

Abstract
Technological advantages in sequencing and proteomics have revealed the remarkable diversity of alternative protein isoforms. Typically, the localization and functions of these isoforms are unknown and cannot be predicted. Also the localization signals leading to particular subnuclear compartments have not been identified and thus, predicting alternative functions due to alternative subnuclear localization is limited only to very few subnuclear compartments. Knowledge of the localization and function of alternative protein isoforms allows for a greater understanding of cellular complexity. In this article, we characterize a short and well-defined signal targeting the bovine papillomavirus type 1 E8/E2 protein to the nuclear matrix. The targeting signal comprises the peptide coded by E8 ORF, which is spliced together with part of the E2 ORF to generate the E8/E2 mRNA. Localization to the nuclear matrix correlates well with the transcription repression activities of E8/E2; a single point mutation directs the E8/E2 protein into the nucleoplasm, and transcription repression activity is lost. Our data prove that adding as few as ˜10 amino acids by alternative transcription/alternative splicing drastically alters the function and subnuclear localization of proteins. To our knowledge, E8 is the shortest known nuclear matrix targeting signal.
Keywords: alternative protein isoform, alternative splicing, alternative transcription, molecular trafficking, nuclear matrix targeting sequence, nuclear organelles, nuclear structures, papillomavirus, subnuclear compartmentalisation
Abbreviations
- BPV1
Bovine Papillomavirus type 1
- CSK
cytoskeleton buffer
- DBD
DNA-binding domain
- FRET
Förster resonance energy transfer
- NM
nuclear matrix
- NMdb
nuclear matrix database
- NMTS
nuclear matrix targeting signal.
Introduction
Protein compartmentalisation inside the cell is an important feature that defines the functions and activities of the protein. Protein compartmentalisation and subcellular localization determines access to interacting partners and post-translational modification machinery and enables the integration of proteins into functional biological networks.1 Mislocalisation and miscompartmentalisation may be detrimental for cells and organisms and have been associated with human diseases.1,2 The eukaryotic cell nucleus contains many different compartments and most nuclear events do not occur uniformly throughout the nucleus. Such events are typically limited to specific, spatially defined sites.3,4
The nuclear matrix (NM) is fibrillar network inside the nucleus that organizes its 3D structure. The NM is one of the most important subnuclear compartments and is targeted by different proteins.5 As in subcellular localization, NM targeting depends on specific sequences known as NM targeting signals (NMTS). The proteins targeted to the NM have been listed in the Nuclear Matrix Database (NMdb) (www.rostlab.org/db/NMPdb).6 The NMdb also contains sequences for NM targeting signals. To date, no clear rules or determinants predicting NM targeting have been published; predictions of NM localization have not been very successful.7 Disruption of NM targeting often leads to changes in protein function, highlighting the relationship between localization and function.8,9
Alternative transcription and alternative splicing generates alternative protein isoforms and is one factor beside others that regulate the subcellular localization and subnuclear compartmentalisation of proteins. Alternative splicing is widely used and is found at least 90% of multiexon genes in humans.10,11 This percentage was believed to be 75% in 2003, and has increased over time.12,13 Alternative transcription initiation is found in 29% of human genes; in 20% of these genes it generates the new first coding exon.14 In silico analysis during the course of the ENCODE project demonstrated that alternative splicing results in changes in subcellular localization in approximately 10% of alternative splicing cases, where it is possible to predict localization.15 However, most subnuclear localization signals or the targeting signals for specific compartments are not well defined and cannot be predicted with high confidence.7 There are increasing list of changes in protein localization due to alternative splicing.16 Alternative splicing has been shown to affect sub-cellular localization and the subnuclear compartmentalisation of viral17 and cellular proteins (ING418,19 and WT120); some of these proteins are related to diseases, such as CIZ1.21 Alternative transcription has been predicted to play a more prominent role compared to alternative splicing in generating diversity at both termini of proteins often coding localization signals, so the alternative localization of protein isoforms is rather underestimated.22
Papillomaviruses are small DNA tumor viruses that encode a limited number of proteins. Alternative transcription and alternative splicing are used to generate functionally diverse viral proteins.23 This also occurs in the E2 family of proteins. The E2 ORF of Bovine Papillomavirus type 1 (BPV1) encodes for the 3 protein isoforms: the full-length E2 protein (E2); E2C (also named E2TR), which is transcribed from an internal promoter; and E8/E2, which is generated by alternative transcription/alternative splicing (Fig. 1A). The E2 protein is a typical transcriptional regulator containing a transactivation domain, a DNA-binding/dimerization domain (DBD) and a “hinge” region between them (Fig. 1A). E2C and E8/E2 contain the “hinge” domain and the DBD but do not have a fully functional transactivation domain (Fig. 1A). The E8/E2 protein is generated by fusing an exon coding 11 aa from the E8 ORF (splice donor at nt 1235 in BPV1 genome) with the “hinge” and the DBD region of the E2 ORF (splice acceptor at nt 3225). All three isoforms contain a NLS located in the DBD and are localized to the cell nucleus.24 The E2 protein functions as a transcriptional regulator of viral gene expression, as a viral replication protein and as a chromatin attachment protein that maintains the stability of viral episomes. The role of the shorter E2 isoforms is not very clear. By competing with E2 for binding to the E2 binding sites (E2BS), these proteins function as repressors of E2 activity. By forming heterodimers with E2, they modulate specific E2 functions.25,26 In this way, the additional E2 isoforms are integral part of viral regulatory networks. Subcellular localization has been described for E2 (mostly chromatin) and E2C (nucleoplasm/nucleosol).27
Figure 1.
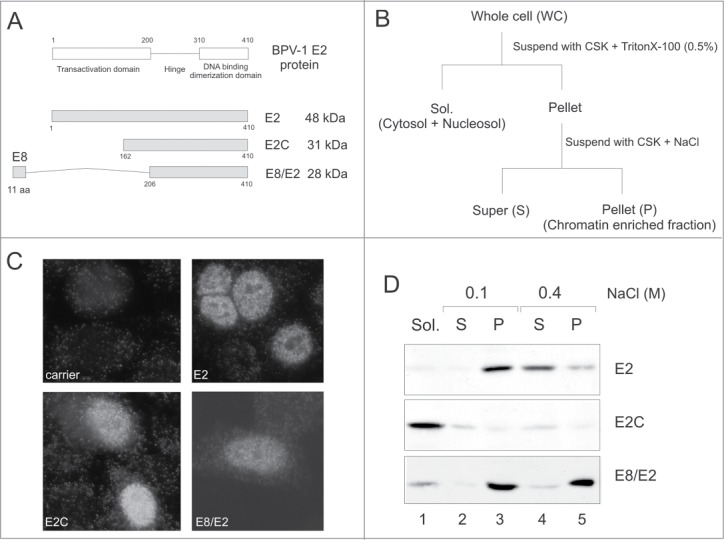
Proteins encoded by the BPV1 E2 ORF have different subnuclear localisations. (A) A schematic representation of the E2 proteins found in BPV1-transformed cells. The location of the E8 ORF with respect to the E2 ORF is not drawn to scale. The ATG of the E8 ORF (at nt 1204) is located 404 bp upstream from the E2 ATG (at nt 2608). (B) The biochemical fractionation protocol. (C) All 3 E2 proteins localized to the cell nucleus in transiently transfected CHO cells in an IF analysis. (D) Western blot of the biochemical fractionation profiles of different E2 proteins in transiently transfected CHO cells.
In this article, we characterize the subnuclear localization of the BPV1 E8/E2 protein. We demonstrate that E8/E2 is found in the insoluble nuclear fraction and this finding shows that all 3 E2 proteins are localized to different subnuclear compartments. Our data indicate that E8/E2 is localized to the NM and that 11 aa from the E8 ORF are required and sufficient for this localization. According to NMdb, this sequence is the shortest NMTS known. The E8 NMTS does not have any sequence similarity to previously characterized NMTS. A single point mutation in the E8 NMTS region directs the E8/E2 protein to the nucleoplasm and loss of E2BS-dependent promoter repression activity also occurred. Therefore, subnuclear miscompartmentalisation resulted in the alteration of protein function.
