

Abstract
Monitoring the progress of DNA molecules through a membrane pore has been postulated as a method for sequencing DNA for several decades. Recently, a nanopore-based sequencing instrument, the Oxford Nanopore MinION, has become available, and we used this for sequencing the Saccharomyces cerevisiae genome. To make use of these data, we developed a novel open-source hybrid error correction algorithm Nanocorr specifically for Oxford Nanopore reads, because existing packages were incapable of assembling the long read lengths (5–50 kbp) at such high error rates (between ∼5% and 40% error). With this new method, we were able to perform a hybrid error correction of the nanopore reads using complementary MiSeq data and produce a de novo assembly that is highly contiguous and accurate: The contig N50 length is more than ten times greater than an Illumina-only assembly (678 kb versus 59.9 kbp) and has >99.88% consensus identity when compared to the reference. Furthermore, the assembly with the long nanopore reads presents a much more complete representation of the features of the genome and correctly assembles gene cassettes, rRNAs, transposable elements, and other genomic features that were almost entirely absent in the Illumina-only assembly.
Most DNA sequencing methods are based on either chemical cleavage of DNA molecules (Maxam and Gilbert 1977) or synthesis of new DNA strands (Sanger et al. 1977), which are used in the majority of today's sequencing routines. In the more common synthesis-based methods, base analogs of one form or another are incorporated into a nascent DNA strand that is labeled either on the primer from which it originates or on the newly incorporated bases. This is the basis of the sequencing method used for most current sequencers, including Illumina, Ion Torrent, and Pacific Biosciences (PacBio) sequencing, and their earlier predecessors (Mardis 2008). Alternatively, it has been observed that individual DNA molecules could be sequenced by monitoring their progress through various types of pores (Kasianowicz et al. 1996; Venkatesan and Bashir 2011) originally envisioned as being pores derived from bacteriophage particles (Sanger et al. 1980). The advantages of this approach include potentially very long and unbiased sequence reads, because neither amplification nor chemical reactions are necessary for sequencing (Yang et al. 2013).
Recently we began testing a sequencing device using nanopore technology from Oxford Nanopore Technologies (ONT) through their early access program (Eisenstein 2012). This device, the MinION, is a nanopore-based device in which pores are embedded in a membrane placed over an electrical detection grid. As DNA molecules pass through the pores, they create measureable alterations in the ionic current. The fluctuations are sequence dependent and thus can be used by a base-calling algorithm to infer the sequence of nucleotides in each molecule (Stoddart et al. 2009; Yang et al. 2013). As part of the library preparation protocol, a hairpin adapter is ligated to one end of a double-stranded DNA sample, while a “motor” protein is bound to the other to unwind the DNA and control the rate of nucleotides passing through the pore (Clarke et al. 2009). Under ideal conditions the leading template strand passes through the pore, followed by the hairpin adapter and then the complement strand. In such a run where both strands are sequenced, a consensus sequence of the molecule can be produced; these consensus reads are termed “2D reads” and have generally higher accuracy than reads from only a single pass of the molecule (“1D reads”).
The ability to generate very long read lengths from a handheld sequencer opens the potential for many important applications in genomics, including de novo genome assembly of novel genomes, structural variation analysis of healthy or diseased samples, or even isoform resolution when applied to cDNA sequencing. However, both the “1D” and “2D” read types currently have a high error rate that limits their direct application to these problems and necessitates a new suite of algorithms. Here we report our experiences sequencing the Saccharomyces cerevisiae (yeast) genome with the instrument, including an in-depth analysis of the data characteristics and error model. We also describe our new hybrid error correction algorithm, Nanocorr, which leverages high-quality short-read MiSeq sequencing to computationally “polish” the long nanopore reads. After error correction, we then de novo assemble the genome using just the error-corrected long reads to produce a very high-quality assembly of the genome with each chromosome assembled into a small number of contigs at very high sequence identity. We further demonstrate that our error correction is nearly optimal: Our results with the error-corrected real data approach those produced using idealized simulated reads extracted directly from the reference genome itself. Finally, we validate these results by error correcting long Oxford Nanopore reads of the E. coli K12 genome sequenced at a different institution and produce an essentially perfect de novo assembly of the genome. As such, we believe our hybrid error correction and assembly approach will be generally applicable to many other sequencing projects.
