

Abstract
Stem cell populations are maintained through self-renewing divisions in which one daughter cell commits to a particular fate while the other retains the multipotent characteristics of its parent. The NUMB, a tumor suppressor, in conjunctions with another tumor suppressor protein p53, preserves this property and acts as a barrier against deregulated expansion of Tumor-associated stem cells. In this context, NUMB-p53 interaction plays a crucial role to maintain the proper homeostasis of both stem cells, as well as differentiated cells. As the molecular mechanism governing the assembly and stability of the NUMB-p53 interaction/complex are poorly understood, we tried to identify the molecule/s govern this process. Using cancer cell lines, tumor-initiating cells (TICs) of liver, the mouse model and clinical samples, we identified that phosphorylations of NUMB destabilize p53 and promotes self-renewal of TICs by pluripotency-associated transcription factor NANOG dependent manner. NANOG phosphorylates NUMB via aPKCζ, through the direct induction of Aurora A kinase (AURKA) and the repression of an aPKCζ inhibitor, LGL-2. By radioactivity based kinase activity assays, we showed that NANOG enhances kinase activities of both AURKA and aPKCζ, an important upstream process for NUMB phosphorylation. Phosphorylation of NUMB by aPKCζ destabilizes the NUMB-p53 interaction, p53 proteolysis and to deregulate self-renewal in TICs.
Conclusion
Posttranslational modification of NUMB by NANOG-AURKA-aPKCζ pathway is an important event in TICs self-renewal and tumorigenesis. Hence, our work identifies the NANOG-NUMB-p53 signaling axis is an important regulatory pathway for TICS event in TICs self-renewal and liver tumorigenesis and suggest a therapeutic strategy by targeting NUMB-phosphorylation. However, further in depth in vivo and clinical studies are warranted to verify this suggestion.
Keywords: NUMB, phosphoNUMB, NANOG, p53, self-renewal, Tumor-initiating Cell
Introduction
Phosphorylation/dephosphorylation of proteins play a crucial role in signal transduction system and regulate complex biological events such as cell proliferation, differentiation and variety of diseases such as diabetes, autoimmune disease, and cancer.1–4 Phosphorylation also plays a crucial role in the regulation of different tumor suppressor proteins including p53, RB, and CDK4.5 Phosphorylation is also considered as a biomarker immunogenic cell death6 and regulation of invasion and migration of cancer cells.7 Recently, it has been shown that TGFβ-induced phosphorylation of Par6, a cell fate determinant, promotes migration and invasion in prostate cancer cells.7
NUMB, a cell fate determinant, and tumor suppressor, frequently observed the loss of expression in breast cancer.8,9 NUMB contains an N-terminal Phosphotyrosine Binding (PTB) domain and a C-terminal proline-rich region (PRR).10 NUMB interacts with p53 through PTB domain.9,10 Genetic deletion or shRNA-mediated depletion of p53 enhances cellular reprogramming to the pluripotent state.11–13 and p53 can directly repress the expression of pluripotency-associated TFs.14 The association of p53 with NUMB promotes the stabilization of p53-NUMB complex, which has been identified independently as a tumor suppressor.15–16 p53 is required to maintain normal stem cells division by interacting directly with the NUMB protein. Thus, p53 along with NUMB serves a critical role in the regulation of pluripotency and stem cell division.
Malignant tumors comprise of heterogeneous cells and at the top of this hierarchy are rare subpopulations of tumor-initiating cells (TICs), which massively and disproportionately contribute to oncogenesis and progression to treatment-refractory disease.17–18 The involvement of NUMB and p53 in multiple facets of stem cell function suggests that the regulation of the assembly or stability of the NUMB-p53 complex may influence TIC-mediated oncogenesis. The recent identification of NUMB-associated oncoproteins that destabilize the NUMB-p53 complex and promote proteolysis of p53 by the MDM2 E3 ubiquitin ligase provides experimental validation for this concept.19 However, the signaling inputs governing the assembly and stability of the NUMB-p53 complex remain poorly defined. Here, we tested if phosphorylation of NUMB destabilization of NUMB-p53 complex and promote self-renewal of TICs. We conclude that NUMB phosphorylation mediated by NANOG and subsequent p53 degradation drive self-renewability and proliferation of TICs, which results in higher oncogenesis.
Materials and Methods
Cells and cell lines
CD133+/CD49f+ murine hepatoma TICs were isolated from Live Tumors.20 We calculated the TICs as per the method of Extreme Limiting Dilution Analysis (ELDA) http://bioinf.wehi.edu.au/software/elda/.
Transformed Liver cell lines (HepG2, and Hep3B) were obtained from ATCC (Manassas, VA). Huh7 is a human hepatoma cell line. Cells were cultured in DMEM with 10% FBS and 1% Penicillin/Streptomycin and were kept in 5% CO2 in an incubator at 37°C.
Human patients’ tissue samples
Forty-five formalin-fixed, paraffin-embedded paired primary HCC tissues or non-tumor (adjacent liver) tissues were obtained from USC Cancer Hospitals and University of Minnesota, Liver Tissue Cell Distribution System, Minneapolis. Detailed methods of collection and clinical and pathological characteristics of the patients were summarized in Supplementary materials and Suppl. Table 1, respectively.