Results
The three E2 proteins localize to different subnuclear compartments
The nuclear localization signal of E2 is mapped to the C-terminal DBD region, which is present in all 3 E2 proteins encoded by the BPV1 genome.28,29 All 3 E2 proteins localized to the cell nucleus of transiently transfected CHO cells, as shown by indirect immunofluorescence analysis (Fig. 1C). The full-length E2 protein displayed a fine dotted pattern - E2C and E8/E2 showed more diffuse signals. Using a biochemical fractionation approach, Kurg et al have previously demonstrated that the full-length E2 protein and E2C distribute differently within the cell nucleus; E2 is associated with nuclear substructures, including cellular chromatin, and E2C localizes to the nucleosol.27 In the present work, we used the same biochemical fractionation approach to determine the subnuclear localization of E8/E2, a second truncated isoform of the E2 protein. We transfected CHO cells with expression plasmids for E2, E2C or E8/E2, and then subjected cells to biochemical fractionation, as depicted in Figure 1B. The presence of E2 proteins in different fractions was analyzed by immunoblotting with E2-specific antibodies that recognized all 3 E2 proteins. As shown on Figure 1D, E2C was extractable from nuclear structures with the non-ionic detergent Triton X-100 (lane 1); E2 and E8/E2 remained in the chromatin-enriched nuclear fraction. E2 was extractable from this fraction with 0.4 M NaCl (lane 4), but E8/E2 was not (lane 5), suggesting that the 2 truncated E2 proteins, E2C and E8/E2, have different nuclear fractionation profiles. Taken together, these data demonstrate that all 3 E2 proteins have different subnuclear compartmentalisation. The full-length E2 protein is localized to the nuclear fraction containing chromatin-associated proteins, E2C is localized to the nucleoplasm, and E8/E2 fractionates with insoluble nuclear substructures.
The E8/E2 protein localizes to the nuclear matrix fraction
The E2C protein localizes to the nucleosol fraction and E2 into chromatin fraction but the precise compartmentalisation of E8/E2 is not known. We examined this phenomenon in more detail and applied a more precise biochemical fractionation protocol, as depicted in Figure 2A. As shown in Figure 2B, the E8/E2 protein remains bound to nuclear substructures after treatment with 0.4 M NaCl (lane 4), after RNase (lane 6) and DNase treatment and remains insoluble after 2 M NaCl extraction (lane 9). Histones where solubilised after DNase treatment (Fig. 4B, Coomassie staining and data not shown). Identical results were obtained in the Cos7 cell-line and in the CHO-derived cell line CHOEBNALT85. Similar localization in different cell-lines indicates the occurrence of a general phenomenon. Therefore, E8/E2 localizes into a salt-, RNase- and DNase-resistant insoluble fraction of the cell nucleus known as the nuclear matrix (NM). Throughout this article, the term “nuclear matrix” is used to refer to this insoluble biochemical fraction.
Figure 2.
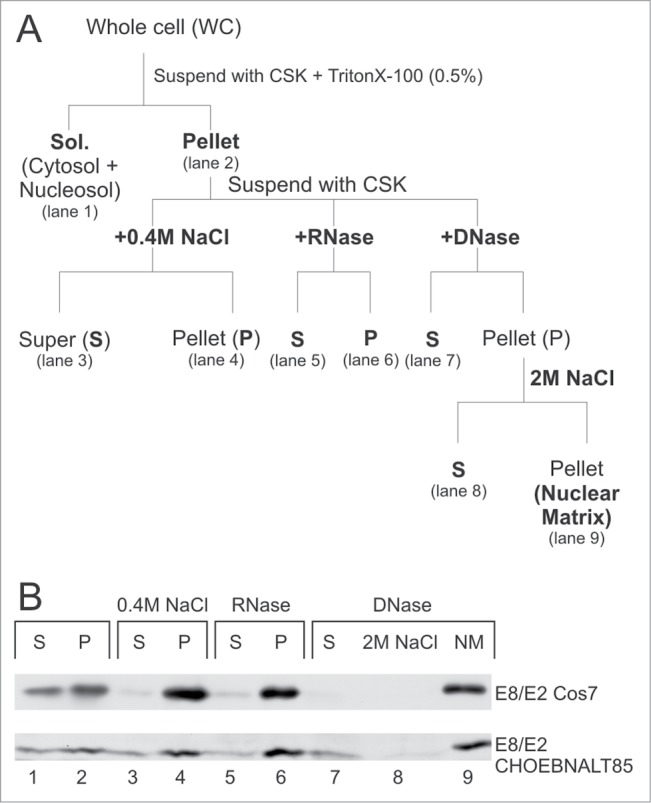
The E8/E2 protein localizes to nuclear matrix (NM) fraction in Cos7 and CHOEBNALT85 cell lines. (A) At the top, the biochemical fractionation scheme is shown. Biochemical fractionation and subsequent protein gel blotting was performed as described in the material and methods section. S and P indicate the supernatant and the pellet fraction, respectively. (B) Fractionation profiles of the respective proteins in different cell lines are shown.
Figure 4.
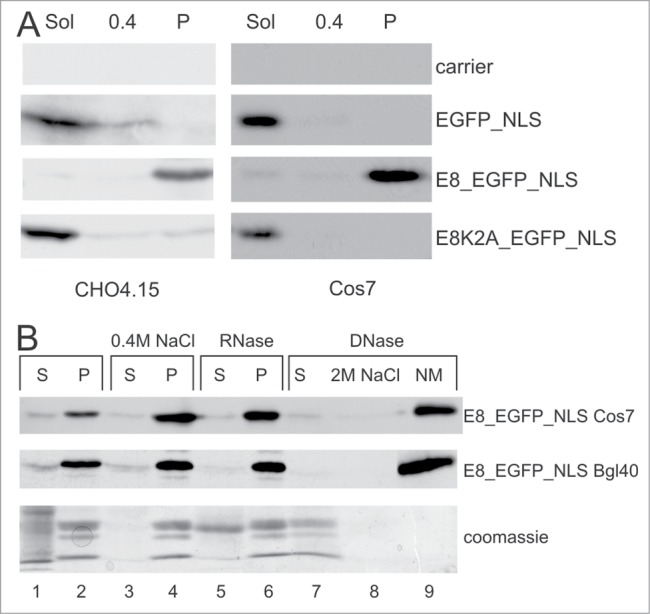
The E8 peptide is sufficient to target a heterologous protein to the NM. (A) Western blotting of the biochemical fractionations from CHO4.15 and Cos7 cells transfected with respective plasmids. Sol - CSK soluble fraction; 0.4 - soluble fraction in CSK + 0.4 M NaCl; P - insoluble fraction in CSK + 0.4 M NaCl. (B) The E8_EGFP_NLS protein localizes to the nuclear matrix (NM) fraction in Cos7 and CHOBgl40 cell lines. Fractionation profiles (as in Fig. 2A) of respective proteins in different cell lines are shown. On the bottom panel, the Coomassie stained gel is shown to verify the efficiency of fractionation. A typical histone pattern appears in lanes 2, 4, 6 and 7. The MW of the strong band in line 5 (marked “RNase S”) corresponds to the MW of RNaseA.
The E8 peptide is required for NM localization
The E2C and E8/E2 isoforms have different biochemical fractionation profiles. Only an 11 aa sequence MKLTVFLRPSR in the N-terminus of the E8/E2 are coded by the E8 ORF (Fig. 1A) and not shared with E2C or E2. To determine whether the retention of E8/E2 in NM fraction is due to the presence of the 11 aa E8 peptide unique to E8/E2, we tested 3 derivatives of E8/E2 in biochemical fractionation experiments. As shown in Figure 3, the removal of the 5′ untranslated leader sequence in the mRNA did not affect protein localization (panel nlE8/E2 in Fig. 3). Deleting the first 15 aa region, what consists of the E8 peptide and 4 extra aa from the N-terminus of E8/E2 resulted in a protein that contains the “hinge” and the DBD region of E2 and localized this protein to the soluble fraction (panel nlE2_HDBD on Fig. 3) similar to E2C. We also tested an E8/E2 protein, where the lysine residue in position 2 of the E8 peptide was mutated to Ala (Lys 2 is the most highly conserved residue among papillomaviruses in this type of E8 peptide) in biochemical fractionation. The corresponding E8/E2_K2A mutant was no longer retained in the NM fraction in the Cos7 and CHO cell lines (panel nlE8/E2_K2A in Fig. 3); the mutant protein instead remained in the soluble fraction.
Figure 3.
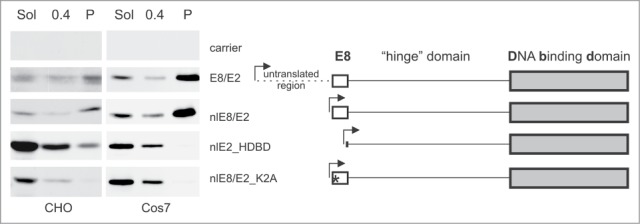
The E8 part of the E8/E2 protein is required for NM targeting. Western blot of biochemical fractions from CHO and Cos7 cells transfected with respective plasmids. The differences between the expression constructs are depicted on the right. The arrowheads mark the transcription start site. The asterisk indicates the point mutation K2A. Sol - CSK soluble fraction, 0.4 - the soluble fraction in CSK + 0.4 M NaCl, P - the insoluble fraction in CSK + 0.4 M NaCl.
These data clearly demonstrate that the E8 peptide (the E8 region of E8/E2) is required for E8/E2 localization to the NM fraction.
