

Abstract
The effectiveness of two new supercharging reagents for producing highly charged ions by electrospray ionization (ESI) from aqueous solutions in which proteins have native structures and reactivities were investigated. In aqueous solution, 2-thiophenone and 4-hydroxymethyl-1,3-dioxolan-2-one (HD) at a concentration of 2% by volume can increase the average charge of cytochrome c and myoglobin by up to 163%, resulting in even higher charge states than those that are produced from water/methanol/acid solutions in which proteins are denatured. The greatest extent of supercharging occurs in pure water, but these supercharging reagents are also highly effective in aqueous solutions containing 200 mM ammonium acetate buffer commonly used in native mass spectrometry (MS). These reagents are less effective supercharging reagents than m-nitrobenzyl alcohol (m-NBA) and propylene carbonate (PC) when ions are formed from water/methanol/acid. The extent to which loss of the heme group from myoglobin occurs is related to the extent of supercharging. Results from guanidine melts of cytochrome c monitored with tryptophan fluorescence show that the supercharging reagents PC, sulfolane and HD are effective chemical denaturants in solution. These results provide additional evidence for the role of protein structural changes in the electrospray droplet as the primary mechanism for supercharging with these reagents in native MS. These results also demonstrate that for at least some proteins, the formation of highly charged ions from native MS is no longer a significant barrier for obtaining structural information using conventional tandem MS methods.
Introduction
Electrospray ionization (ESI) mass spectrometry (MS) is widely used to transfer intact proteins and large macromolecular complexes into the gas phase for structural elucidation and is routinely used for protein identification and quantitation. Solutions containing organic solvents and/or acids are typically used with ESI, owing in part to both stable ion signals and to the high charge-state ions that can be produced. The more highly charged ions produced from denaturing solutions typically have extended gas phase conformations,1–3 and can be efficiently dissociated in tandem MS experiments.4–8 ESI from aqueous solutions in which a protein is in a folded, native-like conformation,9 or native MS,10 has the advantage that information about the stoichiometries11, 12 and presence of ligand binding13, 14 to proteins and protein complexes can be obtained. Native MS has been used to obtain information about assembly pathways of macromolecular complexes15–18 and about subunit interactions.19–21 Aqueous solution-phase structure of folded protein conformations can be studied through hydrogen-deuterium exchange22–26 and covalent labelling techniques, such as oxidative labelling.23, 27–29 The distribution of charge states in ESI can be indicative of the protein conformation in solution. Charge-state distributions from native MS are often narrow and low in charge, whereas charge state distributions in denaturing MS are typically broad and high in charge. Multimodal distributions can indicate the coexistence of multiple protein conformations.30–33
The charge states of ions produced in native MS can be increased using several different methods, often collectively referred to as “supercharging”. Trivalent metal ion supercharging34 uses trivalent metal ion salts in low concentration to produce nonspecific trivalent metal ion adduction, which can result in more than a 50% increase in the maximum charge of protein ions produced by ESI from aqueous solutions.34 Electrothermal supercharging35, 36 uses an elevated spray potential in native MS to unfold proteins in the ESI droplet. This results in charge-state distributions that have nearly the same maximum and average charge as those obtained from conventional denaturing solutions,36 making it the most effective supercharging technique for native solutions reported to date. Supercharging with reagents can be effective at increasing charge from both denaturing37–40 and native41–45 solutions. Typically, a small concentration of supercharging reagent (1–5% by volume) is added to a sample solution. These reagents do not significantly affect protein conformation in solution prior to ESI.24, 44, 46 These reagents all have boiling points higher than that of water and become enriched in the droplet as solvent evaporation occurs.46 These supercharging reagents can cause chemical/thermal denaturation in the ESI droplet,43, 45–47 although these reagents also affect other physical properties, such as the droplet surface tension, that also play a role in charging.39
Many experiments have been done to elucidate factors that affect supercharging in ESI. The supercharging reagent, m-NBA, increases the charge state of native RNase A but decreases the charge state of RNase A with all of its disulfide bonds reduced when these ions are formed from the same aqueous solution.47 The latter protein is a random coil in solution, and the lower surface tension of m-NBA compared to that of water results in less charging. In contrast, the charging of RNase A, which is folded in solution, increases as a result of the supercharging reagents destabilizing the folded form which causes unfolding to occur in the ESI droplet. This shows that the effect of conformational changes can be greater than the effect of droplet surface tension on the extent of protein charging. Less supercharging occurs for proteins that have limited ability to unfold, such as proteins with many disulfide bonds or other chemical cross links.47 Other methods to unfold proteins in ESI droplets have also been demonstrated. Proteins can be made to unfold in ESI droplets by adding gaseous reagents to change droplet pH48, 49 or through rapid mixing experiments using theta glass emitters.50, 51 Protein folding or unfolding processes induced by rapid mixing that occur on the low microseconds time scale of small droplets produced by nanoESI can be readily investigated.50
A large number of factors affect charging in electrospray ionization, and alternate mechanisms for supercharging have been proposed. Supercharging reagents can adduct onto protein ions. More adduction to higher charge states has been observed suggesting that high charge states are formed via a “direct interaction” between the reagents and the proteins.42, 52, 53 Venter and coworkers52 suggested that the large dipole moments of many of these reagents (ranging between 3.96 for dimethyl sulfoxide (DMSO) to 4.35 for sulfolane compared to 1.85 for water) shields adjacent charges on basic sites through solvent reorganization, enabling more charge to be deposited on the protein ions during ESI. However, Donald and coworkers40 investigated a large set of reagents and found no correlation between protein supercharging from denaturing solution and reagent dipole moment. Proton transfer between the protein and the reagents has been suggested as a mechanism for supercharging.42, 54 However, lower charging occurs at low concentration of the supercharging reagent DMSO as a result of compaction of the protein in solution, but supercharging occurs at higher concentrations of DMSO as a result of protein destabilization in solution.46 The effect of reagent concentration on the reduction or increase in charge of the same protein provides strong evidence that proton transfer reactivity does not play a role on supercharging with this reagent.
The greatest extent of charging of protein ions that have been formed from denaturing solutions with supercharging reagents is approximately one in every three residues charged, and ions with this charge density have near-linear structures in the gas phase.3 But supercharging from native solutions has not yet produced comparable highly charged ions. Here, results with two new supercharging reagents, 2-thiophenone and HD, are presented. These reagents produce higher charge states than previously reported reagents and can produce higher charge states than can be formed from solutions containing water/methanol/acid that are typically used to produce high charge states of peptide and protein ions.
