

Abstract
Canine transmissible venereal tumor (CTVT) is a parasitic cancer clone that has propagated for thousands of years via sexual transfer of malignant cells. Little is understood about the mechanisms that converted an ancient tumor into the world's oldest known continuously propagating somatic cell lineage. We created the largest existing catalog of canine genome-wide variation and compared it against two CTVT genome sequences, thereby separating alleles derived from the founder's genome from somatic mutations that must drive clonal transmissibility. We show that CTVT has undergone continuous adaptation to its transmissible allograft niche, with overlapping mutations at every step of immunosurveillance, particularly self-antigen presentation and apoptosis. We also identified chronologically early somatic mutations in oncogenesis- and immune-related genes that may represent key initiators of clonal transmissibility. Thus, we provide the first insights into the specific genomic aberrations that underlie CTVT's dogged perseverance in canids around the world.
Clonally transmissible tumors arise in a single founder case and spread to other members of the same species via allogeneic communication of cancer cells. This phenomenon is known to have evolved independently only twice in mammals—in Tasmanian devils and canines, lineages that diverged >180 million years ago (Meredith et al. 2011). Canine transmissible venereal tumor (CTVT) is a sexually transmitted tumor clone that has continuously proliferated for thousands of years and is now endemic in the canine populations of at least 90 countries (Strakova and Murchison 2014). CTVT typically avoids rejection by the host immune system for months, but is subsequently identified and eliminated in immunocompetent individuals (Yang 1988). Since all CTVT cancers are derived from a single founder tumor, they show strong genetic identity with one another, but are markedly distinct from their transient host (Katzir et al. 1987; Murgia et al. 2006; Murchison et al. 2014). Leveraging this key principle of clonal transmissibility, a recent study characterized genomic elements shared by and unique to two CTVT tumors (Murchison et al. 2014). However, the obvious unavailability of germline DNA from the long-deceased founder animal prevented accurate discrimination between somatically acquired mutations and the genetic variation (the CTVT founder canid's inherited alleles) that was present in the founder canid prior to oncogenesis of the initial tumor. Consequently, only a few candidate somatic drivers of CTVT were identified, and the genomic mechanisms that allow the tumor to thrive in diverse canine hosts remained largely undefined.
To better address these questions, we hypothesized that the founder's inherited alleles could be identified by comparing the CTVT genome against inherited polymorphisms found in whole-genome sequences from a diverse cross section of wild and domesticated modern canids. Furthermore, variants not found in other canids are likely dramatically enriched for somatic mutations, and a subset of these must represent key mediators of CTVT's remarkable behavior. Here, we constructed the most comprehensive existing catalog of canine genomic variation, facilitating the first accurate dissection of the genetics underpinning CTVT biology by examining the somatic mutation landscape.
Results
A previous report on two CTVT tumors leveraged canine dbSNP to identify polymorphic alleles inherited by the CTVT founder (Murchison et al. 2014); however, canine dbSNP only accounts for an average of 32.65% of the germline SNVs found in whole-genome sequencing (WGS) of diverse canids (Fig. 1A). Therefore, published canine polymorphisms are not sufficient for identification of the CTVT founder's inherited alleles. To overcome this limitation, we generated high-coverage WGS (mean 37.9×) for 51 dogs from closed breeding populations and jointly genotyped these with 135 publicly available canine genomes, thereby creating the largest current catalog of genome-wide canine variation representing 186 diverse canids (Supplemental Table S1). Since we also wanted to exclude recurrent systematic sequencing or genotyping errors from downstream analysis of somatic mutations, variant positions that were identified by GATK HaplotypeCaller, but did not pass variant quality score recalibration, were retained for use as a systematic error filter. Together with canine dbSNP (Sherry 2001) and a recently published variation survey (Axelsson et al. 2013), our canine Variation and Systematic Error Catalog (VSEC) consisted of 28.01 million single-nucleotide variants (SNVs), 12.62 million indels, and 31,613 structural variants (SVs) (Supplemental Databases S1–S3). Whereas canine dbSNP contains only SNVs and includes less than one-third of the variants found in the average canid whole-genome sequence, a mean of 99.55% of SNVs, 99.57% of indels, and 95.63% of SVs from any single canid was present in at least one other individual in our newly developed, WGS-derived VSEC catalog (Fig. 1B–D).
Figure 1.

Founder-inherited versus somatic variation in CTVT. (A) Fraction of SNVs from 186 sequenced canids found in canine dbSNP (mean 32.65%). Sequenced canids included the two host dogs, 64 dogs representing 40 modern breeds, 27 dogs representing 12 ancient breeds, 86 outbred canids spanning four continents, and nine wild canids. (B–D) Fraction of SNVs, indels, and SVs found in a single canid that are present in at least one other canid in the panel (means of 99.55%, 99.57%, and 95.63%, respectively). (E) High-quality variants found in both CTVT tumors were compared against our Variant and Systematic Error Catalog (VSEC), which was generated from 186 canid whole-genome sequences. Consensus CTVT variants present in the VSEC catalog were presumed to be alleles inherited by the founder canid. Novel variants were treated as somatic mutations, some of which are crucial to clonal transmissibility. Dog images are from the American Kennel Club (http://www.akc.org).
Due to the unique natural history of CTVT, variants shared by two or more tumors were present in the most recent common ancestor tumor. To leverage this identity by descent phenomenon, we compared high-quality variants shared by the two published CTVT sequences (Murchison et al. 2014) against the VSEC catalog (Fig. 1E). CTVT variants contained in the VSEC catalog were presumed to be the founder's inherited alleles, whereas novel variants were classified as candidate somatic mutations (Kumar et al. 2011). Supplemental Table S2 provides high- and moderate-impact candidate somatic mutations. By these criteria, 75.6% of SNVs, 92.1% of indels, and 17.6% of SVs originated in the founder's germline, whereas the remainder were somatically acquired (Table 1; Supplemental Table S3). These candidate somatic variants were dramatically different from germline alleles but consistent with human tumor mutations for metrics, including transition–transversion ratio (Ti/Tv), nonsynonymous–synonymous ratio (N/S), and conservation at mutated positions (phastCons) (Fig. 2A–C).
