the miRNA genes might still be the obstacle for us to obtain the full-length pri-miRNAs. Thus, there is a need to recruit compensatory high-throughput strategies for reliably defining the transcriptional regions of the miRNA genes. One of the highly efficient approaches is RNA-PET-seq.24-27 RNA-PET-seq is a paired-end tag sequencing method for full-length mRNA analysis based on the HTS platform. Different from RNA-seq that treating the randomly sheared shortgun RNA fragments as sequencing libraries, RNA-PET-seq enables us to simultaneously capture the 5′ and the 3′ ends of the full-length transcripts on a transcriptome-wide scale (Fig. 1). In addition to its ability to quantify the transcription levels of the genes, the most important use of RNA-PET is to demarcate the boundaries of the transcription units.24-27 Since the RNA-PET sequencing libraries are prepared from poly(A)-tailed transcripts, the pri-miRNAs could be included for PET sequencing. Thus, a combinatorial use of the PET tags and the RNA-seq reads could be a high-throughput strategy enabling us to delineate the transcriptional regions of the miRNA genes, and to quantify the abundances of the pri-miRNAs.
SRNA-seq
Different from RNA-seq and RNA-PET, the sRNA-seq libraries originate from the sRNAs (previously defined as the RNAs shorter than 200 nt) extracted from specific biological samples. Since the mature miRNAs and the miRNA*s are ˜21 nt sRNA species in plants,8,9 the sRNA-seq is an indispensible method for quantifying their abundances. Specifically, sequencing of the sRNA samples from specific tissues/organs, or from plants at specific developmental stages, or from plants under specific treatments enables us to investigate the spatio-temporal accumulation patterns of the miRNAs and the miRNA*s. In most cases, relying on the protection by the AGO protein complexes, the stability of the mature miRNAs is much higher than the cognate miRNA*s after their separation from the short duplexes.8,9 Reflecting by sequencing data, the accumulation level of a mature miRNA is usually higher than the miRNA* by several orders of magnitude. Thus, quantification of the abundances of the sRNAs generated from the same precursor could facilitate us to distinguish between the miRNA and the miRNA* (Fig. 1).
As introduced above, once separated from the miRNA:miRNA* duplexes, the miRNA strands are selectively incorporated into the AGO protein complexes for recognizing specific target transcripts. In this consideration, sequencing of the sRNAs associated with the AGO proteins will enable us to detect the accumulation levels of the mature miRNAs in specific AGO silencing complexes. According to the previous study, the 5′ U-started, 21-nt miRNAs are preferentially recruited by the AGO1 protein complex in Arabidopsis.5 However, our statistical result shows that a significant portion of the plant miRNAs registered in miRBase (release 21) do not possess the above sequence features. In Arabidopsis, a total of 427 miRNAs have been included in miRBase, and only 241 (56.44%) start with 5′ U and 324 (75.88%) are 21 nt in length. Only 42.86% (183 out of 427) of the Arabidopsis miRNAs are 5′ U-started and 21 nt in length. Similarly in rice (Oryza sativa), 325 out of 713 (45.58%) registered miRNAs begin with 5′ U, and 383 (53.72%) miRNAs are 21 nt. Only 30.43% (217 out of 713) of the rice miRNAs are 5′ U-started and 21 nt in length. A previous study by Wu et al.28 discovered a new miRNA species of 24 nt in rice. These miRNAs were called long miRNAs (lmiRNAs). Different from the canonical miRNAs, the biogenesis of the lmiRNAs depends on DCL3, and they are incorporated into the AGO4 clade proteins to mediate site-specific DNA methylation.28 More complicatedly, in Arabidopsis, there are 10 AGO proteins, several studies demonstrate that different AGO genes have their specific spatio-temporal expression patterns.29-32 Taken together, by using sRNA-seq data prepared from different AGO proteins, the spatio-temporal AGO loading patterns of the miRNAs along with their sequence features could be deeply investigated.
Based on the biogenesis model of the plant miRNAs, several factors including SE (SERRATE), HYL1, DCL1, HEN1 and AGO1 are required for miRNA processing or for maintaining the miRNA stability (Fig. 1). Moreover, RDRs (RNA-dependent RNA polymerases) and RNA polymerase IV and polymerase V should not engage in miRNA production.8,9,33 Thus, the sRNA-seq data sets prepared from the mutants of SE, HYL1, DCL1, HEN1, AGO1, RDRs and Pol IV/V are useful for the researchers to discriminate miRNAs from the other sRNA species in plants.
To date, huge amounts of sRNA-seq data sets of diverse plant species have been available in the public databases, such as GEO (Gene Expression Omnibus; http://www.ncbi.nlm.nih.gov/geo/),34 Next-Gen Sequence Databases (http://mpss.udel.edu/),35 ASRP (the Arabidopsis Small RNA Project database; http://asrp.danforthcenter.org/),36 the vascular plant small RNA sequencing project database (http://smallrna.udel.edu/),37 and CSRDB (Cereal Small RNA Database; http://sundarlab.ucdavis.edu/smrnas/).38
DsRNA-seq and ssRNA-seq
A single-stranded, stem-loop structure is the prerequisite for precise excision of the miRNA:miRNA* duplex from its precursor by DCL1.8,9 And, this is one of the primary criteria for miRNA annotation.33,39 In this regard, secondary structure profiling is useful for cloning or validating the miRNA genes. Sequencing-based approaches for high-throughput structure mapping have been employed by several previous studies.40-46 These pioneer works showed that the HTS-based approaches were highly efficient for interrogating RNA secondary structures on a global scale.47 Here, we would like to introduce dsRNA-seq and ssRNA-seq by illustrating their use for the studies on miRNA structures.
DsRNA-seq is a novel high-throughput, strand-specific sequencing method developed by Gregory and his colleagues (2010)46 to interrogate intra- and inter-molecular base-paired RNAs in Arabidopsis.46 This method marries classical nuclease-based structure mapping approach48,49 with HTS technology in order to obtain base-pairing information on a transcriptome-wide scale. According to the primary criteria for miRNA annotation in plants, the base-pairing between the miRNA arm and the miRNA* arm of a precursor should be extensive, with only a few mismatches and bulges allowed.33 Thus, the “stem” of a miRNA precursor is a local double-stranded region with high stability. Considering the technical property of dsRNA-seq, the “stem” region of a miRNA precursor could be covered by dsRNA-seq reads (Fig. 1).
SsRNA-seq was also developed by Gregory's group for identification of the single-stranded, unpaired regions of the transcripts. Usually, ssRNA-seq was used in combination with dsRNA-seq for high-throughput probing the RNA structurome of an organism.40,44 The “loop” structure of a miRNA precursor is a locally unpaired region. Theoretically, this region could be identified by ssRNA-seq. Besides, we would like to mention here that after DCL1-mediated processing, the “loop” sequence of a miRNA precursor could also be included in sRNA-seq libraries. Thus, this region could also be covered by sRNA-seq reads. Supportive results were shown in the previous study on the processing of the miRNA precursors of Drosophila melanogaster.50 In that study,50 observed that a portion of sRNA-seq reads could be mapped onto the “loop” regions of certain miRNA precursors.
Taken together, we propose that combinatory use of dsRNA-seq and ssRNA-seq will allow us to probe or validate the hairpin structures of the miRNA precursors through a highly efficient way.
Degradome-seq
There are 3 HTS-based methods for cloning of uncapped transcripts: genome-wide mapping of uncapped and cleaved transcripts (GMUCT),51,52 parallel analysis of RNA ends (PARE),53,54 and degradome sequencing.55,56 All of these methods take advantage of the fact that T4 RNA ligase 1 is utilized for sequencing library construction by ligating an adapter to the 5′ end of the poly(A)-tailed transcript with a free 5′-monophosphate. Thus, the intact mature mRNAs with 5′ caps will not be included for library preparation. These three kinds of sequencing methods integrate the modified 5′ RACE (rapid amplification of cDNA ends) with HTS platforms for mapping the 5′ end of the degraded or cleaved RNA fragments on a transcriptome-wide scale. In this review, the 3 methods are called degradome-seq for simplicity.