Experimental
All mass spectra were acquired using a Thermo LTQ (Linear Trapping Quadrupole) mass spectrometer unless otherwise noted. Ions were formed by nanoelectrospray (nanoESI) from borosilicate capillaries (1.0 mm o.d./0.78 mm i.d., Sutter Instruments, Novato, CA, USA) that were pulled to a tip i.d. of ~1 μm with a Flaming/Brown micropipette puller (Model P-87, Sutter Instruments, Novato, CA, USA). A voltage of ~0.7–1.0 kV was applied to a 0.127 mm diameter platinum wire inserted into the solution in the capillary to initiate nanoESI. The nanoESI potential was adjusted to optimize protein ion signal-to-noise ratios (S/N) for each capillary and was maintained at these low voltages to prevent electrothermal supercharging.35 All other source instrument parameters were constant (inlet capillary temperature = 265 °C, capillary voltage = 35 V, and tube lens voltage = 120 V). Spectra were acquired in triplicate using three different capillaries to account for tip-to-tip variability in the charge-state distributions. Protein solutions at a concentration of 10 μM were prepared from lyophilized powders dissolved in water, 200 mM ammonium acetate, 200 mM ammonium bicarbonate, or denaturing solution (45/54/1 methanol/water/acetic acid) containing different amounts of the supercharging reagents, m-nitrobenzyl alcohol (m-NBA), sulfolane, propylene carbonate (PC), 2-thiophenone, and 4-hydroxymethyl-1,3-dioxolan-2-one (HD).
Guanidine melts of 5 μM equine cytochrome c in water, 200 mM ammonium acetate, and 200 mM ammonium bicarbonate with 0–10% supercharging reagent by volume were performed by monitoring tryptophan fluorescence intensity using a multi-mode microplate reader (Synergy H4 hybrid reader, BioTek, Winooski, VT, USA) in emission acquisition mode with 280 ± 20 nm excitation and 352 ± 10 nm emission wavelengths. Each sample was measured in triplicate in 384-well polystyrene solid black low volume flat bottom microplates (Corning, New York, NY, USA). Cytochrome c unfolding curves were fit to a two-state model U⇄N, where U is the unfolded state and N is the native state of the protein. The free energy of unfolding ΔGN was obtained by fitting the unfolding curve to a sigmoidal plot of the form:
| (Equation 1) |
where I is the fluorescence intensity, A is a normalization constant, R is the gas constant, T is temperature, and m is the linear proportionality constant (average m = 4.0 ± 0.7 kcal/mol/M).44 The uncertainty in the ΔGN values is 0.2 kcal/mol and corresponds to the standard deviation in ΔGN measured for cytochrome c in water, ammonium acetate, and ammonium bicarbonate with no supercharging reagent each measured on three different days. All proteins, salts, solvents, and supercharging reagents were purchased from Sigma (St. Louis, MO, USA) and were used without further purification. The purities of the supercharging reagents are all >98%, with the exception of HD, which is ~90% pure.
Results and Discussion
Supercharging in aqueous solutions
With previously identified supercharging reagents, it has not been possible to produce charge states in native mass spectrometry that are comparable to or higher than those obtained from denaturing solutions containing water, methanol and acid. To illustrate the high charging obtainable with two new supercharging reagents, 2-thiophenone and HD, mass spectra of cytochrome c produced by nanoESI from pure water and with various supercharging reagents were obtained (Figure 1a–f). The charge-state distribution of cytochrome c ions produced by nanoESI from pure water is centered around 8+ (Figure 1a). An increase in average charge occurs with the known supercharging reagents, m-NBA (38%), sulfolane (43%), or PC (28%) when these reagents are used at their optimal concentrations, which is the concentration at which the greatest extent of supercharging is observed without sacrificing spray stability or protein ion signal. Significantly more charging occurs for this protein with either 2% 2-thiophenone or 2% HD (Figure 1e,f). These reagents are structural analogs of the supercharging reagents sulfolane and PC, respectively. The average charge compared to that obtained from pure water increases by ~118% with 2-thiophenone and HD, far exceeding the increases in charge observed with the conventional supercharging reagents. The maximum charge state with 2-thiophenone is 22+ and with HD is 24+. The latter values is the same as the number of basic residues (Arg, Lys, and His) in this protein. The charge state of the most abundant ion increases from 8+ in pure water to 20+ with either of these reagents.
Figure 1.

NanoESI mass spectra of cytochrome c in water (left column), 200 mM ammonium acetate (middle column), and 200 mM ammonium bicarbonate (right column) with no supercharging reagent (a, g, l), 1.5% m-NBA (b, h, m), 5% sulfolane (c, i, n), 5% PC (d, j, o), 2% 2-thiophenone (e), and 2% HD (f, k, p).
Supercharging in denaturing solutions
The effectiveness of these supercharging reagents in denaturing solutions consisting of 45/54/1 methanol/water/acetic acid was evaluated, and the relative extents of supercharging obtained with these reagents is different in denaturing solutions (Figure 2) than in water (Figure 1a–f). The most effective supercharging reagents from denaturing solution are m-NBA and PC (Figure 2b,d). These reagents increase the average charge by ~43% compared to denaturing solution without any reagent, and the most abundant charge state is 23+. 2-thiophenone is the least effective supercharging reagent, increasing the average charge by only 16% with the most abundant charge state the 19+. This relatively poor supercharging with 2-thiophenone is likely due to the low concentration (0.5% by volume), above which the stability of the spray is adversely affected. With HD, there is a 40% increase in average charge with the 21+ the most abundant charge state. HD and 2-thiophenone are the most effective supercharging reagents in aqueous solution, but m-NBA and PC are superior when ions are formed from water/methanol/acid solutions.
Figure 2.