The E8 peptide is sufficient for NM localization
To test whether the E8 peptide is sufficient for localizing heterologous proteins to NM fraction, we fused 13 aa (MKLTVFLRPSRDR) from the N-terminus of the E8/E2 protein to EGFP proteins. All three E2 proteins (E2, E2C and E8/E2) are nuclear proteins containing the NLS in the DNA-binding domain. To target the EGFP fusion proteins to the nucleus of the cell, we fused the NLS motif of the SV40 Large T antigen to the C-terminus of the protein. All of the EGFP derivatives with NLS fusions localized to the nucleus of living cells (data not shown). As shown in Figure 4A, in COS7 cells EGFP_NLS was found mostly in the soluble fraction, consisting of proteins from the cytosol and the nucleosol. E8_EGFP_NLS was found in the insoluble fraction, and E8K2A_EGFP_NLS was found in the soluble fraction (Fig. 4A). Similar results were obtained in the CHO-derived cell line CHO4.15. We also examined the fractionation profile of E8_EGFP_NLS more precisely, as in the case for the E8/E2 protein. As shown in Figure 4B, E8_EGFP_NLS remained insoluble after RNase and DNase treatment; this protein is also resistant to 2 M NaCl extraction. Identical results were obtained in Cos7 and the CHO-derived cell line CHOBgl40. These results demonstrate that the E8 peptide alone is required and sufficient for the localization of proteins to the NM fraction. Therefore, the E8 peptide functions as a NMTS.
We also studied the subnuclear localization of E8_EGFP_NLS and its K2A mutant using fluorescence microscopy before or after extraction with CSK. As shown in Figure 5 there was no difference in localization between E8_EGFP_NLS and its mutant before extraction either in live cell experiments (Fig. 5B-D) or after fixation with PFA following immunofluorescence analysis with anti-GFP antibodies (Fig. 5J-L). Additionally, 3D reconstruction did not reveal any significant differences between the localization of E8_EGFP_NLS and its K2A mutant in live cells or PFA fixed cells at any expression level (data not shown). After CSK treatment the subnuclear localization of E8_EGFP_NLS was similar to that of untreated cells (compare panels B, J and R in Fig. 5 for E8_EGFP_NLS) and no significant differences in subnuclear distribution of the wt protein in treated or untreated cells were discovered by 3D reconstruction (data not shown). The only difference observed that after CSK treatment the faint cytoplasmic signal of E8_EGFP_NLS high expressing cells is lost. At the same time the signal of the EGFP_NLS and the mutant (E8K2A_EGFP_NLS) was not detectable after CSK treatment (Fig. 5 panel S and T). Further treatment of cells with DNase and RNase led to some coalescence of the E8_EGFP_NLS signal in some regions of the nucleus which might be caused by the collapse of the internal nuclear structures as supported also by reduction of nuclear dimensions by few micrometres on z-stack (Fig. 5 Õ).
Figure 5.
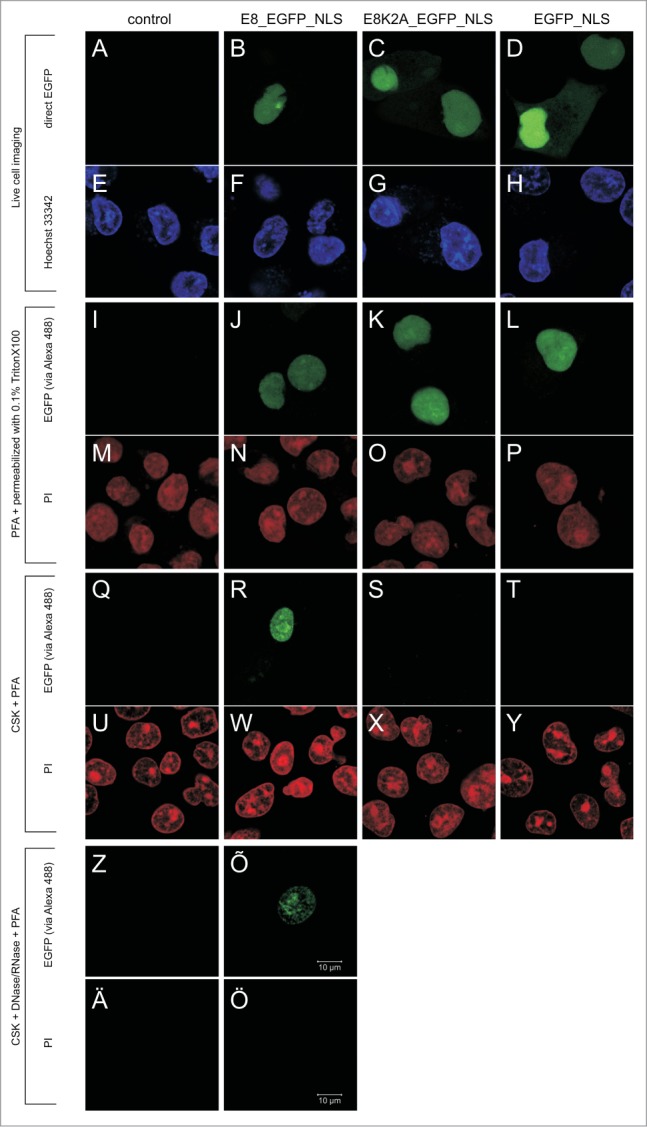
In situ analysis of the resistance of E8_EGFP_NLS to CSK and to DNase/RNase treatments. Localization of E8_EGFP_NLS and its derivatives studied in live cells or in cells fixed, before or after extraction. In columns the cells transfected with indicated plasmid are shown. Panels A-H: live cell imaging; EGFP is visualised by direct fluorescence and nucleic acid is stained with Hoechst 33342. Panels I-P: cells fixed with PFA. Panels Q-Y: cells first extracted with CSK and after that fixed with PFA. Panels Z-Ö: cells extracted with CSK and DNase/RNase and after that fixed with PFA. In fixed cells (panels I-Ö) the EGFP is visualised by immunofluorescence and nucleic acid is stained with propidium iodide (PI). Scale bar corresponding to 10 µm for all panels is shown on panel Õ.
Spatially distinct localization of proteins with intact E8 NMTS and their mutants
Biochemical fractionation and confocal microscopy experiments clearly showed that E8 and its mutant isoforms have different fractionation profiles. This might be due to localization into different subnuclear compartments or to faster koff when residing in the same compartment. To examine the loss of spatial proximity, fluorescence proteins containing the E8 NMTS and its mutant (E8K2A) were used in a FRET assay. The FRET assay allows for the measurement of the proximity of a donor-acceptor pair of fluorescence proteins in living, intact and undisturbed (untreated) cells. We hypothesized that when E8_CFP is expressed with E8_YFP, they both would localize to the NM; the probability that these 2 protein would be close enough in proximity to give a FRET signal is high (Fig. 6A). When one of the fluorescence proteins contains a mutated E8, there are 2 possible scenarios. In the first scenario, the 2 proteins have the same spatial localization but the mutant has a weaker interaction with NM (a faster koff and is extracted more easily). In this case, FRET will be as effective as it is in a homogenous system (E8_CFP + E8_YFP). In the second scenario, the spatial localization of the proteins is distinct (and their co-localization is random); in this scenario, the probability of FRET is much lower. To test this hypothesis, we expressed the respective proteins either alone or in pairs and measured their emission spectra. As shown in Figure 6, the expression of E8_CFP together with E8_YFP results in a clear FRET signal (Fig. 6B and D). The FRET signal for heterologous combinations is much weaker. To illustrate this, the spectra were normalized to the CFP emission maximum (480 nm); the respective results are shown in Figure 6D. A weaker YFP emission signal was observed in heterologous combination with a CFP-specific wavelength (431 nm). This result was not due to an absence of YFP in the cells (Fig. 6C). Emission from YFP (which is excited at YFP specific wavelength 497 nm and measured at 530 nm) is the lowest in the case of E8_CFP+E8_YFP having the highest FRET (Fig. 6C). In case of other combinations the YFP expression level was higher. The spectroscopic concentration of CFP was also lowest in the combination E8_CFP+E8_YFP (Fig. 6B). Therefore, a low FRET signal in heterologous pairs is not due to an absence of fluorescence proteins. As a quantitative measurement we used the ‘FRET ratio’. This ratio measures the difference in YFP emission in the presence of CFP compared to YFP alone when excited at a CFP-specific wavelength (431 nm). The respective values for all pairs in 2 different cell lines are shown in Figure 6E. The E8-E8 pair generated a much stronger FRET signal compared to the heterogeneous pair or the homogenous expression of the mutant E8 protein. All proteins fractionated biochemically as expected from earlier results (only proteins with an intact NMTS localized to the NM; data not shown). These data demonstrate that E8 and its mutants have different spatial localisations in undisturbed living cells.
Figure 6.