Results
Nanopore sequencing of yeast
We chose to sequence the yeast genome with the new nanopore sequencer so that we could carefully measure the accuracy and other data characteristics of the device on a tractable and well-understood genome. Our initial flow cells had somewhat low reliability and throughput but improved substantially over time (Supplemental Fig. S1). This is due to a combination of improvements in chemistry, protocols, instrument software, and shipping conditions. Some runs have produced upwards of 450 Mb of sequencing data per flow cell over a 48-h period. Altogether, we generated more than 195× coverage of the genome with an average read length of 5548 bp but with a long tail extending to a maximum read length of 191,145 bp for a “1D read” and 57,453 bp for a “2D read” (Supplemental Note 3). These reads derived from three separate iterations of the device: R6.0, the earliest version of the device, accounts for ∼11% of the data produced in this study; the R7.0 iteration of the device accounts for ∼49% of the data; and R7.3, the most recent version of this device, accounts for ∼40% of the data produced.
Alignment of the reads to the reference genome using BLAST gave us a deeper analysis of the per base error rate. Of the 361,647 reads produced by our 46 sequencing runs, 44,028 “2D” reads (or ∼56% of the “2D” reads) and 105,771 “1D” reads (about 31% of the “1D” reads) aligned to the reference yeast genome. The remaining reads either mapped to a control sequence used as a spike-in for some experiments (about 8.5% of the reads) or did not show significant similarity to the W303 genome or spike-in sequence, presumably because of insufficient read quality (Supplemental Note 4). The mean identity to the reference of “1D” reads was between 58.8% (R6.0 flow cells) and 64.60% (R7.3 flow cells), while the average “2D” read identity was between 60.96% (R6.0) and 75.39% (R7.3), with many 2D reads exceeding 80% identity (Supplemental Fig. S5A). The overall alignment identities of both 1D and 2D reads are summarized in Figure 1A, which compares both read length and percent identity. Other aligners, including LAST, were also tested and gave comparable results (Supplemental Note 9).
Figure 1.
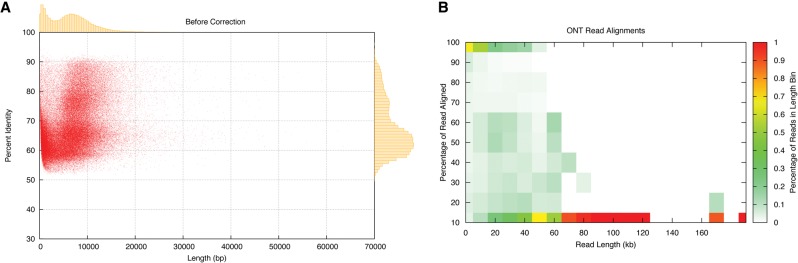
(A) Oxford Nanopore read lengths and accuracy. Scatter plot of read length versus accuracy with marginal histograms summarizing the raw ONT alignments. (B) Heatmap of Oxford Nanopore read lengths and accuracy. Each cell represents a summary of how reads of different lengths align. Each color represents the fraction of reads in a given read-length bin. Maximal alignment efficiency is observed between 10 and ∼40 kb, while fragments longer than 80 kb are virtually unalignable.
Overall read quality is further summarized in Figure 1B, which shows a heatmap of the lengths of the alignments relative to the full length of the reads. On the lower end (<50 kbp), a substantial number (up to 50%) of the reads do not align to the reference in any capacity. However, those that can be aligned have matches that span nearly their entire length. For longer reads (>50 kb), only portions of the reads can be successfully aligned, which suggests that reads are composed of both high- and low-quality segments. However, this local variability in quality does not seem to be position specific, and on average the per-base error rate is consistent across the length of a read (Supplemental Fig. S5B). The very longest reads tend not to be alignable at all, suggesting that the longest reads may be extremely low quality or include other artifacts of the sequencing process.
Evaluating a sample of the aligned reads, the overall coverage distribution approximated a Poisson distribution, although some overdispersion was observed that was better modeled by a negative binomial distribution (Supplemental Fig. S5C). To examine some of the sources of the overdispersion, we also examined the coverage as a function of the GC composition of the genome. Between 20% and 60% GC content, the coverage is essentially uniform, while at higher and lower GC content, the coverage is more variable, partially explaining why some of the regions of the genome lack raw read coverage (Supplemental Fig. S5D).
