

Summary
Both iron overload and iron deficiency have been associated with cardiomyopathy and heart failure, but cardiac iron utilization is incompletely understood. We hypothesized that transferrin receptor (Tfr1) might play a role in cardiac iron uptake and used gene targeting to examine the role of Tfr1 in vivo. Surprisingly, we found that decreased iron, due to inactivation of Tfr1, was associated with severe cardiac consequences. Mice lacking Tfr1 in the heart died in the second week of life with cardiomegaly, poor cardiac function, failure of mitochondrial respiration and ineffective mitophagy. The phenotype could only be rescued by aggressive iron therapy, but it was ameliorated by administration of nicotinamide riboside, an NAD precursor. Our findings underscore the importance of both Tfr1 and iron in the heart, and may inform therapy for patients with heart failure.
Graphical Abstract
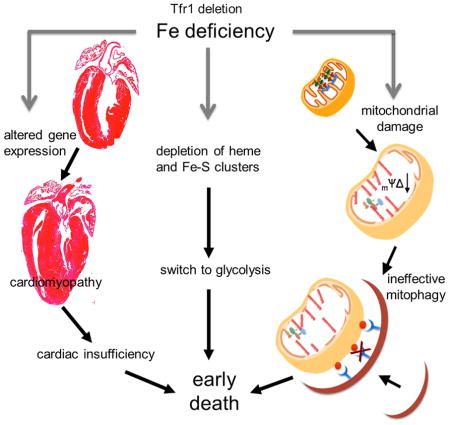
Introduction
Heart failure is a clinical disorder characterized by congestion and decreased functional capacity that, despite current therapy, continues to have a high mortality. While increased iron can cause heart failure in iron overload disorders (Gulati et al., 2014), iron insufficiency is a more common problem. Up to 50% of patients with heart failure are iron deficient, and iron deficiency is associated with poor outcomes (Erbel et al., 2003). Severe iron deficiency causes cardiomyopathy in animals (Medeiros and Beard, 1998; Petering et al., 1990) but previous studies have neither dissociated cardiac iron deficiency from systemic iron deficiency with anemia nor investigated its cellular consequences.
Iron is essential for oxygen transport, oxidative phosphorylation, DNA synthesis and other cellular processes. Iron co-factors -- iron-sulfur (Fe-S) clusters and heme -- are synthesized by mitochondria and necessary for mitochondrial function. Mitochondria are abundant in cardiomyocytes to supply energy for repeated muscle contraction. Mitochondrial failure can lead to increased reactive oxygen species and insufficient ATP. Accordingly, clearance of dysfunctional mitochondria through mitophagy is important for cardiomyocyte maintenance and function (Jimenez et al., 2014).
Transferrin receptor (Tfr1, gene symbol Tfrc) promotes iron uptake through the transferrin (Tf) cycle, by facilitating receptor-mediated endocytosis of iron bound to serum Tf (Hentze et al., 2004). Through targeted gene disruption in mice, we showed that erythroid precursors require Tfr1, but other cells can develop without it (Levy et al., 1999). Tfr1−/− mice appeared normal early in embryogenesis, but died by embryonic day 12.5 with anemia, pericardial effusion, edema and a kinked neural tube. We attributed the pericardial effusion and edema to severe anemia but could not exclude cardiac dysfunction. There were no anatomical defects in the embryonic heart, indicating that heart structures did not require Tfr1 to form. However, we now propose that Tfr1 is critically important for normal heart function.
Mice with profound deficiency of Tf are viable but have severe anemia (Trenor et al., 2000). They accumulate excess iron in non-hematopoietic tissues, including the heart, confirming that many cells can take up non-Tf-bound iron. Chimeric mice generated from blastocysts containing Tfr1−/− embryonic stem cells showed that non-hematopoietic tissues, including the heart, could develop from cells lacking Tfr1 (Ned et al., 2003).
To determine whether Tfr1 is important in the heart, we disrupted floxed Tfr1 in cardiomyocytes using Cre recombinase expressed from a heart-specific promoter. Tfr1-null mice developed early, lethal cardiomyopathy with failure of oxidative phosphorylation and ineffective mitophagy. The abnormalities were prevented by iron supplementation to overwhelm the capacity of serum Tf to bind iron. The lifespan of Tfr1-null mice was prolonged by treatment with nicotinamide riboside (NR), a substrate for NAD production. Our results demonstrate a stringent requirement of cardiomyocytes for Tfr1-mediated iron uptake, and defects in oxidative phosphorylation and mitophagy caused by iron deficiency. They give insight into how isolated cardiac iron deficiency leads to cardiac dysfunction and suggest possible therapeutic approaches for patients with heart failure complicated by iron deficiency.
Results
Tfr1 deficiency causes cardiomyopathy
We inactivated murine Tfr1 in cardiomyocytes by expressing a Myh6-Cre transgene (Agah et al., 1997) to recombine loxP sites flanking exons 3 to 6 (Fig S1A). We confirmed that mutant (Tfr1hrt/hrt) animals expressed little Tfr1 mRNA in heart (Fig S1B) and that Tfr1 was not deleted in other tissues (not shown). Tfr1hrt/hrt mice were born in Mendelian ratios and maintained body weights similar to wild type (WT) littermates (Tfr1fl/fl and Tfr1fl/+ mice, Figs 1A, S1C) but died after several hours of distress by postnatal day 11 (P11) with cardiac hypertrophy (Fig 1B) and elevated heart to body weight ratios, which had developed over time (Fig 1C).
Figure 1. Loss of Tfr1 in cardiomyocytes causes cardiomyopathy.

(A) Tfr1hrt/hrt mice appeared grossly similar to WT at P10.
(B) H&E staining of heart sections at P10, demonstrating cardiomegaly in Tfr1hrt/hrt mice. Scale bars=1 mm.
(C) Tfr1hrt/hrt mice had normal heart to body ratios at P5, but cardiomegaly was apparent at P8 and P10.
(D) Echocardiograms from representative Tfr1hrt/hrt and WT littermates at P5 (top) and P10 (bottom). For each age, upper panel, short axis; lower panel, long axis. Tfr1hrt/hrt mice have markedly impaired cardiac function at P10.
(E) ,(F) Left ventricular diameter and fractional shortening were normal at P5 but abnormal in Tfr1hrt/hrt mice at P10. LVDd=left ventricular diameter in diastole; LVDs=left ventricular diameter in systole.
(G) Representative images of WGA staining for cardiomyocyte morphometrics and quantitation showing Tfr1hrt/hrt cardiomyocyte area similar to WT at P5 (top) and enlarged Tfr1hrt/hrt cardiomyocytes at P10 (bottom). Scale bars=15 μm.
(H) mRNA levels of cardiac hypertrophy biomarkers at P5 and P10 as described in the text.
Data are presented as means ± SEM. Sample size (n) is indicated. *p < 0.05; **p < 0.01; ***p<0.001 by one-way ANOVA. See also Figure S1.
Echocardiography of Tfr1hrt/hrt mice was normal at P5 but showed left ventricular dilatation and decreased fractional shortening at P10 (Fig 1D-F), indicating compromised cardiac performance. Wheat germ agglutinin staining showed normal Tfr1hrt/hrt cardiomyocyte size at P5 but enlarged cardiomyocytes at P10, consistent with hypertrophy (Fig 1G). At P5 mRNA encoding one biomarker for cardiac hypertrophy, Nppb, was increased, but Acta1, another biomarker, was decreased (Fig 1H). However, at P10, all cardiac hypertrophy biomarkers examined (Nppa, Nppb, Myh7, Acta1) were significantly increased. Thus, deletion of Tfr1 in cardiomyocytes leads to dilated cardiomyopathy over the first 10 days of life.