Table 1.
Panel of 186 canid genome sequences and functional annotations efficiently highlight high-priority somatic mutations
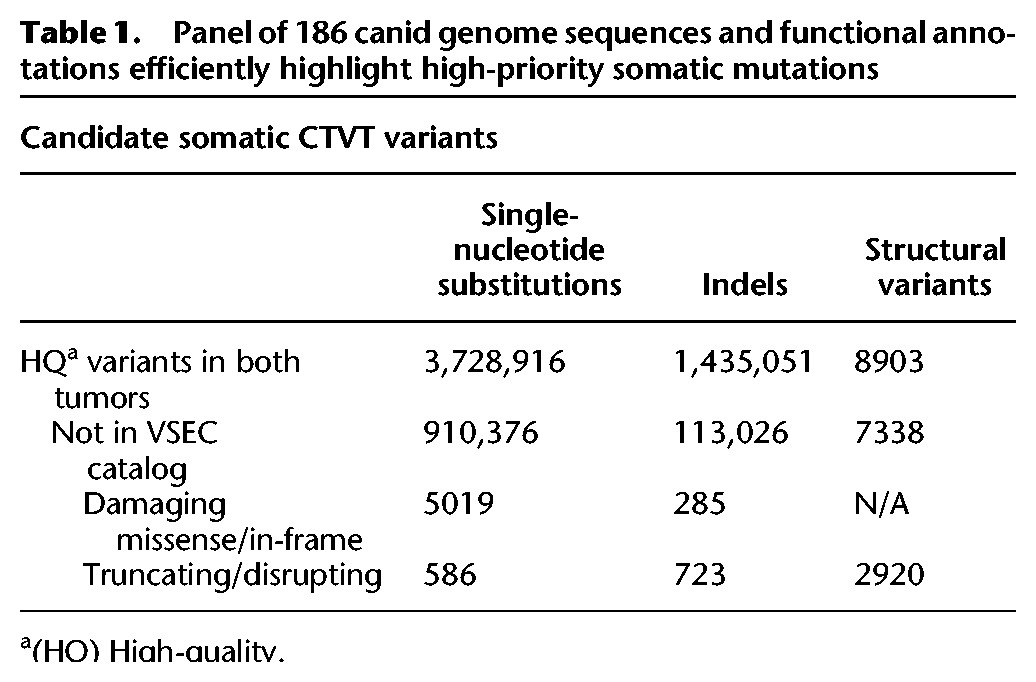
Figure 2.

Candidate somatic mutations are highly enriched for true somatic mutations. (A–C) The transition–transversion (Ti:Tv) ratio, nonsynonymous–synonymous (N:S) ratio, and four-way PhastCons score at missense substitution sites for CTVT somatic variants were statistically significant outliers (two-sided Grubb's test, critical value of Z for 188 observations = 3.59) compared to both the canid WGS panel and the CTVT founder's inherited alleles. (D–F) Substitution profiles in their trinucleotide sequence context for germline variation averaged across the 186 canids (D), the CTVT founder's putative inherited alleles (E), and the candidate CTVT somatic mutations (F). Germline variants have more C>T and T>C variants than other substitutions, but no dramatic overrepresentation of surrounding sequence contexts. In contrast, the CTVT candidate somatic mutations are enriched for C>T transitions, especially TCC>TTC (15.9%) and CCC>CTC (6.0%). Notably, the enrichment of T>C substitutions is also absent from the CTVT candidate somatic mutations. For all three plots, the base substitutions are indicated by color and displayed relative to the reference pyrimidine base (i.e., C>A and G>T transversions are shown together). Each bar conveys the fraction of variants for which the indicated substitution occurs in the context of the adjacent bases in the reference sequence. For example, the leftmost bar represents the percentage of mutations that are ACA>AAA transversions.
We also investigated substitution signatures within their trinucleotide sequence context. The putative founder-inherited alleles were indistinguishable from germline variants in modern canids, with C>T and T>C substitutions occurring somewhat more frequently than other variants (Fig. 2D,E). The candidate somatic mutations had a strikingly different signature that did not include overrepresentation of T>C variants, but instead showed a dramatic enrichment for TCC>TTC and CCC>CTC substitutions. As previously observed (Murchison et al. 2014), we found that dinucleotide substitutions were dominated by CC>TT and GG>AA substitutions (Supplemental Fig. S1). Both the trinucleotide and dinucleotide substitution patterns observed in CTVT also occur in human melanomas and have been linked to UV radiation exposure (Alexandrov et al. 2013).
Examination of gene-specific N/S highlighted 16 genes with strong enrichment for somatic nonsynonymous substitutions (Z > 3.0) when compared to high coverage individuals within the VSEC (Supplemental Table S4). These genes are involved in cell adhesion (CHL1), extracellular cohesion (COL11A1), cytoskeletal structure and function (CDC42BPA, MACF1), cell membrane structure (HSPG2), caspase and apoptotic signal transduction (CARD6, MADD), and chromatin organization/histone modification (NIPBL, ASH1L, KMT2A), as well as genes expressed almost exclusively in specific tissues such as testis (RNF17) or muscle (UCP3). Each gene contained at least one mutation with the most severe SIFT (Ng and Henikoff 2001) tolerance score (<0.05), suggesting the imposition of a significant molecular burden during tumor evolution.