Hybrid error correction and de novo assembly
To demonstrate the utility of the long reads, we attempted to assemble the yeast genome de novo using the Celera Assembler, which can assemble low-error-rate reads up to 500 kbp long. However, when raw nanopore reads were given to the assembler, not one single contig was assembled, and it became apparent that error correction was critical to the success of the assembly. Consequently, we developed a novel algorithm called Nanocorr to error correct the reads prior to de novo assembly or other purposes. Nanocorr uses a hybrid strategy for error correction, using high-quality MiSeq short reads to error correct the long but highly erroneous nanopore reads. It follows the design of hybrid error correction pipelines for PacBio long-read sequencing (Koren et al. 2012), although in our testing none of the available algorithms were capable of utilizing the nanopore reads. For example, the HGAP error correction algorithm for PacBio reads produced 2318 reads (0.18× coverage), while the hybrid PacBio/Illumina error correction algorithm PacbioToCA produced only 167 reads (0.06×). We were therefore motivated to develop an entirely new algorithm.
Briefly, Nanocorr begins by aligning the short MiSeq reads to the long nanopore reads using the BLAST sequence aligner. This produced a mix of correct, near-full-length alignments, along with false or partial alignments of the short reads. To separate these types of alignments, Nanocorr uses a dynamic programming algorithm based on the longest-increasing-subsequence (LIS) problem to select the optimal set of short read alignments that span each long read. The consensus reads are then calculated using a finite state machine of the most commonly observed sequence transitions using the open source algorithm pbdagcon (Fig. 3A, see below; Chin et al. 2013). Overall, we found that this process increased the percent identity from an average of 67% for uncorrected reads from flow cell iterations R6.0–R7.3 to >97% (Fig. 2B; Supplemental Fig. S6A). The error-corrected long reads can be used for any purpose, especially de novo genome assembly.
Figure 3.

NG-graph of a simulated perfect read assembly, the corrected Oxford Nanopore assembly, and an Illumina-only assembly. The curve extends the common N50 metric to trace the contig size such that the top x% of the genome is assembled into contigs this size or larger. The Oxford Nanopore assembly is substantially more contiguous across the entire size spectrum and is far closer to the perfect read assembly than the Illumina-only assembly. Notably, the N50 contig length for the Oxford Nanopore-based assembly is 678 kbp compared to ∼60 kbp for the Illumina assembly and is quite close to the 811 kbp perfect read assembly N50.
Figure 2.
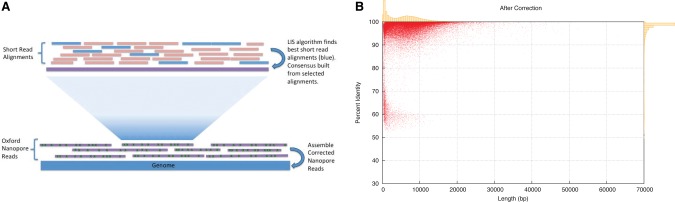
(A) Nanocorr workflow. Short high-identity reads are aligned to raw ONT reads. The best overlapping set is determined by the LIS algorithm, and a consensus sequence of these alignments is built using pbdagcon. Error-corrected reads can then be assembled using a long-read assembler. (B) Post-Nanocorr correction read length and accuracy. Scatter plot with marginal histograms summarizing the percent identity of reads after correction for W303. Average identity before correction is ∼68% for all iterations of flow cells, while the average post-correction identity was >97%.
After error correction, we selected the set of reads that were >4 kb in length from the three highest yielding flow cells (Supplemental Note 10). This brought us to our target of ∼20× coverage of the genome for de novo assembly. We then assembled those reads with the Celera Assembler, which follows an overlap-layout-consensus approach without decomposing the long reads into k-mers as is used in de Bruijn graph assemblers. This produced an assembly consisting of 108 nonredundant contigs with an N50 size of 678 kbp and requiring only a few contigs to span each chromosome (Supplemental Fig. F6B). Upon alignment to the reference sequence, we found that >99% of the reference genome aligned to our assembly and the per-base accuracy of our assembly was >99.78%. Furthermore, after polishing the assembly with the algorithm Pilon (Walker et al. 2014), the per-base identity was further improved to 99.88%. We investigated the residual differences and found that the majority of differences between the nanopore assembly and the S288C reference genome resides in repetitive regions, especially long repetitive regions and homopolymer sequences, while the accuracy of gene sequences was >99.9%. The assembly also has substantially better resolution of the genome compared to an assembly of the MiSeq reads on their own, which has a contig N50 size of only 59 kbp. The nanopore-based assembly is more than an order of magnitude more contiguous across all cutoffs in the contig length distribution (Fig. 3).