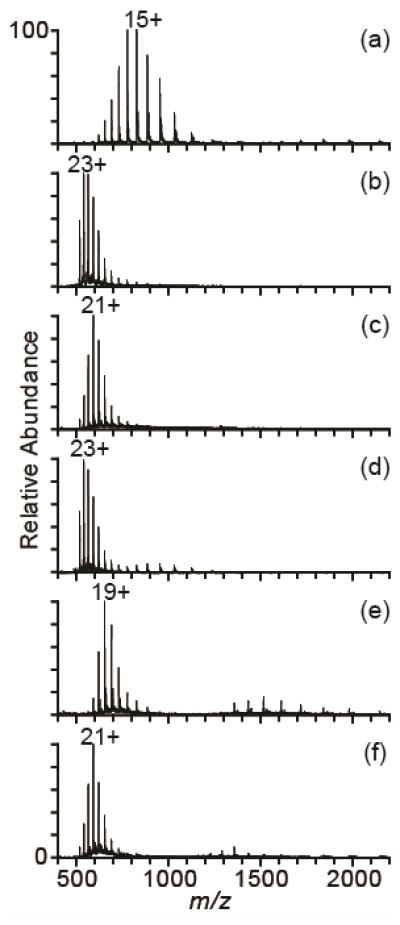
NanoESI mass spectra of cytochrome c in denaturing solution (45/54/1 methanol/water/acetic acid) with no supercharging reagent (a), 5% m-NBA (b), 10% sulfolane (c), 15% PC (d), 0.5% 2-thiophenone (e), and 5% HD (f).
A key finding is that the more highly charged ions can be produced with the supercharging reagents 2-thiophenone or HD in water than can be produced under typical denaturing conditions using water/methanol/acid solutions! Supercharging with 2-thiophenone or HD from water (Figure 1e,f) produces average and maximum charge states that are ~20% higher than those obtained from denaturing solutions without supercharging reagents (Figure 2a). Moreover, the most abundant charge state is 20+ with these reagents in water compared to 15+ from a denaturing solution. Electrothermal supercharging from aqueous ammonium bicarbonate solutions produces charge-state distributions with similar extents of charging to those obtained from denaturing solution.36 The data with the two new supercharging reagents demonstrates for the first time that charging greater than that obtainable from denaturing solution without supercharging reagents can be obtained from aqueous solutions with either 2-thiophenone or HD.
Effect of supercharging reagent concentration
The extent of supercharging depends on the reagent concentration.37, 38, 40, 42, 44–47 A 2% concentration was found to be the optimal concentration for both 2-thiophenone and for HD in aqueous solutions. The average charge decreases at higher concentrations (Figure S1). For cytochrome c in water, the average charge decreases from 17.8 ± 1.1+ to 14.6 ± 1.0+ when the concentration of HD is increased from 2% to 3%. A similar decrease in charge occurs with PC in denaturing solutions above its optimal concentration of 15%.40 At HD concentrations higher than 2%, a significant fraction of the ion signal corresponds to cytochrome c dimer. Increasing the HD concentration from 2% to 3% increases the dimer population from 21 ± 1% to 54 ± 1% of the total protein ion abundance. This increasing prevalence of dimer population with increasing reagent concentration suggests that the supercharging reagent could be affecting the protein conformation in the ESI droplet, which can increase protein aggregation. At a concentration of 2% 2-thiophenone or HD, a significant amount of chemical noise due to cluster formation and adduction to the protein ions occurs when a quadrupole-time-of-flight (Q-TOF) mass spectrometer is used (Figure S2). This instrument has softer source conditions compared to the Thermo LTQ.35 Thus, a lower volume of reagent should be used when supercharging with these two new reagents on instruments with gentle source conditions for optimal protein ion signal.
Supercharging in buffered solutions
Buffers are typically used in native MS to increase ionic strength and mitigate pH changes, both of which can affect the native structures of proteins and protein complexes. To test the relative effectiveness of these supercharging reagents to increase the charge of protein ions formed from buffered solutions, 10 μM cytochrome c ions with the same concentration of reagents were formed by nanoESI from aqueous solutions with 200 mM ammonium acetate or 200 mM ammonium bicarbonate (Figure 1g–p). No spectra were obtained with 2-thiophenone in these ammonium buffer solutions because the electrospray was unstable. The average charge obtained for each supercharging reagent in 200 mM aqueous ammonium acetate is about 11% lower than that obtained with the same reagent in pure water. The only exception is sulfolane, for which there is a slight increase in charge. The average charge of cytochrome c produced from solutions containing HD and ammonium acetate is 15.4 ± 0.1+. This average charge is higher than that produced from a denaturing solution (14.9 ± 0.3+) and corresponds to an increase in average charge of ~123% compared to ammonium acetate without any supercharging reagent. In contrast, there is only an increase of ~57% on average for the other reagents. These increases in average charge are similar to those observed from water, suggesting that the denaturing strength of these reagents is not significantly different in pure water and ammonium acetate buffer.
In striking contrast to results in water and aqueous 200 mM ammonium acetate, supercharging with any of these reagents is ineffective in 200 mM ammonium bicarbonate. The charge-state distributions are all centered near 7+ with or without supercharging reagent, and the average charge state is nearly the same except for HD, for which the average charge is slightly lower. These data show that more highly charged ions can be produced from solutions with low buffer concentration and that ammonium acetate is the preferred buffer.
Supercharging and noncovalent complexes
The supercharging reagents, sulfolane and DMSO, are chemical denaturants that destabilize the native structures of proteins.44, 46 In addition, sulfolane and m-NBA can disrupt noncovalent interactions and cause partial or complete dissociation of protein-protein complexes.43–45 The extent to which the new supercharging reagents, 2-thiophenone and HD, disrupt noncovalent interactions compared to the standard supercharging reagents was evaluated by measuring mass spectra of myoglobin (Figure 3). The charge-state distributions of holo- and apo-myoglobin (highlighted in red) produced by nanoESI out of aqueous solutions are centered around the 8+ and 9+ charge states (Figure 3a,g,l), and holo-myoglobin is the most abundant form of these ions. An increase in charge is obtained with m-NBA (77%), sulfolane (29%), or PC (29%) in aqueous solutions. The dominant form of the protein is apo-myoglobin, not holo-myoglobin, with these reagents. In contrast, the average charge with the new supercharging reagents, 2-thiophenone and HD, is much greater. The average charge is 163% and 138% higher with 2-thiophenone and HD, respectively, and the maximum charge state increases from 11+ to 28+. The maximum charge state is close to the number of basic residues (32) in this protein. The average charge is about 10% greater than that produced from denaturing solution (18.9 ± 0.3+ for apo-myoglobin), and the maximum charge state is the same as that obtained from denaturing solution (28+). Apo-myoglobin is the dominant form of the protein observed with HD, and apo-myoglobin composes 100% of the protein ion population with 2-thiophenone.
Figure 3.

NanoESI mass spectra of myoglobin in water (left column), 200 mM ammonium acetate (middle column), and 200 mM ammonium bicarbonate (right column) with no supercharging reagent (a, g, l), 1.5% m-NBA (b, h, m), 5% sulfolane (c, i, n), 5% PC (d, j, o), 2% 2-thiophenone (e), and 2% HD (f, k, p). Apo-myoglobin is labeled with red circles.