Tumorigenicity studies in NOG mice
NOD/Shi-scid, IL-2Rγ null (NOG) mice were obtained from the Central Institute for Experimental Animals (Kanagawa, Japan) and housed under pathogen-free conditions in accordance with approved Institutional Animal Care and Use Committee protocols. Liver TICs (5 × 104) were re-suspended in 100 μL of 50% Matrigel (BD Biosciences) in PBS and injected subcutaneously into the dorsal hind flanks of anesthetized mice as previously described.21 Tumor size was measured with calipers, and the volume was calculated according to the formula V = (a × b2)/2, where ‘a’ represents the largest and ‘b’ the smallest superficial diameters.21
Statistical Analysis
Statistical significance was estimated by unpaired, two-tailed Student’s t test. Bars represent the mean ± S.D. For all figures, statistical significance is represented by asterisks above each column: *, P < 0.05. TIC frequency was calculated from tumor formation titration experiments using the limit function of the statmod package in the R-statistical software suite. For each tumor marker, the percent of staining and intensity of staining, as well as the product of the two (IRS), were presented with dot plots. Paired t-tests were used to compare the marker expression levels between tumor vs. non-tumor tissues. Statistical analyzes were performed using STATA software (version 11.0; StataCorp LP College Station, TX).22
Results
NUMB phosphorylations are positively correlated with NANOG level in the tumor-initiating cells and clinical tissues
As an attempt towards identifying the phosphorylation status of NUMB under different level of NANOG, if any, we performed immunoblot analysis in tumor-initiating cells (TICs). We employed a two-way approach where NANOG was either knocked-down or overexpressed in TICs. After 48h post-transfection, NANOG, NUMB, and pNUMB levels were analyzed. Though NUMB levels were maintained, phospho-NUMB (pNUMB) levels were observed to be reduced in NANOG-knocked-down cells and increased in NANOG-overexpressed cells (Fig. 1A). These data suggest that NANOG modulates the phosphorylation levels of NUMB. We next determine the levels of pNUMB vis a vis NANOG levels in human clinical liver specimens of matched normal and cancer samples (clinicopathological factors are listed in Suppl. Table 1) by immunofluorescence analysis. In general, the staining was stronger in cancer tissues than in normal tissues (Fig. 1Bi). A significant difference was found in mean immunoreactivity score (IRS) (p<0.001) between tumor vs. non-tumor tissues (Fig. 1Bii). For the mean, median and range of difference in IRS between tumor vs. non-tumor tissues were presented in Table 1. For the distribution of the percent of staining, intensity of staining, and IRS for each tumor marker were given in Suppl. Fig 1A–B. Taken together, these data show that levels of pNUMB increases with increasing NANOG levels.
Figure 1. NUMB phosphorylations and p53 levels are linked to NANOG level and in the Tumor Initiating Cells (TICs) and Clinical Tissues.

(A) Immunoblot analysis of lysates prepared from patient HCC tissue specimens and matched, non-cancerous tissues using the indicated antisera. (Bi) Representative photomicrographs showing the expression of NANOG and pNUMB (Ser-265) in human HCCs and adjacent non-cancerous tissues as assessed by an immunofluorescence assay. The merged lane shows the co-localization of 2 proteins. DAPI was used as a nuclear staining control. (Bii) Comparison of Immuno Reactivity Score (IRS, the product of a percent of staining and intensity of staining) in tumor vs. non-tumor tissues. (C) Immunoblot analysis of lysates prepared from human hepatocytes stably expressed or constitutively active TLR4 (caTLR4). The lysates were analyzed using the indicated antisera. (D) Immunoblot analysis of lysates prepared from patient HCC tissue specimens and matched, non-cancerous tissues using the indicated antisera. (Ei) Photomicrographs represent immunostaining of NANOG and p53 in liver specimens of liver cancer patients.
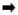 Arrows indicate staining for respective protein. Magnifications X20 and X40. (Eii) Comparison of Immuno Recativity Score (IRS, product of percent of staining and intensity of staining) in tumor vs. non-tumor tissues. (F) Figure represents the level of NANOG and p53 proteins in NANOG transfected Huh7 cells as assessed by immunoblot analysis.
Arrows indicate staining for respective protein. Magnifications X20 and X40. (Eii) Comparison of Immuno Recativity Score (IRS, product of percent of staining and intensity of staining) in tumor vs. non-tumor tissues. (F) Figure represents the level of NANOG and p53 proteins in NANOG transfected Huh7 cells as assessed by immunoblot analysis.
Table 1.