To evaluate the effectiveness of the hybrid error correction and assembly algorithm, we also computed a “reference-based” assembly of the nanopore reads by extracting sequences from the reference as “perfect reads” where the nanopore reads aligned. Interestingly, assembling these “perfect reads” leads to nearly the same results: The contig N50 was, at best, 811 kbp for the reference assembly compared to 678 kbp for the Nanocorr-corrected reads. This highlights that the remaining contig breaks in the Nanocorr assembly were due to the sequence composition and repeat structure of the genome and, to a much lesser degree, the small amount of residual error after correction (Supplemental Note 8). Finally, to evaluate the minimum amount of raw coverage needed to achieve these results, we computed 46 separate assemblies using the top N most productive flow cells. We find that the best result was achieved by using the data from just the top three flow cells, representing ∼30× raw coverage of the genome (Supplemental Note 10).
We sought to observe the differences in biological insights that could be obtained by the analysis of genome assemblies with different degrees of underlying contiguity. Aligning the Illumina and Oxford Nanopore/Illumina hybrid assemblies against the reference yeast genome allowed us to evaluate how well the two assemblies represented the various classes of annotated genomic features. While both the Illumina-only and nanopore-based assemblies could correctly assemble short genomic features, the nanopore-based assembly was able to substantially outperform the Illumina-only assembly of the longest genomic features (Fig. 4). In particular, rRNAs (averaging 1393 bp), gene cassettes (averaging 2951 bp), telomeres (averaging 4396 bp), and transposable elements (averaging 3201 bp) were substantially better represented in the nanopore assembly and nearly completely absent from the Illumina-only assembly. Only the very longest repeats in the genome, such as the 20-kbp telomeric repeats, remain unresolved in the Oxford Nanopore assembly and become fragmented in both assemblies as well as the reference-based assembly. The MiSeq assembly slightly outperforms for “binding site” features, although these are binding sites within the telomeric repeats that were not well assembled by either technology.
Figure 4.

Genomic features assembly by Oxford Nanopore and Illumina sequencing. Quantification of different annotated genomic features assembled completely by the Nanopore and Illumina/MiSeq-only assembly relative to the complete S288C reference annotation. The Nanopore-based assembly produces an assembly with many more of the longer and repetitive features assembled compared to the Illumina/MiSeq-only assembly.
E. coli K12 error correction and assembly
In order to validate the utility of this workflow, we also error corrected and de novo assembled the Oxford Nanopore reads generated by Quick et al. (2014) of E. coli K12 using the same approach (Supplemental Note 7). In this experiment, a total of 145× Oxford Nanopore read coverages of the genome was error corrected with the Nanocorr pipeline using 30× Illumina MiSeq coverage to improve the average identity to >99%. This time, only reads >7 kb in length, representing ∼28× coverage of the genome, were used in the assembly. The final result was an essentially perfect single 4.6-Mbp chromosome length contig with >99.99% identity. In contrast, the Illumina-only assembly produced an assembly with hundreds of contigs and a contig N50 size of only 176 kbp.
Discussion
The results of this study indicate that the Oxford Nanopore sequence data currently have substantial errors (∼5% to 40% error) and a high proportion of reads that completely fail to align (∼50%). This is likely due to the challenges of the signal processing the ionic current measurements (Schreiber et al. 2013) as well as the challenges inherent in any type of single-molecule sequencing. Oxford Nanopore has indicated that the pores are more than a single base in height so that the ionic signal measurements are not of individual nucleotides but of ∼5 nt at a time. Consequently, the base calling must individually recognize at least 45 = 1024 possible states of ionic current for each possible 5-mer. We also observed the potential for some bias in the signal processing and base caller, particularly for homopolymers (Supplemental Fig. S4). Despite the limitations of this early phase device, there has been notable improvement over the course of this program, and well-performing flow cells of the current iteration (R7.3 at the time this publication was written) can generate upwards of 400 Mb on a single run. Continuing this improvement of yields with future generations of the technology would obviously add considerably to the utility of the system.