With 200 mM ammonium acetate, there is a slight decrease in average charge for all supercharging reagents compared to the results with these reagents in pure water, again with the exception of sulfolane, for which there is a slight increase in average charge. The charge-state distribution with HD is shifted to significantly higher charge compared to that obtained with the other reagents. The average charge with HD in 200 mM ammonium acetate (18.3 ± 0.2+) is similar to that out of denaturing solution (18.9 ± 0.3+). For all reagents except PC, which shows little supercharging, apo-myoglobin is the most abundant protein species.
With ammonium bicarbonate, there is very little increase in charge with any supercharging reagent, and similar to cytochrome c, there is a decrease in the average charge with HD in this buffer. All charge-state distributions are centered around the 8+ or 9+ charge states, and holo-myoglobin is the most abundant form of myoglobin, with the exception of m-NBA.
MS evidence for protein conformational changes in ESI droplet
Apo-myoglobin is formed in solution as a result of unfolding of the F-helix in the native structure of holo-myoglobin and subsequent loss of the non-covalently bound heme group.55, 56 The percentage of apo-myoglobin observed in all spectra in Figure 3 as a function of the average charge of all myoglobin species in a given mass spectrum is shown in Figure 4. These data show a trend of increasing fraction of apo-myoglobin with increasing charging obtained with the supercharging reagents. This suggests that the high charge states formed with supercharging reagents are a result of chemical destabilization of the native protein structure, which results in protein conformational changes in the ESI droplet and the formation of apo-myoglobin by loss of the heme. A decrease in supercharging as well as a decrease in protein complex dissociation in the buffered solutions suggests that the stability of the protein increases in these buffers, and that denaturation by the supercharging reagents is less effective. Buffers, particularly phosphate buffers, are routinely used in biology to stabilize the native forms of proteins, and some proteins and protein complexes require a certain ionic strength or essential salts in order to be in their active state or to assemble.57–61 A similar effect was reported for a much larger complex, the homotetramer concanavalin A, where less supercharging with m-NBA occurs with increasing ammonium acetate concentration.45 The buffer capacity increases with higher concentration, and this reduces pH changes in the ESI droplet during droplet evaporation that might also destabilize the protein structure during supercharging. This buffer capacity is highest for ammonium bicarbonate at neutral pH, and the least supercharging occurs for this buffer. The decrease in average charge with HD in ammonium bicarbonate for both cytochrome c and myoglobin may be a result of surface tension effects. HD has a lower surface tension than water (44 ± 3 dynes/cm versus 72 dynes/cm, respectively).62 A droplet consisting of a substantial fraction of HD can hold less charge than a droplet of pure water, which can lead to lower charging in the absence of protein conformational changes.39, 47 The other reagents in this study also have lower surface tensions than water,62, 63 and the surface tension effects may be obscured by conformational changes to the protein. It has been shown that conformational effects with proteins can result in significantly larger differences in charging than surface tension effects.47
Figure 4.
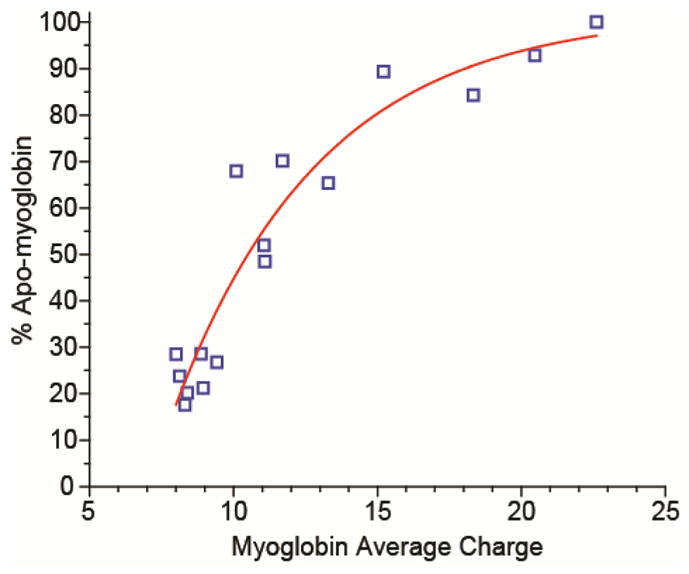
The average charge of myoglobin (using an intensity weighted average of both holo- and apo-myoglobin) plotted versus the percentage of the total protein ion signal that is apo-myoglobin under all of the solution conditions shown in Figure 3.
Fluorescence evidence for protein conformational changes in ESI droplets
DMSO, sulfolane, and 4-vinyl-1,3-dioxolan-2-one were previously shown to be effective chemical denaturants in solution.44, 46, 63, 64 To obtain additional evidence for the role of supercharging reagents on destabilizing protein conformation in solution, the effects of buffer and supercharging reagents on the stability of cytochrome c was investigated with guanidine HCl melts using tryptophan fluorescence to measure protein unfolding. When cytochrome c is in a native conformation, the single tryptophan (residue 59) is in close proximity to the heme group, which is covalently bound at residues 14 and 17, and tryptophan fluorescence is entirely quenched by the heme group.65 When unfolded, the tryptophan residue is on average further away from the heme, and tryptophan fluorescence occurs.65 Thus, tryptophan fluorescence can be used as a probe of cytochrome c unfolding in solution. Guanidine melts were performed with between 0% and 10% by volume of PC, sulfolane, and HD in water, 200 mM ammonium acetate, and 200 mM ammonium bicarbonate. No fluorescence experiments were performed with m-NBA because this reagent absorbs strongly up to ~400 nm, so no tryptophan fluorescence is observed with m-NBA in solution. Experiments were not done with 2-thiophenone owing to a reaction between the reagent and guanidine that results in a black precipitate.
An example of the fluorescence data for guanidine melts with propylene carbonate in water is shown in Figure 5. With increasing propylene carbonate concentration, less guanidine is required to unfold the protein (Figure 5), indicating that PC destabilizes the native form of cytochrome c relative to the unfolded form. The Gibbs free energies of protein folding, ΔGN, are obtained from these data (eq. 1) and show that ΔGN becomes less negative, increasing from −6.1 kcal/mol to −4.0 kcal/mol, when the PC concentration increases from 0% to 10%. This result clearly demonstrates the extent to which PC destabilizes the native form of the protein in solution.