Cardiac iron deficiency
The canonical function of Tfr1 is to supply iron to meet cellular needs. We measured non-heme cardiac iron to determine whether loss of Tfr1 resulted in iron deficiency. While tissue iron concentration increased over time in WT animals, it was decreased at birth and did not change substantially in Tfr1hrt/hrt hearts (Fig 2A–C). The total iron concentration at P10 was also decreased in Tfr1hrt/hrt mice (Fig 2D). Fe-S clusters are synthesized from non-heme iron, and the amounts of enzymes Dpyd and Ppat decrease when Fe-S clusters are not available (Stehling et al., 2013). Both proteins were deficient in Tfr1hrt/hrt hearts at P7 to P10 (Fig 2E, S1D), consistent with compromised Fe-S cluster biogenesis due to iron deficiency or mitochondrial dysfunction. We conclude that Tfr1 is important for iron uptake by cardiomyocytes.
Figure 2. Iron deficiency and Fe-S cluster insufficiency in Tfr1hrt/hrt mice.

(A–C) Non-heme iron levels in WT and mutant heart at P0 (A), P5 (B) and P10 (C).
(D) Total iron concentration at P10.
(E) Decreased Fe-S cluster proteins Dpyd and Ppat in hearts from Tfr1hrt/hrt mice; Rpl19 control. Ages and genotypes are shown at the top.
Data are presented as means ± SEM. Sample size (n) is indicated. ***p<0.001 by one-way ANOVA.
Metabolic changes associated with cardiomyopathy
Mitochondria from Tfr1hrt/hrt hearts were slightly abnormal at P5, but severely disrupted and enlarged at P10 (Fig 3A). Fe-S clusters and heme are required by most complexes of the electron transport chain (ETC) (Xu et al., 2013). We immunoblotted for proteins that are labile when ETC complexes are not assembled properly. At P5 complex II was decreased in Tfr1hrt/hrt hearts and complex IV was increased, but the other ETC complexes did not differ from controls (Fig 3B). At P10 complexes I to IV were all diminished in Tfr1hrt/hrt hearts (Fig 3C). Activity of complex II was decreased at P5 (Fig 3D), and activities of complexes I to IV were all markedly decreased at P10 in Tfr1hrt/hrt hearts (Fig 3E) at P10. However, complex V, which does not contain iron, appeared unchanged at both ages, and its activity was not decreased in Tfr1hrt/hrt hearts at P10 (Fig S2A). Expression of mitochondria-encoded mRNA for Polrmt, Nd4, Cytb and Cox3 was similar to WT at P5 (not shown), but by P10 all were decreased in Tfr1hrt/hrt hearts (Fig 3F), suggesting fewer mitochondria or mitochondria incapable of normal gene expression.
Figure 3. Abnormal mitochondrial morphology and function in hearts from Tfr1hrt/hrt mice.

(A) Electron micrographs comparing mitochondria in WT and Tfr1hrt/hrt hearts. Tfr1hrt/hrt mitochondria were slightly abnormal at P5 (top) but markedly enlarged and disrupted at P10 (bottom). Scale bars=500 nm.
(B) Representative protein levels for ETC complexes by immunoblot at P5 using complex V as the standard.
(C) Representative protein levels for ETC complexes by immunoblot at P10 using complex V as the standard.
(D) Enzymatic activity of complex II of ETC from P5 Tfr1hrt/hrt and WT littermates
(E) Enzymatic activity of complexes I to IV of ETC from P10 Tfr1hrt/hrt and WT littermates.
(F) Relative mRNA levels of Polrmt and mitochondria-encoded genes at P10.
(G) Relative mRNA levels of PGC1-α (Ppargc1a) and PGC1-β (Ppargc1b) at P10.
(H) Relative mRNA levels of PPARα (Ppara), Rxr gamma (Rxrg), and fatty acid transport protein (Fatp1) at P10.
Data are presented as means ± SEM. Sample size (n) is indicated. *p < 0.05; **p < 0.01; ***p<0.001 by one-way ANOVA. See also Figure S2.
We profiled mRNA expression in Tfr1hrt/hrt hearts at P10 [results deposited online (Xu and Andrews, 2015)] and looked for patterns using Gene Set Enrichment Analysis (Mootha et al., 2003; Subramanian et al., 2005). Genes downregulated in the mutants were significantly associated with PPAR (particularly PPARα) and PGC1-α signaling, myogenesis, insulin signaling and cardiomyopathy. Upregulated genes included hypoxia-inducible targets, Myc targets and glycolytic enzymes.
We confirmed decreased expression of PGC1-α (Ppargc1a) and PGC1-β (Ppargc1b) mRNA in Tfr1hrt/hrt hearts (Fig 3G) as well as PGC1-α protein (Fig S2B). PGC1-α controls transcription of a suite of nuclear genes to induce mitochondrial biogenesis (Lehman et al., 2000; Wu et al., 1999b). Mice deficient in PGC1-α in the heart develop cardiomyopathy, similar to our mutant mice (Arany et al., 2005). Our results suggested impaired ability to induce mitochondrial biosynthesis.
Cardiomyopathy is associated with a switch to fetal-like metabolism, with glucose, rather than fatty acids, as the preferred energy source (van Bilsen et al., 1998). The switch has been attributed to decreased activity of PPARα (Ppara) (Barger et al., 2000), which forms a heterodimer with Rxr. Expression of Ppara and Rxrg was decreased in Tfr1-null hearts, as was fatty acid transport protein (Fatp1/Slc27a1, Fig 3H). These changes were not apparent earlier at P5. Interestingly, Ppara expression is induced by the histone demethylase Kdm3a (Okada et al., 2010), which requires iron (Yamane et al., 2006). Ppara and other genes induced by Kdm3a, Ucp2, and Acadm, had decreased mRNA levels at P10 but were not decreased at P5 (not shown). We hypothesize that iron deficiency caused decreased Ppara expression, contributing to the metabolic switch.
At P10 we observed increased mRNA expression of hypoxia-inducible genes (Fig 4A) and glycolytic enzymes (Fig 4B). Of the glycolytic enzymes, only Pfkl was slightly increased at P5 (not shown). Iron is a cofactor for hydroxylases that cause HIFα transcriptional factors to be inactivated, suggesting that iron deficiency could explain induction of hypoxia-inducible genes (Kaelin and Ratcliffe, 2008). In addition, Myc, which induces expression of glycolytic enzymes, was upregulated (Figs 4C, S3). However, glycolysis cannot meet energy needs of cardiomyocytes, which depend on mitochondrial respiration. Apoptosis was increased in Tfr1hrt/hrt hearts, consistent with severe mitochondrial dysfunction, but only at P10 and not earlier at P5 (Fig 4D). These results suggest that iron deficiency leads to mitochondrial insufficiency, metabolic changes and increased apoptosis, contributing to cardiomyocyte hypertrophy and cardiac dysfunction.
Figure 4. Metabolic changes and increased apoptosis in hearts from Tfr1hrt/hrt mice at P10.

(A) Relative mRNA levels of transcripts induced by hypoxia.
(B) Relative mRNA levels of transcripts encoding enzymes of glycolysis.
(C) Representative protein levels for Myc by immunoblot at P10.