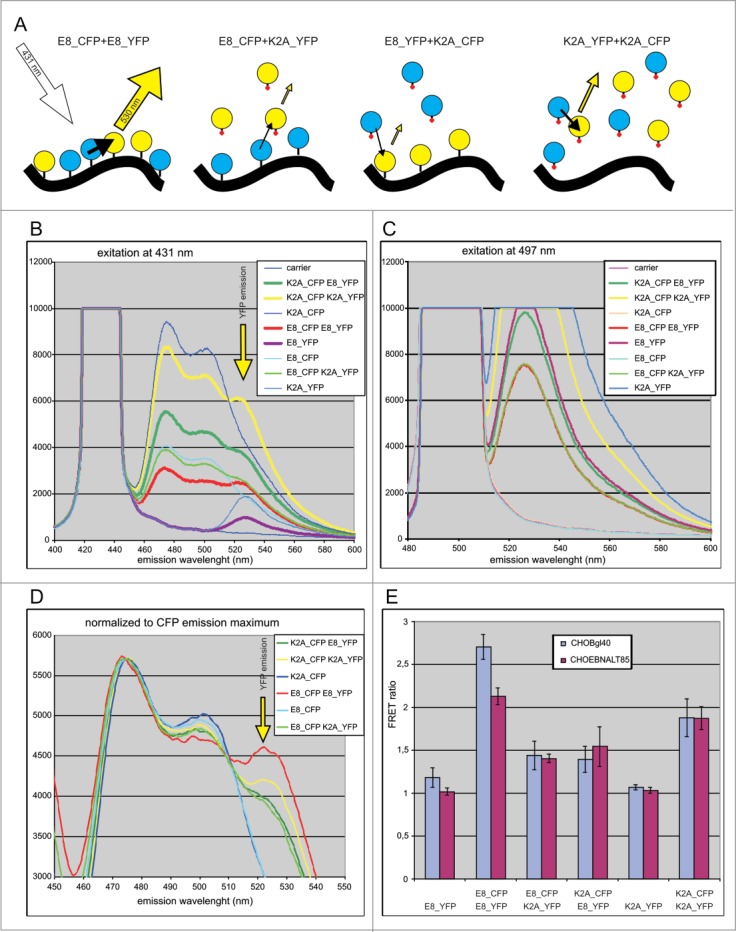
Examination of the colocalisation of E8 and its mutant in vivo in undisturbed cells by FRET analysis. (A) Schema for the FRET assay. Emission from different combinations of proteins when excited at the CFP-specific wavelength (431 nm). Yellow arrows correspond to emission at the YFP-specific wavelength (530 nm), and black arrows indicate FRET. The size of the arrows corresponds to emission or FRET intensity. Circles in cyan or yellow correspond to CFP and YFP, respectively. The tittle on each circle is the wild-type E8 peptide, and the tittle with a red dot is the mutant K2A. The black solid line represents the NM. (B) Emission spectra of the cell suspension when excited at the CFP-specific wavelength (431 nm). (C) Emission spectra of the cell suspension when excited at the YFP-specific wavelength (497 nm). (D) Emission spectra when excited at the CFP-specific wavelength and normalized to the CFP emission maximum (475 nm). (E) The FRET ratio shows how the YFP emission differs in the presence of CFP compared to YFP alone when excited at the CFP-specific wavelength in CHOBgl40 and CHOEBNALT85 cell lines.
Localization to the NM correlates with E8/E2 transcriptional repression activities
Transcriptional repression activity seems to be quite common for papillomavirus E8/E2 proteins; for the BPV1 E8/E2 proteins transcriptional repression activity was described in 1989.30 Transcriptional repression is the only functional activity reported for BPV1 E8/E2 to date. We tested E8/E2 and its derivatives for transcriptional repression activity using a dual luciferase system. As shown in Figure 7, the E8/E2 and nlE8/E2 proteins repress the native BPV1 promoter (P89) and a heterologous promoter containing an E2 BS in a dose-dependent manner in CHO cells. No repressive effects were observed for nlE2_HDBD and nlE8E2_K2A proteins missing a functional NMTS (although the expression level of proteins with a defective NMTS was higher, data not shown). Similar results were obtained in HeLa cell lines (data not shown). The repressive effect was E2 DNA binding dependent as the E8/E2 mutant defective in DNA binding (K339A)31 was not able to repress transcription (data not shown). Repression is also E2 BS-dependent, as a SV40 promoter without an E2 BS was not repressed by E8/E2 (data not shown). This transcriptional repression activity correlates well with the subnuclear compartmentalisation of E8/E2.
Figure 7.
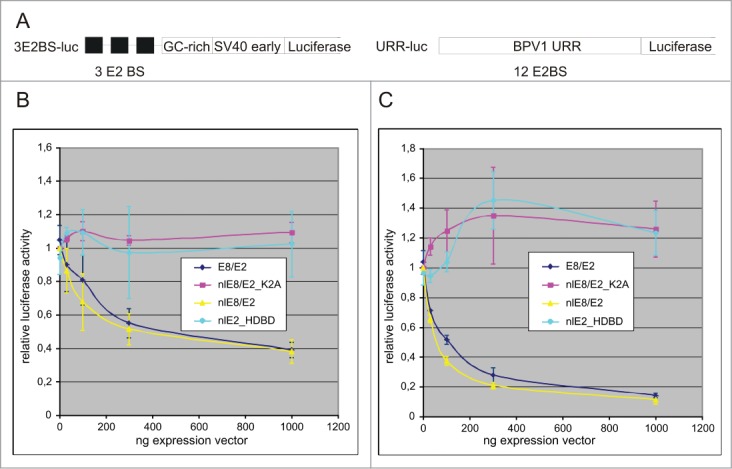
Only the NM targeting competent of E8/E2 can repress transcription from reporters containing an E2 binding site. (A) Schematic representation of the E2-dependent transcriptional reporter plasmid 3E2BS-luc with 3 E2BS, a GC-rich region and an SV40early promoter cloned upstream of the reporter gene. A second reporter plasmid, URR-luc, contains a native BPV1 promoter (P89, alias P2) with 12 E2 BS and a luciferase reporter gene. (B) Transcriptional repression of a reporter construct with a heterologous promoter is dependent on BPV1 E8/E2 and its derivatives. (C) Transcriptional repression of a reporter construct with a native BPV1 promoter is dependent on BPV1 E8/E2 and its derivatives.
Discussion
Two decades ago, the BPV1 E8/E2 protein was characterized as a transcriptional repressor; however, the mechanism of this repression is not yet known. The transcripts corresponding to the E8/E2 protein are also described for HPV1,32 HPV5,33 HPV11,34 HPV16,35 HPV18,36 HPV31,37 HPV3338 and SfPV39 (previously named CRPV1).40 Our bioinformatics analysis indicates that almost all mammalian PVs have the ability to encode an E8/E2 protein (Puustusmaa and Abroi, manuscript in preparation). The most highly studied PV E8/E2 is from HPV31. The HPV31 E8/E2 protein represses transcription and extra-chromosomal replication; many interactions responsible for these repressive functions are described.41-43 However, the HPV31 E8 and BPV1 E8 proteins belong to different E8 clades (Puustusmaa and Abroi, manuscript in preparation). In addition, the subcellular localization of HPV31 E8/E2 has not yet been studied. According to sequence similarity, PV E8 peptides can be classified into 2 major and one minor clade (Puustusmaa and Abroi, manuscript in preparation). One major clade includes α-papillomaviruses (like HPV11, 16, 18, 31 and 33). The other major clade includes almost all other mammalian PVs. Corresponding to the viral taxon of these 2 major E8 clades, these groups diverged more than 100 million years ago.44 Therefore, the functions of one type of E8 proteins cannot be assumed for other types of E8 proteins without experimental verification. Beside BPV1 E8/E2, the only functionally characterized E8 in this clade is CRPV E8/E2.39 This transcriptional repressor is required to efficiently complete the viral life cycle.45 In this study, we demonstrated that proper subnuclear localization to the NM is required for the efficient repression of transcription by BPV-1 E8/E2. All three BPV1 E2 protein isoforms have different subnuclear localization. This is in agreement with the different functions of these isoforms in transcription; E2 is a strong activator, E2C is a weak activator and E8/E2 is a transcriptional repressor.46,30
Throughout this study, we examined the subnuclear localization of E8/E2 protein in different cell lines and at 2 principally different conditions. In the Cos cell line, expression plasmids replicate and the expression level of E8_EGFP_NLS and E8/E2 is homogenous according to FACS analysis (data not shown). In the CHO cell line (and its derivatives), the expression plasmid does not replicate and the expression level of E8_EGFP_NLS and E8/E2 is very heterogeneous, as was shown previously for E2.47 According to FACS analyses, expression of these proteins varied by over 2 orders of magnitude between individual cells in the same sample (data not shown). To verify that localization of nlE2_HDBD and nlE8/E2_K2A is correct and not determined by expression levels, we titrated decreasing amounts of expression plasmids down to the detection limit. The same fractionation profile was observed when the least amount of plasmids was used (data not shown). Therefore, E8/E2 and its mutant have different fractionation profiles.