Comparision of immunoreactivity score as measured by immunofluorescence (IRS, product of percent of positive cells and staining intensity) between Tumor vs. Non-Tumor tissues
| Marker | Non-Tumor | Tumor | Difference (Tumor vs. non-Tumor) |
|---|---|---|---|
| pNUMB | 0.76, 1 (0, 4)1 | 3.7, 3 (1, 9)1 | 3.0, 3 (−1, 9) |
| NANOG | 0.76, 1 (0, 4)1 | 3.9, 3 (1, 9)1 | 3.2, 2 (−3, 9) |
| LGL2 | 4.4, 4 (2, 9)1 | 1.2, 1 (0, 4)1 | −3.2, −3 (−8, 1) |
| AURKA | 0.87, 1 (0, 4)1 | 4.2, 4 (1, 8)1 | 3.3, 3 (−1, 8) |
Mean, Median (Range)
Tumor suppressor p53 levels decrease with the increase of NANOG levels in normal, tumor cells and human clinical tissues
As we showed, pNUMB is linked with the levels of NANOG and NUMB has been shown to interact with p53,9 we next investigated if an increase in the levels of NANOG could have any effect on p53 levels. For this purpose, cultured human hepatocytes engineered to express a constitutively active form of Toll-like receptor 4 (caTLR4), an oncogene associated with HCC induction and induces NANOG expression, exhibited increased levels of pNUMB and reduced levels of p53 (Fig. 1C). To validate these data, we carried out immunoblot analysis in human HCC specimens or matched, non-cancerous liver tissue. In the clinical specimens, we found that, in HCC tissues, elevated expression of NANOG corresponded closely with increased phosphorylation of NUMB (Ser 265) and reduced levels of p53 (Fig. 1D). Next, we investigated the relation between NANOG and p53 in the clinical specimens by immunostaining. As shown in Fig. 1Ei–ii, we observed an inverse relationship between two proteins, where p53 expression was highest in normal tissues and NANOG in cancerous tissues. For the mean, median, and range of difference in IRS between tumor vs. non-tumor tissues were presented in Table 2. For the distribution of the percent of staining, intensity of staining, and IRS for each tumor marker were presented in Suppl. Fig 1C–D. A similar trend with decreased p53 levels was observed with the forced expression of NANOG in a time-dependent manner and maximum decline was observed at 72 h after the second transfection at 36 h in human hepatic cancer cell line (Fig. 1F), substantiating the data from the clinical samples and primary hepatocytes.
Table 2.
Comparision of immunoreactivity score as measured by DAB staining (IRS, product of percent of positive cells and staining intensity) between Tumor vs. Non-Tumor tissues
| Marker | Non-Tumor | Tumor | Difference (Tumor vs. non-Tumor) |
|---|---|---|---|
| NANOG | 0.96, 1 (0, 4)1 | 4.5, 4 (1, 9)1 | 3.5, 3 (−2, 9) |
| p53 | 4.9, 6 (2, 12)1 | 0.91, 1 (0, 4)1 | −4.0, −4 (−11, 2) |
Mean, Median (Range)
Consequences of post-translational modification of NUMB by NANOG in TICs and tumor cells: Mechanism of action
NUMB plays a crucial role against the deregulated expansion of tumor-associated cells, and it has been shown that decreased NUMB function correlates with increased cancer growth.8–9 Due to the NANOG mediated increase in the phosphorylation of NUMB and decrease in the level of its interacting protein p53, we hypothesized that this observed effect might be due to (i) modulation aPKCζ-Aurora A Kinase (AURKA) pathway, an upstream pathway for NUMB phosphorylation by NANOG in TICs and Tumor cells and (ii). Phosphorylation of NUMB by NANOG destabilizes NUMB-p53 complex, leading to p53 proteolysis. To test our hypothesis, we conduct following studies.
(i) Modulation of aPKCζ-AURKA kinases by NANOG: Upstream kinases for NUMB phosphorylation
Previous research showed that aPKCζ phosphorylates NUMB and AURKA plays an upstream molecular role in this pathway.23
(a) NANOG activates aPKCζ by repressing LGL-2
Since NANOG was observed to regulate the phosphorylation level of NUMB, we sought to determine if NANOG modulates the expression and kinase activity of aPKCζ. For that purpose, we overexpressed NANOG in TICs and analyzed the level of total aPKCζ and the active phosphorylated form of aPKCζ. We observed that enforced expression of NANOG in CD133+ TICs stimulated the activation of aPKCζ, as indicated by increased steady-state levels of its phosphorylated, catalytically active form (p-aPKCζ) detected by immunoblotting of lysates (Fig. 2A). Next, we determine the link through which NANOG directs activation of aPKCζ, we performed chromatin immunoprecipitation followed by deep sequencing (ChIP-seq) of NANOG-associated DNA sequences in murine liver TICs and matched CD133− control cells (manuscript in preparation). Profiling of NANOG-associated sequences in CD133+ and CD133− cells revealed a unique pattern of enrichment immediately upstream of transcription start sites in CD133+ cells, which was not observed in CD133− controls (Fig. 2B), in line with previous observations that NANOG is selectively induced in CD133+ TICs and drives TIC proliferation. Genome-wide mapping of NANOG-enriched sites identified multiple candidate transcriptional effectors specific to CD133+ TICs. These included the LGL-2 tumor suppressor protein, which restrains aPKCζ activity by suppressing the formation of an active PAR-3/PAR-6/aPKCζ complex. Enforced expression of NANOG in Huh7 cells resulted in a dosage-dependent inhibition of endogenous LGL-2 transcripts, as determined by qRT-PCR (Fig. 2Ci), and decreased levels of LGL-2 protein detected by immunoblotting (Fig. 2Cii). Strikingly, expression of NANOG also potently suppressed the activity of a human LGL-2 reporter, while NANOG depletion resulted in a 75% increase in expression from this construct (p < 0.05). A promoter lacking a 75-bp segment that carries two putative NANOG sequences motifs (−227 to +327 relative to the TSS) was refractory to this increase in activity upon depletion of NANOG (Fig. 2D).