While short-read sequencers in general have lower error rates and, to date, have become the standard approach of genomics, short reads are not sufficient to generate long continuous assemblies of complex genomes. To this day, the reference human genome remains incomplete as do the reference genomes for most higher species, especially plants. Long reads are necessary to span repetitive elements and other complex sequences to generate high-quality, highly contiguous assemblies. Currently, there are limited methods for generating adequately long reads. Synthetic long reads can be generated on existing short-read platforms using barcoding approaches such as those employed by Illumina's TruSeq Synthetic Long-Read approach (formerly Moleculo) and the new 10X Genomics platform; however, these approaches still rely on the existing short-read infrastructure. Alternatively, true long reads can be generated by the Pacific Bioscience System and now the Oxford Nanopore MinION.
Improving the contiguity of a genome assembly enables more detailed study of its biological content and function in every aspect. Genes will more often be correctly assembled along with their flanking sequences, enabling deeper study of regulatory elements. Longer reads will also resolve more repetitive sequences as well, especially transposable elements, high-copy genes, segmental duplications, and centromeric/telomeric repeats that are difficult to assemble with short reads. Finally, high-quality assemblies are also essential to study high-level genome structures such as the evolution and synteny of entire chromosomes across species. Even in genome resequencing, short reads can be problematic, with some (perhaps many) structural variants unresolved, obscuring the true gene content of a member of a species or obscuring clinically relevant structural variants in an affected individual (Chaisson et al. 2015).
Modern genome assemblers are not equipped to natively handle reads with error rates above a few percent. Consequently, before the Oxford Nanopore reads can be used for de novo assembly they must first be error corrected. These general strategies are helpful for other single-molecule, long-read sequences such as those from Pacific Biosciences, although existing algorithms were not capable of resolving the Oxford Nanopore errors (Lee et al. 2014; Chaisson et al. 2015). We successfully developed a new hybrid error correction approach that can improve the average per base identity of the Oxford Nanopore reads from 65% across all flow cell iterations to >97% and generates highly contiguous and complete assemblies given sufficient coverage and read lengths. Using the error-corrected data, we were able to fully reconstruct an entire microbial genome and produce a highly contiguous assembly of yeast that had many important genomic features that were almost entirely lost in the Illumina-only assembly. This work has demonstrated how single-molecule, long-read data generated by the Oxford MinION can be successfully used to compliment short-read data to create highly contiguous genome assemblies, paving the way for essentially any laboratory to create perfect or high-quality reference sequences for their microbial or small eukaryotic projects using a handheld long-read sequencer.
Methods
Yeast growth
An aliquot of yeast strain W303 was obtained from Dr. Gholson Lyon (CSHL). Four-milliliter cultures in 15-mL Falcon tubes of yeast were grown in YPD overnight at 32°C to ∼1 × 108 cells. The cells were purified using the Gentra Puregene Yeast/Bacteria kit (Qiagen). DNA was stored at −20°C for no more than 7 d prior to use.
Library preparation
Oxford Nanopore
Purified DNA was sheared to 10-kb or 20-kb fragments using a Covaris g-tube (Covaris). Four micrograms of purified DNA in 150 µL of deionized water was loaded into a g-tube and spun at 6000 rpm in an Eppendorf 5424 for 120 sec (10 kb) or 4200 rpm for 120 sec (20 kb). All DNA was further purified by adding 0.4× AMPure beads. A twisted Kim wipe was used to remove all visible traces of ethanol from the walls of the tube. The beads were allowed to air dry and DNA was eluted into 30 µL of deionized water.
R6.0 and R7.0 preparation
The DNA concentration was measured with a Qubit fluorometer and an aliquot was diluted up to 80 µL. Five microliters of CS DNA (Oxford Nanopore) was added, and the DNA was end-repaired using the NEBNext End Repair Module (NEB). The DNA was purified with AMPure beads and eluted in 25.2 µL of deionized water. DNA A-tailing was performed with the NEBNext dA-Tailing module (NEB).