The different localization profiles for EGFP fused E8 peptide and its mutant were further verified by FRET assay. Using a FRET assay, we demonstrate with high confidence that the localization of E8 and its mutant were indeed spatially distinct in their native environment in undisturbed cells. Compared to biochemical fractionation, the FRET assay is more direct. In addition, the resolution of the FRET assay is about one order of magnitude better than co-localization experiments using confocal microscopy. Experiments with fluorescent proteins also revealed that the E8 peptide is not targeting the proteins to compartments where proteins are completely misfolded and oligomerised. Due to high spatial resolution and non-invasive nature, the FRET assay can be used for in vivo analysis of other sub-assemblies as well. Having a working FRET pair of 2 proteins conjugated with fluorophores enables to study the presence of mutant proteins in sub-assemblies (especially when the sub-assembly is not soluble and thus cannot be easily studied using biochemical methods). We would expect that FRET, caused by tight co-localization of the same proteins species (or signal peptides) with different fluorophores, presume specific conditions like di- or multimeric nature of binding partner or a binding to “surface like” entity.
Viruses are an important part of the biosphere and have long been used as important tools in molecular biology.48,49 The discovery of first the NLS in the SV40 LT protein is one well known example in the field of localization signals. In this article, we described for the first time a well-defined NMTS used by BPV1 E8/E2. Our data demonstrate that the first 13 aa (MKLTVFLRPSRDR) at the N-terminus in E8/E2 are necessary and sufficient to relocate the proteins to the NM and therefore work as a NMTS. According to the Database of Nuclear Matrix Proteins (NMPdb) www.rostlab.org/db/NMPdb, this sequence is the shortest NMTS known to date.6 As a short and well-defined sequence, the E8 NMTS allows us to study the interactions and mechanisms that determine NM localization. It also helps us understand the rules for subnuclear compartmentalisation and study the architecture and dynamics of NM. Expanding the repertoire of characterized functional motifs also helps us to improve the automated annotation of sequences in the current post-genomic era and helps us to connect sequence space with functional space.50
In E8/E2, the addition of less as ∼10 aa to the protein alters its subnuclear localization and function. Recently, a similar phenomenon has been described for NF90 (differences in 13 aa occur due to alternative splicing).51 This finding indicates that this may be a rather general phenomenon. In larger proteins, these isoforms may not be separated by gel-electrophoresis and different isoforms may remain undetected. If alternative protein isoforms constitute only a small fraction of the total protein and are localized to different subcellular compartments, their differential localization cannot be annotated or observed. The localization and functions of different isoforms can therefore remain hidden to researchers, except in cases where this question is addressed directly. Understanding the localization and compartmentalisation of different isoforms is crucial for our understanding of the biological roles of these proteins. Further study into the localization and functioning of different isoforms allows us to better understanding the rules for subnuclear compartmentalisation. Complementary set of methods (microscopy, biochemistry, FRET etc) will help to perform that.
Materials and Methods
Cell culture and plasmids
A Chinese hamster ovary cell line (CHO) was maintained in Ham's F12 medium supplemented with 10% foetal calf serum. Transfections into CHO cells were performed as described previously.52 The CHO derivatives CHO4.15 and CHOBgl40 are described in Piirsoo et al.53 As a well-transfected suspension cell line, we used the IcosageneCellFactory cell line CHOEBNALT85 (described in patents EP1851319 and US 7790446 B2) derived from CHO-S cells (Invitrogen Corporation). This cell line expresses the EBV EBNA1 protein and the mouse polyomavirus large T antigen. The pCGE2, pCGE2C and pCGE8/E2 plasmids have been described previously in Ustav 1991.52 Reporter plasmids for the transcription assay (3E2BS-Luc, URR-Luc, pRL-TK) have been described previously.25,54 The reporter plasmid 3E2BS-Luc contains 3 E2BS, 3 21-bp GC-rich repeats, and an enhancerless simian virus 40 early promoter in front of the firefly luciferase gene. URR-Luc contains the BPV1 upstream regulatory region (URR; nucleotides 7476 to 94 from the BPV1 circular genome including a P2 promoter, also named the P89 promoter). The plasmid nlE2_HDBD was generated from pCGE2 by opening the plasmid with PaeI, removing the 3′ overhang with T4 DNA polymerase, cutting with Tth111I, filling overhanging ends with Klenow fragment and religating. Therefore, the protein starts with the first 3 aa from the E2 ORF followed by aa 211 in E2 and the remainder of the protein (E2delta 4-210). The nlE8/E2 and nlE8/E2_K2A proteins were constructed by PCR. pCGE8/E2 was used as a template, with the forward primer E8N (GGA AGA TCT AGA GCC ATG AAG CTA ACC GTG) for the wt, E8K2A (A TCC GCT AGC GCC ATG GCG CTA ACC GTG TTC) for the mutant and KpnRev (GTC CAC CGG TAC CGT GCC) as a reverse primer for both constructs. The PCR products were cut using XbaI (wt) or NheI (mutant) and KpnI and cloned into pCGE2 opened with XbaI and KpnI.
All cloned plasmids where verified by sequencing. All of the plasmids in the E8 series (pCGE8/E2, nlE8/E2, nlE8/E2_K2A, and nlE2_HDBD) contain an identical 3′ UTR up to the BamHI site, located at 4450 bp in the BPV1 genome. EGFP_NLS was derived from pEGFP-C1 by inserting the annealed oligonucleotides NLS_1 (GATCTGATATCCCCAAGAAGAAACGCAAAGTTAGCGCTA) and NLS_2 (GATCTAGCGCTAACTTTGCGTTTCTTCTTGGGGATATCA) into the SV40 LT NLS between the BamHI and BglII sites. To generate E8_EGFP_NLS, the E8 region of pCGE8/E2 was amplified by PCR using the oligos E8N and KpnRev. The PCR product was digested with MboI, blunted, and digested with XbaI. The digested fragment (MboI-XbaI) was inserted into pEGFP-N1 between the NheI and SmaI sites. The native ATG of GFP was mutated to ATC to eliminate the internal initiation of translation. To introduce NLS, the sequence between BsrGI and EagI was replaced with a BsrGI-EagI fragment originating from EGFP_NLS. The final plasmid contained 13 aa from the N-terminus of E8/E2 followed by GFP without native ATG and with SV40 LT NLS at the C-terminus. At the amino acid level the introduced sequence is MKLTVFLRPSRDR/dppvat/I, where 13 aa from E8/E2 fused to EGFP are in capital letters, 11 aa from E8 ORF are in bold, small letters denote aa coded by polylinker and EGFP initiator methionine was mutated to Ile (in capital letter). The E8K2A_EGFP_NLS construct was generated using the same strategy. The oligo E8K2A was used in place of E8N; NheI was used in place of XbaI. As a result, lysine in position 2 was mutated to alanine.
The YFP_NLS and CFP_NLS constructs were homologous to EGFP_NLS, starting from pYFP-C1 and pCFP-C1, respectively. The E8_CFP, E8_YFP, E8K2A_CFP and E8K2A_YFP constructs were derived from E8_EGFP_NLS and E8K2A_EGFP_NLS. The sequences between AgeI and BsrGI in these plasmids were replaced with the sequences of the fluorescence proteins pYFP-C1 or pCFP-C1. The native ATG start codons in the fluorescence genes were mutated to ATC to eliminate internal initiations of translation. None of the fusion constructs contain a native ATG in the fluorescence gene, and all of the fluorescence proteins contain an identical NLS in the C-terminus.
Subcellular fractionation
Chromatin fractionation with Triton X-100 was performed as described previously55 with modifications. CHO cells transfected with expression plasmids were treated with PBS-3 mM EDTA, collected by centrifugation (2000 rpm, ∼400 g, 3 min RT), washed with ice-cold PBS, and centrifuged again. Cells (∼2 × 106) were resuspended in 100 μl of cytoskeleton buffer (CSK), which contained 10 mM piperazine-N,N′-bis(2-ethanesulfonic acid) at pH 6.8, 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 1 mM EGTA supplemented with leupeptin and aprotinin (5 ng/μl), 0.5 mM phenylmethylsulfonyl fluoride, and 1 mM dithiothreitol (DTT). Triton X-100 (0.5%, vol/vol) was added, and the sample was incubated for 15 min at 0°C on ice. Soluble and insoluble fractions were separated in a microcentrifuge (7500 rpm, ˜5400 g, 3 min). The pellet was washed at 0°C with ice-cold CSK buffer. The insoluble fraction was further incubated with CSK buffer containing either 0.1 or 0.4 M NaCl for 20 min on ice. The insoluble fraction was resuspended in CSK buffer. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer was added to the soluble and insoluble fractions, and these fractions were subjected to SDS-PAGE and immunoblotting. The CHO cells were transfected by electroporation (1000 μF, 230 V) with 250 ng pCGE2, 1250 ng pCGE2C or 1250 ng pCGE8/E2 (for Fig. 1) or with 2 μg of each plasmids used in Figure 3. The CHO4.15 cell line was transfected using the same parameters as the CHO cell line; 1 μg of expression plasmid was used in Figure 4. Cos7 cells were transfected by electroporation (1000 μF, 180 V); 50 ng of expression plasmid was used in all experiments.