(D) TUNEL staining for apoptosis at both P10 and P5. Top row without DAPI staining of nuclei; bottom row with DAPI staining. Vertical pairs of panels from left to right: negative control, positive control, WT and Tfr1hrt/hrt at P10 and P5 respectively. Bright green fluorescent nuclei represent apoptotic cells; scale bars=100 μm. Results are quantified on the right; data are presented as means ± SEM.
Sample size (n) is indicated;*p < 0.05; **p < 0.01; ***p<0.001 by one-way ANOVA. See also Figure S3.
Interruption of mitophagy
Mitochondrial damage should activate mitophagy to clear dysfunctional organelles and recover iron for re-use. Glycolytic enzyme Hk2 promotes autophagy during energy deprivation (Roberts et al., 2014) and was not upregulated at P5 (not shown) but was substantially induced at P10 (Figs 4B, 5A, S4A). Both isoforms of Rcan1, which also induces mitophagy (Ermak et al., 2012) and protects against apoptosis due to hypoxia (Yan et al., 2014), were upregulated in P10 Tfr1hrt/hrt hearts (Figs 5B, S4B,C).
Figure 5. Altered expression of molecules involved in autophagy and mitophagy in hearts from Tfr1hrt/hrt mice.

Multiple autophagy- and mitophagy-related genes were examined in P10 (A,B,D,E,H–J), P8 (C,G), and P5 (F) heart samples for protein levels, as indicated in each panel. Differences suggested stimulation of autophagy but failure to complete autophagy in Tfr1hrt/hrt hearts. Sample sizes for WT and Tfr1hrt/hrt not shown in the figure panels: (H) 14 WT, 6 Tfr1hrt/hrt mice; (I) 3 WT and 5 Tfr1hrt/hrt mice for Cisd2; 5 mice each for Beclin1; (J) 5–6 mice of each genotype except for Atg4B (11 mice) and Atg3 (16 mice).
(K) Lysosomal cathepsin D (Ctsd) and its cleaved intermediate were elevated in hearts from Tfr1hrt/hrt mice, indicating normal lysosomal function.
(L) Lpin1 mRNA was decreased in hearts from Tfr1hrt/hrt mice.
(M) Optineurin (Optn) mRNA was increased in hearts from Tfr1hrt/hrt mice.
Data are presented as means ± SEM. Sample size (n) is indicated; *p < 0.05; **p < 0.01; ***p<0.001 by one-way ANOVA; n.s., not significant. See also Figure S4.
We characterized mitophagy using tissue because neonatal mouse cardiomyocytes cannot be cultured efficiently without contaminating cells. Expression of putative mitophagy receptors, Nix (Bnip3l) and Fundc1 (Liu et al., 2012; Novak et al., 2010), was decreased in Tfr1hrt/hrt hearts at P8 and P10 (Figs 5C,D, S4D,E). Ulk1, which phosphorylates Fundc1 to clear damaged mitochondria (Wu et al., 2014), was also decreased (Figs 5C,D, S4D,F). Bnip3, a homolog of Nix, was markedly increased in mutant hearts as early as P5 (Figs 5E, S4G and not shown). Bnip3 triggers opening of the mitochondrial permeability transition pore and loss of mitochondrial membrane potential (Regula et al., 2002), but may not function as a mitophagy receptor. Bnip3 is induced by hypoxia-inducible factors (Bruick, 2000), consistent with upregulation of other hypoxia-inducible genes. Overall, our results suggested that molecules important for cargo recognition were deficient.
Map1lc3 (LC3) and Gabarap are also involved in the cargo recognition step of autophagy. LC3-II, a phosphatidylethanolamine (PE)-conjugated form of LC3, increases during active autophagy, but was decreased in heart samples from Tfr1hrt/hrt mice at P5 and decreased more at P8 and P10 (Figs 5F–H, S4H–J,L). Gabarap-II was also decreased at P10 (Figs 5H, S4K).
We evaluated other proteins involved in early steps of autophagy. Cisd2 (Naf-1), an FeS cluster protein associated with the mitochondrial membrane, is depleted in Tfr1hrt/hrt mice (Figs 5I, S4M). Deficiency of Cisd2 should promote autophagy by liberating Beclin-1 from Bcl2 (Chang et al., 2010). Beclin-1 levels were similar in mutant and WT hearts (Figs 5I, S4N) but we could not assess its activity. Atg16L, involved early in formation of the phagophore, was increased in P10 Tfr1hrt/hrt hearts (Figs 5J, S4J). Atg10, which was decreased (Figs 5J, S4O), is an E2-like enzyme involved in both Atg12-Atg5 conjugation and LC3 conjugation to PE (Nemoto et al., 2003). While LC3-II was decreased (Figs 5F–H, S4H–J,L), Atg12-Atg5 was increased in mutant hearts (Figs 5J, S4J). The Atg12-Atg5 complex forms before conjugation of LC3 (Geng et al., 2008). Knockdown of LC3 or Gabarap leads to maintenance of the Atg12-Atg5 complex (Weidberg et al., 2010), consistent with our observations. Atg4b cleaves the carboxyl termini of LC3 and Gabarap to expose their lipidation sites, but it also de-lipidates both proteins. Overexpression of Atg4b thus inhibits membrane localization and PE conjugation of LC3 (Tanida et al., 2004). Atg4b was increased in Tfr1hrt/hrt hearts (Figs 5J, S4O), possibly contributing to decreased levels of LC3-II and Gabarap-II. Atg7, an E1-like enzyme involved in the development of autophagosomes, and Atg3, an E2-like enzyme for the LC3/Gabarap conjugation system, were both increased in Tfr1hrt/hrt mice (Figs 5J, S4J). These results suggest that mutant heart cells were attempting to initiate mitophagy, but key proteins involved in cargo recognition were deficient.
Sqstm1 (p62) links the phagophore to cargo. During autophagic flux, lysosomal enzymes degrade p62. The amount of p62 was similar to WT at P5 (Fig S4H) but it was increased in Tfr1hrt/hrt hearts at P8 and P10 (Figs 5G,J, S4I,J), even though p62 mRNA was not increased (Figure S4P), suggesting that p62 was not degraded. We examined cathepsin D (Ctsd), an indicator for lysosomal function. Both forms of Ctsd were increased in hearts from Tfr1hrt/hrt mice (Figs 5K, S4Q), showing that lysosomes were functioning. Consistent a possible defect in cargo recognition, these results suggested that a mitophagy step prior to autophagosome-lysosome fusion was impaired. Lipin1 (Lpin1) enhances transcription regulated by PPAR-α and PGC-1α (Finck et al., 2006) and controls autophagic clearance in skeletal muscle (Zhang et al., 2014). Lpin1 mRNA was decreased in hearts from Tfr1hrt/hrt mice (Fig 5L), possibly contributing both to the metabolic switch and the interruption of mitophagy. Ndrg1 is induced by iron depletion (Le and Richardson, 2004) and upregulated in Tfr1hrt/hrt hearts (Fig 4A). Overexpression of Ndrg1 suppresses LC3-II accumulation and autophagosome formation (Kachhap et al., 2007; Sahni et al., 2014). Atg9 delivers membrane components to developing autophagosomes (Puri et al., 2014). Atg9 was increased in Tfr1hrt/hrt hearts (Figs 5J, S4O). Optn, also important in autophagosome maturation (Tumbarello et al., 2012), was induced in Tfr1hrt/hrt hearts (Fig 5M). These results reinforce the idea that autophagy/mitophagy was generally stimulated, but cargo recognition was defective in Tfr1hrt/hrt hearts.