Previous analysis of these tumors identified more than 3 million candidate somatic SNVs (Murchison et al. 2014), which even after adjustment for missed germline variants overestimated the tumor-specific changes due to misclassification of the founder's inherited alleles as somatic changes. Filtering with the VSEC catalog in place of dbSNP, we found that CTVT tumors shared 910,376 high-confidence somatic substitutions, an order of magnitude more than even the most mutated human cancers, but 66% fewer than the previous analysis of these tumors (Alexandrov et al. 2013; Murchison et al. 2014). Functional annotation highlighted 586 high-confidence truncating SNVs, 723 frameshift indels, and 2920 SVs that span at least one exon (Table 1), which together affect 2247 genes (Supplemental Table S2). Due to this very large number of likely damaging somatic mutations, we used Gene Set Enrichment Analysis (Subramanian et al. 2005) to identify pathways with the greatest enrichment for high-impact somatic disruptions. For genes affected by somatic truncating SNVs, frameshift indels, or exon-spanning SVs, the most enriched pathway was the “Reactome immune system” with a reported P-value of 2.27 × 10−25 (Supplemental Table S5).
Looking more closely at immune-related pathways, we observed candidate somatic mutations spanning all aspects of somatic cell participation in immunosurveillance (Supplemental Table S6). This included candidates in the self-antigen presentation pathway, such as ERAP1, which trims peptides for binding in antigen presentation molecules (Saric et al. 2002; York et al. 2002), and each component of the transporter associated with antigen processing (TAP1, TAP2, TAPBP), which facilitates peptide localization and loading into self-antigen presentation molecules (Supplemental Table S2; Spies et al. 1992). Candidate somatic mutations were also enriched among genes involved in the initiation and execution of apoptosis (Fig. 3A) and maintenance of genome integrity (Supplemental Table S2).
Figure 3.

Somatic mutations shared by both CTVT tumors lend insight into the biology of clonal transmissibility. (A) Somatic mutations impact all aspects of pathways involved in immunosurveillance. (B) Seven hundred twenty-eight chromosome-to-chromosome translocations were shared by both tumors. (C) Recent LOH events are characterized by complete homozygosity of both alleles inherited by the CTVT founder and somatic SNVs, as in this example on Chromosome 6. (D) Older LOH events have elevated inherited SNV homozygosity, but low somatic mutation homozygosity, since SNVs that predate the LOH event are homozygous, but their relative contribution has been diminished by subsequent heterozygous mutations, as observed on Chromosome 7. Plots for all chromosomes can be found in Supplemental Figure S2.
We also used the CTVT and canid genomes to investigate the somatic genomic architecture of CTVT at base-pair resolution. CTVT tumors worldwide share a highly rearranged, aneuploid genome with 57–59 chromosomes compared to 78 in normal canids, although copy number analysis and chromosome painting indicate that much of the genome content is preserved (Thomas et al. 2009; Murchison et al. 2014). Our base pair resolution analyses uncovered the specific manifestation of these structural rearrangements, including 7338 deletions, duplications, inversions, and translocations that underlie the unusual shared karyotype of CTVT tumors (Fig. 3B). In addition, we also found evidence for at least 247 potential gene fusions that may contribute to loss-of-function and aberrant gene action (Supplemental Table S7).
We leveraged the unbalanced nature of somatic loss-of-heterozygosity (LOH) events to establish a putative chronology for a subset of the somatic mutations. The regional homozygosity fraction (HF; regional homozygous variants/regional total variants) for both somatic mutations and founder-inherited alleles is 1.0 after such a LOH event. Since the germline genetic variation from the lost copy of the chromosome cannot be replaced, the HF of founder-inherited alleles must remain at approximately 1.0, irrespective of subsequent tumor evolution. In contrast, somatic HF in such regions declines over time due to accrual of additional, heterozygous post-LOH somatic mutations. This phenomenon allows the establishment of chronology for somatic mutations in LOH-affected regions—homozygous somatic mutations likely originated prior to the LOH event, whereas heterozygous mutations arose afterward. Furthermore, since somatic mutation HF declines as a function of the mutation rate and elapsed time, this metric allows estimation of the relative age of different LOH events. On this basis, we defined regions that underwent copy-neutral LOH in the distant past by identifying 1-Mb regions with >85% homozygosity among alleles inherited by the founder, but <50% homozygosity among somatic mutations (Fig. 3C,D; Supplemental Fig. S2). The full list of such potentially ancient somatic mutations can be found in Supplemental Tables S8, S9.
Leveraging our diverse sampling of existing canine variation to accurately discriminate candidate somatic mutations from putative founder-inherited variation enabled us to show the CTVT founder's relationship to contemporary breeds, as well as to pinpoint the geographic origin of this ancient canine. To place the founder's inferred, reconstructed genome in the context of contemporary variation, we constructed a maximum likelihood phylogeny using 1.3 million informative founder-inherited alleles. The observed high bootstrap support confidently places the founder as a post-domestication canid, and phylogenetic concordance was highest with contemporary Arctic spitz breeds (Fig. 4A; Supplemental Fig. S3), which agrees with the earlier SNP-chip-based analysis of these same tumors (Murchison et al. 2014). To dissect this result, we used principal components analysis to explicitly model ancestry differences among Arctic spitz breed dogs and the CTVT genotypes. For principal components 1 and 2, the CTVT tumors clustered nearest the Alaskan malamutes (Fig. 4B).
Figure 4.

Dissection of the rebuilt founder canid's genome. (A) Maximum likelihood phylogeny of canine variation catalog using putative founder-inherited variant positions with clades colored by monophyly. The position of the CTVT germline genomes indicate that the founder was a post-wolf canine with closest similarity to a contemporary Arctic spitz breed. Observed acceleration on the CTVT branch is due to the use of all positions determined to vary from reference in the CTVT germline, therefore ensuring the proportion of these variant characters exceeds all other genomes. For the unrooted maximum likelihood tree with bootstrap support, see Supplemental Figure S3. (B) We used 135,833 SNPs from the Illumina HD chip and genotypes extracted from the tumor WGS to perform principal components analysis on the CTVT tumors and dogs from three Arctic spitz breeds. The CTVT founder was likely most genetically similar to the modern Alaskan malamute.