Biochemical fractionation of the nuclear matrix
The classical nuclear matrix biochemical fractionation procedure was performed as follows: transfected cells in a 10 cm dish were washed twice with PBS, collected with 3 mM EDTA/PBS and centrifuged at 45 g for 3 min at RT. Cells were resuspended in 100 μl CSK buffer, incubated for 10 min on ice and centrifuged at 8000 rpm (6200 g) for 3 min at 4°C in a microcentrifuge. Pellets were resuspended in 200 μl CSK buffer and divided into 4 aliquots of 50 μl (1-4). The four samples were treated as follows: 1) CSK control; 2) NaCl was added to a final concentration of 0.4 M and incubated for 20 min on ice; 3) RNase A was added to final concentration of 0.1 μg/μl and incubated for 5 min at RT. (NH4)2SO4 was added to a final concentration of 0.25 M; 4) DNase was added to a final concentration of 20 u/μl and incubated for 5 min at RT. (NH4)2SO4 was added to a final concentration of 0.25 M. Cells were centrifuged at 8000 rpm (6200 g) for 3 min at 4°C to separate the soluble and insoluble fractions. Pellets from the DNase treatment were fractionated further by resuspension in CSK buffer containing 2 M NaCl. Samples were centrifuged again and the nuclear matrix pellet was solubilised in 8 M urea. SDS-PAGE loading buffer was added to all biochemical fractions, and samples were subjected to immunoblotting. In general, our procedure follows the classical NM preparation protocol described in56 and depicted in Figure 2A. The amount of expression plasmid was 50 ng for the Cos7 cell lines, 1 μg of E8_EGFP_NLS for the CHOBgl40 cell line and 1 μg of pCGE8E2 for the CHOEBNALT85 cell line.
Immunoblotting analysis
For immunoblotting analysis, proteins were separated by SDS-PAGE and transferred using a semidry blotting method to a polyvinylidene difluoride membrane (Millipore Corp.). Membranes were incubated with the anti-E2 antibodies 1E4 and 1H10 (1 ng/μl).57 This probe recognized all 3 E2 species. An anti-mouse horseradish peroxidase-conjugated antibody (LabAs Ltd., Tartu, Estonia) was used according to the manufacturers' recommendations. EGFP and its derivatives were detected using a polyclonal anti-GFP antibody58 and with an anti-rabbit horseradish peroxidase-conjugated antibody (LabAs Ltd., Tartu, Estonia). Detection was performed using an ECL detection kit (Amersham Pharmacia Biotech) following the manufacturers' manuals.
Transcription assay
For the transcription assay, CHO cells were cotransfected with 50 ng of pRL-TK and 125 ng of 3E2BS-Luc or URR-Luc luciferase reporter plasmid. Increasing concentrations of E8/E2 or its derivative expression plasmids were also cotransfected. Cells were lysed 24 h after transfection. Luciferase assays were performed using a Dual Luciferase Assay Kit (Promega) in a GloMax 20/20 luminometer (Promega) according to the manufacturer's instructions. Firefly luciferase luminescence values (for 3E2BS-Luc or URR-Luc) were divided by the Renilla luciferase luminescence values (for pRL-TK) from the same transfection to normalize for differences in transfection efficiency and to measure the specific effects of E2BSs.
Immunofluorescence
For immunofluorescence analysis, CHO cells where transfected by electroporation (1000 μF, 230 V) with 250 ng pCGE2, 1250 ng pCGE2C and 1250 ng pCGE8/E2 (these amounts of plasmids lead to equal expression levels on a western blot). Cells were fixed 48 h after transfection with methanol:acetone (1:1) for 10 min. at -20°C. Immunofluorescence was performed as described in59. A mixture of antibodies 3E8 and 5H4 (2.5 ng/µl each), which recognized all 3 E2 species57 was used as a primary antibody. An Alexa Fluor 488-conjugated goat anti-mouse antibody from Invitrogen was used as a secondary antibody (1:2000 dilution). The results were analyzed with an Olympus BX41 fluorescence microscope equipped with DX70 CCD camera.
For in situ fractionation analysis the transfected Cos7 cells were grown on 8-well format coverglasses (Lab-TekTM Chambered Coverglass w/cvr, Thermo Scientific) for 20 hours. Adherent cells for immunofluorescence analysis were washed with PBS. For non-extracted cells 3% PFA in PBS was added for 10 min, and cells were washed with PBS. Before immunofluorescence analyses these cells were permeabilised with PBS/0.1% Triton X-100 for 2 min. For extracted cells CSK was added for 10 min. After that CSK was gently removed and replaced with a new volume of CSK or with 20 u/μl DNase and 0.1 μg/μl RNase in CSK and incubated for 10 min. In case of DNase/RNase treatment (NH4)2SO4 was added to final concentration of 0.25 M for 5 min. After extraction the wells were washed once with PBS following fixation with 3% PFA in PBS for 10 min and another wash with PBS. For immunofluorescence the cells were first incubated for 30 min with blocking solution (20 µg/ml BSA in TBS/0.1% Tween20). The anti-GFP antibody58 was used as a primary antibody and anti-rabbit AlexaFluor488 from Invitrogen as a secondary antibody both added in 20 µg/ml BSA/PBS/0.1% Tween20 and incubated for 1 hour each. After secondary antibody incubation cells were washed once with PBS. Cells were mounted in slow-fade solution supplemented with 4 ng/µl of propidium iodide. All procedures were done at room temperature.
For live cell imaging Hoechst 33342 was added 10 min before microscopy to growth medium at a final concentration of 10 µg/ml. Live cell imaging was done at 37°C in 5% CO2. Fluorescence was visualized using confocal laser scanning microscope LSM710 (Zeiss). Images were obtained with 63× lens and analyzed by ZEN2011 software.
FRET assay
For FRET assays, cells (CHOBgl40 or CHOEBNALT85) were transfected alone or in a pairwise manner with 1 μg E8_CFP or E8_YFP, 0.3 μg of K2A_CFP or K2A_YFP, 0.2 μg ECFPN1_CNLS or EYFPN1_CNLS. The GFP region of the E8_EGFP_NLS and K2A_EGFP constructs was changed in both cases to YFP and CFP; the respective plasmids contain E8 or the E8K2A peptide at the N-terminus followed by CFP or YFP. All of proteins used in the FRET assay contain an NLS in the C-terminus (as in E8_EGFP_NLS). As with E8_EGFP_NLS, all proteins localized to the cell nucleus (data not shown). We chose the amount of plasmid to obtain comparable spectroscopic concentration of fluorescence proteins (measured by fluorimeter) and comparable percentage of transfected cells (measured as % of YFP expressing cells in FACS analysis, depending on transfection ∼25% for CHOBgl40 and 40% for CHOEBNALT85 cell lines). Cells were collected ∼48 h after transfection. Cells from suspension cultures of CHOEBNALT85 were pelleted and resuspended in PBS; CHO cells where collected with PBS/EDTA (3 mM), pelleted, resuspended in PBS and stored on ice until spectra were recovered. The recovery of the cell population spectra was measured using a Hitachi F-4500 fluorescence spectrophotometer with an excitation at 431 nm (for CFP) or 497 nm (for YFP) with the following parameters: ScanMode - emission; DataMode - fluorescence; scan speed at 2400 nm/min; excitation slit of 10.0 nm; emission slit of 5.0 nm and PMT of 950 V. The emission was measured between 400 and 700 nm; specific emission values of 480 nm for CFP and 530 nm for YFP were used. The ‘FRET ratio’ (FR) is the fractional increase in YFP emission (at 530 nm) due to FRET (due to presence of CFP) when excited with a CFP-specific wavelength (431 nm). FR was essentially calculated as FR=FRET431/530/YFP431/530 (see Figure S1). The terms in bold are experimental conditions where one of the fluorochromes or both (FRET) are expressed. The values for CFP, YFP and FRET are measured in different transfections. Therefore, the respective values were normalized to a spectral concentration of fluorochromes to take the number of cells and the transfection efficiency into account according to formula:
YFP - only YFP or its fusion were transfected; CFP - only CFP or its fusion were transfected; FRET - CFP and YFP (or their fusions) were transfected. Subscript indicates the excitation wavelength (431 or 497) or emission/data collection wavelength (480 or 530). See Figure S1 for an illustration and explanation.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank the University of Tartu Department of Biochemistry, and prof. Juhan Sedman for the use of a fluorescence spectrometer. The Icosagen Cell Factory OÜ for use of the CHOEBNALT85 cell line and company contact person Kerttu Murumets for assistance with this cell line. We also thank Eve Toomsoo and Kadri Õunap from the Core Facility of Institute of Technology for an introduction and help with confocal microscopy.