One of the major applications of degradome-seq is to identify the truncated transcripts resulting from endonucleolytic cleavages guided by miRNAs or other sRNAs in plants.54-58 The degradome-seq signatures could be used for mapping the slicing sites resided within the miRNA or sRNA binding regions on specific target transcripts (Fig. 1).
Here, we would like to introduce the novel utilities of degradome-seq. One of the utilities is to detect the DCL1-mediated cleavage remnants produced during miRNA processing (Fig. 1). As illustrated in our previous work,59 poly(A)-tailed, uncapped fragments generated from pri-miRNAs after the first cropping by DCL1 could be included in the degradome-seq libraries. Thus, the degradome-seq signatures mapped to the slicing sites of DCL1 (usually mapped to the 3′ and/or 5′ ends of mature miRNAs and/or miRNA*s) could facilitate us for identification or validation of pri-miRNAs recognizable by DCL1. Recently, a degradome-seq-based approach called SPARE (specific parallel amplification of 5′ RNA ends) was developed to detect the miRNA processing intermediates with a high-throughput purpose in plants.60 However, the limitation of the SPARE method is the requirement of designing a mixture of specific primers against the already known miRNA precursors for reverse transcription. Another novel use of degradome-seq for plant miRNA research is to investigate the self-regulation of the miRNA precursors by the encoded miRNAs or miRNA*s. According to the criteria for miRNA annotation, the miRNA:miRNA* duplexes resided within the “stem” regions of the miRNA precursors are extensively base-paired with a few mismatches and bulges.33 On the other hand, the miRNA–target interactions in plants rely on the high sequence complementarity between the miRNAs and the target binding sites on the transcripts.8,9 Thus, we proposed that in some cases, the miRNA precursors could be recognized by their encoded mature miRNAs based on the highly complementary miRNA*-coding regions for cleavages.59 Degradome signals supporting the self-regulation of the miRNA genes, such as miR172b in Arabidopsis, have been observed in a previous study.54
The utilities of the HTS methods for plant miRNA research: Special cases
Brief introduction of the HTS methods useful for plant miRNA research has been provided above. Here, we would like to present some special cases based on the reported studies and our preliminary results.
Processing modes of the miRNA precursors inferred from degradome-seq data
As introduced above, the poly(A)-tailed processing intermediates of pri-miRNAs could be detected by degradome-seq in plants.59,60 Here, as illustrated in Figure 2, we proposed that degradome-seq signatures could be utilized for investigation of the processing modes of the miRNA precursors. To date, 2 modes of DCL1-mediated processing have been reported in plants, i.e. the canonical “base-to-loop” processing (Fig. 2B)8,9 and the “loop-to-base” processing (Fig. 2A).61,62 For the “base-to-loop” processing, only the first step of DCL1-mediated cropping could produce poly(A)-tailed cleavage remnants, which could be detected by degradome-seq. Theoretically, the degradome signatures mapped to the 5′ end of the 5′-armed miRNA (or miRNA*) and the 3′ end of the 3′-armed miRNA* (or miRNA) could be obtained (Fig. 2B). For the “loop-to-base” processing, the degradome signals mapped to the 3′ end of the 5′-armed miRNA (or miRNA*) and the 5′ end of the 3′-armed miRNA* (or miRNA) could be detected for the first step of DCL1-mediated processing. Moreover, the degradome signatures mapped to the 3′ end of the 3′-armed miRNA (or miRNA*) could be detected for the second round of DCL1-mediated cropping (Fig. 2A).
Figure 2.
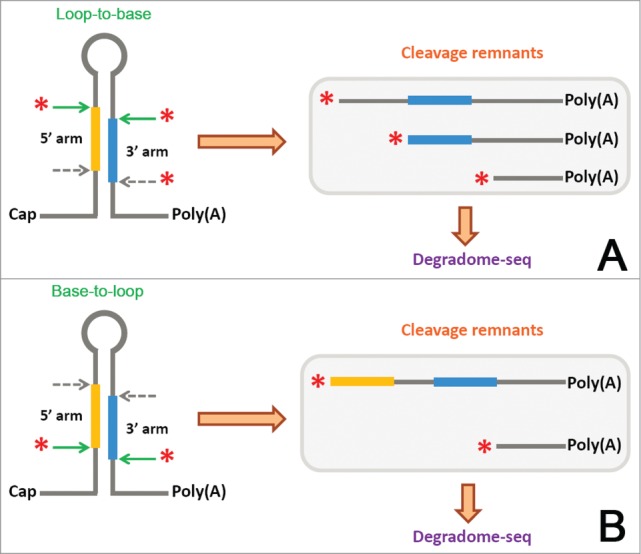
The processing modes of the microRNA precursors could be partially inferred from degradome sequencing (degradome-seq) data. (A) Schematic diagram of DCL1-mediated “loop-to-base” [first cleave on the “loop” side (green arrows), and then on the “base” side (gray arrows)] processing of a pri-miRNA (primary microRNAs). The poly(A)-tailed cleavage remnants generated from the first and the second steps of DCL1-mediated cropping could be detected by degradome-seq. (B) Schematic diagram of DCL1-mediated “base-to-loop” [first cleave on the “base” side (green arrows), and then on the “loop” side (gray arrows)] processing of a pri-miRNA. The poly(A)-tailed processing intermediates generated from the first cleavages on the “base” side could be detected by degradome-seq.
Search for potential RNA editing sites influencing miRNA abundances and miRNA–target interactions
RNA editing was defined as the site-specific alterations on RNA sequences, which include insertions or deletions of nucleotides and base conversions. RNA editing is an effective strategy employed by both animals and plants for post-transcriptional regulation of gene expression.63-68 Different from animals, RNA editing performed by pentatricopeptide repeat (PPR) family proteins in plants is restricted to mitochondrial and plastid transcripts based on the previous reports.67-70 However, recent research progresses point to the potential involvement of RNA editing in the modifications of plant nuclear transcripts.59,71 More interestingly, in both animals and plants, RNA modifications were discovered on the miRNA precursors and the mature miRNAs.59,71-73 Considering the fact that the featured “stem-loop” structures are critical for processing of the miRNA precursors, RNA editing might alter the conformation and the stability of the secondary structures of the miRNA precursors, thus affecting their processing efficiency and the miRNA abundances. Besides, RNA editing occurred on mature miRNAs or their target transcripts could have a great impact on the stability of miRNA–target interactions (Fig. 1). In this regard, high-throughput methods for the identification of potential editing sites on the miRNA precursors, the mature miRNAs and their targets are required for plant miRNA research.
In most of the previous studies, the sRNA-seq reads that could not be perfectly mapped onto the corresponding genomes were generally considered to be caused by technological sequencing errors, and would not be included in the following analyses. However, these discarded “error” reads were utilized by Ebhardt et al.71 for study on the post-transcriptional modifications of tRNAs and mature miRNAs in plants.71 They hypothesized that some of the “error” reads might have their biological origins. The discrepancies between the “error” reads and the genomic sequences could originate from post-transcriptional modifications. In contrast to the random occurrences of technical sequencing errors, the site-specific modifications or editing events should be repeatedly observed at single sites with much higher frequencies.71 In other words, a reliable editing site should be supported by a certain number of sRNA-seq reads with a fixed mismatch site compared to the genomic site (Fig. 1). As a result, many candidate editing sites were discovered on both the tRNAs and the mature miRNAs. Interestingly, tissue-specific editing of miRNAs was observed. And, some modifications at the 5′ ends of the miRNAs were demonstrated to be capable of altering the AGO complex preference of the edited miRNAs.71
In human and mouse, RNA editing was experimentally identified on the precursors of miRNA22, and was predicted to have significant implications for the biogenesis and function of this miRNA.72 The involvement of RNA editing in the miRNA maturation pathway in metazoans was summarized by Ohman73. But, how about plants? In our previous study, RNA-seq data was used to search for the potential editing sites on the nuclear transcripts.74 Some candidate editing sites were discovered on the miRNA precursors in Arabidopsis. Consistent with the notion proposed in animals,72 transformation of the secondary structures of certain precursors (such as ath-MIR845) caused by RNA editing was predicted to be implicated in DCL1-mediated processing.