Discussion
We created the largest existing catalog of canine genetic variation and compared it to two CTVT tumors, thereby isolating CTVT's somatic changes from the genetic variation that was present in the founder canid. As demonstrated by our comparison of one sequenced canid against all others, our VSEC catalog encompasses the vast majority of modern canine genetic variation and should therefore contain most CTVT founder-inherited alleles (Fig. 1B–D). Independent of this study, the VSEC catalog will be integral to the identification of breed-specific and shared genomic variation between breeds, the investigation of other canine cancers, as well as rare diseases. It represents the most exhaustive breadth of canine genomic sampling, combined with the deepest sequencing for individuals within closed-breeding populations of dogs.
Despite our unrivaled survey of canine genomic variation, we note that our approach misclassifies founder-inherited alleles that are not represented in our panel of modern canids, instead defining these as somatic changes acquired since tumor inception. Since the misclassified alleles amount to private variation in the founder, we estimated the magnitude of this effect using the observed level of private variation among the 186 sequenced canids. The globally ascertained wolves in our WGS panel harbored the most private variation, presumably reflecting the genetic bottleneck of domestication. If the CTVT founder had a high level of private variation equal to modern wolves, we projected that the true number of CTVT somatic variants would be ∼3.7% lower than the number of candidate somatic mutations (Supplemental Fig. S4). It remains possible that the ancient founder canid had a level of private variation that vastly outstripped the private genetic diversity of modern wolves. However, this scenario is unlikely because the Ti/Tv, N/S, and phastCons profiles of the candidate somatic mutations are divergent from the values observed during the course of evolution in other organisms (Yang and Nielsen 1998; Yang and Yoder 1999), as well as from the germline variants of all 186 sequenced canids (Fig. 2A–C). Instead, the candidate somatic mutations mirror trends observed for somatic mutations in human tumors. Furthermore, the somatic mutations exhibited a completely different substitution profile from germline alleles, including a much lower T>C substitution rate than the inherited variants, accounting for 14.6% and 33.5%, respectively (Fig. 2D–F). Somatic mutations also demonstrated a predilection for C>T substitutions and CC>TT dinucleotide substitutions (Fig. 2D–F; Supplemental Fig. S1), a signature of UV exposure in human melanomas (Alexandrov et al. 2013; Murchison et al. 2014). Together, the low rate of private variation in even the most divergent canids, significant differences in mutation metrics, and recapitulation of trends found in human tumors, but not natural evolution suggest that although residual founder-inherited alleles are present, they are uncommon among the candidate somatic mutations. Based on these lines of evidence and paired with the fact that the protracted natural history of CTVT implies an elevated somatic mutation burden, we inferred that variants not found in other canids are highly enriched for somatic mutations. Accordingly, these variants were evaluated for prospective contributions to clonal transmissibility.
Our analysis revealed candidate somatic mutations disrupting every step of the adaptive immune safeguards that typically detect and destroy allografted cells (Fig. 3A; Supplemental Table S6). Normally, T-cells scrutinize cytosolic peptide fragments presented in the context of self-antigen presentation molecules. CTVT mutations block generation of antigenic peptides, prevent their transport into the endoplasmic reticulum, and inhibit their loading into self-antigen presentation molecules. In addition to dozens of somatic mutations affecting ubiquitination and protein cleavage, CTVT has a deleterious missense substitution in ERAP1, which trims peptide fragments for ideal fit in antigen presentation molecules (Saric et al. 2002; York et al. 2002). Truncating or predicted deleterious mutation candidates affect all three components of the transporter associated with antigen processing (TAP1, TAP2, TAPBP), which imports cytosolic protein fragments into the endoplasmic reticulum and facilitates antigen presentation molecule loading (Supplemental Table S2; Spies et al. 1992). This collection of mutations limits antigenic peptide fragment availability (Van Kaer et al. 1992; Saric et al. 2002; York et al. 2002); and together, these overlapping somatic mutations likely underlie the observation that CTVT has low surface MHC class I (Murgia et al. 2006) and prevent host T-cells from initiating an immune response.
In addition to preventing recognition by the host, CTVT also likely disrupts immune rejection via somatic mutations in numerous initiators and executors of apoptosis (Fig. 3A). Among other mutations (Supplemental Table S6), both tumors harbor predicted deleterious mutations in CFLAR, IGF2R, DAPK1, DAPK2, FADD, TNFRSF1A, TRADD, and TRAF2, genes that inhibit the ability of T and NK cells to induce apoptosis via either cytolytic granules or death receptors. For example, IGF2R can facilitate the entry of granzyme into cells, while FADD recruits and cleaves CASP8 upon activation of the FAS or TNF receptors (Imai et al. 1999; Motyka et al. 2000). Apoptotic pathways converge at caspase 3, an executioner protease that activates DNA- and mitochondria-damaging death substrates (Darmon et al. 1995; Enari et al. 1996). In CTVT, a homozygous Chromosome 16 to 9 translocation in the second intron of CASP3 dislodges the gene's transcription start site and 5′ UTR, likely preventing expression of this key effector of apoptosis. The tumor also has mutations in many other genes that modulate the balance of pro- and anti-apoptotic signaling (Fig. 3A).
Broadly, we conclude that CTVT avoids immune-mediated destruction via a combination of mechanisms used by other malignancies. For example, allografted murine tumors with low self-antigen presentation due to TAP1 suppression do not elicit rejection in immunocompetent mice (Shankaran et al. 2001), and the same strategy is deployed via epigenetic means in Tasmanian devil facial tumor (Siddle et al. 2013). In addition, most cancers disrupt the balance between survival and apoptotic signaling. Although many tumors use a small subset of the immune evasion repertoire apparently at work in CTVT, this tumor is remarkable for the redundant, comprehensive combination of mutations that continues to facilitate its unparalleled horizontal transmissibility. These functionally overlapping mutations are likely the manifestation of the thousands of years and unique evolutionary pressures that have molded this transmissible allograft.