Figure 2. NANOG modulates LGL2 and aPKCζ, a NUMB kinase, expression and activity.

(A) NANOG overexpression promotes phosphorylation of aPKCζ. (B) Read density for NANOG ChIP-seq libraries at the Lgl-2 locus in CD133+ TICs and CD133− controls. A red bar representing a significantly enriched NANOG binding site detected at the Lgl-2 locus is shown. Representations of the annotated mRNA and coding sequencing (CDS) for Lgl-2 are shown at the top. (Ci) NANOG represses LGL-2 expression as measured by qRT-PCR. Error bars represent the SD from at least three independent biological replicates (*P < 0.05). (Cii) NANOG inhibits expression levels of LGL-2 protein. (D) Silencing of NANOG transcriptionally activates LGL-2 promoter. Huh7 cells stably overexpressing NANOG or scrambled shRNA, or sh-NANOG were transduced with the LGL-2 promoter sequences placed upstream of a firefly luciferase reporter gene. Promoter activity is displayed as relative light units (RLU) normalized to the activity of cotransfected Renilla luciferase. Error bars represent the SD from at least three independent biological replicates (**P < 0.05). (Ei–ii) The histogram represents the effect of NANOG-silenced and -overexpression of the kinase activity of aPKCζ in Huh7 cells (Ei) and TICs (Eii). (Fi–ii) The histogram represents the effect of Lipopolysaccharides on aPKCζ kinase activity Huh7 cells (Fi) and TICs (Fii). Each bar in the histogram represents mean ± SD of three independent experiments, * represents P < 0.05.
Since we observed that NANOG suppresses LGL-2, we next asked if NANOG modulates the kinase activities aPKCζ in Huh7 cells and TICs. We employed a two-way approach where NANOG was either suppressed or over-expressed in human hepatocellular cancer cell line Huh7 and TICs (Fig. 2Ei–ii). NANOG-silenced cells showed significantly lower activities of the kinase in comparison to scrambled shRNA-transfected cells (Fig. 2Ei–ii). On the contrary, NANOG-overexpressing cells exhibited significantly increased the rate of activities (Fig. 2Ei–ii). These data established the influence of NANOG on the activities of the kinase in Huh7 and TICs. It has been known that TLR4 is the upstream molecule of NANOG and plays a crucial role in liver carcinogenesis.24 We also observed TLR4 activates phosphorylations of NUMB (Fig 1C). For this reason, we treated both cancer and TICs with Lipopolysaccharide (LPS), a known inducer of NANOG.24 As assessed by kinase activity assays, the treatment of LPS increased the activities of the enzymes in a concentration-dependent manner (Fig. 2Fi–ii). These data suggested that the LPS increased the activities of the kinases in both hepatic cancer cells and TICs.
(b) NANOG acts as a transcriptional activator of AURKA
To determine the mechanism through which NANOG directs activation of aPKCζ, in silico analysis of profiling data identified multiple candidate transcriptional effectors unique to CD133+ cells. These included the AURKA, which acts as an upstream activating kinase of aPKCζ. In Silico, data analysis revealed that AURKA mRNA increases in CD133+ cells, NANOG mRNA levels also increase in a similar manner in CD133+ cells (iPS cells). No such relationship was observed between AURKA and NANOG mRNA levels in CD133− cells (Suppl. Fig. 2C). We used the multiple sequence alignment algorithm Clustal. We searched the human and mouse AURKA genes and associated promoter-proximal sequences for matches to the consensus target motifs recognized by NANOG. Using this in silico approach, we identified three conserved NANOG-binding sites located within 1 kb upstream of the TSS (Fig. 3A). To validate the interaction of NANOG at the AURKA promoter, we performed ChIP-qPCR using NANOG-specific antisera or isotype-matched control IgG in several human liver cancer cell lines (Huh7, Hep3B and HepG2) harboring either non-targeting, scrambled shRNA, or stably expressing a NANOG-targeting shRNA. The choice of these human hepatocellular carcinoma cells was also based on the fact that these cells demonstrate differential p53 status. HepG2 cells express wild p53. Huh7 cells express mutated p53 and Hep3B cells are negative for p53. Therefore, these three cell lines constitute a panel of diverse cellular models for hepatocellular carcinoma based on p53. Through this approach, we confirmed the specific association of NANOG at multiple positions within 1kb upstream of the AURKA transcription start site (TSS), with peak enrichment occurring in the region −757 to −374 bp upstream of the TSS in all these cells. These data, further suggest that NANOG occupies AURKA promoter irrespective of p53 status. To investigate the potential functional significance of NANOG in the regulation of AURKA expression, we used a series of three promoter-reporter constructs in which defined segments of the human AURKA promoter were fused upstream of a firefly luciferase reporter gene. These reporter vectors or the luciferase backbone vector were transfected into Huh7 hepatocellular carcinoma cells stably overexpressing NANOG or stably transduced with a lentivirus shRNA to silence NANOG. Strikingly, expression of NANOG potently induced the AURKA reporter containing a 1486 bp sequence encompassing upstream regulatory and promoter sequences, while NANOG depletion resulted in an 85% decrease in expression from this construct (Fig. 3B, p<0.05). The promoter sequence that harbored mutations in either the first or second candidate NANOG interaction motifs (−757 and −374) exhibited sharply attenuated expression in Huh7 cells (Fig. 3C). We also observed that NANOG and LPS modulate the activities of AURK kinase activity in both cancer cell lines and TICs as we observed with aPKCζ (Fig. 3D–E).