Blunt/TA ligase (NEB) was added to the A-tailed library along with 10 µL of the adapter mix (ONT) and 10 µL of HP adapter (ONT). The reaction was allowed to incubate at 25°C for 15 min. The DNA was purified with 0.4× of AMPure beads. After removal of supernatant, the beads were washed 1× with 150 µL Wash Buffer (ONT). After the supernatant was removed, the beads were briefly spun down and then repelleted and the remaining supernatant was removed. A twisted Kim wipe was used to remove all traces of Wash Buffer from the wall of the tube. The DNA was resuspended in 25 µL of Elution Buffer (ONT).
The DNA was quantified using a Qubit to estimate the total ng of genomic + CS DNA in the final library. Ten microliters of tether (ONT) was added to the ligated library and allowed to incubate at room temperature for 10 min. Fifteen microliters of HP motor was then added and allowed to incubate for 30 min or overnight.
Between 5 and 250 ng of the presequencing library was diluted to 146 µL in EP Buffer (ONT), and 4 µL of Fuel Mix (ONT) was added to the sequencing mix. The library was immediately loaded onto a flow cell.
R7.3 preparation
The DNA concentration was measured with a Qubit fluorometer and an aliquot was diluted up to 80 µL. The DNA was end-repaired using the NEBNext End Repair Module (NEB). The DNA was purified with AMPure beads and eluted in 25.2 µL of deionized water. DNA A-tailing was performed with the NEBNext dA-Tailing module (NEB).
Blunt/TA ligase (NEB) was added to the A-tailed library along with 10 µL of the adapter mix (ONT) and 2 µL of HP adapter (ONT). The reaction was allowed to incubate at 25°C for 15 min. The DNA was purified with 10 µL of His-tag Dynabeads (Life Technologies) suspended in 100 µL of 2× Wash Buffer (ONT). After removal of supernatant, the beads were washed 2× with 250 µL of 1× Wash Buffer (ONT). After the supernatant was removed, the beads were briefly spun down and then repelleted and the remaining supernatant was removed. A twisted Kim wipe was used to remove all traces of Wash Buffer from the wall of the tube. The DNA was resuspended in 25 µL of Elution Buffer (ONT). The DNA was quantified using a Qubit fluorometer to estimate the total ng of genomic DNA in the final library.
Between 5 and 250 ng of the presequencing library was diluted to 146 µL in EP Buffer (ONT), and 4 µL of Fuel Mix (ONT) was added to the sequencing mix. The library was immediately loaded onto a flow cell.
Libraries were sequenced using the MinION device for between 48 and 72 h. Whenever possible, DNA was handled with a wide-bore, low-bind pipette tip. Mixing of DNA with reagents was done by flicking or preferably pipetting with a wide-bore tip. All tubes used were Protein LoBind (Eppendorf). All material loaded onto a flow cell was loaded using a 1000-µL pipette. Deviations from this protocol for each flow cell can be found in Supplemental Methods.
MiSeq
One microgram of yeast DNA purified using the Gentra Puregene Yeast/Bacteria kit (Qiagen) was prepared using a TruSeq PCR-Free kit (Illumina). The insert size was 350 bp with a paired-end 250 bp run.
Flow cell disposition
Flow cells were received on ice and immediately stored at 4°C. Ideally, within 3 d, each flow cell was QC'd with the minKnow software and the number of available pores was recorded. The flow cells with 400 available pores or more were generally considered “good” and used first. Immediately prior to library loading, the flow cell was removed from the 20°C refrigerator and flushed with 150 µL of EP Buffer (ONT). The flow cell was allowed to incubate at room temperature for 10 min, followed by a second EP flush and incubation.
For flow cells that were washed prior to the addition of an additional library, the flow cells were washed with 150 µL of Solution A (ONT) followed by a 10-min room-temperature incubation. One hundred and fifty microliters for solution B (ONT) was then added, and the flow cells were stored at 4°C until use. Prior to use, the washed flow cells were flushed with EP Buffer (ONT) as previously described.
Read alignment and error characteristics
Yield-over-time data extraction, individual flow cell statistics calculation, and FASTA/FASTQ generation were all performed using poretools (Loman and Quinlan 2014). Plots were generated using R (ggplot2) and gnuplot. Overall accuracy was calculated by aligning the raw Oxford Nanopore reads to the W303 PacBio assembly using BLAST version 2.2.30+ with the following parameters: -reward 5 -penalty -4 -gapopen 8 -gapextend 6 -task blastn -dust no -evalue 1 × 10−10.