Through careful analysis of the numerous read mapping inconsistencies between CTVT and the CanFam3 genome within the context of contemporary variation, our work specifically identifies the location and nature of genome-wide somatic structural aberrations (Supplemental Table S2). Somatic mutations in DNA repair and genome stability genes likely contributed to the observed genomic disarray in CTVT, as we found truncating or predicted damaging changes in ATM, BRCA1, BRCA2, MRE11A, MLH1, PMS1, RAD21, and TP53 (Supplemental Table S2). This instability likely leads to the previously reported copy-neutral somatic loss LOH events (Thomas et al. 2009; Murchison et al. 2014), which result from single-copy loss of a chromosome segment and concomitant duplication of the other copy. This phenomenon initially causes all inherited and acquired variants in the affected region to become homozygous. The unbalanced nature of some structural rearrangements enabled us to identify some early somatic mutations in genes essential to proper cellular function, such as TP53, CASP3, TAP2, and CDKN2A/B, which have well-described roles in oncogenesis and immune evasion (Supplemental Tables S8, S9). Although the homozygous deletion of CDKN2A/B was one of the four putative somatic drivers of CTVT identified in previous analysis of these tumors (Murchison et al. 2014), our ability to specify additional mutations (Supplemental Tables S8, S9) and place the timing of these changes during early tumor evolution underscores the major role that these additional genetic alterations may have played in oncogenesis. These and other early mutations must have enabled the founder tumor to rapidly proliferate and escape detection by new hosts, initiating the transition from ancient tumor to infectious allograft.
In light of our discovery that CTVT has multiple somatic mutations in DNA repair genes including MLH1, a mismatch repair mediator associated with somatic hypermutation (Zhao et al. 2014), modeling CTVT's mutation rate is problematic using only two genomes. Nevertheless, our significantly reduced number of putative somatic SNVs, paired with an increased mutation rate commensurate with multiple DNA repair defects and UV radiation exposure, implies a much more recent origin for CTVT than the previous estimate of 11,000 yr (Murchison et al. 2014).
Even given CTVT's origin within a more recent time frame, this allograft represents a unique organism that defies current taxonomic classification. Originating as a canine cell, the ameiotic acquisition of mutations via Muller's ratchet (Muller 1964) promoted accumulation of some mutations deleterious to normal mammalian cells and others advantageous to self-propagation via inter-organismal transmissibility. In the process, the tumor was transformed from a multicellular cancer to a loosely organized unicellular colony. As an infectious allograft, it has adopted strategies similar to intercellular obligate parasites (Wijayawardena et al. 2013), even subsuming mitochondria from its host (Rebbeck et al. 2011). In spite of these many mechanisms at work, our phylogenetic inference firmly placed the founder canine after the emergence of Canis lupus familiaris (Fig. 4A; Supplemental Fig. S3), and further investigation pointed to the Alaskan malamute as the closest modern canid to the CTVT founder (Fig. 4B).
Though CTVT's unusual life history complicates dissection of its genome, our survey of variation in modern canids facilitated accurate enrichment of somatic mutations. Our comprehensive catalog can be readily utilized to investigate the genomic characteristics underlying other manifestations of canine cancer, as well as rare disease phenotypes. By leveraging it against two CTVT tumor genomes, we reveal critical and early somatic mutations that likely contributed to the rise of clonal transmissibility. Furthermore, key survival strategies manifest as redundant, high-impact somatic mutations spanning immunosurveillance pathways. These observations exemplify the myriad adaptations that transformed a single canine tumor into a parasitic, globally distributed, clonally transmissible cell lineage. After millennia of molding in this crucible of selection, CTVT has evolved into an unrivaled laboratory for studying the tumor–host arms race, as well as immunomodulating therapeutics that unbalance this struggle.
Methods
Canine genome sequencing and data aggregation
Whole-genome sequences were generated for 51 individuals belonging to closed-breed populations using the Illumina TruSeq DNA PCR-Free Protocol (Cat.# FC-121-3001). Libraries were constructed with fragments of 300–500 bp and sequenced on the Illumina HiSeq 2000 platform using 100-bp paired-end parameters. To fully explore the documented variation in canines, sequences for all canine genomes available at the onset of this study that were sequenced on a contemporary Illumina platform were also obtained via BioProject accessions from the Sequence Read Archive or the European Nucleotide Archive (Supplemental Table S1).
SNV and indel identification and filtering
To construct the genome for each individual, paired-end sequences were aligned to the CanFam3 reference genome using the BWA 0.7.10 MEM algorithm (Li and Durbin 2009) and sorted with SAMtools 0.1.19 (Li et al. 2009). Putative PCR duplicate reads were annotated with PicardTools 1.119 (https://github.com/broadinstitute/picard) and not used for subsequent variant or copy number detection. Local realignment around documented and novel insertion–deletion events was performed using published indel data (Axelsson et al. 2013) and base quality recalibration using dbSNP and positions from the Illumina Canine HD chip data as training sets with GATK 3.2-2 (DePristo et al. 2011). Putative SNVs were identified in each sample and genotyped across all 188 samples simultaneously using GATK HaplotypeCaller in GVCF mode (Van der Auwera et al. 2013). Variant quality score recalibration (VQSR) was executed separately for SNVs and indels using GATK best practices recommendations with the same training sets as implemented in the indel realignment and base quality recalibration steps. We identified 28.01 million raw SNV positions and 12.62 million raw indel positions, and all that were not private to the CTVT genome were retained for use in the variation and systematic error catalog (Supplemental Databases S1–S2). For downstream variant analysis of individual genomes, we retained only those that passed the 99.0 tranche for VQSR, with VQSlod ≤0.0594 for SNVs and VQSlod ≤9.7328 for indels. The VQSR status for SNV and indel positions is indicated in Supplemental Databases S1–S2.
We then determined how many variants from one canid were found in at least one other individual in the panel. For each canid, we used VCFtools (Danecek et al. 2011) to extract variants that passed VQSR and had a genotype quality greater than 20, and these variants were then filtered against a list of positions with a variant of the same type of any quality in any other canid. We allowed indel start positions to differ by ±1 bp to account for the decreased precision of indel localization. After establishing that the vast majority of variants present in one canid can be found in at least one other in our diverse panel (Fig. 1B–D), we applied the same approach to the two CTVT tumors.