Rescue of Tfr1hrt/hrt mice
To test whether cardiac iron repletion could rescue Tfr1hrt/hrt mice, we administered iron dextran at P3 to supersaturate Tf and induce non-Tf-bound iron uptake. This prolonged survival, but Tfr1hrt/hrt mice still died at 4 to 5 weeks with severe cardiomegaly. We confirmed that the hearts had assimilated iron by immunoblotting for Fe-S cluster proteins. In contrast to untreated Tfr1hrt/hrt mice, Dpyd and Ppat levels were similar at P10 in Tfr1hrt/hrt and WT mice treated with iron dextran (Fig S5A). Proteins representing ETC complexes were also similar at P10 (Fig S5B). At that time Tfr1hrt/hrt mice and WT littermates had similar heart to body weight ratios (Fig S5C). To try to improve the rescue, we administered a second dose of iron dextran at P7. The onset of cardiomyopathy was further delayed and Tfr1hrt/hrt mice survived up to 13 weeks.
However, ETC complexes were already decreased in hearts from doubly treated Tfr1hrt/hrt mice at 6–8 weeks of age (Fig S5D) and the hearts were already enlarged (Fig S5E). Together, these results indicated that iron-treated Tfr1hrt/hrt mice assimilated and used supplemental iron to survive beyond their usual lifespan, but they eventually showed abnormalities in mitochondrial ETC complexes and autophagy-related proteins (not shown) similar to untreated Tfr1hrt/hrt mice at P10.
We hypothesized that the heart might require continuous iron uptake, and that iron administered early might no longer be available for utilization. To sustain elevated plasma iron concentrations, we took advantage of hemojuvelin knockout (Hjv−/−) mice, which persistently have increased non-Tf-bound iron (Huang et al., 2005). We generated Tfr1hrt/hrt;Hjv−/− mice in which Tfr1 was deleted in the heart and Hjv was deleted globally. These mice also died at P11, similar to Tfr1hrt/hrt mice. However, it takes time for Hjv−/− mice to accumulate iron. We therefore treated Tfr1hrt/hrt;Hjv−/− and control mice with iron dextran at P3 to support the animals until the Hjv mutation caused elevated iron levels. With this strategy, the Tfr1hrt/hrt;Hjv−/− mice were healthy when sacrificed at 12 months and had heart to body weight ratios (Fig S5F) similar to Tfr1fl/fl ;Hjv−/− controls.
We confirmed that this protocol restored cardiomyocyte iron by immunoblotting for Dypd and ferritin at 10–11 weeks (Fig 6A). Markers for ETC complexes I to IV were indistinguishable between Tfr1hrt/hrt;Hjv−/− mice and controls (Fig 6B). Autophagy-related proteins LC3-II, p62, Fundc1, Nix, Ulk-1 and Cisd2, showed no significant differences (Fig 6C–H). Collectively, these results indicate that the Tfr1hrt/hrt mutant phenotype is primarily attributable to a defect in iron assimilation, and iron deficiency results in cardiac hypertrophy, mitochondrial dysfunction and interruption of mitophagy. Further experiments will be needed to fully characterize the defect in mitophagy. Importantly, it appears that the heart is highly sensitive to iron deprivation due to inactivation of Tfr1 and requires a continuous source of iron to function normally.
Figure 6. Rescue of Tfr1hrt/hrt mice with continuous iron.

Iron overload achieved by Fe dextran administration to mutant animals and WT littermates at P3 and an Hjv−/− hemochromatosis background provided sufficient iron to fully rescue Tfr1hrt/hrt mice.
(A) Dpyd and H-ferritin levels at 10 weeks.
(B) Proteins representative of ETC complexes at 10 weeks.
(C–H) Markers of autophagy, as indicated in the panels, at 10 weeks of age.
Data are presented as means ± SEM. Sample size (n) is indicated; n.s., not significant by one-way ANOVA. See also Figure S5.
Treatment with nicotinamide riboside
Mitochondrial dysfunction can cause a decreased NAD/NADH ratio and inactivation of sirtuin deacetylases (Nunnari and Suomalainen, 2012). A decreased NAD/NADH ratio might block signals for mitochondrial biogenesis while also causing defective mitophagy (Fang et al., 2014). Furthermore, in the absence of Sirt1, LC3-II is decreased and p62 accumulates (Hu et al., 2003), similar to what we observed in Tfr1hrt/hrt mice. We speculated that augmentation of NAD levels might modify the mutant phenotype.
We noted dramatic induction of mRNA encoding nicotinamide riboside kinase 2 (Nmrk2/Itgb1bp3, Fig 7A) and the Slc3a2/Slc7a5 transport system for the NAD precursor tryptophan (Boado et al., 1999) (Fig 7B), along with decreased expression of ADP-ribosyltransferases (Fig 7C), suggesting that mutant cardiomyocytes were trying to increase cellular NAD levels. Mitochondria from Tfr1hrt/hrt hearts showed increased lysine acetylation (Fig 7D), consistent with increased acetylation or decreased mitochondrial sirtuin deacetylase activity due to decreased mitochondrial NAD.
Figure 7. Transient rescue of Tfr1hrt/hrt mice by treatment with NR.

(A) mRNA encoding Nmrk2/Itgb1bp3 was massively increased in Tfr1hrt/hrt mice.
(B) mRNAs encoding Slc3a2 and Slc7a5, components of the uptake system for tryptophan, an NAD precursor, were increased in Tfr1hrt/hrt mice.
(C) mRNAs encoding ADP-ribosyltransferases Art1, Art4 and Art5 were markedly decreased in Tfr1hrt/hrt mice.
(D) Proteins from mitochondria isolated from Tfr1hrt/hrt heart showed increased lysine acetylation.
(E) Administration of NR, an NAD precursor and Nmrk2 substrate, extended the lifespan of Tfr1hrt/hrt mice for up to 5 days.
(F) Levels of UPRMT mRNAs in hearts from WT and Tfr1hrt/hrt mice that were untreated (control group, on left) or treated with NR (right). NR treatment appears to have blunted the UPRMT response.
(G) p62 protein levels in hearts from WT and Tfr1hrt/hrt mice that were untreated or treated with NR.
Data are presented as means ± SEM. P-values for (A) to (C) and (F) were determined by one-way ANOVA. Sample size (n) is indicated; P-value for (E) was determined by Logrank test as described in Supplemental Data; P-value for (G) was determined by two-way ANOVA followed by Bonferroni correction; *p < 0.05; **p<0.01; ***p<0.001.
Nicotinamide riboside (NR) can be phosphorylated by Nmrk proteins to induce NAD production, activating sirtuins and mitochondrial biogenesis (Chi and Sauve, 2013). Considering that Nmrk2 (Itgb1bp3) was induced in Tfr1hrt/hrt hearts (Fig 7A), we hypothesized that NR might benefit the mutant animals. We administered NR and observed up to 50% prolongation of lifespans of Tfr1hrt/hrt mice (Fig 7E), indicating that NR could ameliorate the phenotype.
NR might improve the NAD/NADH ratio by increasing NAD. We attempted to measure NAD/NADH ratios without and with NR treatment, but our results from tissue were unreliable. Alternatively, NR may enhance the mitochondrial unfolded protein response [UPRMT; (Khan et al., 2014)]. Although not apparent at P5 (not shown), we observed increased mRNA for multiple genes associated with the UPRMT in P10 hearts including Atf4, Lonp1, Ddit3 and Fgf21 (Fig 7F). In contrast to the earlier report (Khan et al., 2014), NR treatment was associated with decreased expression of UPRMT mRNAs (Fig 7F). Furthermore, NR alleviated the accumulation of p62 in the Tfr1hrt/hrt hearts (Fig 7G). It appears that NR improves mitochondrial quality or enhances mitophagy in Tfr1hrt/hrt hearts, but understanding its beneficial effect will require more work.