Re-examination of the current miRNA registries
In the face of the increasingly discovered miRNAs, maintaining the quality of the miRNA annotations have become a significant challenge. However, accurate annotations are essentially important for functional studies on the miRNAs. Recent improvements in the confidence of miRNA annotations in miRBase (release version 20) were achieved by employing sRNA-seq data.75 To obtain a list of “high confidence” miRNAs, Griffiths-Jones and his co-worker mapped reads of publically available sRNA-seq data sets onto the already registered miRNA precursors, and adopted the following criteria to extract miRNA genes with high confidence: (1) for both the miRNA- and the miRNA*-coding regions (Griffiths-Jones and his coworker recommended to rename them as 5′-armed and 3′-armed miRNAs) of a precursor, at least 10 reads without mismatches must be mapped to each region; (2) the most abundant reads from the 2 regions could form a duplex with 0 to 4 nt overhangs at their 3′ ends; (3) for each miRNA-coding region, at least 50% of the mapped reads should have the same 5′ end; (4) the predicted hairpin structure of the precursor must have a folding free energy lower than −0.2 kcal/mol/nt; (5) for both the 5′-armed and the 3′-armed miRNAs, at least 60% of the bases of the mature sequences should be paired in the predicted hairpin structure. Obviously, the sRNA-seq- and structure-based criteria could improve the accuracy of miRNA annotations.
As introduced in the previous sections, the processing intermediates of the miRNA precursors could be detected by degradome-seq.59,60 Besides, dsRNA-seq could be utilized to validate the base-paired “stem” regions. In this consideration, we proposed that in addition to sRNA-seq, dsRNA-seq and degradome-seq might be useful for re-examination of the miRNA registries. For illustration, we performed a case study on the miRBase-registered (release 20) miRNAs of Arabidopsis thaliana by using dsRNA-seq data [GSM575243 and GSM575244 retrieved from GEO were the gifts from a previous study46] and degradome-seq data (AxIDT, AxIRP, AxSRP, Col, ein5, TWF and Tx4F were obtained from Next-Gen Sequence Databases, and GSM278333 was retrieved from GEO). A total of 337 mature miRNAs on 298 precursors were re-examined. First, we mapped the dsRNA-seq and degradome-seq reads onto the 298 precursors, and the perfectly mapped reads were retained. At first glance, 211 (62.61%) out of 337 mature miRNA-coding regions on 116 (38.93%) out of 298 precursors were covered or partially covered by dsRNA-seq reads, supporting the great potential of miRNA-coding regions to form well-paired, double-stranded “stem” structures (Fig. 3; Table S1 and Data S1). Degradome signals could be detected at 5′ and/or 3′ ends of 209 (62.02%) mature miRNA-coding regions on 122 precursors (40.94%), supporting the processing of these precursors into mature miRNAs by DCL1. A total of 167 (49.55%) mature miRNA-coding regions on 91 precursors (30.54%) were supported by both dsRNA-seq and degradome-seq data (Fig. 3; Table S1). Additionally, 44 mature miRNAs were supported by dsRNA-seq but not by degradome-seq data, and 42 miRNAs were supported by degradome-seq but not by dsRNA-seq data (Fig. 3; Table S1). For the 76 “high confidence” mature miRNAs in miRBase (release 20) which were annotated based on sRNA HTS data, 56 (73.68%) were supported by both dsRNA-seq and degradome-seq data (Fig. 3). For the remaining “high confidence” mature miRNAs, 8 were supported by dsRNA-seq, 9 were evidenced by degradome-seq data, and 3 were not supported by dsRNA-seq or degradome-seq data. Besides, we noticed that 81 mature miRNAs (24.04%) on 67 precursors (22.48%) were not supported by dsRNA-seq and degradome-seq data, and were also not included in the list of “high confidence” miRNAs in miRBase. These weak candidates need experimental re-examination. From this point of view, both dsRNA-seq and degradome-seq data might be used to increase the reliability of the “high confidence” miRNAs annotated solely based on sRNA-seq data. In other words, both the dsRNA-seq data and the degradome-seq data could be integrated into the current sRNA-seq data-based approach for annotating “high confidence” miRNA registries. Notably, in the above case study, the 2 dsRNA-seq data sets originated from the unopened flower buds of Arabidopsis. Six degradome-seq libraries were prepared from inflorescences, and only one library was from the Arabidopsis seedlings. From this point of view, we suspect that the ratio of dsRNA-seq- or degradome-seq-supported miRNAs might be underestimated, especially for the miRNA genes abundantly accumulated in the non-floral organs.
Figure 3.
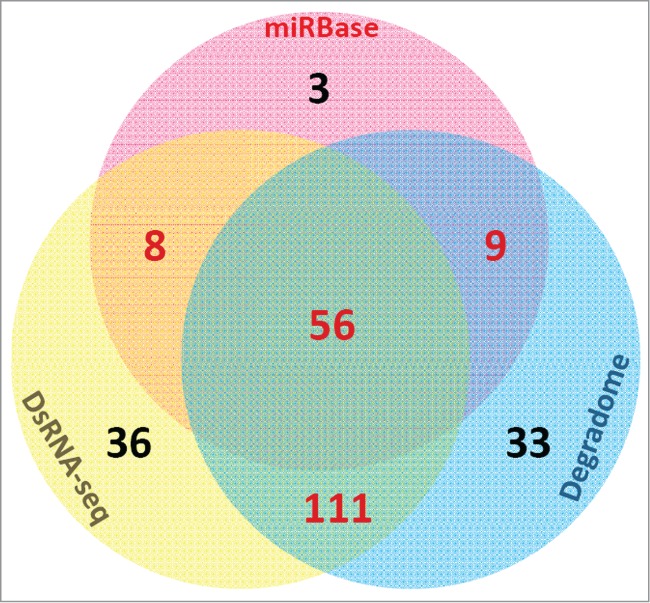
The numbers of the miRBase-registered (release 20) mature microRNAs of Arabidopsis thaliana supported by double-stranded RNA sequencing (dsRNA-seq) reads and degradome signatures, and those annotated as “high confidence” ones by miRBase are shown in the Venn diagram.