High scoring segment pairs were filtered using the LIS algorithm and a scoring function that penalizes overlaps while maximizing alignment lengths and accuracy. Overall accuracy was calculated by averaging the percent identity of all of the filtered HSPs derived from all of the reads. Error rate over the read length was calculated by taking the HSPs from a sampling of 1000 random reads in the data set with read lengths between 9 and 10 kb. The identity was calculated for 100-bp sliding windows over the length of the alignment and averaged over all of the alignments.
Read correction and assembly
Raw reads were extracted from the h5 files generated by the base caller. Only independent reads, one per molecule, were corrected for assembly. Because a channel can produce three reads of the same molecule, reads were chosen in order of their expected accuracy: 2D or the 1D template to represent each DNA fragment. As part of the Nanocorr algorithm, 30× coverage of 300-bp paired-end MiSeq data was then aligned to the nanopore reads using BLASTN with the following parameters: -reward 5 -penalty -4 -gapopen 8 -gapextend 6 -task blastn -dust no -evalue 1 × 10−10.
Nanopore reads to which no MiSeq reads aligned were excluded from the process. The Nanocorr algorithm then filters the alignments by first removing those contained within a larger alignment, and then an LIS Dynamic Programming algorithm was applied using a scoring scheme to minimize the overlaps in the alignments. The filtered set of alignments was then used to build a consensus using ‘pbdagcon’ (Chin et al. 2013) (https://github.com/PacificBiosciences/pbdagcon.git).
The error-corrected nanopore reads were then assembled using Celera Assembler version 8.2β (http://wgs-assembler.sourceforge.net/). Redundant contigs, representing individual nanopore reads with higher rates of residual errors, were then identified using ‘blastclust’ (which is part of the BLAST executable package found at http://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE_TYPE=BlastDocs&DOC_TYPE=Download). This algorithm identifies sequences that align to the interior of another longer sequence, using the parameters: -b F -p F -e F -L 0.80 -S 60 -W 14.
Finally, the nonredundant contigs were then ‘polished’ using the Pilon algorithm that revises the consensus sequence using the alignments of the MiSeq reads to the newly assembled contigs.
Alignments and dotplots were generated using ‘nucmer’ and ‘mummerplot’ from the MUMmer version 3.23 package (Kurtz et al. 2004).
Feature quantification
Each assembly was aligned to the S288C reference genome using nucmer from the MUMmer version 3.23 package. Alignments were filtered using the command delta-filter -1, also from the MUMmer 3.23 package to find the best nonredundant set of contigs. The nonredundant set of alignments was intersected with the feature coordinates from the S288C annotation obtained from the Saccharomyces Genome Database using BEDTools (Quinlan and Hall 2010) command intersectBed with the parameters : -u –wa –f 1.0. The features that were fully contained in an alignment were included in the tally seen in Figure 4.
Data access
The sequencing data generated in this study have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) under accession number SRP055987. The assemblies have been submitted to NCBI GenBank (http://www.ncbi.nlm.nih.gov/genbank/) under accession number LIUU00000000. The version described in this paper is version LIUU01000000. The Nanocorr software is open source and available at https://github.com/jgurtowski/nanocorr and also in the Supplemental Material.
Competing interest statement
W.R.M. has participated in Illumina sponsored meetings over the past four years and has received travel reimbursement and an honorarium for presenting at these events. Illumina had no role in decisions relating to the study/work to be published, data collection, analysis of data, and the decision to publish. W.R.M. has participated in Pacific Biosciences sponsored meetings over the past three years and received travel reimbursement for presenting at these events. W.R.M. is a founder and shared holder of Orion Genomics, which focuses on plant genomics and cancer genetics. W.R.M. is an SAB member for RainDance Technologies, Inc. S.G. has participated in an Oxford Nanopore sponsored meeting in 2015 and received travel reimbursement for presenting at this event. Oxford Nanopore had no role in decisions relating to the study/work to be published, data collection, analysis of data, and the decision to publish.
Supplementary Material
Acknowledgments
This project was supported in part by National Science Foundation awards DBI-1350041, IOS-1032105; National Institutes of Health award R01-HG006677 to M.C.S.; and Cancer Center Support grant CA045508. Funding for this study was provided by a grant from T. and V. Stanley. We thank Oxford Nanopore for affording us the opportunity to participate in the MinION early access program (MAP). In particular, we thank Clive Brown, James Brayer, and all the members of the technical support staff for their support and assistance during this research. Finally, we thank all the members of the MAP community for their ongoing insight and dedication into the novel device.