Discussion
Iron overload has long been known to cause cardiomyopathy, but relatively little was understood about the molecular consequences of cardiac iron deficiency. Iron deficiency has been implicated in the pathogenesis of heart failure, even in the absence of frank anemia, but previous animal models of cardiomyopathy have involved systemic iron deficiency (Medeiros and Beard, 1998; Petering et al., 1990), which also causes anemia, confounding the interpretation of the role of iron in the heart. In the course of elucidating how cardiomyocytes assimilate iron, we developed a mouse mutant that allowed us to examine the consequences of isolated cardiac iron deficiency. We observed that mice lacking Tfr1 in the heart died from early onset cardiac hypertrophy, caused by iron deficiency and associated with mitochondrial failure. There was induction of a protective mitophagy response, as expected, but failure to complete mitophagy due to inhibition of cargo recognition or another step early in the mitophagy pathway.
The iron deficit in Tfr1hrt/hrt hearts led to abnormal mitochondrial morphology and ETC function, similar to frataxin-deficient mice with impaired Fe-S cluster biogenesis (Puccio et al., 2001). Gene expression results suggested impaired mitochondrial biogenesis. A link between iron and mitochondrial biogenesis was reported previously by Rensvold et al., who screened for genes induced by overexpression of PGC-1α in muscle cells (Rensvold et al., 2013). They observed induction of Tfr1 mRNA, suggesting that iron uptake is stimulated when mitochondria are needed. They showed that iron deprivation causes a reversible decrease in expression of nuclear genes encoding ETC proteins, which are regulated by PGC-1α. We similarly observed decreased mRNA levels of Ndufab1, Ndufb2, Ndufb4, Ndufs4, Sdh, Cox7b2, Cox15, Atp5e and Atp5g (not shown) along with genes encoding PGC-1α and PGC-1β and PGC-1α protein. Our observations support the conclusion that regulation of mitochondrial biogenesis is functionally linked to Tfr1 and cellular iron homeostasis. Tfr1 has also been linked to mitochondrial biogenesis in osteoclasts, where iron uptake stimulated expression of PGC-1β, while iron chelation blunted it (Ishii et al., 2009).
Tfr1hrt/hrt cardiomyocytes increased expression of all enzymes of glycolysis, as well as other hypoxia-inducible genes. These changes may be attributable, in part, to stabilization of HIFα proteins when the iron supply is insufficient for function of hydroxylases that normally modify HIFα proteins to cause their degradation and inactivation. Additionally, many upregulated genes are targets of Myc, which was increased in Tfr1hrt/hrt hearts. Myc regulates genes to promote iron uptake (Wu et al., 1999a), and Tfr1 itself is a Myc transcriptional target (O'Donnell et al., 2006). Increased expression of Myc is an appropriate response to cellular iron deficiency, but futile in the absence of Tfr1.
Myc also contributes to mitochondrial biogenesis (Karamanlidis et al., 2013). More than one third of nucleus-encoded mitochondrial genes are Myc targets, and mitochondrial mass generally correlates with Myc expression. Myc is induced in cardiomyocytes in response to stress, and assists in the metabolic shift from fatty acid oxidation to glucose oxidation, as observed in our mice, by inducing glycolytic enzymes and downregulating fatty acid oxidation enzymes by inhibiting PGC-1α expression.
While induction of glycolytic enzymes makes sense in Tfr1hrt/hrt cardiomyocytes with failing mitochondria, increased expression of Myc and decreased expression of PGC-1α might provide contradictory signals for mitochondrial biogenesis. Gomes recently reported that PGC-1α and Myc function in distinct pathways, responding to different cues (Gomes et al., 2013). Decreased expression of mitochondria-encoded proteins can result from decreased deacetylase activity, as might occur as a result of an NAD deficit. Decreased deacetylase activity leads to stabilization of Hifα proteins, and Hif-1α inhibits Myc-induced mitochondrial gene expression. In parallel, it leads to decreased PGC-1α activity.
Mitophagy is also impaired in Tfr1hrt/hrt cardiomyocytes. Many proteins involved in stimulating mitophagy are induced, but levels of cargo recognition-related proteins Bnip3l (Nix), Fundc1, Ulk1, LC3-II, Gabarap-II and Atg10 are depressed and p62 accumulates in spite of normal lysosomal function. These changes, along with decreased expression of Lpin1 and increased expression of Atg4b and Ndrg1, might explain the defect in mitophagy progression. However, it is also possible that mitophagy is interrupted due to decreased availability of NAD to activate Sirt1, and consequent increased acetylation of autophagy-related proteins as previously reported (Hu et al., 2003). Similar to our results, Sirt1−/− mice exhibited abnormal cardiac mitochondria and increased accumulation of p62.
Normally, autophagy and mitophagy replete cellular nutrients, particularly at times of stress. The failure of mitophagy in Tfr1hrt/hrt mice suggests that damaged mitochondria, which are rich in iron, cannot be effectively broken down to recover iron for re-use. This would set up a vicious cycle, in which iron deficiency compromises mitochondrial integrity, but new iron is not available for new mitochondrial biogenesis. We speculate that this explains why the phenotype of Tfr1hrt/hrt mice is so severe. Iron is present in the mutant cardiomyocytes, but may not be effectively used, exacerbating cellular distress.
We showed that the phenotype of our Tfr1hrt/hrt mice could be corrected through aggressive and ongoing iron supplementation, confirming that iron deficiency is the root cause for the abnormalities. Nonetheless, treatment with NR prolonged the lifespan of our mice. The response to NR is reminiscent of other studies where it increases NAD, sirtuin activity and mitochondrial biogenesis (Canto et al., 2012; Cerutti et al., 2014). NR was previously reported to stimulate the UPRMT (Khan et al., 2014), but we observed the opposite. An increased NAD/NADH ratio and activation of Sirt1 also lead to induction of autophagy (Hu et al., 2003; Huang et al., 2003). We found that NR treatment alleviated the accumulation of p62, suggesting improvement of autophagy/mitophagy in Tfr1hrt/hrt hearts. Regardless of its mechanism of action, catastrophic failure of the ETC, due to iron deficiency, cannot be overcome, and the benefits of NR are transient.
Our findings in mice provide mechanistic support for the known benefit of iron treatment in heart failure accompanied by iron deficiency. We showed that iron is not only necessary for cardiac function, but also must be continuously available. Furthermore, our findings suggest that NR, a potent, NAD-enhancing form of vitamin B3, might provide added benefit in patients with cardiac iron deficiency.
Experimental Procedures
See also Supplemental Experimental Procedures
Animals
We crossed 129/SvEv mice bearing a floxed Tfrc allele (Chen et al., 2015) with transgenic C57BL/6 mice expressing Cre recombinase under the control of the α-MyHC promoter (Agah et al., 1997). We backcrossed with 129/SvEv mice for more than 10 generations and after the initial phenotypic characterization all studies used mice with a homogeneous 129/SvEv background. Animals were genotyped by PCR using genomic DNA from toe clips (Mizutani et al., 2002). Primers for Tfrc alleles, Cre and Hjv alleles are described in Supp Table 1.