Uncovering novel miRNA species and novel miRNA–target interactions
In recent years, atypical miRNA species such as mirtrons76-84 and reverse complementary miRNA genes85 were identified based on computational predictions, sRNA-seq data and further experimental validations. Besides, based on sRNA-seq and degradome-seq data, we previously proposed that in plants, the intronic regions of the pre-mRNAs (precursors of mRNAs) might be targeted by specific miRNAs for cleavages. The 3′ cleaved remnants might be converted to double-stranded RNAs for phased sRNA production through an RDR-, DCL−dependent pathway. Some of the phased sRNAs could be recruited by AGO1 silencing complexes for target binding and cleavages.86
As introduced above, the miRNAs play an important regulatory role in gene expression. On the other hand, the accumulation of the miRNAs should be tightly controlled at both transcriptional and post-transcriptional levels. Recent reports in plants and animals showed that the activities of the miRNAs could be modulated after their incorporation into the AGO-associated silencing complexes. In animals, certain long non-coding RNAs were identified to act as competing endogenous RNAs (ceRNAs) to compete with the genuine targets for the binding of specific miRNAs.40,87 Thus, a portion of functional miRNAs could be sequestered by the ceRNAs, adding another layer of post-transcriptional regulation of the miRNA activities. These ceRNAs were also called miRNA decoys or sponges, indicating their ability of trapping mature miRNAs. Additionally, another kind of non-coding RNAs called circular RNAs (circRNAs) was recently discovered to be capable of serving as miRNA sponges.88 More interestingly, in addition to the non-coding RNAs, an mRNA sharing a common miRNA recognition site with the other mRNAs could compete with the other target mRNAs for miRNA binding, resulting in the diluted interactions between the miRNAs and the other target mRNAs. Thus, certain mRNAs possess a coding-independent role in modulating the miRNA activities.89-92 The endogenous, miRNA sponge-like transcripts also exist in plants. The first case was discovered in phosphate signaling.93,94 IPS1 (INDUCED BY PHOSPHATE STARVATION 1), a phosphate starvation-responsive gene, encodes a non-coding transcript containing a motif with high sequence complementarity to miR399. The AGO1-associated silencing complex guided by miR399 could bind to the IPS1 transcript, but could not perform target cleavage due to the 3-nucleotide mismatches at the expected cleavage site. In this case, the non-cleavable IPS1 transcript could efficiently sequester miR399, preventing miR399 from excessive cleavages of PHO2 transcripts, another target of this miRNA. This kind of regulation was called target mimicry, and the non-cleavable transcripts like IPS1 was named as target mimics. Subsequently, bioinformatics predictions showed that natural target mimic sites might be widespread on both the protein-coding transcripts and the long non-coding RNAs in plants.95,96 The mechanism of target mimicry enabled the researchers to introduce manually designed mimics into the plants for investigation of the biological outputs through loss-of-function of specific miRNAs.96,97 It is an efficient way for the study on the biological functions of a specific miRNA or even a miRNA family. Similarly, in animals, manually introduced sponges were utilized for functional studies on certain miRNAs.98 Notably, a novel kind of sponges called anti-miRs is becoming a strong candidate for miRNA-based therapeutics.43,99-107 The anti-miRs are chemically modified oligonucleotides developed to modulate the activities of specific miRNAs. A recent study carried out by Hogan and his colleagues108 demonstrated that the anti-miR physically associated with the AGO protein complex in the context of the cognate target miRNA both in vitro and in vivo. The targeted miRNA was stable and continued to be associated with the AGO protein. Their results indicate that the anti-miR could specifically associate with the AGO-bound miRNA, and inhibit the miRNA from binding to the target mRNA, leading to the increased level of the targeted transcript.108
Based on the above mentioned observation that the manually introduced anti-miRs could stably exist in the AGO protein complexes with the target miRNAs, we questioned whether natural anti-miRs exist in plants. To partially address the question, we did a tentative search for the natural anti-miR candidates in AGO1 protein complexes of Arabidopsis. First, the mature miRNAs of Arabidopsis were retrieved from miRBase (release 20). The sRNA-seq data sets generated from AGO1-associated sRNA population of Arabidopsis were obtained from GEO. Specifically, 4 HTS data sets contributed by a previous study109 were utilized: GSM707682 (AGO1-associated sRNA in flowers), GSM707683 (AGO1-associated sRNA in leaves), GSM707684 (AGO1-associated sRNA in roots) and GSM707685 (AGO1-associated sRNA in seedlings). Then, an in-house script was developed to search for the anti-miR candidates highly complementary to the miRNAs. To ensure the stability of the in vivo interactions, at most 3 mismatches between the anti-miR candidates and the specific miRNAs were allowed. Besides, both the anti-miRs and the target miRNAs should be highly enriched in AGO1, thus increasing the possibility of anti-miR–miRNA interactions in AGO1. The following criterion was adopted to extract the anti-miR–miRNA interacting pairs enriched in AGO1: for an anti-miR–miRNA pair, the accumulation levels of both the anti-miR and the miRNA should be higher than 5 RPM [to allow cross-library comparison, the normalized read count (in RPM, reads per million) of a sRNA from a specific data set was calculated by dividing the raw count of this sRNA by the total counts of the data set, and then multiplied by 106] in the AGO1 data set, and their levels in the AGO1 data set should be 3 times or higher than in the control data set [GSM707678 (control_flowers), GSM707679 (control_leaves), GSM707680 (control_roots) and GSM707681 (control_seedlings)].
As a result, several well-paired anti-miR–miRNA interactions were identified to be enriched in at least one of the AGO1 data sets (since this is not a paper for presenting full primary research results, we will just show examples of identified anti-miR candidates below). After mapping these anti-miR candidates to the Arabidopsis genome retrieved from TAIR (The Arabidopsis Information Resource; release 10; http://www.arabidopsis.org/),110 we surprisingly observed that most of the anti-miR candidates originated from the miRBase-registered pre-miRNAs. Interestingly, these anti-miRs located on the miRNA*- or isomiR*-coding regions opposite to the miRNA-coding regions on the stem-loop structured pre-miRNAs (Fig. S1). The miRNA*s, named as the passenger strands, were generally considered to subject to degradation after the incorporation of the mature miRNAs into the AGO1-associated silencing complexes in plants. However, recent studies revealed some important biological functions of miRNA*s in both plants and animals.44,58,111-114 Here, based on the results obtained from our sRNA-seq-based bioinformatics analysis, we proposed that certain miRNA*s or isomiR*s could act as anti-miRs to inhibit the cognate miRNAs from binding the target transcripts, thus reducing the activities of the miRNAs. It is based on the following evidences: (1) According to the biogenesis model of the plant miRNAs, after DCL1-mediated 2-step processing, miRNA and its miRNA*, forming as a short RNA duplex, were exported to the cytoplasm for loading into the AGO1 complex. Thus, the miRNA*s are the natural ideal anti-miRs with high sequence complementarity to the cognate miRNAs. (2) Our results showed that certain anti-miR candidates originated from the miRNA*- or isomiR*-coding regions, together with the corresponding miRNAs, were highly enriched in AGO1 in Arabidopsis (Fig. S1). Thus, the high accumulation levels of both anti-miRs and the target miRNAs increase the likelihood that the anti-miRs and the miRNAs form interaction pairs in AGO1. However, in-depth experimental studies are needed for exploration and confirmation of the genuine anti-miRs in planta.
Taken together, based on the above case study, we would like to emphasize the utility of HTS data in identification of novel miRNA species and novel miRNA–target interactions.
Concluding Remarks
The plant miRNAs are already known key players in gene regulation. Although their biogenesis and action modes have been well depicted,8,9 new features are being uncovered and added onto the miRNAs encoded by the precursors encompassing stem-loop structures.10,11 Research on the discovery and the regulatory activities of the miRNA species will be greatly accelerated by integrative use of diverse sequencing approaches developed from HTS platform. In this review, we introduced several HTS methods valuable for plant miRNA studies, including RNA-seq, RNA-PET-seq, sRNA-seq, dsRNA-seq, ssRNA-seq and degradome-seq. By performing case studies, we presented a more sensible view on the utilities of sRNA-seq, dsRNA-seq and degradome-seq data for re-examination of the current miRNA registries, and uncovering novel sRNA species. Summarily, we hope that this review could serve as a reference material for researchers interested in plant miRNA biology.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank all the publicly available data sets and the scientists behind them.
Funding
This work was funded by the National Natural Science Foundation of China [31100937], Zhejiang Provincial Natural Science Foundation of China [LY15C060006], the Starting Grant funded by Hangzhou Normal University to Yijun Meng [2011QDL60].