Figure 3. NANOG acts as a transcriptional activator of AURKA and modulates kinase activity.

(A) ChIP-qPCR demonstrates direct association of NANOG at the AURKA promoter. Huh7, HepG2 and Hep3B cells stably expressing NANOG or harboring a non-targeting, scrambled shRNA, or NANOG-targeting shRNA were subjected to Chromatin immunoprecipitation using ChIP grade NANOG antisera or isotype-matched control IgG. Immunoprecipitated chromatin was analyzed by qPCR using primer sets designed to amplify the indicated regions. (B) AURKA promoter reporter assay. Huh7 cells were transfected with the indicated promoter-reporter constructs. (C) Mutation of NANOG-binding sites reduces levels of transactivation. Promoter activity is expressed as relative light units (RLU) normalized to the activity of cotransfected Renilla luciferase. Error bars represent the SD from at least three independent biological replicates (*P < 0.05). (Di–ii) Figures represent the effect of NANOG-silenced and overexpression of the kinase activity of AURKA in Huh7 cells (Di) and TICs (Dii). (Ei–ii) The histogram represents the effect of Lipopolysaccharides (LPS) on AURKA activity Huh7 cells (Ei) and TICs (Eli). Each bar in the histogram represents mean ± SD of three independent experiments, * represents P < 0.05.
(C) Validation in clinical human samples
To validate data, we carried out immunfluorescence analysis in HCC specimens or matched normal liver tissue. In the clinical specimens, we observed that an inverse relationship with NANOG and LGL2 and a positive correlation with AURKA by immunfluorescence (Fig. 4A–B). A significant difference was found in mean IRS for all markers (p<0.001) between tumor vs. non-tumor tissues (Fig. 4C). For the mean, median and range of difference in IRS between tumor vs. non-tumor tissues were presented in Table 1. Collectively, these findings demonstrate a concerted transcriptional activation of AURKA, as well as the repression of human LGL-2 by NANOG. Finally, we also examined the direct affect of AURKA level on NUMB expression and phosphorylation in both CD133+ and CD133− cells. As shown in the Fig. 4Dii, AURKA overexpression in both cells increased the phosphorylation of NUMB without affecting the NUMB levels. The result indicates that AURKA plays an important role in NUMB phosphorylation.
Figure 4. Validation of AURKA and LGL-2 expression in clinical samples.

(A–B) Representative photomicrographs showing the expression of NANOG, AURKA, LGL2 and pNUMB (Ser-265) in human HCCs and adjacent non-cancerous tissues as assessed by an immunofluorescence assay. The merged lane shows the co-localization of 2 proteins. DAPI was used as a nuclear staining control. (C) Comparison of Immuno Reactivity Score (IRS, the product of a percent of staining and intensity of staining) in tumor vs. non-tumor tissues p<0.05, n=45. (D) CD133+ and CD133− cells were transfected with empty vector or AURKA expression vectors and were resolved by SDS-PAGE followed by immunoblotting using the indicated antibodies.
(ii) Phosphorylation of NUMB by NANOG destabilizes NUMB-p53 complex, leading to p53 proteolysis
(a) Phosphorylation of NUMB dstabilize NUMB-p53 complex
Phosphorylation of NUMB by the aPKCζ abolishes its association with the NUMB-associated proteins α-Adaptin, Integrin-β, and Par-3,23 prompting us to consider whether such a mode of regulation extends to the NUMB-p53 interaction. Further, NUMB shields p53 from MDM2-mediated proteolysis and is required for the correct execution of cell division in stem cells.8 For this reason, we sought to determine whether the elevated level of pNUMB due to the increased expression of NANOG and can impact the stability of the NUMB-p53 complex. While MYC-tagged p53 was efficiently co-immunoprecipitated with endogenous NUMB, co-expression of NANOG destabilized this interaction (Fig. 5Ai). These findings suggest that NANOG acts as an important player to destabilize the NUMB-p53 complex. As aPKCζ phosphorylates NUMB, we sought to examine if aPKCζ expression dissociates p53 from NUMB. As expected, expression of a myristoylated, constitutively active forms of aPKCζ (CA-aPKCζ) dislodged MYC-p53 from its association with NUMB in a dosage-dependent manner (Fig. 5Aii). Accordingly, steady-state levels of MYC-p53 were diminished in the absence of Nutlin-3 (10 μM), a selective small-molecule inhibitor of MDM2, upon expression of CA-aPKCζ while a dominant-negative allele of aPKCζ increased the level of co-expressed MYC-p53 (Fig. 5B). Expression of NUMB variants harboring alanine substitutions in the three serine residues that serve as the primary aPKCζ phosphorylation sites (Flag-NUMB-3A)23 stabilized its association with MYC-p53, while a mutant form of NUMB harboring the phosphomimetic residue aspartic acid in these positions (Flag-NUMB-3D) displayed sharply reduced binding to MYC-p53 (Fig. 5Ci). Expression of Flag-NUMB-3A rescued p53 levels in the presence of elevated NANOG while Flag-NUMB-3D failed to do so (Fig. 5Cii).