Figure 5.
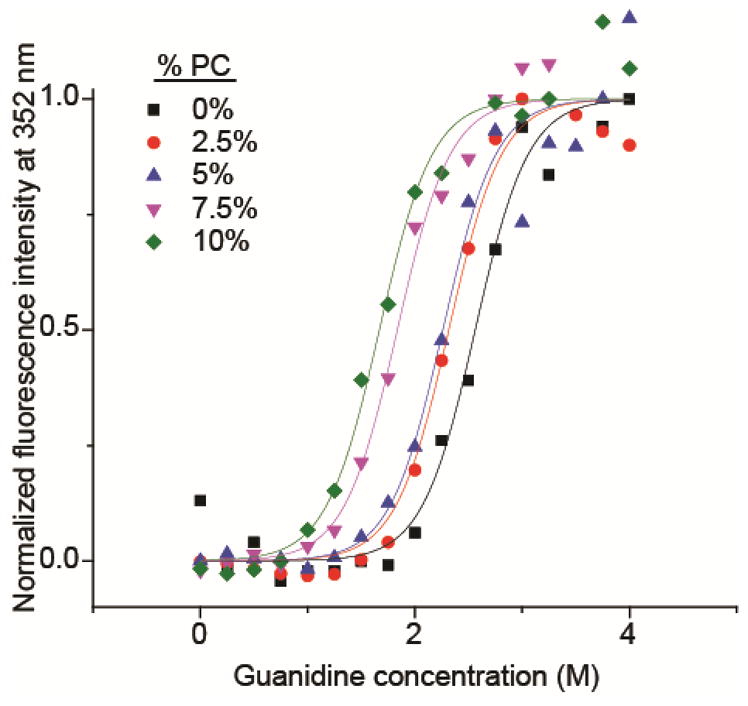
Guandine melts of cytochrome c measured by tryptophan fluorescence. Data is normalized in the plot so that the maximum of the sigmoidal fit (from Equation 1) is defined as one. Guanidine concentration was increased in increments of 0.25 M, and PC concentration was increased in increments of 2.5% by volume.
ΔGN as a function of supercharging reagent concentration for the three supercharging reagents under all buffer conditions is shown in Figure 6a–c, and all of the data is overlaid in Figure 6d. The data are fit with lines, the slope of which corresponds to the denaturing strength of a supercharging reagent under the given buffer conditions. For example, PC has a denaturing strength of 1.8 kcal/mol/M in both water and 200 mM ammonium acetate, and the ΔGN values show that the stability of cytochrome c is the same in ammonium acetate as in pure water. In 200 mM ammonium bicarbonate with PC, the ΔGN values are all more negative than in ammonium acetate and pure water, even without any supercharging reagent present. This indicates that ammonium bicarbonate stabilizes cytochrome c against denaturation in solution both with and without the supercharging reagent. This significant stabilization observed with ammonium bicarbonate may be due in part to ammonium bicarbonate’s high buffer capacity and the fact that its highest buffering capacity is around pH 7, a pH at which cytochrome c is folded.66 By contrast, ammonium acetate has very poor buffer capacity at this pH, and buffers instead around pH 5, closer to the pH at which equine cytochrome c starts to unfold (pH 3) in the absence of other denaturants.67 The pH of 6 M guanidine HCl in water is 4.5, so the sample solution acidifies as the guanidine concentration increases. In all three buffer conditions, the denaturing strength of PC is about the same ~1.8 kcal/mol/M, indicating that the buffer does not affect the destabilizing effect of the supercharging reagent on the native form of the protein.
Figure 6.

Free energy of folding (ΔGN) of cytochrome c as a function of supercharging reagent concentration for PC (a), sulfolane (b), HD (c), and all three reagents (d). The free energy of folding was calculated from guanidine melt data fit to Equation 1.
The denaturing strength of sulfolane is 1.9 to 2.0 kcal/mol/M in pure water and ammonium acetate and is similar to that of PC. These values are similar to the previously measured denaturing strength of sulfolane for myoglobin in Tris buffer (1.5 ± 0.1 kcal/mol/M).46 In ammonium bicarbonate, the denaturing strength is 0.9 kcal/mol/M, indicating that sulfolane is a less effective chemical denaturant in ammonium bicarbonate, and the protein is stabilized in this buffer both with and without sulfolane.
HD has a denaturing strength of about 1.8, 1.1 and 1.3 kcal/mol/M in water, ammonium acetate and ammonium bicarbonate, respectively. These results are consistent with those of sulfolane that show that the effectiveness of these chemical denaturants can depend on the buffer concentration and identity.
Stability of native proteins in solution and supercharging
The fluorescence data provide compelling evidence that the structure of cytochrome c is unaffected by the supercharging reagents in the original ESI solutions. With 10% PC, the structure of cytochrome c as monitored by fluorescence is unaffected even with ~0.5 M guanidine (Figure 5). Similarly, there is no measurable change in protein structure with up to 10% sulfolane or 10% HD without guanidine. In native supercharging, the optimal concentrations of these reagents is less than 10% (5%, 5%, and 2% for PC, sulfolane, and HD, respectively). These data indicate that the structure of cytochrome c is not significantly disrupted in the presence of these supercharging reagents in the solutions prior to ESI in native supercharging experiments.
A comparison of the mass spectra in Figure 1 and these ΔGN data reveals a correlation between lower solution-phase stability of the native form of the protein and more effective supercharging. The charge-state distributions with PC and sulfolane are similar both in water and ammonium acetate (Figure 1), which is consistent with the similar denaturing ability of these two reagents from both of these solutions (Figure 6). For HD, the ΔGN and denaturing strength is slightly lower in ammonium acetate than water, which is reflected in the mass spectra where there is a more substantial decrease in the most abundant charge state from 20+ to 16+ compared to that observed for PC or sulfolane. With ammonium bicarbonate, there is no supercharging observed with any reagent, consistent with the high stability of the folded form of the protein in all solutions containing ammonium bicarbonate. All mass spectra are centered around the 7+ charge state, and all ΔGN plots with ammonium bicarbonate are similar (Figure 6d). This qualitative correlation between the bulk solution-phase studies and the mass spectra indicate that conformational effects play a large role in the supercharging phenomenon.