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website
References
- 1.Lee TI, Young RA. Transcription of eukaryotic protein-coding genes. Annu. Rev. Genet. 2000; 34:77-137; PMID:11092823; http://dx.doi.org/ 10.1146/annurev.genet.34.1.77 [DOI] [PubMed] [Google Scholar]
- 2.Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, Kim VN. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004; 23:4051-60; PMID:15372072; http://dx.doi.org/ 10.1038/sj.emboj.7600385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xie Z, Allen E, Fahlgren N, Calamar A, Givan SA, Carrington JC. Expression of Arabidopsis MIRNA genes. Plant Physiol 2005; 138:2145-54; PMID:16040653; http://dx.doi.org/ 10.1104/pp.105.062943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vaucheret H. Plant ARGONAUTES. Trends Plant Sci 2008; 13:350-58; PMID:18508405; http://dx.doi.org/ 10.1016/j.tplants.2008.04.007 [DOI] [PubMed] [Google Scholar]
- 5.Mi S, Cai T, Hu Y, Chen Y, Hodges E, Ni F, Wu L, Li S, Zhou H, Long C, et al.. Sorting of small RNAs into Arabidopsis argonaute complexes is directed by the 5′ terminal nucleotide. Cell 2008; 133:116-27; PMID:18342361; http://dx.doi.org/ 10.1016/j.cell.2008.02.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic Acids Res 2008; 36:D154-158; PMID:17991681; http://dx.doi.org/ 10.1093/nar/gkm952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baumberger N, Baulcombe DC. Arabidopsis ARGONAUTE1 is an RNA Slicer that selectively recruits microRNAs and short interfering RNAs. Proc Natl Acad Sci U S A 2005; 102:11928-33; PMID:16081530; http://dx.doi.org/ 10.1073/pnas.0505461102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones-Rhoades MW, Bartel DP, Bartel B. MicroRNAs and their regulatory roles in plants. Annu Rev Plant Biol 2006; 57:19-53; PMID:16669754; http://dx.doi.org/ 10.1146/annurev.arplant.57.032905.105218 [DOI] [PubMed] [Google Scholar]
- 9.Voinnet O. Origin, biogenesis, and activity of plant microRNAs. Cell 2009; 136:669-87; PMID:19239888; http://dx.doi.org/ 10.1016/j.cell.2009.01.046 [DOI] [PubMed] [Google Scholar]
- 10.Lauressergues D, Couzigou JM, Clemente HS, Martinez Y, Dunand C, Becard G, Combier JP. Primary transcripts of microRNAs encode regulatory peptides. Nature 2015; 520:90-93; PMID:25807486; http://dx.doi.org/ 10.1038/nature14346 [DOI] [PubMed] [Google Scholar]
- 11.Waterhouse PM, Hellens RP. Plant biology: Coding in non-coding RNAs. Nature 2015; 520:41-42; PMID:25807488; http://dx.doi.org/ 10.1038/nature14378 [DOI] [PubMed] [Google Scholar]
- 12.Chen X. Small RNAs and their roles in plant development. Annu Rev Cell Dev Biol 2009; 25:21-44; PMID:19575669; http://dx.doi.org/ 10.1146/annurev.cellbio.042308.113417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Han H, Nutiu R, Moffat J, Blencowe BJ. SnapShot: High-throughput sequencing applications. Cell 2011; 146:1044, 1044 e1041–1042; http://dx.doi.org/ 10.1016/j.cell.2011.09.002 [DOI] [PubMed] [Google Scholar]
- 14.Imelfort M, Edwards D. De novo sequencing of plant genomes using second-generation technologies. Brief Bioinform 2009; 10:609-18; PMID:19933209; http://dx.doi.org/ 10.1093/bib/bbp039 [DOI] [PubMed] [Google Scholar]
- 15.Jain M. Next-generation sequencing technologies for gene expression profiling in plants. Brief Funct Genomics 2012; 11:63-70; PMID:22155524; http://dx.doi.org/ 10.1093/bfgp/elr038 [DOI] [PubMed] [Google Scholar]
- 16.Lister R, Gregory BD, Ecker JR. Next is now: new technologies for sequencing of genomes, transcriptomes, and beyond. Curr Opin Plant Biol 2009; 12:107-18; PMID:19157957; http://dx.doi.org/ 10.1016/j.pbi.2008.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Metzker ML. Sequencing technologies – the next generation. Nat Rev Genet 2010; 11:31-46; PMID:19997069; http://dx.doi.org/ 10.1038/nrg2626 [DOI] [PubMed] [Google Scholar]
- 18.Ozsolak F, Milos PM. RNA sequencing: advances, challenges and opportunities. Nat Rev Genet 2011; 12:87-98; PMID:21191423; http://dx.doi.org/ 10.1038/nrg2934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simon SA, Zhai J, Nandety RS, McCormick KP, Zeng J, Mejia D, Meyers BC. Short-read sequencing technologies for transcriptional analyses. Annu Rev Plant Biol 2009; 60:305-33; PMID:19575585; http://dx.doi.org/ 10.1146/annurev.arplant.043008.092032 [DOI] [PubMed] [Google Scholar]
- 20.Thudi M, Li Y, Jackson SA, May GD, Varshney RK. Current state-of-art of sequencing technologies for plant genomics research. Brief. Funct. Genomics 2012; 11:3-11; PMID:22345601; http://dx.doi.org/ 10.1093/bfgp/elr045 [DOI] [PubMed] [Google Scholar]
- 21.Varshney RK, Nayak SN, May GD, Jackson SA. Next-generation sequencing technologies and their implications for crop genetics and breeding. Trends Biotechnol 2009; 27:522-30; PMID:19679362; http://dx.doi.org/ 10.1016/j.tibtech.2009.05.006 [DOI] [PubMed] [Google Scholar]
- 22.Graveley BR. Molecular biology: power sequencing. Nature 2008; 453:1197-98; PMID:18580940; http://dx.doi.org/ 10.1038/4531197b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meng Y, Shao C, Gou L, Jin Y, Chen M. Construction of MicroRNA- and MicroRNA*-mediated regulatory networks in plants. RNA Biol 2011; 8:1124-48; PMID:21955495; http://dx.doi.org/ 10.4161/rna.8.6.17743 [DOI] [PubMed] [Google Scholar]
- 24.Fullwood MJ, Wei CL, Liu ET, Ruan Y. Next-generation DNA sequencing of paired-end tags (PET) for transcriptome and genome analyses. Genome Res 2009; 19:521-32; PMID:19339662; http://dx.doi.org/ 10.1101/gr.074906.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ng P, Wei CL, Sung WK, Chiu KP, Lipovich L, Ang CC, Gupta S, Shahab A, Ridwan A, Wong CH, et al.. Gene identification signature (GIS) analysis for transcriptome characterization and genome annotation. Nat Methods 2005; 2:105-11; PMID:15782207; http://dx.doi.org/ 10.1038/nmeth733 [DOI] [PubMed] [Google Scholar]
- 26.Peters BA, Velculescu VE. Transcriptome PETs: a genome's best friends. Nat Methods 2005; 2:93-94; PMID:15782204; http://dx.doi.org/ 10.1038/nmeth0205-93 [DOI] [PubMed] [Google Scholar]
- 27.Ruan X, Ruan Y, Genome wide full-length transcript analysis using 5′ and 3′ paired-end-tag next generation sequencing (RNA-PET). Methods Mol Biol 2012; 809:535-62; PMID:22113299 [DOI] [PubMed] [Google Scholar]
- 28.Wu L, Zhou H, Zhang Q, Zhang J, Ni F, Liu C, Qi Y. DNA methylation mediated by a microRNA pathway. Mol Cell 2010; 38:465-75; PMID:20381393; http://dx.doi.org/ 10.1016/j.molcel.2010.03.008 [DOI] [PubMed] [Google Scholar]
- 29.Mallory A, Vaucheret H. Form, function, and regulation of ARGONAUTE proteins. Plant Cell 2010; 22:3879-89; PMID:21183704; http://dx.doi.org/ 10.1105/tpc.110.080671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mallory AC, Vaucheret H. ARGONAUTE 1 homeostasis invokes the coordinate action of the microRNA and siRNA pathways. EMBO Rep 2009; 10:521-26; PMID:19343050; http://dx.doi.org/ 10.1038/embor.2009.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nonomura K, Morohoshi A, Nakano M, Eiguchi M, Miyao A, Hirochika H, Kurata N. A germ cell specific gene of the ARGONAUTE family is essential for the progression of premeiotic mitosis and meiosis during sporogenesis in rice. Plant Cell 2007; 19:2583-94; PMID:17675402; http://dx.doi.org/ 10.1105/tpc.107.053199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmid M, Davison TS, Henz SR, Pape UJ, Demar M, Vingron M, Scholkopf B, Weigel D, Lohmann JU. A gene expression map of Arabidopsis thaliana development. Nat Genet 2005; 37:501-506; PMID:15806101; http://dx.doi.org/ 10.1038/ng1543 [DOI] [PubMed] [Google Scholar]