Funding
This work was supported by the ETF6903 from Estonian Science Foundation to AA, SF0180175s08 and IUT20-27 from Estonian Research Council and by the European Regional Development Fund through Centre of Excellence in Chemical Biology 3.2.0101.08-0017.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Hung MC, Link W. Protein localization in disease and therapy. J Cell Sci 2011; 124:3381-92; PMID:22010196; http://dx.doi.org/ 10.1242/jcs.089110 [DOI] [PubMed] [Google Scholar]
- 2.Wang X, Li S. Protein mislocalization: Mechanisms, functions and clinical applications in cancer. Biochim Biophys Acta 2014; 1846:13-25; PMID:24709009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Misteli T. Concepts in nuclear architecture. Bioessays 2005; 27:477-87; PMID:15832379; http://dx.doi.org/ 10.1002/bies.20226 [DOI] [PubMed] [Google Scholar]
- 4.Misteli T. Beyond the sequence: cellular organization of genome function. Cell 2007; 128:787-800; PMID:17320514; http://dx.doi.org/ 10.1016/j.cell.2007.01.028 [DOI] [PubMed] [Google Scholar]
- 5.Albrethsen J, Knol JC, Jimenez CR. Unravelling the nuclear matrix proteome. J Proteomics 2009; 72:71-81; PMID:18957335; http://dx.doi.org/ 10.1016/j.jprot.2008.09.005 [DOI] [PubMed] [Google Scholar]
- 6.Mika S, Rost B. NMPdb: Database of Nuclear Matrix Proteins. Nucleic Acids Res 2005; 33:D160-3; PMID:15608168; http://dx.doi.org/ 10.1093/nar/gki132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Han GS, Yu ZG, Anh V, Krishnajith AP, Tian YC. An ensemble method for predicting subnuclear localizations from primary protein structures. PLoS One 2013; 8:e57225; PMID:23460833; http://dx.doi.org/ 10.1371/journal.pone.0057225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zaidi SK, Javed A, Pratap J, Schroeder TM, Westendorf JJ, Lian JB, van Wijnen AJ, Stein GS, Stein JL. Alterations in intranuclear localization of Runx2 affect biological activity. J Cell Physiol 2006; 209:935-42; PMID:16972259; http://dx.doi.org/ 10.1002/jcp.20791 [DOI] [PubMed] [Google Scholar]
- 9.Seo J, Lozano MM, Dudley JP. Nuclear matrix binding regulates SATB1-mediated transcriptional repression. J Biol Chem 2005; 280:24600-9; PMID:15851481; http://dx.doi.org/ 10.1074/jbc.M414076200 [DOI] [PubMed] [Google Scholar]
- 10.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature 2008; 456:470-6; PMID:18978772; http://dx.doi.org/ 10.1038/nature07509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 2008; 40:1413-5; PMID:18978789; http://dx.doi.org/ 10.1038/ng.259 [DOI] [PubMed] [Google Scholar]
- 12.Johnson JM, Castle J, Garrett-Engele P, Kan Z, Loerch PM, Armour CD, Santos R, Schadt EE, Stoughton R, Shoemaker DD. Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science 2003; 302:2141-4; PMID:14684825; http://dx.doi.org/ 10.1126/science.1090100 [DOI] [PubMed] [Google Scholar]
- 13.Elliott DJ. Illuminating the Transcriptome through the Genome. Genes (Basel) 2014; 5:235-53; PMID:24705295; http://dx.doi.org/ 10.3390/genes5010235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jacox E, Gotea V, Ovcharenko I, Elnitski L. Tissue-specific and ubiquitous expression patterns from alternative promoters of human genes. PLoS One 2010; 5:e12274; PMID:20806066; http://dx.doi.org/ 10.1371/journal.pone.0012274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tress ML, Martelli PL, Frankish A, Reeves GA, Wesselink JJ, Yeats C, Olason PI, Albrecht M, Hegyi H, Giorgetti A, et al.. The implications of alternative splicing in the ENCODE protein complement. Proc Natl Acad Sci U S A 2007; 104:5495-500; PMID:17372197; http://dx.doi.org/ 10.1073/pnas.0700800104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kelemen O, Convertini P, Zhang Z, Wen Y, Shen M, Falaleeva M, Stamm S. Function of alternative splicing. Gene 2013; 514:1-30; PMID:22909801; http://dx.doi.org/ 10.1016/j.gene.2012.07.083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murata K, Hayashibara T, Sugahara K, Uemura A, Yamaguchi T, Harasawa H, Hasegawa H, Tsuruda K, Okazaki T, Koji T, et al.. A novel alternative splicing isoform of human T-cell leukemia virus type 1 bZIP factor (HBZ-SI) targets distinct subnuclear localization. J Virol 2006; 80:2495-505; PMID:16474156; http://dx.doi.org/ 10.1128/JVI.80.5.2495-2505.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsai KW, Tseng HC, Lin WC. Two wobble-splicing events affect ING4 protein subnuclear localization and degradation. Exp Cell Res 2008; 314:3130-41; PMID:18775696; http://dx.doi.org/ 10.1016/j.yexcr.2008.08.002 [DOI] [PubMed] [Google Scholar]
- 19.Englert C, Vidal M, Maheswaran S, Ge Y, Ezzell RM, Isselbacher KJ, Haber DA. Truncated WT1 mutants alter the subnuclear localization of the wild-type protein. Proc Natl Acad Sci U S A 1995; 92:11960-4; PMID:8618823; http://dx.doi.org/ 10.1073/pnas.92.26.11960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Larsson SH, Charlieu JP, Miyagawa K, Engelkamp D, Rassoulzadegan M, Ross A, Cuzin F, van Heyningen V, Hastie ND. Subnuclear localization of WT1 in splicing or transcription factor domains is regulated by alternative splicing. Cell 1995; 81:391-401; PMID:7736591; http://dx.doi.org/ 10.1016/0092-8674(95)90392-5 [DOI] [PubMed] [Google Scholar]
- 21.Dahmcke CM, Büchmann-Møller S, Jensen NA, Mitchelmore C. Altered splicing in exon 8 of the DNA replication factor CIZ1 affects subnuclear distribution and is associated with Alzheimer's disease. Mol Cell Neurosci 2008; 38:589-94; PMID:18583151; http://dx.doi.org/ 10.1016/j.mcn.2008.05.007 [DOI] [PubMed] [Google Scholar]
- 22.Shabalina SA, Ogurtsov AY, Spiridonov NA, Koonin EV. Evolution at protein ends: major contribution of alternative transcription initiation and termination to the transcriptome and proteome diversity in mammals. Nucleic Acids Res 2014; 42:7132-44; PMID:24792168; http://dx.doi.org/ 10.1093/nar/gku342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jia R, Zheng ZM. Regulation of bovine papillomavirus type 1 gene expression by RNA processing. Front Biosci (Landmark Ed) 2009; 14:1270-82; PMID:19273129; http://dx.doi.org/ 10.2741/3307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hubbert NL, Schiller JT, Lowy DR, Androphy EJ. Bovine papilloma virus-transformed cells contain multiple E2 proteins. Proc Natl Acad Sci U S A 1988; 85:5864-8; PMID:2842752; http://dx.doi.org/ 10.1073/pnas.85.16.5864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kurg R, Tekkel H, Abroi A, Ustav M. Characterization of the functional activities of the bovine papillomavirus type 1 E2 protein single-chain heterodimers. J Virol 2006; 80:11218-25; PMID:16943289; http://dx.doi.org/ 10.1128/JVI.01127-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kurg R, Uusen P, Sepp T, Sepp M, Abroi A, Ustav M. Bovine papillomavirus type 1 E2 protein heterodimer is functional in papillomavirus DNA replication in vivo. Virology 2009; 386:353-9; PMID:19232665; http://dx.doi.org/ 10.1016/j.virol.2009.01.025 [DOI] [PubMed] [Google Scholar]
- 27.Kurg R, Sild K, Ilves A, Sepp M, Ustav M. Association of bovine papillomavirus E2 protein with nuclear structures in vivo. J Virol 2005; 79:10528-39; PMID:16051845; http://dx.doi.org/ 10.1128/JVI.79.16.10528-10539.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Skiadopoulos MH, McBride AA. The bovine papillomavirus type 1 E2 transactivator and repressor proteins use different nuclear localization signals. J Virol 1996; 70:1117-24; PMID:8551571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Allikas A, Ord D, Kurg R, Kivi S, Ustav M. Roles of the hinge region and the DNA binding domain of the bovine papillomavirus type 1 E2 protein in initiation of DNA replication. Virus Res 2001; 75:95-106; PMID:11325464; http://dx.doi.org/ 10.1016/S0168-1702(01)00219-2 [DOI] [PubMed] [Google Scholar]