For iron rescue experiments, mice were injected IP with 5mg of Uniferon® 100 (25 μl) at P3, or at both P3 and P7. For NR rescue experiments, mice were injected IP with NR in PBS as previously described (Yang et al., 2007) at 750 mg/kg daily from P5. Volume was based on body weight, with less than 100 μl at postnatal day 10. Control animals were injected with PBS only.
Histology
4μm paraffin sections were stained with hematoxylin and eosin for light microscopy. Cardiomyocyte dimensions were measured by digital morphometry of paraffin-embedded myocardial cross-sections stained with Alexa Fluor® 594 WGA (Life Technologies, Invitrogen) using ImageJ image processing. Apoptotic cells were identified and quantified using the In Situ Cell Death Detection Kit (Roche) on paraffin sections according to the manufacturer’s instructions. Quantitation was done by ImageJ using three consecutive sections for each mouse.
Echocardiography
Echocardiography was performed without anesthesia on age-, sex- and body weight-matched mice by an investigator blinded to genotypes. Left ventricular dimensions and fractional shortening were calculated based on echocardiography data as described previously (Esposito et al., 2000).
Electron microscopy
Hearts were removed, rinsed in cold Krebs-Henseleit buffer (Krebs and Henseleit, 1932), and immersed in 5% glutaraldehyde buffer. Samples were prepared for transmission electron microscopy and imaged by Duke Research Electron Microscopy Services.
Tissue iron analysis
Heart non-heme iron was measured as described previously (Levy et al., 1999; Torrance and Bothwell, 1980).
Analysis of ETC complexes I–V
Tissues were homogenized in 250 mM sucrose, 40 mM potassium chloride, 1 mM EGTA, 1 mg/ml fatty acid free BSA, 20 mM Tris-HCl (pH 7.2) and homogenates were centrifuged at 600 x g for 10 min at 4°C. Steady st ate activities of enzyme complexes I-IV in the supernatant were determined as previously described (Janssen et al., 2007; Spinazzi et al., 2011). For complex V, crude mitochondria were collected and activity was determined as previously described (Kirby et al., 2007).
Preparation of mitochondrial lysates and acetyl-lysine analysis
Mouse hearts were homogenized (40–50 strokes) in 15 volumes of ice-cold homogenization buffer [320 mM sucrose, 50 mM KH2PO4 (pH 7.4), 10 mM Tris-HCl (pH 7.4), 1 mM EDTA] in the presence of protease inhibitors, phosphatase inhibitors (Roche), and deacetylase inhibitors (2.5 μM Trichostatin A, 5 mM nicotinamide, 5 mM sodium butyrate) using glass homogenizers. Crude mitochondria were isolated by differential centrifugation. Homogenates were centrifuged at 1600 rpm for 10 min at 4°C. The supernatant was centrifuged again at 1600 rpm for 10 min at 4°C. The supernatant was subsequently centrifuged at 10,000 x g for 10 min at 4°C. The pellet was collected, rinsed with 1 ml homogenization buffer and centrifuged at 10,000 x g again for 10 min at 4°C. The pellet was resuspended in 100 μl 20 mM HEPES (pH 7.4), 150 mM NaCl, and 1% Triton-X-100 with protease, phosphatase and deacetylase inhibitors. Western blot analysis was performed with 70 μg protein per lane. Antibodies recognizing acetyl-lysine were from Cell Signaling Technology.
Statistical analysis
One-way ANOVA was performed for comparisons between two means. Two-way ANOVA followed by Bonferroni post-hoc was performed for multiple comparisons. Survival analysis was performed using Logrank (see Supplemental Experimental Procedures). Dr. Kingshuk Roy Choudhury (Duke Department of Biostatistics and Bioinformatics) assisted with statistical analyses. P < 0.05 was considered statistically significant.
Supplementary Material
Highlights.
Transferrin receptor 1 mediates iron uptake in the heart
Cardiac iron deficit leads to metabolic changes and ineffective mitophagy
Cardiomyocytes require continuous iron availability
Nicotinamide riboside ameliorates the effects of cardiac iron deficit
Acknowledgments
We thank John Shelton at UT Southwestern for preparing heart tissue sections, Kingshuk Roy Choudhury for assistance with statistical analysis, the Duke Microarray Core facility (supported by NIH P30 CA014236) for microarray data ascertainment, management and analysis, Geoff Pitt and Matt Hirschey for advice, Michael Schneider for use of Myh6-Cre mice and members of the Andrews laboratory for helpful discussions. This work was supported by R01 DK089705 to NCA and HL56687 to HAR.
Footnotes
Accession number
Microarray data have been deposited as NCBI GSE68745
Author Contributions
WX and NCA designed experiments and wrote the manuscript. Most experiments were carried out by WX. TB performed initial characterization of the mutant mice. LM and HAR performed electrocardiography and calculations of heart function. AAS provided nicotinamide riboside and advice.
Disclosures
HAR is a scientific cofounder and consultant for Trevena Inc., a company that is developing G protein-coupled receptor targeted drugs. AAS has intellectual property related to methods of production of nicotinamide riboside and receives royalties from a commercial license to Chromadex Inc. for this intellectual property. AAS is also a consultant for and co-founder of Metro Mid-Atlantic Biotech, LLC.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agah R, Frenkel PA, French BA, Michael LH, Overbeek PA, Schneider MD. Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J Clin Invest. 1997;100:169–179. doi: 10.1172/JCI119509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arany Z, He H, Lin J, Hoyer K, Handschin C, Toka O, Ahmad F, Matsui T, Chin S, Wu PH, et al. Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 2005;1:259–271. doi: 10.1016/j.cmet.2005.03.002. [DOI] [PubMed] [Google Scholar]
- Barger PM, Brandt JM, Leone TC, Weinheimer CJ, Kelly DP. Deactivation of peroxisome proliferator-activated receptor-alpha during cardiac hypertrophic growth. J Clin Invest. 2000;105:1723–1730. doi: 10.1172/JCI9056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boado RJ, Li JY, Nagaya M, Zhang C, Pardridge WM. Selective expression of the large neutral amino acid transporter at the blood-brain barrier. Proc Natl Acad Sci U S A. 1999;96:12079–12084. doi: 10.1073/pnas.96.21.12079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruick RK. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc Natl Acad Sci U S A. 2000;97:9082–9087. doi: 10.1073/pnas.97.16.9082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, Fernandez-Marcos PJ, Yamamoto H, Andreux PA, Cettour-Rose P, et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012;15:838–847. doi: 10.1016/j.cmet.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerutti R, Pirinen E, Lamperti C, Marchet S, Sauve AA, Li W, Leoni V, Schon EA, Dantzer F, Auwerx J, et al. NAD(+)-Dependent Activation of Sirt1 Corrects the Phenotype in a Mouse Model of Mitochondrial Disease. Cell Metab. 2014;19:1042–1049. doi: 10.1016/j.cmet.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang NC, Nguyen M, Germain M, Shore GC. Antagonism of Beclin 1-dependent autophagy by BCL-2 at the endoplasmic reticulum requires NAF-1. EMBO J. 2010;29:606–618. doi: 10.1038/emboj.2009.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen AC, Donovan A, Ned-Sykes R, Andrews NC. A non-canonical role of transferrin receptor 1 is essential for intestinal homeostasis. Proc Natl Acad Sci U S A. 2015 doi: 10.1073/pnas.1511701112. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi Y, Sauve AA. Nicotinamide riboside, a trace nutrient in foods, is a vitamin B3 with effects on energy metabolism and neuroprotection. Curr Opin Clin Nutr Metab Care. 2013;16:657–661. doi: 10.1097/MCO.0b013e32836510c0. [DOI] [PubMed] [Google Scholar]