The genome sequence of CTVT represents a complex mixture of entities: contamination from the canine host, systematic errors, alleles inherited by the founder, lineage-specific somatic mutations, and earlier somatic mutations that must be the essential drivers of clonal transmissibility. Since CTVT is most remarkable for its persistence as a transmissible somatic cell lineage, our variant filtering approach was designed to enrich for somatic mutations shared by both tumors. Accordingly, we aggressively sought to remove both systematic errors and polymorphic alleles inherited by the founder. Among putative somatic mutations, we also excluded lineage-specific variants and those with lower quality in one or both tumors. To accomplish these goals, we used the quality and depth thresholds described above to identify high-confidence variants shared by both tumors. These variants were filtered against the type-specific catalogs of positions with variants of any quality in any of the 186 modern canids (Supplemental Databases S1–S3). Shared CTVT variants that are in our VSEC database were likely inherited by the founder, though this set also contains host contamination and systematic false positives. Novel variants were evaluated as somatic mutations, although these also contain variants inherited by the ancient founder but not present in our panel of modern canids. Putative somatic mutations within repetitive elements were also removed (http://hgdownload.cse.ucsc.edu/goldenPath/canFam3/database/rmsk.txt.gz). Remaining somatic mutations were annotated with the Variant Effect Predictor (McLaren et al. 2010) and evaluated for potential contributions to clonal transmissibility.
Genome-wide and gene-specific substitution ratios
Utilizing the variant effect prediction annotations for missense and nonsense (nonsynonymous [N]), as well as synonymous (S) mutations, genome-wide and gene-specific totals for N and S were tallied. The N/S ratio was calculated for each protein-coding gene using the Ensembl v79 gene models (Flicek et al. 2014) for both putative somatic and founder-inherited variants as well as a panel of 20 high coverage canids (Supplemental Table S4; Nei and Gojobori 1986). Average values across the canid panel were compared to those of putative somatic variants from CTVT to highlight genes with enrichment for nonsynonymous changes as well as severely deleterious SIFT (Ng and Henikoff 2001) scores (<0.05), indicating untolerated and repeated loss of normal function (Ostrow et al. 2014).
Structural variant identification and filtering
DELLY v0.5.5 was used to detect deletions (DEL), tandem duplications (DUP), inversions (INV), and translocations (TRA) in CTVT tumors and the normal canid genomes (Rausch et al. 2012). Raw DELLY calls were filtered for size <1 kb or/and when either SV breakpoint fell within 100 kb from the start or end of the canine chromosome. Candidate SVs were merged into a single event when both breakpoints were ±200 bp for INV, DEL, DUP, and ±500 bp for TRA. SVs were also filtered if SR < 1 and PE < 5 or PE < 1 and SR < 5. We identified SVs found in both CTVT tumors and then filtered these against all SVs present in any of the canid genomes.
For somatic TRA in CTVT, we identified SVs in tumor 79T and imposed quality filters and then subtracted against all variants present in any of the canid genomes. Since we sought TRA shared by both tumors, we extracted reads within 1 kb of the 79T TRA breakpoints for both 24T, merged these reads with 50 million properly mapped read pairs, and used the procedures described above.
We used the depth-based copy number algorithm CNVnator v0.3 to identify genomic regions with homozygous deletions or duplications in the CTVT tumor genomes (Abyzov et al. 2011). After CNVs were called with a bin size of 400 bp, we removed low-confidence calls by filtering events with (1) E-value > 0.01; (2) >50% of reads with mapping quality zero; or (3) gap in the reference sequence constituting >35% of the region. We retained deletions with a normalized read depth (RD) ≤0.6 for the control genomes and ≤0.25 for the union of the CTVT tumors. For duplications, we kept those with a normalized RD ≥1.4 for the control genomes and ≥2 for the union of the CTVT tumors. Finally, we filtered the union of the CTVT tumor CNVs against all CNVs present in any of the canid control genomes. Chromosome-to-chromosome translocation events were plotted in Circos (Krzywinski et al. 2009).
Gene set enrichment analysis
Among the somatic mutation candidates, all stop-gained, stop-lost, frameshift, and SVs that disrupted an exon were evaluated with the online version of Gene Set Enrichment Analysis (http://www.broadinstitute.org/gsea/msigdb/annotate.jsp). Somatic mutation candidates were compared against the “canonical pathways” gene sets, and a stringent false discovery rate of 0.0001 was specified. The top 100 most significantly mutated pathways for these protein-altering variants and disruptive SVs can be found in Supplemental Table S5. The most enriched pathway was the “Reactome Immune System,” and this pathway was further investigated.
Identification of early somatic mutations
CTVT genotypes were assigned to founder-inherited alleles and somatic mutations. All positions where either host canid had variant alleles were excluded to avoid conflation of host contamination with CTVT founder-inherited alleles. Genotype assignments were corrected for host contamination using variant allele fraction (VAF), defined as alternate allele depth divided by total depth. Based on the observed VAF distribution for founder-inherited alleles (Supplemental Fig. S5), SNVs with VAF between 0.25 and 0.55 were called heterozygous, whereas variants with VAF > 0.65 were called homozygous. Variants outside these ranges could not be reliably assigned to either state, and therefore were not included in this analysis.
Regional homozygosity fraction (HF) (number of homozygous alternate variants divided by total variants) was calculated independently for founder-inherited alleles and somatic mutations in 1-Mb sliding windows with 250-kb step size across the genome. LOH regions were defined as intervals with germline HF > 0.85, and the subset of these regions with somatic HF < 0.50 were designated as ancient LOH events on the basis that half of the variants arose after the LOH event. Variants with depth less than 10 were excluded from the sliding window analysis. Homozygous SNVs and indels with depth greater than 20, as well as all SVs in likely ancient LOH regions, were identified and evaluated. The mean depth at the remaining positions was 105.0 and 45.6 for CTVT 24T and CTVT 79T, respectively.