The ΔGN values are very similar for all three supercharging reagents in pure water (Figure 6d), yet the extent of supercharging differs (Figure 1b–f), with HD producing more charging than either PC or sulfolane. This indicates that in addition to the intrinsic denaturing ability of these reagents in solution, other factors play a role in the extent of supercharging observed. Some of these factors may be reagent solubility and boiling point, which influence to what extent and how quickly the reagent concentrates in the ESI droplet and the extent to which droplet heating occurs. For example, the boiling points of PC and sulfolane are similar (285 °C for sulfolane and 240 °C for PC) and are much lower than that of HD (354 °C).62 Because the boiling points of PC and sulfolane are lower than that of HD, the rate of concentration in the ESI droplet will be less, and this may produce less supercharging with these reagents than occurs with HD. Moreover, there will be less evaporative cooling with HD, and this may lead to higher ESI droplet temperatures that also promote protein unfolding. PC has a solubility limit of 17% in water, whereas sulfolane and HD are completely miscible with water. Because they are completely miscible, higher concentrations of both sulfolane and HD can occur in the evaporating droplet, consistent with more supercharging observed with sulfolane and HD than with PC. The presence of m-NBA has been shown previously to increase droplet lifetimes by inhibiting droplet evaporation,68 and may lead to increased protein unfolding as a result of lower evaporative cooling and longer times for unfolding to occur. Additional factors, such as surface tension, likely play a role as well in the relative effectiveness of these supercharging reagents.
Conclusion
Electrospray ionization in combination with two new supercharging reagents, 2-thiophenone and HD, can produce more highly charged ions from aqueous solutions in which proteins have native conformations than obtained from more traditional solutions consisting of water/methanol/acid in which proteins are unfolded. Supercharging with these new reagents in native mass spectrometry produces significantly more than a two-fold increase in average and maximum charge, and these reagents are about twice as effective at native MS supercharging than m-NBA, sulfolane, and PC. Both m-NBA and PC are still the most effective reagents for increasing protein ion charge from denaturing solution. More loss of the heme from myoglobin occurs with increasing supercharging, indicating that supercharging is a result of protein conformational changes in the electrospray droplet. The supercharging reagents PC, sulfolane and HD are effective chemical denaturants in solution, and the extent of supercharging observed with these reagents is related to the denaturing capability of the reagent and the stabilities of proteins in different buffers. Combined, these results provide compelling evidence that the primary mechanism of supercharging with these reagents in native mass spectrometry is their effectiveness at destabilizing native protein structure, resulting in unfolding of the protein in the ESI droplet.
These results demonstrate that it is possible to keep proteins in solutions in which they have native structures and reactivities, yet produce more highly charged ions than is possible with conventional solutions in which proteins are denatured. The highly charged ions produced with these reagents in native MS are almost certainly as unfolded in the gas phase as comparably charged ions produced by other methods used to form highly charged ions, such as ESI from solutions consisting of water, methanol, and acetic acid in which proteins are denatured. This should make it possible to combine the advantages of native mass spectrometry with the capabilities of tandem mass spectrometry to obtain extensive structural information on the highly charged ions that can be produced with these reagents. This should be particularly advantageous for top-down H/D exchange methods for deducing information about protein conformations and dynamics. Continuous H/D exchange can be monitored without the need for proteolysis and denaturing conditions necessary for producing high charge states.24 Because the lifetime of the droplet in which protein denaturation occurs can be less than 27 μs,69 the potential for back-exchange in solution is eliminated.
Supplementary Material
Table 1.
Average charge of cytochrome c in water, 200 mM ammonium acetate (AA), 200 mM ammonium bicarbonate (ABC) and denaturing solution (45/54/1 methanol, water, acetic acid by volume) with supercharging reagents added at the optimal concentration for maximal charging.
| Water | AA | ABC | Denaturing | ||
|---|---|---|---|---|---|
| No Additive | 8.2 ± 0.3+ | 6.9 ± 0.1+ | 7.4 ± 0.2+ | No Additive | 14.9 ± 0.3+ |
| 1.5% m-NBA | 11.3 ± 0.5+ | 10.8 ± 0.1+ | 7.6 ± 0.2+ | 5% m-NBA | 21.6 ± 0.4+ |
| 5% sulfolane | 11.7 ± 0.2+ | 12.9 ± 0.1+ | 7.4 ± 0.1+ | 10% sulfolane | 20.4 ± 0.2+ |
| 5% PC | 10.5 ± 0.2+ | 8.9 ± 0.4+ | 7.7 ± 0.1+ | 15% PC | 21.1 ± 0.3+ |
| 2% 2-thiophenone | 18.5 ± 0.1+ | -- | -- | 0.5% 2-thiophenone | 17.3 ± 0.1+ |
| 2% HD | 17.8 ± 1.1+ | 15.4 ± 0.1+ | 6.9 ± 0.1+ | 5% HD | 20.8 ± 0.1+ |
Table 2.
Average charge of myoglobin in water, 200 mM ammonium acetate (AA), and 200 mM ammonium bicarbonate (ABC) with supercharging reagents added at the optimal concentration for maximal charging. Holo-myoglobin values are without parentheses and apo-myoglobin values are within parentheses.
| Water | AA | ABC | |
|---|---|---|---|
| No Additive | 8.6 ± 0.5+ (8.9 ± 0.8+) |
8.3 ± 0.1+ (7.5 ± 0.1+) |
8.5 ± 0.4+ (7.9 ± 0.5+) |
| 1.5% m-NBA | 15.5 ± 1.5+ (15.2 ± 0.9+) |
12.1 ± 0.3+ (11.5 ± 0.3+) |
9.5 ± 0.5+ (10.4 ± 1.0+) |
| 5% sulfolane | 11.2 ± 0.5+ (10.9 ± 0.3+) |
14.1 ± 1.4+ (12.9 ± 1.1+) |
8.9 ± 0.1+ (8.7 ± 0.1+) |
| 5% PC | 10.9 ± 0.5+ (11.3 ± 0.7+) |
8.4 ± 0.3+ (8.0 ± 0.5+) |
9.5 ± 0.3+ (9.1 ± 0.2+) |
| 2% 2-thiophenone | -- (22.6 ± 0.2+) |
-- -- |
-- -- |
| 2% HD | 19.5 ± 2.2+ (20.5 ± 0.1+) |
18.5 ± 0.2+ (18.3 ± 0.2+) |
8.2 ± 0.1+ (7.6 ± 0.1+) |
Acknowledgments
The authors thank the National Institutes of Health (Grant No. R01GM097357) and the National Science Foundation (Graduate Research Fellowship for CAC; Grant No. DGE1106400) for financial support, and the Kuriyan Lab at the University of California, Berkeley for use of their fluorometer.