- Erbel PJ, Card PB, Karakuzu O, Bruick RK, Gardner KH. Structural basis for PAS domain heterodimerization in the basic helix--loop--helix-PAS transcription factor hypoxia-inducible factor. Proc Natl Acad Sci U S A. 2003;100:15504–15509. doi: 10.1073/pnas.2533374100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermak G, Sojitra S, Yin F, Cadenas E, Cuervo AM, Davies KJ. Chronic expression of RCAN1-1L protein induces mitochondrial autophagy and metabolic shift from oxidative phosphorylation to glycolysis in neuronal cells. J Biol Chem. 2012;287:14088–14098. doi: 10.1074/jbc.M111.305342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito G, Santana LF, Dilly K, Cruz JD, Mao L, Lederer WJ, Rockman HA. Cellular and functional defects in a mouse model of heart failure. Am J Physiol Heart Circ Physiol. 2000;279:H3101–3112. doi: 10.1152/ajpheart.2000.279.6.H3101. [DOI] [PubMed] [Google Scholar]
- Fang EF, Scheibye-Knudsen M, Brace LE, Kassahun H, Sengupta T, Nilsen H, Mitchell JR, Croteau DL, Bohr VA. Defective Mitophagy in XPA via PARP-1 Hyperactivation and NAD(+)/SIRT1 Reduction. Cell. 2014;157:882–896. doi: 10.1016/j.cell.2014.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finck BN, Gropler MC, Chen Z, Leone TC, Croce MA, Harris TE, Lawrence JC, Jr, Kelly DP. Lipin 1 is an inducible amplifier of the hepatic PGC-1alpha/PPARalpha regulatory pathway. Cell Metab. 2006;4:199–210. doi: 10.1016/j.cmet.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Geng J, Baba M, Nair U, Klionsky DJ. Quantitative analysis of autophagy-related protein stoichiometry by fluorescence microscopy. J Cell Biol. 2008;182:129–140. doi: 10.1083/jcb.200711112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JS, Wrann CD, Hubbard BP, et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155:1624–1638. doi: 10.1016/j.cell.2013.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulati V, Harikrishnan P, Palaniswamy C, Aronow WS, Jain D, Frishman WH. Cardiac involvement in hemochromatosis. Cardiology in Review. 2014;22:56–68. doi: 10.1097/CRD.0b013e3182a67805. [DOI] [PubMed] [Google Scholar]
- Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell. 2004;117:285–297. doi: 10.1016/s0092-8674(04)00343-5. [DOI] [PubMed] [Google Scholar]
- Hu X, Qiu J, Grafe MR, Rea HC, Rassin DK, Perez-Polo JR. Bcl-2 family members make different contributions to cell death in hypoxia and/or hyperoxia in rat cerebral cortex. Int J Dev Neurosci. 2003;21:371–377. doi: 10.1016/s0736-5748(03)00089-3. [DOI] [PubMed] [Google Scholar]
- Huang FW, Pinkus JL, Pinkus GS, Fleming MD, Andrews NC. A mouse model of juvenile hemochromatosis. J Clin Invest. 2005;115:2187–2191. doi: 10.1172/JCI25049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Li Z, Yang Z. Roles of ischemia and hypoxia and the molecular pathogenesis of post-burn cardiac shock. Burns. 2003;29:828–833. doi: 10.1016/s0305-4179(03)00204-3. [DOI] [PubMed] [Google Scholar]
- Ishii KA, Fumoto T, Iwai K, Takeshita S, Ito M, Shimohata N, Aburatani H, Taketani S, Lelliott CJ, Vidal-Puig A, et al. Coordination of PGC-1beta and iron uptake in mitochondrial biogenesis and osteoclast activation. Nature Med. 2009;15:259–266. doi: 10.1038/nm.1910. [DOI] [PubMed] [Google Scholar]
- Janssen AJ, Trijbels FJ, Sengers RC, Smeitink JA, van den Heuvel LP, Wintjes LT, Stoltenborg-Hogenkamp BJ, Rodenburg RJ. Spectrophotometric assay for complex I of the respiratory chain in tissue samples and cultured fibroblasts. Clin Chem. 2007;53:729–734. doi: 10.1373/clinchem.2006.078873. [DOI] [PubMed] [Google Scholar]
- Jimenez RE, Kubli DA, Gustafsson AB. Autophagy and mitophagy in the myocardium: therapeutic potential and concerns. Br J Pharmacol. 2014;171:1907–1916. doi: 10.1111/bph.12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kachhap SK, Faith D, Qian DZ, Shabbeer S, Galloway NL, Pili R, Denmeade SR, DeMarzo AM, Carducci MA. The N-Myc down regulated Gene1 (NDRG1) Is a Rab4a effector involved in vesicular recycling of E-cadherin. PLoS One. 2007;2:e844. doi: 10.1371/journal.pone.0000844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin WG, Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- Karamanlidis G, Lee CF, Garcia-Menendez L, Kolwicz SC, Jr, Suthammarak W, Gong G, Sedensky MM, Morgan PG, Wang W, Tian R. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab. 2013;18:239–250. doi: 10.1016/j.cmet.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan NA, Auranen M, Paetau I, Pirinen E, Euro L, Forsstrom S, Pasila L, Velagapudi V, Carroll CJ, Auwerx J, et al. Effective treatment of mitochondrial myopathy by nicotinamide riboside, a vitamin B3. EMBO Mol Med. 2014;6:721–731. doi: 10.1002/emmm.201403943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby DM, Thorburn DR, Turnbull DM, Taylor RW. Biochemical assays of respiratory chain complex activity. Meth Cell Biol. 2007;80:93–119. doi: 10.1016/S0091-679X(06)80004-X. [DOI] [PubMed] [Google Scholar]
- Krebs HA, Henseleit K. Untersuchungen über die Harnstoffbildung im Tierkörper. Hoppe-Seyler's Zeitschrift für Physiol Chemie. 1932;210:33–66. [Google Scholar]
- Le NT, Richardson DR. Iron chelators with high antiproliferative activity up-regulate the expression of a growth inhibitory and metastasis suppressor gene: a link between iron metabolism and proliferation. Blood. 2004;104:2967–2975. doi: 10.1182/blood-2004-05-1866. [DOI] [PubMed] [Google Scholar]
- Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. 2000;106:847–856. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy JE, Jin O, Fujiwara Y, Kuo F, Andrews NC. Transferrin receptor is necessary for development of erythrocytes and the nervous system. Nat Genet. 1999;21:396–399. doi: 10.1038/7727. [DOI] [PubMed] [Google Scholar]
- Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14:177–185. doi: 10.1038/ncb2422. [DOI] [PubMed] [Google Scholar]
- Medeiros DM, Beard JL. Dietary iron deficiency results in cardiac eccentric hypertrophy in rats. Proc Soc Exp Biol Med. 1998;218:370–375. doi: 10.3181/00379727-218-44306. [DOI] [PubMed] [Google Scholar]