Figure 5. NANOG triggers destabilization of the NUMB-p53 complex through aPKCζ-mediated phosphorylation of NUMB. NANOG induces the proteolysis of p53 in TICs, and its expression is inversely related to p53 in cells and clinical samples.

(Ai) CD133+ TICs were transfected with empty vector or the indicated combinations of MYC-p53 and NANOG, followed by exposure to Nutlin-3 for 24h. Cell lysates were immunoprecipitated using anti-NUMB antibody and analyzed by SDS-PAGE and immunoblotting. (Aii) TICs were transfected with MYC-p53 and either empty vector or increasing amounts of CA-aPKCζ. Following exposure to Nutlin-3, lysates were prepared and subjected to immunoprecipitation using an anti-NUMB antibody. (B) Murine TICs were transfected with MYC-p53 together with empty vector, dominant-negative aPKCζ (dn-aPKCζ), constitutively active, aPKCζ (CA-aPKCζ). Lysates were resolved by SDS-PAGE and analyzed by immunoblotting. (Ci) (Upper panel) Structures of wild type, unphosphorylatable mutant NUMB-3A and phosphomimetic mutant of NUMB-3D. PTB: phosphotyrosine-binding domain, PRR: proline-rich region. (Ci, lower panel) Interaction of MYC-p53 with Flag-NUMB variants. TICs were transfected with empty vector or NANOG, then analyzed by SDS-PAGE and immunoblotting. Flag-NUMB or the indicated phosphorylation site variants of Flag-NUMB were expressed alone or with MYC-p53, followed by lysis and immunoprecipitation using an anti-MYC antibody. Proteins were resolved by SDS-PAGE and analyzed by immunoblotting. Lysates represent 10% of the input volume used in the immunoprecipitation. (Cii) TICs were transfected with empty vector or NANOG along with the indicated variants of Flag-NUMB, followed by lysis and immunoblotting. (Di–ii) CD133+/CD49f+ murine liver TICs were stably transduced with pG13-luc (Di) or p21-luc (Dii), in the absence or presence of p53 and with increasing amounts of NANOG expression vector. Promoter activity is expressed as relative light units (RLU) normalized to the activity of cotransfected Renilla luciferase. Error bars represent the SD from at least three independent biological replicates (**P< 0.05). (Ei) Lysates prepared from CD133+/CD49f+ TICs transfected with p53, NANOG, or both expression vectors were resolved by SDS-PAGE followed by immunoblotting using the indicated antibodies. (Eii) In vitro proteolysis assay. Purified, recombinant GST-p53 was incubated for 30 minutes in buffer alone (lane 1) or in equal amounts of lysates prepared from TICs transfected with either empty vector or NANOG at the indicated ratios. (F) TICs were transfected with NANOG or empty vector, followed by exposure to Nutlin-3 (10μM) or vehicle for 24h. Cell lysates were analyzed by immunoblotting using an anti-p53 antibody. Ponceau S stain (bottom) serves as a loading control.
As NANOG acts as an upstream molecule for NUMB phosphorylation pathway, we investigated if NANOG modulates p53 trans-activation activity. To monitor p53 activity, we employed reporter constructs containing either the pG13 promoter, which carries thirteen tandem p53-responsive elements, or the native p21 promoter, both of which confer p53-dependent expression of firefly luciferase (Fig 5Di–ii). As expected, expression of p53 in CD133+ TICs stimulated the expression of luciferase from each construct, and exposure of cells to genotoxic agent doxorubicin (100 ng/ml) further augmented p53 trans-activation (Fig. 5Di). Strikingly, co-expression of NANOG resulted in dosage-dependent decreases in luciferase activity (Fig. 5DiI). Immunoblot analysis showed that lysates prepared from TICs transfected with p53 in presence or absence of recombinant NANOG revealed a sharp decrease in p53 levels (Fig. 5Ei). Next, we examined the mechanism by which NANOG antagonizes p53, purified GST-p53 in cytoplasmic lysates prepared from TICs transfected with either empty vector or NANOG expressing vector were incubated. Strikingly, GST-p53 underwent rapid proteolysis upon incubation in lysates prepared from NANOG-overexpressing TICs while no such effect was observed using control lysates (Fig. 5Eii). The MDM2 oncoprotein, a RING domain-containing E3 ubiquitin ligase, is a central regulator of p53 stability, prompting us to examine whether it is required for the NANOG-mediated destabilization of p53. Although cellular levels of MDM2 and its upstream regulator p19 ARF were unchanged following the enforced expression of NANOG (data not shown), exposure of TICs to Nutlin-3, blocked the destabilization of p53 by NANOG (Fig. 5F), suggesting that NANOG stimulates the catalytic activity of MDM2 towards p53.