- 30.Choe J, Vaillancourt P, Stenlund A, Botchan M. Bovine papillomavirus type 1 encodes two forms of a transcriptional repressor: structural and functional analysis of new viral cDNAs. J Virol 1989; 63:1743-55; PMID:2538655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ustav E, Ustav M, Szymanski P, Stenlund A. The bovine papillomavirus origin of replication requires a binding site for the E2 transcriptional activator. Proc Natl Acad Sci U S A 1993; 90:898-902; PMID:8381536; http://dx.doi.org/ 10.1073/pnas.90.3.898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Palermo-Dilts DA, Broker TR, Chow LT. Human papillomavirus type 1 produces redundant as well as polycistronic mRNAs in plantar warts. J Virol 1990; 64:3144-9; PMID:2159571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sankovski E, Männik A, Geimanen J, Ustav E, Ustav M. Mapping of betapapillomavirus human papillomavirus 5 transcription and characterization of viral-genome replication function. J Virol 2014; 88:961-73; PMID:24198410; http://dx.doi.org/ 10.1128/JVI.01841-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rotenberg MO, Chow LT, Broker TR. Characterization of rare human papillomavirus type 11 mRNAs coding for regulatory and structural proteins, using the polymerase chain reaction. Virology 1989; 172:489-97; PMID:2552659; http://dx.doi.org/ 10.1016/0042-6822(89)90191-8 [DOI] [PubMed] [Google Scholar]
- 35.Doorbar J, Parton A, Hartley K, Banks L, Crook T, Stanley M, Crawford L. Detection of novel splicing patterns in a HPV16-containing keratinocyte cell line. Virology 1990; 178:254-62; PMID:2167553; http://dx.doi.org/ 10.1016/0042-6822(90)90401-C [DOI] [PubMed] [Google Scholar]
- 36.Kurg R, Uusen P, Vosa L, Ustav M. Human papillomavirus E2 protein with single activation domain initiates HPV18 genome replication, but is not sufficient for long-term maintenance of virus genome. Virology 2010; 408:159-66; PMID:20940072; http://dx.doi.org/ 10.1016/j.virol.2010.09.010 [DOI] [PubMed] [Google Scholar]
- 37.Stubenrauch F, Hummel M, Iftner T, Laimins LA. The E8E2C protein, a negative regulator of viral transcription and replication, is required for extrachromosomal maintenance of human papillomavirus type 31 in keratinocytes. J Virol 2000; 74:1178-86; PMID:10627528; http://dx.doi.org/ 10.1128/JVI.74.3.1178-1186.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Snijders PJ, van den Brule AJ, Schrijnemakers HF, Raaphorst PM, Meijer CJ, Walboomers JM. Human papillomavirus type 33 in a tonsillar carcinoma generates its putative E7 mRNA via two E6* transcript species which are terminated at different early region poly(A) sites. J Virol 1992; 66:3172-8; PMID:1313922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jeckel S, Loetzsch E, Huber E, Stubenrauch F, Iftner T. Identification of the E9/E2C cDNA and functional characterization of the gene product reveal a new repressor of transcription and replication in cottontail rabbit papillomavirus. J Virol 2003; 77:8736-44; PMID:12885893; http://dx.doi.org/ 10.1128/JVI.77.16.8736-8744.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bernard HU, Burk RD, Chen Z, van Doorslaer K, zur Hausen H, de Villiers EM. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology 2010; 401:70-9; PMID:20206957; http://dx.doi.org/ 10.1016/j.virol.2010.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fertey J, Ammermann I, Winkler M, Stöger R, Iftner T, Stubenrauch F. Interaction of the papillomavirus E8-E2C protein with the cellular CHD6 protein contributes to transcriptional repression. J Virol 2010; 84:9505-15; PMID:20631145; http://dx.doi.org/ 10.1128/JVI.00678-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ammermann I, Bruckner M, Matthes F, Iftner T, Stubenrauch F. Inhibition of transcription and DNA replication by the papillomavirus E8-E2C protein is mediated by interaction with corepressor molecules. J Virol 2008; 82:5127-36; PMID:18353941; http://dx.doi.org/ 10.1128/JVI.02647-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Powell ML, Smith JA, Sowa ME, Harper JW, Iftner T, Stubenrauch F, Howley PM. NCoR1 mediates papillomavirus E8;E2C transcriptional repression. J Virol 2010; 84:4451-60; PMID:20181716; http://dx.doi.org/ 10.1128/JVI.02390-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shah SD, Doorbar J, Goldstein RA. Analysis of host-parasite incongruence in papillomavirus evolution using importance sampling. Mol Biol Evol 2010; 27:1301-14; PMID:20093429; http://dx.doi.org/ 10.1093/molbev/msq015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cladel NM, Hu J, Balogh KK, Christensen ND. Differences in methodology, but not differences in viral strain, account for variable experimental outcomes in laboratories utilizing the cottontail rabbit papillomavirus model. J Virol Methods 2010; 165:36-41; PMID:20036285; http://dx.doi.org/ 10.1016/j.jviromet.2009.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lace MJ, Ushikai M, Yamakawa Y, Anson JR, Ishiji T, Turek LP, Haugen TH. The truncated C-terminal E2 (E2-TR) protein of bovine papillomavirus (BPV) type-1 is a transactivator that modulates transcription in vivo and in vitro in a manner distinct from the E2-TA and E8ˆE2 gene products. Virology 2012; 429:99-111; PMID:22551766; http://dx.doi.org/ 10.1016/j.virol.2012.03.020 [DOI] [PubMed] [Google Scholar]
- 47.Abroi A, Ilves I, Kivi S, Ustav M. Analysis of chromatin attachment and partitioning functions of bovine papillomavirus type 1 E2 protein. J Virol 2004; 78:2100-13; PMID:14747575; http://dx.doi.org/ 10.1128/JVI.78.4.2100-2113.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abroi A, Gough J. Are viruses a source of new protein folds for organisms? - Virosphere structure space and evolution. Bioessays 2011; 33:626-35; PMID:21633962; http://dx.doi.org/ 10.1002/bies.201000126 [DOI] [PubMed] [Google Scholar]
- 49.Enquist LW, Editors of the Journal of Virology. Virology in the 21st century. J Virol 2009; 83:5296-308; PMID:19297504; http://dx.doi.org/ 10.1128/JVI.00151-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Casadio R, Martelli PL, Pierleoni A. The prediction of protein subcellular localization from sequence: a shortcut to functional genome annotation. Brief Funct Genomic Proteomic 2008; 7:63-73; PMID:18283051; http://dx.doi.org/ 10.1093/bfgp/eln003 [DOI] [PubMed] [Google Scholar]
- 51.Viranaicken W, Gasmi L, Chaumet A, Durieux C, Georget V, Denoulet P, Larcher JC. L-Ilf3 and L-NF90 traffic to the nucleolus granular component: alternatively-spliced exon 3 encodes a nucleolar localization motif. PLoS One 2011; 6:e22296; PMID:21811582; http://dx.doi.org/ 10.1371/journal.pone.0022296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ustav M, Stenlund A. Transient replication of BPV-1 requires two viral polypeptides encoded by the E1 and E2 open reading frames. EMBO J 1991; 10:449-57; PMID:1846806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Piirsoo M, Ustav E, Mandel T, Stenlund A, Ustav M. Cis and trans requirements for stable episomal maintenance of the BPV-1 replicator. EMBO J 1996; 15:1-11; PMID:8598191 [PMC free article] [PubMed] [Google Scholar]
- 54.Ilves I, Mäemets K, Silla T, Janikson K, Ustav M. Brd4 is involved in multiple processes of the bovine papillomavirus type 1 life cycle. J Virol 2006; 80:3660-5; PMID:16537635; http://dx.doi.org/ 10.1128/JVI.80.7.3660-3665.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Okuno Y, McNairn AJ, den Elzen N, Pines J, Gilbert DM. Stability, chromatin association and functional activity of mammalian pre-replication complex proteins during the cell cycle. EMBO J 2001; 20:4263-77; PMID:11483529; http://dx.doi.org/ 10.1093/emboj/20.15.4263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.He DC, Nickerson JA, Penman S. Core filaments of the nuclear matrix. J Cell Biol 1990; 110:569-80; PMID:2307700; http://dx.doi.org/ 10.1083/jcb.110.3.569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kurg R, Parik J, Juronen E, Sedman T, Abroi A, Liiv I, Langel U, Ustav M. Effect of bovine papillomavirus E2 protein-specific monoclonal antibodies on papillomavirus DNA replication. J Virol 1999; 73:4670-7; PMID:10233926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tamberg N, Lulla V, Fragkoudis R, Lulla A, Fazakerley JK, Merits A. Insertion of EGFP into the replicase gene of Semliki Forest virus results in a novel, genetically stable marker virus. J Gen Virol 2007; 88:1225-30; PMID:17374766; http://dx.doi.org/ 10.1099/vir.0.82436-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abroi A, Kurg R, Ustav M. Transcriptional and replicational activation functions in the bovine papillomavirus type 1 E2 protein are encoded by different structural determinants. J Virol 1996; 70:6169-79; PMID:8709243 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.