Maximum likelihood phylogeny
To place the germline-specific variants into an evolutionary context within our catalog of variation, we extracted putative founder-inherited alleles at the 1,380,310 SNV positions where the host dogs had no evidence for variation. We also included two wild canids, golden jackal and Andean fox. Homozygotes were coded by their respective nucleotide, and heterozygotes by the IUPAC code for the respective 2-base combination. Calls falling below the VQSlod threshold were coded as unknown “N.” Positions determined to be invariant were excluded. The resulting multialignment was partitioned by chromosome and evaluated for an appropriate nucleotide substitution model in 10 kb subpartitions using ModelTest (Posada 2006). Each intrachromosomal subpartition with an identical substitution model was concatenated, and the final data converted to Phylip format with Perl. Using the phylogentic supercomputing resource CIPRES, a comprehensive maximum likelihood analysis was executed using ExaML for 10 regular treespace searches and RAxML for 150 rapid bootstrap pseudoreplicates per search (Stamatakis and Aberer 2013; Stamatakis 2014). AutoMRE was used to determine bootstrapping significance, and ascertainment bias correction was implemented to account for the absence of invariant sites. For regular searches, RAxML was used to generate a starting tree using maximum parsimony from a random number seed and ExaML for the search. For bootstrapping, 160 computer cores were used per run of RAxML. Phylogenetic tree was rerooted to the wild-canid clade (Golden Jackal), and monophyletic groups were color coded to indicate unique bipartitions.
Principal component analysis
DNA samples from 10 Siberian Huskies and 10 Alaskan Malamutes were genotyped using the Illumina CanineHD BeadChip following standard protocols (Illumina). Genotypes from 10 Greenland Sledge Dogs were obtained from previously published data (Vaysse et al. 2011). A total of 135,833 SNPs were retained after comparison to the CTVT tumor sequence. All three data sources were combined and analyzed using EIGENSTRAT (Price et al. 2006). Significance of each PC was evaluated using Tracy-Widom statistics (Patterson et al. 2006).
Data access
Sequencing data for each canid and CTVT genome have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra/) under BioProject accession numbers PRJEB7734–PRJEB7736 and PRJNA288568. The genotype data for 10 Alaskan malamutes and 10 Siberian huskies have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE70454. Germline structural variants from the panel of diverse canids and somatic variants from the tumors have been deposited in NCBI's database of genomic structural variation (dbVar; http://www.ncbi.nlm.nih.gov/dbvar) under accession number nstd115 (ftp://ftp.ncbi.nlm.nih.gov/pub/dbVar/data/Canis_lupus/by_study/nstd115_Decker_ et_al_2015/). Somatic and germline SNVs and indels for CTVT have been integrated into the Canine Annotation track hub at the UCSC Genome Browser (https://genome.ucsc.edu) under “CTVT Variation.”
Supplementary Material
Acknowledgments
We thank the dog owners for donating blood samples to research. We thank Adam Boyko for additional information about published canid genomes. We also thank the National Intramural Sequencing Program, the Next Generation Sequencing Platform of the University of Bern, Wayne Pfeiffer at the San Diego Supercomputer Center with support from NSF, and the Biowulf Linux cluster at the National Institutes of Health. This work was supported by the Intramural Program of the National Human Genome Research Institute (E.A.O., B.D., B.W.D., M.R., E.K., D.M.K., H.G.P., E.S.C.), the Intramural Program of the National Cancer Institute (A.H.L.), the National Institutes of Health–Cambridge Scholars Program (B.D.), and UC2CA148149 from the National Cancer Institute (M.J.H., J.M.T., R.R., J.J.C., awarded to J.M.T.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.190314.115.
Freely available online through the Genome Research Open Access option.
References
- Abyzov A, Urban AE, Snyder M, Gerstein M. 2011. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res 21: 974–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, et al. 2013. Signatures of mutational processes in human cancer. Nature 500: 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelsson E, Ratnakumar A, Arendt ML, Maqbool K, Webster MT, Perloski M, Liberg O, Arnemo JM, Hedhammar A, Lindblad-Toh K. 2013. The genomic signature of dog domestication reveals adaptation to a starch-rich diet. Nature 495: 360–364. [DOI] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, et al. 2011. The variant call format and VCFtools. Bioinformatics 27: 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darmon AJ, Nicholson DW, Bleackley RC. 1995. Activation of the apoptotic protease CPP32 by cytotoxic T-cell-derived granzyme B. Nature 377: 446–448. [DOI] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43: 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enari M, Talanian RV, Wong WW, Nagata S. 1996. Sequential activation of ICE-like and CPP32-like proteases during Fas-mediated apoptosis. Nature 380: 723–726. [DOI] [PubMed] [Google Scholar]
- Flicek P, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fitzgerald S, et al. 2014. Ensembl 2014. Nucleic Acids Res 42: D749–D755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y, Kimura T, Murakami A, Yajima N, Sakamaki K, Yonehara S. 1999. The CED-4-homologous protein FLASH is involved in Fas-mediated activation of caspase-8 during apoptosis. Nature 398: 777–785. [DOI] [PubMed] [Google Scholar]
- Katzir N, Arman E, Cohen D, Givol D, Rechavi G. 1987. Common origin of transmissible venereal tumors (TVT) in dogs. Oncogene 1: 445–448. [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. 2009. Circos: an information aesthetic for comparative genomics. Genome Res 19: 1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, White TA, MacKenzie AP, Clegg N, Lee C, Dumpit RF, Coleman I, Ng SB, Salipante SJ, Rieder MJ, et al. 2011. Exome sequencing identifies a spectrum of mutation frequencies in advanced and lethal prostate cancers. Proc Natl Acad Sci 108: 17087–17092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. 2010. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics 26: 2069–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith RW, Janečka JE, Gatesy J, Ryder OA, Fisher CA, Teeling EC, Goodbla A, Eizirik E, Simão TLL, Stadler T, et al. 2011. Impacts of the Cretaceous Terrestrial Revolution and KPg extinction on mammal diversification. Science 334: 521–524. [DOI] [PubMed] [Google Scholar]