Author contributions: S.G. performed data analysis, library preparation, managed flow cells, and was the MAP lead. J.G. performed data analysis, developed Nanocorr, and performed library preparation. S.E. and P.D. performed library preparation. M.C.S. assisted in data analysis and in the overall design of the project. W.R.M. developed the overall design of the study and assisted with library preparation. S.G., J.G., M.C.S., and W.R.M. wrote the manuscript. All authors reviewed and approved the final manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.191395.115.
References
- Chaisson MJ, Huddleston J, Dennis MY, Sudmant PH, Malig M, Hormozdiari F, Antonacci F, Surti U, Sandstrom R, Boitano M, et al. 2015. Resolving the complexity of the human genome using single-molecule sequencing. Nature 517: 608–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin CS, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, Clum A, Copeland A, Huddleston J, Eichler EE, et al. 2013. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods 10: 563–569. [DOI] [PubMed] [Google Scholar]
- Clarke J, Wu HC, Jayasinghe L, Patel A, Reid S, Bayley H. 2009. Continuous base identification for single-molecule nanopore DNA sequencing. Nat Nanotechnol 4: 265–270. [DOI] [PubMed] [Google Scholar]
- Eisenstein M. 2012. Oxford Nanopore announcement sets sequencing sector abuzz. Nat Biotechnol 30: 295–296. [DOI] [PubMed] [Google Scholar]
- Kasianowicz JJ, Brandin E, Branton D, Deamer DW. 1996. Characterization of individual polynucleotide molecules using a membrane channel. Proc Natl Acad Sci 93: 13770–13773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S, Schatz MC, Walenz BP, Martin J, Howard JT, Ganapathy G, Wang Z, Rasko DA, McCombie WR, Jarvis ED, et al. 2012. Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nat Biotechnol 30: 693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. 2004. Versatile and open software for comparing large genomes. Genome Biol 5: R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Gurtowski J, Yoo S, Marcus S, McCombie WR, Schatz M. 2014. Error correction and assembly complexity of single molecule sequencing reads. bioRxiv 10.1101/006395. [DOI] [Google Scholar]
- Loman NJ, Quinlan AR. 2014. Poretools: a toolkit for analyzing nanopore sequence data. Bioinformatics 30: 3399–3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardis ER. 2008. Next-generation DNA sequencing methods. Annu Rev Genomics Hum Genet 9: 387–402. [DOI] [PubMed] [Google Scholar]
- Maxam AM, Gilbert W. 1977. A new method for sequencing DNA. Proc Natl Acad Sci 74: 560–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick J, Quinlan AR, Loman NJ. 2014. A reference bacterial genome dataset generated on the MinION portable single-molecule nanopore sequencer. Gigascience 3: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger F, Nicklen S, Coulson AR. 1977. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci 74: 5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger F, Coulson AR, Barrell BG, Smith AJ, Roe BA. 1980. Cloning in single-stranded bacteriophage as an aid to rapid DNA sequencing. J Mol Biol 143: 161–178. [DOI] [PubMed] [Google Scholar]
- Schreiber J, Wescoe ZL, Abu-Shumays R, Vivian JT, Baatar B, Karplus K, Akeson M. 2013. Error rates for nanopore discrimination among cytosine, methylcytosine, and hydroxymethylcytosine along individual DNA strands. Proc Natl Acad Sci 110: 18910–18915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoddart D, Heron AJ, Mikhailova E, Maglia G, Bayley H. 2009. Single-nucleotide discrimination in immobilized DNA oligonucleotides with a biological nanopore. Proc Natl Acad Sci 106: 7702–7707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesan BM, Bashir R. 2011. Nanopore sensors for nucleic acid analysis. Nat Nanotechnol 6: 615–624. [DOI] [PubMed] [Google Scholar]
- Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo CA, Zeng Q, Wortman J, Young SK, et al. 2014. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9: e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Liu R, Xie H, Hui Y, Jiao R, Gong Y, Zhang Y. 2013. Advances in nanopore sequencing technology. J Nanosci Nanotechnol 13: 4521–4538. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.