Phosphorylation of NUMB promotes self-renewal in TIC
Our findings suggest that NANOG stimulates the aPKCζ-dependent phosphorylation of NUMB, resulting in the destabilization of the NUMB-p53 complex and degradation of p53 (Fig. 6A). Next, we sought to determine the functional significance of NANOG-mediated, aPKCζ-dependent phosphorylation of NUMB in TIC self-renewal, by carrying out a colony formation assay in which TICs stably expressing NANOG in the absence or presence of NUMB-3A or NUMB-3D. We found that a stable expression of NUMB-3A efficiently suppressed NANOG-induced self-renewal and colony formation while NUMB-3D failed to do so (Fig. 6Bi–ii). Exposure of NANOG-overexpressing TICs to a small molecule inhibitor of AURKA (i-Aura), a kinase that can act upstream of aPKCζ,23 also potently inhibited colony formation in these TICs, indicating that aPKCζ-mediated NUMB phosphorylation promotes self-renewal of NANOG-expressing TICs. We next asked if the pro-growth role of NUMB phoshorylation is anyhow affect the CD133 expression, a cell surface marker used to distinguish TICs and non-TICs. The frequency of CD133+ cells as measured by Flow cytometry using the anti-CD133, did not significantly change among the groups (Suppl. Fig 2D). These data also suggest that NUMB phosphorylation does not influence the CD133 expression, and the pro-growth role is due higher self-renewal of the TICs.
Figure 6. aPKCζ-dependent phosphorylation of the NUMB-p53 complex promotes self-renewal and tumorigenesis.

(A) Conceptual model of NANOG-mediated oncogenesis in TICs. (Bi–ii) Methylcellulose colony formation assay. TICs stably expressing the indicated combinations of NANOG and Flag-NUMB were seeded in low adhesion methylcellulose media and cultured for one week, then harvested and reseeded for a total of four platings. The number of colonies that formed for each cell type is indicated. (C) Tumor formation titration assay. TICs (102–105) stably expressing the indicated transgenes or lentivirus shRNAs were implanted subcutaneously into the dorsal hind flanks of NOG mice and tumor growth was monitored for 60 days. Tumors greater than 25 mm3 and which exhibited growth progression during the course of the study were scored as positive. (Di) Tumor growth kinetics. TICs were implanted subcutaneously into NOG mice, and tumor volumes were measured on the indicated days. (Dii) Representative photomicrographs showing the expression of CD133 in cancer cells derived xenograft and TICs derived xenograft tissues as assessed by an immunostaining.
Arrows indicate staining for respective protein. Magnifications X20 and X40. (E) Postulated mechanism of self-renewal ability through NANOG-AURKA-pNUMB pathways.
Phosphorylation of NUMB promotes Tumorigenesis in Mouse Model
To assess the impact of NANOG-induced, aPKCζ-mediated NUMB phosphorylation on tumor initiation, we carried out tumor-initiation titration studies in which defined numbers of TICs (102-5×105) were implanted subcutaneously into immuno-compromised NOG (NOD/Shi-scid/IL-2Rγ null) mice and monitored for tumor formation over the course of a 30-day period. While implantation of as few as 100 NANOG-overexpressing TICs resulted in rapid tumor formation (2/4 mice), co-expression of NUMB-3A but not NUMB-3D reversed this potent tumourigenicity such that a minimum of 104 TICs were required for tumor formation (Fig. 6C). In accordance with findings from the in vitro colony formation assays, treatment of mice with i-Aurka (10 mg/kg) potently suppressed NANOG-induced tumorigenesis (Fig. 6Di). Next, we asked if the pro-growth role of NUMB phoshorylation is anyhow affecting the CD133 expression in vivo. We did not observe any difference of CD133 staining among the groups. Although, we observed a significant difference on CD133 staining pattern between tissues from tumor cells derived xenografts and TICs derived xenografts (Fig. 6Dii). These data further suggest that NUMB phosphorylation does not influence the CD133 expression, and the pro-growth role is due higher self-renewal of the TICs.
Discussion
The success of cancer treatment is dependent on the cellular death by apoptosis, a process in which p53 plays a significant role. It has been known that increasing p53 sensitizes cancer cells and deletion or mutation cause therapy resistance.25 The cellular level of p53 is controlled by MDM2, which is in turn regulated by NUMB, as NUMB inactivates the E3 ubiquitin ligase MDM2, which prevents p53 ubiquitylation and degradation.8 It has been observed that NUMB directly interacts with p53, a primary control switch of cell cycle progression whose function is frequently impaired in different cancers.8,9 Here, we demonstrate for the first time that the NUMB interacts with p53 mainly in the non-phosphorylated form and phosphorylation of its Ser residues by aPKCζ dissociates it from p53. We observed that higher NANOG expression leads to increased NUMB-phosphorylation, and subsequently decreased p53 levels. Our findings reveal a post-transcriptional mechanism of NUMB-p53 complex destabilization mediated by NANOG through AURKA-aPKCζ kinases.
Expression of NANOG is associated with poor clinical outcomes in a broad array of cancers20,26 and is implicated in the deregulated expansion of tumor cells. Earlier, it was observed that LGL2 inhibits NUMB phosphorylation, whereas AURKA promotes NUMB phosphorylation during cell division to ensure asymmetric segregation.27 The involvement of NANOG as both a positive regulator of AURKA and transcriptional repressor of LGL-2 suggests a mechanistic link between the pluripotency, cell polarity and the control of self-renewability. Further, we demonstrated that lipopolysaccharides (LPS) treatment, a known inducer of NANOG via TLR4 receptor,24 activates both AURKA and aPKCζ activities. These data prove that TLR4-NANOG signaling pathway is involved in NUMB phosphorylation. Our findings add a layer of complexity to this regulatory relationship, revealing that AURKA also acts to antagonize p53 indirectly through the activation of aPKCζ, resulting in the destabilization of the NUMB-p53 complex.