- 33.Meyers BC, Axtell MJ, Bartel B, Bartel DP, Baulcombe D, Bowman JL, Cao X, Carrington JC, Chen X, Green PJ, et al.. Criteria for annotation of plant MicroRNAs. Plant Cell 2008; 20:3186-90; PMID:19074682; http://dx.doi.org/ 10.1105/tpc.108.064311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barrett T, Troup DB, Wilhite SE, Ledoux P, Rudnev D, Evangelista C, Kim IF, Soboleva A, Tomashevsky M, Marshall KA, et al.. NCBI GEO: archive for high-throughput functional genomic data. Nucleic Acids Res 2009; 37:D885-890; PMID:18940857; http://dx.doi.org/ 10.1093/nar/gkn764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakano M, Nobuta K, Vemaraju K, Tej SS, Skogen JW, Meyers BC. Plant MPSS databases: signature-based transcriptional resources for analyses of mRNA and small RNA. Nucleic Acids Res 2006; 34:D731-735; PMID:16381968; http://dx.doi.org/ 10.1093/nar/gkj077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gustafson AM, Allen E, Givan S, Smith D, Carrington JC, Kasschau KD. ASRP: the Arabidopsis Small RNA Project Database. Nucleic Acids Res 2005; 33:D637-640; PMID:15608278; http://dx.doi.org/ 10.1093/nar/gki127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Montes RA, de Fatima Rosas-Cardenas F, De Paoli E, Accerbi M, Rymarquis LA, Mahalingam G, Marsch-Martinez N, Meyers BC, Green PJ, de Folter S. Sample sequencing of vascular plants demonstrates widespread conservation and divergence of microRNAs. Nat Commun 2014; 5:3722; PMID:24759728; http://dx.doi.org/ 10.1038/ncomms4722 [DOI] [PubMed] [Google Scholar]
- 38.Johnson C, Bowman L, Adai AT, Vance V, Sundaresan V. CSRDB: a small RNA integrated database and browser resource for cereals. Nucleic Acids Res 2007; 35:D829-833; PMID:17169981; http://dx.doi.org/ 10.1093/nar/gkl991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ambros V, Bartel B, Bartel DP, Burge CB, Carrington JC, Chen X, Dreyfuss G, Eddy SR, Griffiths-Jones S, Marshall M, et al.. A uniform system for microRNA annotation. RNA 2003; 9:277-79; PMID:12592000; http://dx.doi.org/ 10.1261/rna.2183803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cesana M, Cacchiarelli D, Legnini I, Santini T, Sthandier O, Chinappi M, Tramontano A, Bozzoni I. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 2011; 147:358-69; PMID:22000014; http://dx.doi.org/ 10.1016/j.cell.2011.09.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ding Y, Tang Y, Kwok CK, Zhang Y, Bevilacqua PC, Assmann SM. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature 2014; 505:696-700; PMID:24270811; http://dx.doi.org/ 10.1038/nature12756 [DOI] [PubMed] [Google Scholar]
- 42.Kertesz M, Wan Y, Mazor E, Rinn JL, Nutter RC, Chang HY, Segal E. Genome-wide measurement of RNA secondary structure in yeast. Nature 2010; 467:103-07; PMID:20811459; http://dx.doi.org/ 10.1038/nature09322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lu Y, Xiao J, Lin H, Bai Y, Luo X, Wang Z, Yang B. A single anti-microRNA antisense oligodeoxyribonucleotide (AMO) targeting multiple microRNAs offers an improved approach for microRNA interference. Nucleic Acids Res 2009; 37:e24; PMID:19136465; http://dx.doi.org/ 10.1093/nar/gkn1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Okamura K, Phillips MD, Tyler DM, Duan H, Chou YT, Lai EC. The regulatory activity of microRNA* species has substantial influence on microRNA and 3′ UTR evolution. Nat Struct Mol Biol 2008; 15:354-63; PMID:18376413; http://dx.doi.org/ 10.1038/nsmb.1409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Underwood JG, Uzilov AV, Katzman S, Onodera CS, Mainzer JE, Mathews DH, Lowe TM, Salama SR, Haussler D. FragSeq: transcriptome-wide RNA structure probing using high-throughput sequencing. Nat Methods 2010; 7:995-1001; PMID:21057495; http://dx.doi.org/ 10.1038/nmeth.1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zheng Q, Ryvkin P, Li F, Dragomir I, Valladares O, Yang J, Cao K, Wang LS, Gregory BD. Genome-wide double-stranded RNA sequencing reveals the functional significance of base-paired RNAs in Arabidopsis. PLoS Genet 2010; 6:e1001141; PMID:20941385; http://dx.doi.org/ 10.1371/journal.pgen.1001141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Westhof E, Romby P. The RNA structurome: high-throughput probing. Nat Methods 2010; 7:965-67; PMID:21116245; http://dx.doi.org/ 10.1038/nmeth1210-965 [DOI] [PubMed] [Google Scholar]
- 48.Fischer RL, Goldberg RB. Structure and flanking regions of soybean seed protein genes. Cell 1982; 29:651-660; PMID:6288266; http://dx.doi.org/ 10.1016/0092-8674(82)90181-7 [DOI] [PubMed] [Google Scholar]
- 49.Walker TA, Johnson KD, Olsen GJ, Peters MA, Pace NR. Enzymatic and chemical structure mapping of mouse 28S ribosomal ribonucleic acid contacts in 5.8S ribosomal ribonucleic acid. Biochemistry 1982; 21:2320-29; PMID:7093191; http://dx.doi.org/ 10.1021/bi00539a008 [DOI] [PubMed] [Google Scholar]
- 50.Berezikov E, Robine N, Samsonova A, Westholm JO, Naqvi A, Hung JH, Okamura K, Dai Q, Bortolamiol-Becet D, Martin R, et al.. Deep annotation of Drosophila melanogaster microRNAs yields insights into their processing, modification, and emergence. Genome Res 2011; 21:203-15; PMID:21177969; http://dx.doi.org/ 10.1101/gr.116657.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gregory BD, O'Malley RC, Lister R, Urich MA, Tonti-Filippini J, Chen H, Millar AH, Ecker JR. A link between RNA metabolism and silencing affecting Arabidopsis development. Dev Cell 2008; 14:854-66; PMID:18486559; http://dx.doi.org/ 10.1016/j.devcel.2008.04.005 [DOI] [PubMed] [Google Scholar]
- 52.Willmann MR, Berkowitz ND, Gregory BD. Improved genome-wide mapping of uncapped and cleaved transcripts in eukaryotes–GMUCT 2.0. Methods 2014; 67:64-73; PMID:23867340; http://dx.doi.org/ 10.1016/j.ymeth.2013.07.003 [DOI] [PubMed] [Google Scholar]
- 53.German MA, Luo S, Schroth G, Meyers BC, Green PJ. Construction of Parallel Analysis of RNA Ends (PARE) libraries for the study of cleaved miRNA targets and the RNA degradome. Nature Protoc 2009; 4:356-62; http://dx.doi.org/ 10.1038/nprot.2009.8 [DOI] [PubMed] [Google Scholar]
- 54.German MA, Pillay M, Jeong DH, Hetawal A, Luo S, Janardhanan P, Kannan V, Rymarquis LA, Nobuta K, German R, et al.. Global identification of microRNA-target RNA pairs by parallel analysis of RNA ends. Nature Biotechnol 2008; 26:941-46; http://dx.doi.org/ 10.1038/nbt1417 [DOI] [PubMed] [Google Scholar]
- 55.Addo-Quaye C, Eshoo TW, Bartel DP, Axtell MJ. Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr Biol 2008; 18:758-62; PMID:18472421; http://dx.doi.org/ 10.1016/j.cub.2008.04.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li YF, Zheng Y, Addo-Quaye C, Zhang L, Saini A, Jagadeeswaran G, Axtell MJ, Zhang W, Sunkar R. Transcriptome-wide identification of microRNA targets in rice. Plant J 2010; 62:742-59; PMID:20202174; http://dx.doi.org/ 10.1111/j.1365-313X.2010.04187.x [DOI] [PubMed] [Google Scholar]