References
- 1.Shelimov KB, Clemmer DE, Hudgins RR, Jarrold MF. J Am Chem Soc. 1997;119:2240–2248. [Google Scholar]
- 2.Wyttenbach T, Bowers MT. J Phys Chem B. 2011;115:12266–12275. doi: 10.1021/jp206867a. [DOI] [PubMed] [Google Scholar]
- 3.Going CC, Williams ER. Anal Chem. 2015;87:3973–3980. doi: 10.1021/acs.analchem.5b00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iavarone AT, Paech K, Williams ER. Anal Chem. 2004;76:2231–2238. doi: 10.1021/ac035431p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iavarone AT, Williams ER. Anal Chem. 2003;75:4525–4533. doi: 10.1021/ac034144i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rožman M, Gaskell SJ. Rapid Commun Mass Spectrom. 2012;26:282–286. doi: 10.1002/rcm.5330. [DOI] [PubMed] [Google Scholar]
- 7.Hogan JM, McLuckey SA. J Mass Spectrom. 2003;38:245–256. doi: 10.1002/jms.458. [DOI] [PubMed] [Google Scholar]
- 8.Madsen JA, Brodbelt JS. J Am Soc Mass Spectrom. 2009;20:349–358. doi: 10.1016/j.jasms.2008.10.018. [DOI] [PubMed] [Google Scholar]
- 9.Chowdhury SK, Katta V, Chait BT. J Am Chem Soc. 1990;112:9012–9013. [Google Scholar]
- 10.Heuvel RHHvd, Heck AJR. Curr Opin Chem Biol. 2004;8:519–526. doi: 10.1016/j.cbpa.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 11.Benesch JLP, Robinson CV. Curr Opin Struct Biol. 2006;16:245–251. doi: 10.1016/j.sbi.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 12.Snijder J, van de Waterbeemd M, Damoc E, Denisov E, Grinfeld D, Bennett A, Agbandje-McKenna M, Makarov A, Heck AJR. J Am Chem Soc. 2014;136:7295–7299. doi: 10.1021/ja502616y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.El-Hawiet A, Kitova EN, Arutyunov D, Simpson DJ, Szymanski CM, Klassen JS. Anal Chem. 2012;84:3867–3870. doi: 10.1021/ac3005082. [DOI] [PubMed] [Google Scholar]
- 14.Robinson CV, Chung EW, Kragelund BB, Knudsen J, Aplin RT, Poulsen FM, Dobson CM. J Am Chem Soc. 1996;118:8646–8653. [Google Scholar]
- 15.Kintzer AF, Thoren KL, Sterling HJ, Dong KC, Feld GK, Tang II, Zhang TT, Williams ER, Berger JM, Krantz BA. J Mol Biol. 2009;392:614–629. doi: 10.1016/j.jmb.2009.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Painter AJ, Jaya N, Basha E, Vierling E, Robinson CV, Benesch JLP. Chem Biol. 2008;15:246–253. doi: 10.1016/j.chembiol.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 17.Sharon M, Witt S, Glasmacher E, Baumeister W, Robinson CV. J Biol Chem. 2007;282:18448–18457. doi: 10.1074/jbc.M701534200. [DOI] [PubMed] [Google Scholar]
- 18.Remaut H, Rose RJ, Hannan TJ, Hultgren SJ, Radford SE, Ashcroft AE, Waksman G. Mol Cell. 2006;22:831–842. doi: 10.1016/j.molcel.2006.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krishnaswamy SR, Williams ER, Kirsch JF. Protein Sci. 2006;15:1465–1475. doi: 10.1110/ps.062083406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boeri Erba E, Barylyuk K, Yang Y, Zenobi R. Anal Chem. 2011;83:9251–9259. doi: 10.1021/ac201576e. [DOI] [PubMed] [Google Scholar]
- 21.Quintyn Royston S, Yan J, Wysocki Vicki H. Chem Biol. 2015;22:583–592. doi: 10.1016/j.chembiol.2015.03.019. [DOI] [PubMed] [Google Scholar]
- 22.Wales TE, Engen JR. Mass Spectrom Rev. 2006;25:158–170. doi: 10.1002/mas.20064. [DOI] [PubMed] [Google Scholar]
- 23.Pan Y, Piyadasa H, O’Neil JD, Konermann L. J Mol Biol. 2012;416:400–413. doi: 10.1016/j.jmb.2011.12.052. [DOI] [PubMed] [Google Scholar]
- 24.Sterling HJ, Williams ER. Anal Chem. 2010;82:9050–9057. doi: 10.1021/ac101957x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abzalimov RR, Kaplan DA, Easterling ML, Kaltashov IA. J Am Soc Mass Spectrom. 2009;20:1514–1517. doi: 10.1016/j.jasms.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tuma R, Coward LU, Kirk MC, Barnes S, Prevelige PE., Jr J Mol Biol. 2001;306:389–396. doi: 10.1006/jmbi.2000.4383. [DOI] [PubMed] [Google Scholar]
- 27.Xu G, Chance MR. Chem Rev. 2007;107:3514–3543. doi: 10.1021/cr0682047. [DOI] [PubMed] [Google Scholar]
- 28.Gau BC, Sharp JS, Rempel DL, Gross ML. Anal Chem. 2009;81:6563–6571. doi: 10.1021/ac901054w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smedley JG, Sharp JS, Kuhn JF, Tomer KB. Biochemistry. 2008;47:10694–10704. doi: 10.1021/bi800533t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mirza UA, Cohen SL, Chait BT. Anal Chem. 1993;65:1–6. doi: 10.1021/ac00049a003. [DOI] [PubMed] [Google Scholar]
- 31.Loo JA, Loo RRO, Udseth HR, Edmonds CG, Smith RD. Rapid Commun Mass Spectrom. 1991;5:101–105. doi: 10.1002/rcm.1290050303. [DOI] [PubMed] [Google Scholar]
- 32.Konermann L, Douglas DJ. Biochemistry. 1997;36:12296–12302. doi: 10.1021/bi971266u. [DOI] [PubMed] [Google Scholar]
- 33.Frimpong AK, Abzalimov RR, Eyles SJ, Kaltashov IA. Anal Chem. 2007;79:4154–4161. doi: 10.1021/ac0704098. [DOI] [PubMed] [Google Scholar]