- Mizutani A, Furukawa T, Adachi Y, Ikehara S, Taketani S. A zinc-finger protein, PLAGL2, induces the expression of a proapoptotic protein Nip3, leading to cellular apoptosis. J Biol Chem. 2002;277:15851–15858. doi: 10.1074/jbc.M111431200. [DOI] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- Ned RM, Swat W, Andrews NC. Transferrin receptor 1 is differentially required in lymphocyte development. Blood. 2003;102:3711–3718. doi: 10.1182/blood-2003-04-1086. [DOI] [PubMed] [Google Scholar]
- Nemoto T, Tanida I, Tanida-Miyake E, Minematsu-Ikeguchi N, Yokota M, Ohsumi M, Ueno T, Kominami E. The mouse APG10 homologue, an E2-like enzyme for Apg12p conjugation, facilitates MAP-LC3 modification. J Biol Chem. 2003;278:39517–39526. doi: 10.1074/jbc.m300550200. [DOI] [PubMed] [Google Scholar]
- Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, Rogov V, Lohr F, Popovic D, Occhipinti A, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010;11:45–51. doi: 10.1038/embor.2009.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145–1159. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell KA, Yu D, Zeller KI, Kim JW, Racke F, Thomas-Tikhonenko A, Dang CV. Activation of transferrin receptor 1 by c-Myc enhances cellular proliferation and tumorigenesis. Mol Cell Biol. 2006;26:2373–2386. doi: 10.1128/MCB.26.6.2373-2386.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada Y, Tateishi K, Zhang Y. Histone demethylase JHDM2A is involved in male infertility and obesity. J Androl. 2010;31:75–78. doi: 10.2164/jandrol.109.008052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petering DH, Stemmer KL, Lyman S, Krezoski S, Petering HG. Iron deficiency in growing male rats: a cause of development of cardiomyopathy. Ann Nutr Metab. 1990;34:232–243. doi: 10.1159/000177592. [DOI] [PubMed] [Google Scholar]
- Puccio H, Simon D, Cossee M, Criqui-Filipe P, Tiziano F, Melki J, Hindelang C, Matyas R, Rustin P, Koenig M. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat Genet. 2001;27:181–186. doi: 10.1038/84818. [DOI] [PubMed] [Google Scholar]
- Puri C, Renna M, Bento CF, Moreau K, Rubinsztein DC. ATG16L1 meets ATG9 in recycling endosomes: additional roles for the plasma membrane and endocytosis in autophagosome biogenesis. Autophagy. 2014;10:182–184. doi: 10.4161/auto.27174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regula KM, Ens K, Kirshenbaum LA. Inducible expression of BNIP3 provokes mitochondrial defects and hypoxia-mediated cell death of ventricular myocytes. Circ Res. 2002;91:226–231. doi: 10.1161/01.res.0000029232.42227.16. [DOI] [PubMed] [Google Scholar]
- Rensvold JW, Ong SE, Jeevananthan A, Carr SA, Mootha VK, Pagliarini DJ. Complementary RNA and protein profiling identifies iron as a key regulator of mitochondrial biogenesis. Cell Rep. 2013;3:237–245. doi: 10.1016/j.celrep.2012.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts DJ, Tan-Sah VP, Ding EY, Smith JM, Miyamoto S. Hexokinase-II positively regulates glucose starvation-induced autophagy through TORC1 inhibition. Mol Cell. 2014;53:521–533. doi: 10.1016/j.molcel.2013.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahni S, Bae DH, Lane DJ, Kovacevic Z, Kalinowski DS, Jansson PJ, Richardson DR. The metastasis suppressor, N-myc downstream-regulated gene 1 (NDRG1), inhibits stress-induced autophagy in cancer cells. J Biol Chem. 2014;289:9692–9709. doi: 10.1074/jbc.M113.529511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinazzi M, Casarin A, Pertegato V, Ermani M, Salviati L, Angelini C. Optimization of respiratory chain enzymatic assays in muscle for the diagnosis of mitochondrial disorders. Mitochondrion. 2011;11:893–904. doi: 10.1016/j.mito.2011.07.006. [DOI] [PubMed] [Google Scholar]
- Stehling O, Mascarenhas J, Vashisht AA, Sheftel AD, Niggemeyer B, Rosser R, Pierik AJ, Wohlschlegel JA, Lill R. Human CIA2A-FAM96A and CIA2B-FAM96B integrate iron homeostasis and maturation of different subsets of cytosolic-nuclear iron-sulfur proteins. Cell Metab. 2013;18:187–198. doi: 10.1016/j.cmet.2013.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanida I, Ueno T, Kominami E. Human light chain 3/MAP1LC3B is cleaved at its carboxyl-terminal Met121 to expose Gly120 for lipidation and targeting to autophagosomal membranes. J Biol Chem. 2004;279:47704–47710. doi: 10.1074/jbc.M407016200. [DOI] [PubMed] [Google Scholar]
- Torrance JD, Bothwell TH. Tissue iron stores. In: Cook JD, editor. Methods in Hematology. New York: Churchill Livingstone Press; 1980. pp. 104–109. [Google Scholar]
- Trenor CC, 3rd, Campagna DR, Sellers VM, Andrews NC, Fleming MD. The molecular defect in hypotransferrinemic mice. Blood. 2000;96:1113–1118. [PubMed] [Google Scholar]
- Tumbarello DA, Waxse BJ, Arden SD, Bright NA, Kendrick-Jones J, Buss F. Autophagy receptors link myosin VI to autophagosomes to mediate Tom1-dependent autophagosome maturation and fusion with the lysosome. Nat Cell Biol. 2012;14:1024–1035. doi: 10.1038/ncb2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bilsen M, van der Vusse GJ, Reneman RS. Transcriptional regulation of metabolic processes: implications for cardiac metabolism. Pflugers Arch. 1998;437:2–14. doi: 10.1007/s004240050739. [DOI] [PubMed] [Google Scholar]
- Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V, Elazar Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 2010;29:1792–1802. doi: 10.1038/emboj.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu KJ, Polack A, Dalla-Favera R. Coordinated regulation of iron-controlling genes, H-ferritin and IRP2, by c-MYC. Science. 1999a;283:676–679. doi: 10.1126/science.283.5402.676. [DOI] [PubMed] [Google Scholar]
- Wu W, Tian W, Hu Z, Chen G, Huang L, Li W, Zhang X, Xue P, Zhou C, Liu L, et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 2014;15:566–575. doi: 10.1002/embr.201438501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999b;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- Xu W, Andrews NC. Expression data from wildtype and Tfr1 heart KO mice (NCBI, GSE68745) 2015. [Google Scholar]
- Xu W, Barrientos T, Andrews NC. Iron and copper in mitochondrial diseases. Cell Metab. 2013;17:319–328. doi: 10.1016/j.cmet.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamane K, Toumazou C, Tsukada Y, Erdjument-Bromage H, Tempst P, Wong J, Zhang Y. JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell. 2006;125:483–495. doi: 10.1016/j.cell.2006.03.027. [DOI] [PubMed] [Google Scholar]
- Yan L, Yang H, Li Y, Duan H, Wu J, Qian P, Li B, Wang S. Regulator of calcineurin 1–1L protects cardiomyocytes against hypoxia-induced apoptosis via mitophagy. J Cardiovasc Pharmacol. 2014;64:310–317. doi: 10.1097/FJC.0000000000000121. [DOI] [PubMed] [Google Scholar]
- Yang T, Chan NY, Sauve AA. Syntheses of nicotinamide riboside and derivatives: effective agents for increasing nicotinamide adenine dinucleotide concentrations in mammalian cells. J Med Chem. 2007;50:6458–6461. doi: 10.1021/jm701001c. [DOI] [PubMed] [Google Scholar]
- Zhang P, Verity MA, Reue K. Lipin-1 regulates autophagy clearance and intersects with statin drug effects in skeletal muscle. Cell Metab. 2014;20:267–279. doi: 10.1016/j.cmet.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.