- 57.Folkes L, Moxon S, Woolfenden HC, Stocks MB, Szittya G, Dalmay T, Moulton V. PAREsnip: a tool for rapid genome-wide discovery of small RNA/target interactions evidenced through degradome sequencing. Nucleic Acids Res 2012; 40:e103; PMID:22467211; http://dx.doi.org/ 10.1093/nar/gks277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zheng Y, Li YF, Sunkar R, Zhang W. SeqTar: an effective method for identifying microRNA guided cleavage sites from degradome of polyadenylated transcripts in plants. Nucleic Acids Res. 2012; 40:e28; PMID:22140118; http://dx.doi.org/ 10.1093/nar/gkr1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Meng Y, Gou L, Chen D, Wu P, Chen M. High-throughput degradome sequencing can be used to gain insights into microRNA precursor metabolism. J Exp Bot 2010; 61:3833-37; PMID:20643809; http://dx.doi.org/ 10.1093/jxb/erq209 [DOI] [PubMed] [Google Scholar]
- 60.Schapire AL, Bologna NG, Moro B, Zhai J, Meyers BC, Palatnik JF. Construction of Specific Parallel Amplification of RNA Ends (SPARE) libraries for the systematic identification of plant microRNA processing intermediates. Methods 2013; 64:283-91; PMID:24018204; http://dx.doi.org/ 10.1016/j.ymeth.2013.08.032 [DOI] [PubMed] [Google Scholar]
- 61.Bologna NG, Mateos JL, Bresso EG, Palatnik JF. A loop-to-base processing mechanism underlies the biogenesis of plant microRNAs miR319 and miR159. EMBO J 2009; 28:3646-56; PMID:19816405; http://dx.doi.org/ 10.1038/emboj.2009.292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li Y, Li C, Ding G, Jin Y. Evolution of MIR159/319 microRNA genes and their post-transcriptional regulatory link to siRNA pathways. BMC Evol Biol 2011; 11:122; PMID:21569383; http://dx.doi.org/ 10.1186/1471-2148-11-122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bass BL. RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem 2002; 71:817-46; PMID:12045112; http://dx.doi.org/ 10.1146/annurev.biochem.71.110601.135501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cattaneo R. Different types of messenger RNA editing. Annu Rev Genet 1991; 25:71-88; PMID:1725953; http://dx.doi.org/ 10.1146/annurev.ge.25.120191.000443 [DOI] [PubMed] [Google Scholar]
- 65.Gott JM, Emeson RB. Functions and mechanisms of RNA editing. Annu Rev Genet 2000; 34:499-531; PMID:11092837; http://dx.doi.org/ 10.1146/annurev.genet.34.1.499 [DOI] [PubMed] [Google Scholar]
- 66.Hoopengardner B. Adenosine-to-inosine RNA editing: perspectives and predictions. Mini Rev Med Chem 2006; 6:1213-16; PMID:17100632; http://dx.doi.org/ 10.2174/138955706778742812 [DOI] [PubMed] [Google Scholar]
- 67.Maier RM, Zeltz P, Kossel H, Bonnard G, Gualberto JM, Grienenberger JM. RNA editing in plant mitochondria and chloroplasts. Plant Mol. Biol 1996; 32:343-65; PMID:8980487; http://dx.doi.org/ 10.1007/BF00039390 [DOI] [PubMed] [Google Scholar]
- 68.Shikanai T. RNA editing in plant organelles: machinery, physiological function and evolution. Cell Mol Life Sci 2006; 63:698-708; PMID:16465445; http://dx.doi.org/ 10.1007/s00018-005-5449-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hayes ML, Hanson MR. Identification of a sequence motif critical for editing of a tobacco chloroplast transcript. RNA 2007; 13:281-88; PMID:17158709; http://dx.doi.org/ 10.1261/rna.295607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tillich M, Funk HT, Schmitz-Linneweber C, Poltnigg P, Sabater B, Martin M, Maier RM. Editing of plastid RNA in Arabidopsis thaliana ecotypes. Plant J 2005; 43:708-15; PMID:16115067; http://dx.doi.org/ 10.1111/j.1365-313X.2005.02484.x [DOI] [PubMed] [Google Scholar]
- 71.Ebhardt HA, Tsang HH, Dai DC, Liu Y, Bostan B, Fahlman RP. Meta-analysis of small RNA-sequencing errors reveals ubiquitous post-transcriptional RNA modifications. Nucleic Acids Res 2009; 37:2461-70; PMID:19255090; http://dx.doi.org/ 10.1093/nar/gkp093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Luciano DJ, Mirsky H, Vendetti NJ, Maas S. RNA editing of a miRNA precursor. RNA 2004; 10:1174-77; PMID:15272117; http://dx.doi.org/ 10.1261/rna.7350304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ohman M. A-to-I editing challenger or ally to the microRNA process. Biochimie 2007; 89:1171-76; PMID:17628290; http://dx.doi.org/ 10.1016/j.biochi.2007.06.002 [DOI] [PubMed] [Google Scholar]
- 74.Meng Y, Chen D, Jin Y, Mao C, Wu P, Chen M. RNA editing of nuclear transcripts in Arabidopsis thaliana. BMC Genomics 2010; 11(Suppl 4):S12; PMID:21143795; http://dx.doi.org/ 10.1186/1471-2164-11-S4-S12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 2014; 42:D68-73; PMID:24275495; http://dx.doi.org/ 10.1093/nar/gkt1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Berezikov E, Chung WJ, Willis J, Cuppen E, Lai EC. Mammalian mirtron genes. Mol Cell 2007; 28:328-36; PMID:17964270; http://dx.doi.org/ 10.1016/j.molcel.2007.09.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Berezikov E, Liu N, Flynt AS, Hodges E, Rooks M, Hannon GJ, Lai EC. Evolutionary flux of canonical microRNAs and mirtrons in Drosophila. Nat Genet 2010; 42:6-9; author reply 9–10; PMID:20037610; http://dx.doi.org/ 10.1038/ng0110-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chung WJ, Agius P, Westholm JO, Chen M, Okamura K, Robine N, Leslie CS, Lai EC. Computational and experimental identification of mirtrons in Drosophila melanogaster and Caenorhabditis elegans. Genome Res 2011; 21:286-300; PMID:21177960; http://dx.doi.org/ 10.1101/gr.113050.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Curtis HJ, Sibley CR, Wood MJ. Mirtrons, an emerging class of atypical miRNA. Wiley Interdiscip Rev RNA 2012; 3:617-32; PMID:22733569; http://dx.doi.org/ 10.1002/wrna.1122 [DOI] [PubMed] [Google Scholar]
- 80.Joshi PK, Gupta D, Nandal UK, Khan Y, Mukherjee SK, Sanan-Mishra N. Identification of mirtrons in rice using MirtronPred: a tool for predicting plant mirtrons. Genomics 2012; 99:370-75; PMID:22546559; http://dx.doi.org/ 10.1016/j.ygeno.2012.04.002 [DOI] [PubMed] [Google Scholar]
- 81.Ladewig E, Okamura K, Flynt AS, Westholm JO, Lai EC. Discovery of hundreds of mirtrons in mouse and human small RNA data. Genome Res 2012; 22:1634-45; PMID:22955976; http://dx.doi.org/ 10.1101/gr.133553.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Meng Y, Shao C. Large-scale identification of mirtrons in Arabidopsis and rice. PLoS One 2012; 7:e31163; PMID:22348048; http://dx.doi.org/ 10.1371/journal.pone.0031163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Okamura K, Hagen JW, Duan H, Tyler DM, Lai EC. The mirtron pathway generates microRNA-class regulatory RNAs in Drosophila. Cell 2007; 130:89-100; PMID:17599402; http://dx.doi.org/ 10.1016/j.cell.2007.06.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sibley CR, Seow Y, Saayman S, Dijkstra KK, El Andaloussi S, Weinberg MS, Wood MJ. The biogenesis and characterization of mammalian microRNAs of mirtron origin. Nucleic Acids Res 2012; 40:438-48; PMID:21914725; http://dx.doi.org/ 10.1093/nar/gkr722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shao C, Ma X, Xu X, Wang H, Meng Y. Genome-wide identification of reverse complementary microRNA genes in plants. PLoS One 2012; 7:e46991; PMID:23110057; http://dx.doi.org/ 10.1371/journal.pone.0046991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Meng Y, Shao C, Ma X, Wang H. Introns targeted by plant microRNAs: a possible novel mechanism of gene regulation. Rice 2013; 6:8; http://dx.doi.org/ 10.1186/1939-8433-6-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ebert MS, Sharp PA. Emerging roles for natural microRNA sponges. Curr Biol 2010; 20:R858-861; PMID:20937476; http://dx.doi.org/ 10.1016/j.cub.2010.08.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, Kjems J. Natural RNA circles function as efficient microRNA sponges. Nature 2013; 495:384-88; PMID:23446346; http://dx.doi.org/ 10.1038/nature11993 [DOI] [PubMed] [Google Scholar]