- 34.Flick T, Williams ER. J Am Soc Mass Spectrom. 2012;23:1885–1895. doi: 10.1007/s13361-012-0463-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sterling HJ, Cassou CA, Susa AC, Williams ER. Anal Chem. 2012;84:3795–3801. doi: 10.1021/ac300468a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cassou CA, Sterling HJ, Susa AC, Williams ER. Anal Chem. 2013;85:138–146. doi: 10.1021/ac302256d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iavarone AT, Jurchen JC, Williams ER. J Am Soc Mass Spectrom. 2000;11:976–985. doi: 10.1016/S1044-0305(00)00169-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iavarone AT, Jurchen JC, Williams ER. Anal Chem. 2001;73:1455–1460. doi: 10.1021/ac001251t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Iavarone AT, Williams ER. J Am Chem Soc. 2003;125:2319–2327. doi: 10.1021/ja021202t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Teo CA, Donald WA. Anal Chem. 2014;86:4455–4462. doi: 10.1021/ac500304r. [DOI] [PubMed] [Google Scholar]
- 41.Lomeli SH, Yin S, Loo RRO, Loo JA. J Am Soc Mass Spectrom. 2009;20:593–596. doi: 10.1016/j.jasms.2008.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lomeli SH, Peng IX, Yin S, Loo RRO, Loo JA. J Am Soc Mass Spectrom. 2010;21:127–131. doi: 10.1016/j.jasms.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sterling HJ, Williams ER. J Am Soc Mass Spectrom. 2009;20:1933–1943. doi: 10.1016/j.jasms.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sterling HJ, Daly MP, Feld GK, Thoren KL, Kintzer AF, Krantz BA, Williams ER. J Am Soc Mass Spectrom. 2010;21:1762–1774. doi: 10.1016/j.jasms.2010.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sterling HJ, Kintzer AF, Feld GK, Cassou CA, Krantz BA, Williams ER. J Am Soc Mass Spectrom. 2012;23:191–200. doi: 10.1007/s13361-011-0301-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sterling HJ, Prell JS, Cassou CA, Williams ER. J Am Soc Mass Spectrom. 2011;22:1178–1186. doi: 10.1007/s13361-011-0116-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sterling HJ, Cassou CA, Trnka MJ, Burlingame AL, Krantz BA, Williams ER. Phys Chem Chem Phys. 2011;13:18288–18296. doi: 10.1039/c1cp20277d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kharlamova A, McLuckey SA. Anal Chem. 2010;83:431–437. doi: 10.1021/ac1027319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kharlamova A, Prentice BM, Huang TY, McLuckey SA. Anal Chem. 2010;82:7422–7429. doi: 10.1021/ac101578q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mortensen DN, Williams ER. Anal Chem. 2015;87:1281–1287. doi: 10.1021/ac503981c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fisher CM, Kharlamova A, McLuckey SA. Anal Chem. 2014;86:4581–4588. doi: 10.1021/ac500721r. [DOI] [PubMed] [Google Scholar]
- 52.Douglass K, Venter A. J Am Soc Mass Spectrom. 2012;23:489–497. doi: 10.1007/s13361-011-0319-1. [DOI] [PubMed] [Google Scholar]
- 53.Chingin K, Xu N, Chen H. J Am Soc Mass Spectrom. 2014;25:928–934. doi: 10.1007/s13361-014-0887-y. [DOI] [PubMed] [Google Scholar]
- 54.Ogorzalek Loo R, Lakshmanan R, Loo J. J Am Soc Mass Spectrom. 2014;25:1675–1693. doi: 10.1007/s13361-014-0965-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dasmeh P, Kepp KP. PLoS ONE. 2013;8:e80308. doi: 10.1371/journal.pone.0080308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Awad ES, Deranleau DA. Biochemistry. 1968;7:1791–1795. doi: 10.1021/bi00845a025. [DOI] [PubMed] [Google Scholar]
- 57.Watt SJ, Urathamakul T, Schaeffer PM, Williams NK, Sheil MM, Dixon NE, Beck JL. Rapid Comm Mass Spectrom. 2007;21:132–140. doi: 10.1002/rcm.2818. [DOI] [PubMed] [Google Scholar]
- 58.Batchelor JD, Doucleff M, Lee C-J, Matsubara K, De Carlo S, Heideker J, Lamers MH, Pelton JG, Wemmer DE. J Mol Biol. 2008;384:1058–1075. doi: 10.1016/j.jmb.2008.10.024. [DOI] [PubMed] [Google Scholar]
- 59.Smith SP, Barber KR, Dunn SD, Shaw GS. Biochemistry. 1996;35:8805–8814. doi: 10.1021/bi952698c. [DOI] [PubMed] [Google Scholar]
- 60.Bonazza K, Rottensteiner H, Schrenk G, Fiedler C, Scheiflinger F, Allmaier G, Turecek P, Friedbacher G. Anal Bioanal Chem. 2015;407:6051–6056. doi: 10.1007/s00216-015-8778-z. [DOI] [PubMed] [Google Scholar]
- 61.Wagner R, Gonzalez DH, Podesta FE, Andreo CS. Eur J Biochem. 1987;164:661–666. doi: 10.1111/j.1432-1033.1987.tb11177.x. [DOI] [PubMed] [Google Scholar]
- 62.Royal Society of Chemistry ChemSpider. [accessed August 13, 2015]; http://www.chemspider.com/
- 63.Zenaidee MA, Donald WA. Analyst. 2015;140:1894–1905. doi: 10.1039/c4an02338b. [DOI] [PubMed] [Google Scholar]
- 64.Hamdy O, Julian R. J Am Soc Mass Spectrom. 2012;23:1–6. doi: 10.1007/s13361-011-0284-8. [DOI] [PubMed] [Google Scholar]
- 65.Tsong TY. J Biol Chem. 1974;249:1988–1990. [PubMed] [Google Scholar]
- 66.Goto YC, Linda J, Fink, Anthony L. Proc Natl Acad Sci USA. 1990;87:573–577. [Google Scholar]
- 67.Goto Y, Hagihara Y, Hamada D, Hoshino M, Nishii I. Biochemistry. 1993;32:11878–11885. doi: 10.1021/bi00095a017. [DOI] [PubMed] [Google Scholar]
- 68.Grimm RL, Beauchamp JL. J Phys Chem A. 2010;114:1411–1419. doi: 10.1021/jp907162w. [DOI] [PubMed] [Google Scholar]
- 69.Mortensen DN, Williams ER. Anal Chem. 2014;86:9315–9321. doi: 10.1021/ac502545r. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.