- Motyka B, Korbutt G, Pinkoski MJ, Heibein JA, Caputo A, Hobman M, Barry M, Shostak I, Sawchuk T, Holmes CF, et al. 2000. Mannose 6-phosphate/insulin-like growth factor II receptor is a death receptor for granzyme B during cytotoxic T cell–induced apoptosis. Cell 103: 491–500. [DOI] [PubMed] [Google Scholar]
- Muller HJ. 1964. The relation of recombination to mutational advance. Mutat Res 106: 2–9. [DOI] [PubMed] [Google Scholar]
- Murchison EP, Wedge DC, Alexandrov LB, Fu B, Martincorena I, Ning Z, Tubio JMC, Werner EI, Allen J, De Nardi AB, et al. 2014. Transmissible dog cancer genome reveals the origin and history of an ancient cell lineage. Science 343: 437–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murgia C, Pritchard JK, Kim SY, Fassati A, Weiss RA. 2006. Clonal origin and evolution of a transmissible cancer. Cell 126: 477–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Gojobori T. 1986. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol 3: 418–426. [DOI] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. 2001. Predicting deleterious amino acid substitutions. Genome Res 11: 863–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrow SL, Barshir R, DeGregori J, Yeger-Lotem E, Hershberg R. 2014. Cancer evolution is associated with pervasive positive selection on globally expressed genes. PLoS Genet 10: e1004239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson N, Price AL, Reich D. 2006. Population structure and eigenanalysis. PLoS Genet 2: e190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posada D. 2006. ModelTest Server: a web-based tool for the statistical selection of models of nucleotide substitution online. Nucleic Acids Res 34: W700–W703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. 2006. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38: 904–909. [DOI] [PubMed] [Google Scholar]
- Rausch T, Zichner T, Schlattl A, Stütz AM, Benes V, Korbel JO. 2012. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28: i333–i339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebbeck CA, Leroi AM, Burt A. 2011. Mitochondrial capture by a transmissible cancer. Science 331: 303. [DOI] [PubMed] [Google Scholar]
- Saric T, Chang SC, Hattori A, York IA, Markant S, Rock KL, Tsujimoto M, Goldberg AL. 2002. An IFN-γ–induced aminopeptidase in the ER, ERAP1, trims precursors to MHC class I–presented peptides. Nat Immunol 3: 1169–1176. [DOI] [PubMed] [Google Scholar]
- Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, Schreiber RD. 2001. IFNγ and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 410: 1107–1111. [DOI] [PubMed] [Google Scholar]
- Sherry ST. 2001. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29: 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddle HV, Kreiss A, Tovar C, Yuen CK, Cheng Y, Belov K, Swift K, Pearse AM, Hamede R, Jones ME, et al. 2013. Reversible epigenetic down-regulation of MHC molecules by devil facial tumour disease illustrates immune escape by a contagious cancer. Proc Natl Acad Sci 110: 5103–5108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spies T, Cerundolo V, Colonna M, Cresswell P, Townsend A, DeMars R. 1992. Presentation of viral antigen by MHC class I molecules is dependent on a putative peptide transporter heterodimer. Nature 355: 644–646. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30: 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A, Aberer AJ. 2013. Novel parallelization schemes for large-scale likelihood-based phylogenetic inference. In 2013 IEEE 27th international symposium on parallel and distributed processing (IPDPS), pp. 1195–1204, IEEE, Piscataway, NJ. [Google Scholar]
- Strakova A, Murchison EP. 2014. The changing global distribution and prevalence of canine transmissible venereal tumour. BMC Vet Res 10: 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas R, Rebbeck C, Leroi AM, Burt A, Breen M. 2009. Extensive conservation of genomic imbalances in canine transmissible venereal tumors (CTVT) detected by microarray-based CGH analysis. Chromosome Res 17: 927–934. [DOI] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. 2013. From fastQ data to high-confidence variant cells: the Genome Analysis Toolkit Best Practices pipeline. Curr Protoc Bioinformatics 11: 11.10.1–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Kaer L, Ashton-Rickardt PG, Ploegh HL, Tonegawa S. 1992. TAP1 mutant mice are deficient in antigen presentation, surface class I molecules, and CD4−8+ T cells. Cell 71: 1205–1214. [DOI] [PubMed] [Google Scholar]
- Vaysse A, Ratnakumar A, Derrien T, Axelsson E, Rosengren Pielberg G, Sigurdsson S, Fall T, Seppälä EH, Hansen MS, Lawley CT, et al. 2011. Identification of genomic regions associated with phenotypic variation between dog breeds using selection mapping. PLoS Genet 7: e1002316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijayawardena BK, Minchella DJ, DeWoody JA. 2013. Hosts, parasites, and horizontal gene transfer. Trends Parasitol 29: 329–338. [DOI] [PubMed] [Google Scholar]
- Yang TJ. 1988. Immunobiology of a spontaneously regressive tumor, the canine transmissible venereal sarcoma (review). Anticancer Res 8: 93–95. [PubMed] [Google Scholar]
- Yang Z, Nielsen R. 1998. Synonymous and nonsynonymous rate variation in nuclear genes of mammals. J Mol Evol 46: 409–418. [DOI] [PubMed] [Google Scholar]
- Yang Z, Yoder AD. 1999. Estimation of the transition/transversion rate bias and species sampling. J Mol Evol 48: 274–283. [DOI] [PubMed] [Google Scholar]
- York IA, Chang SC, Saric T, Keys JA, Favreau JM, Goldberg AL, Rock KL. 2002. The ER aminopeptidase ERAP1 enhances or limits antigen presentation by trimming epitopes to 8–9 residues. Nat Immunol 3: 1177–1184. [DOI] [PubMed] [Google Scholar]
- Zhao H, Thienpont B, Yesilyurt BT, Moisse M, Reumers J, Coenegrachts L, Sagaert X, Schrauwen S, Smeets D, Matthijs G, et al. 2014. Mismatch repair deficiency endows tumors with a unique mutation signature and sensitivity to DNA double-strand breaks. Elife 3: e02725. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.