- 89.Arvey A, Larsson E, Sander C, Leslie CS, Marks DS. Target mRNA abundance dilutes microRNA and siRNA activity. Mol Syst Biol 2010; 6:363; PMID:20404830; http://dx.doi.org/ 10.1038/msb.2010.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 2010; 465:1033-38; PMID:20577206; http://dx.doi.org/ 10.1038/nature09144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sumazin P, Yang X, Chiu HS, Chung WJ, Iyer A, Llobet-Navas D, Rajbhandari P, Bansal M, Guarnieri P, Silva J, et al.. An extensive microRNA-mediated network of RNA-RNA interactions regulates established oncogenic pathways in glioblastoma. Cell 2011; 147:370-81; PMID:22000015; http://dx.doi.org/ 10.1016/j.cell.2011.09.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tay Y, Kats L, Salmena L, Weiss D, Tan SM, Ala U, Karreth F, Poliseno L, Provero P, Di Cunto F, et al.. Coding-independent regulation of the tumor suppressor PTEN by competing endogenous mRNAs. Cell 2011; 147:344-57; PMID:22000013; http://dx.doi.org/ 10.1016/j.cell.2011.09.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chitwood DH, Timmermans MC. Target mimics modulate miRNAs. Nat Genet 2007; 39:935-36; PMID:17660806; http://dx.doi.org/ 10.1038/ng0807-935 [DOI] [PubMed] [Google Scholar]
- 94.Franco-Zorrilla JM, Valli A, Todesco M, Mateos I, Puga MI, Rubio-Somoza I, Leyva A, Weigel D, Garcia JA, Paz-Ares J. Target mimicry provides a new mechanism for regulation of microRNA activity. Nat Genet 2007; 39:1033-37; PMID:17643101; http://dx.doi.org/ 10.1038/ng2079 [DOI] [PubMed] [Google Scholar]
- 95.Meng Y, Shao C, Wang H, Jin Y. Target mimics: an embedded layer of microRNA-involved gene regulatory networks in plants. BMC Genomics 2012; 13:197; PMID:22613869; http://dx.doi.org/ 10.1186/1471-2164-13-197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wu HJ, Wang ZM, Wang M, Wang XJ. Widespread long noncoding RNAs as endogenous target mimics for microRNAs in plants. Plant Physiol 2013; 161:1875-84; PMID:23429259; http://dx.doi.org/ 10.1104/pp.113.215962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Todesco M, Rubio-Somoza I, Paz-Ares J, Weigel D. A collection of target mimics for comprehensive analysis of microRNA function in Arabidopsis thaliana. PLoS Genet 2010; 6:e1001031; http://dx.doi.org/ 10.1371/journal.pgen.1001031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ebert MS, Sharp PA. MicroRNA sponges: progress and possibilities. RNA 2010; 16:2043-50; PMID:20855538; http://dx.doi.org/ 10.1261/rna.2414110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cheng AM, Byrom MW, Shelton J, Ford LP. Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res 2005; 33:1290-97; PMID:15741182; http://dx.doi.org/ 10.1093/nar/gki200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Eckstein F. The versatility of oligonucleotides as potential therapeutics. Expert Opin Biol Ther 2007; 7:1021-34; PMID:17665991; http://dx.doi.org/ 10.1517/14712598.7.7.1021 [DOI] [PubMed] [Google Scholar]
- 101.Hammond SM. MicroRNA therapeutics: a new niche for antisense nucleic acids. Trends Mol Med 2006; 12:99-101; PMID:16473043; http://dx.doi.org/ 10.1016/j.molmed.2006.01.004 [DOI] [PubMed] [Google Scholar]
- 102.Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature 2005; 438:685-89; PMID:16258535; http://dx.doi.org/ 10.1038/nature04303 [DOI] [PubMed] [Google Scholar]
- 103.Stelzer Y, Sagi I, Benvenisty N. Involvement of parental imprinting in the antisense regulation of onco-miR-372-373. Nat Commun 2013; 4:2724; PMID:24201333; http://dx.doi.org/ 10.1038/ncomms3724 [DOI] [PubMed] [Google Scholar]
- 104.Stenvang J, Kauppinen S. MicroRNAs as targets for antisense-based therapeutics. Expert. Opin. Biol. Ther. 2008; 8:59-81; PMID:18081537; http://dx.doi.org/ 10.1517/14712598.8.1.59 [DOI] [PubMed] [Google Scholar]
- 105.Mullokandov G, Baccarini A, Ruzo A, Jayaprakash AD, Tung N, Israelow B, Evans MJ, Sachidanandam R, Brown BD. High-throughput assessment of microRNA activity and function using microRNA sensor and decoy libraries. Nat. Methods 2012; 9:840-46; PMID:22751203; http://dx.doi.org/ 10.1038/nmeth.2078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Xie J, Ameres SL, Friedline R, Hung JH, Zhang Y, Xie Q, Zhong L, Su Q, He R, Li M, et al.. Long-term, efficient inhibition of microRNA function in mice using rAAV vectors. Nat Methods 2012; 9:403-09; PMID:22388288; http://dx.doi.org/ 10.1038/nmeth.1903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhang Y, Roccaro AM, Rombaoa C, Flores L, Obad S, Fernandes SM, Sacco A, Liu Y, Ngo H, Quang P, et al.. LNA-mediated anti-miR-155 silencing in low-grade B-cell lymphomas. Blood 2012; 120:1678-86; PMID:22797699; http://dx.doi.org/ 10.1182/blood-2012-02-410647 [DOI] [PubMed] [Google Scholar]
- 108.Hogan DJ, Vincent TM, Fish S, Marcusson EG, Bhat B, Chau BN, Zisoulis DG. Anti-miRs Competitively Inhibit microRNAs in Argonaute Complexes. PLoS One 2014; 9:e100951; PMID:24992387; http://dx.doi.org/ 10.1371/journal.pone.0100951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wang H, Zhang X, Liu J, Kiba T, Woo J, Ojo T, Hafner M, Tuschl T, Chua NH, Wang XJ. Deep sequencing of small RNAs specifically associated with Arabidopsis AGO1 and AGO4 uncovers new AGO functions. Plant J. 2011; 67:292-304; PMID:21457371; http://dx.doi.org/ 10.1111/j.1365-313X.2011.04594.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Huala E, Dickerman AW, Garcia-Hernandez M, Weems D, Reiser L, LaFond F, Hanley D, Kiphart D, Zhuang M, Huang W, et al.. The Arabidopsis Information Resource (TAIR): a comprehensive database and web-based information retrieval, analysis, and visualization system for a model plant. Nucleic Acids Res 2001; 29:102-105; PMID:11125061; http://dx.doi.org/ 10.1093/nar/29.1.102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1141.Devers EA, Branscheid A, May P, Krajinski F. Stars and symbiosis: microRNA- and microRNA*-mediated transcript cleavage involved in arbuscular mycorrhizal symbiosis. Plant Physiol 2011; 156:1990-2010; PMID:21571671; http://dx.doi.org/ 10.1104/pp.111.172627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Meng Y, Shao C, Wang H, Chen M. The regulatory activities of plant microRNAs: a more dynamic perspective. Plant Physiol. 2011; 157:1583-1595; PMID:22003084; http://dx.doi.org/ 10.1104/pp.111.187088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Packer AN, Xing Y, Harper SQ, Jones L, Davidson BL. The bifunctional microRNA miR-9/miR-9* regulates REST and CoREST and is downregulated in Huntington's disease. J Neurosci 2008; 28:14341-46; PMID:19118166; http://dx.doi.org/ 10.1523/JNEUROSCI.2390-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yang JS, Phillips MD, Betel D, Mu P, Ventura A, Siepel AC, Chen KC, Lai EC. Widespread regulatory activity of vertebrate microRNA* species. RNA 2011; 17; 312-26; PMID:21177881; http://dx.doi.org/ 10.1261/rna.2537911 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.