In conclusion, we demonstrate for the first time the functional cooperation among NANOG, NUMB, and p53 involved in the development of liver cancer through the proliferation of TICs. We have identified a mechanism of regulation of the NUMB-p53 interaction and have shown that AURKA-aPKCζ mediated Ser phosphorylation of NUMB plays a crucial role in self-renewal and liver tumorigenesis. It has been demonstrated that NUMB was phosphorylated by aPKCζ on three Ser residues near the PTB domain to which p53 binds.10 We demonstrated that upon phosphorylation by aPKCζ; NUMB lost its ability to bind p53. Since phosphorylated NUMB is incapable of binding p53, these results in enhanced p53 ubiquitination, likely by MDM2,28 and the subsequent degradation. NUMB-bound p53 is resistant to ubiquitination and degradation, leading to increased sensitivity to iAURK. Our findings with patient tumor specimens also suggest that, in tumors that exhibit elevated expression of NANOG, treatment with inhibitors of AURKA warrant consideration as a potential useful therapeutic strategy. In this direction, further in depth in vivo and clinical studies are warranted to verify this suggestion and are currently under investigation in our laboratory. In a nutshell, we observed that phospho-dependent disengagement of the NUMB-p53 tumor suppressor complex led to the destabilization of p53 and deregulated self-renewal in TICs. Our findings are summarized in a model presented in Figure 6E that depicts how NUMB phosphorylation, which is mediated by NANOG, destabilize the NUMB-p53 complex and subsequent higher self-renewal of TICs. Thus, NANOG-NUMB-p53 signaling axis might be an important regulatory pathway for TICs event in TICs self-renewal and live tumorigenesis and suggest a therapeutic strategy by targeting NUMB-phosphorylation.
Supplementary Material
Acknowledgments
Financial Support:
This work was supported by grants from the National Cancer Institute (P01 CA123228) and the National Institute on Alcohol Abuse and Alcoholism (R01 AA018857, P50 AA11999). The work is also supported in part by award number P30CA014089 from the National Cancer Institute. P50AA11999 (Animal Core, Morphology Core, and Pilot Project Program), R24AA012885 (Non-Parenchymal Liver Cell Core). D.F. was supported by a postdoctoral scholarship from the California Institute of Regenerative Medicine. This research is also supported by a Research Scholar Grant, RSG-12-177-01-MPC and pilot funding (IRG-58-007-48) from American Cancer Society. Imaging study was performed by the Cell and Tissue Imaging Core of the USC Research Center for Liver Diseases (P30 DK048522).
Tissue pathological slide preparation was performed by Ms. Moli Chen of Norris Comprehensive Cancer Center. We also thank Prof. Dennis Strand (Johannes Gutenberg University, Germany) and Prof. Yoshiaki Ishigatsubo (Yokohama City University, Japan) for generously providing the reporter plasmids for LGL2 and AURKA promoter, respectively. FACS analysis was performed by the Flow Cytometry Core at USC Keck Medical Center and Cell Isolation Core, Research Center for Liver Diseases. Statistical analysis was performed by Dr. Susan Groshen and Ms. Lingyun Ji in Norris Comprehensive Cancer Center Biostatistics Core. Confocal microscopy services were provided by the Cell and Tissue Imaging Core of the USC Research Center for Liver Diseases, NIH grants P30 DK048522 and S10 RR022508. Confocal microscopy services were provided by the Cell and Tissue Imaging Core of the USC Research Center for Liver Diseases, NIH grants P30 DK048522 and S10 RR022508.
List of Abbreviations
- TICS
Tumor-initiating cells
- LPS
Lipopolysaccharide
- AURKA
Aurora A kinase
- aPKCζ
atypical protein kinaseCζ
- ChIP
Chromatin Immunprecipitation
- IRS
Immunoreactivity score
- TLR4
Toll-like receptor 4
- HCC
Hepatocellular carcinoma tumor specimens
Footnotes
Disclosures of Potential Conflict of Interest: Authors have nothing to disclose.
Authors’ Contributions
Conception and design: H. R. Siddique, D. Feldman and K. Machida
Development of methodology: H. R. Siddique, D. Feldman, and K. Machida
Acquisition of data: D. Feldman, H. R. Siddique, K. Machida, and C. Chen
Analysis and interpretation of data: V. Punj, D. Feldman, H.R. Siddique and K. Machida
Writing and revision of the manuscript: H. R. Siddique, D. Feldman, V. Punj, and K. Machida.
Administrative, technical, or material support: H. Tokumitsu
Study supervision: K. Machida
References
- 1.Hunter T. Signaling-2000 and beyond. Cell. 2000;100:113–27. doi: 10.1016/s0092-8674(00)81688-8. [DOI] [PubMed] [Google Scholar]
- 2.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–25. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 3.Cuesta N, Martín-Cófreces NB, Murga C, van Santen HM. Receptors, signaling networks, and disease. Sci Signal. 2011;4:mr3. doi: 10.1126/scisignal.2001687. [DOI] [PubMed] [Google Scholar]
- 4.Cohen P. The twentieth century struggle to decipher insulin signalling. Nat Rev Mol Cell Biol. 2006;7:867–73. doi: 10.1038/nrm2043. [DOI] [PubMed] [Google Scholar]
- 5.Boulikas T. The phosphorylation connection to cancer. Int J Oncol. 1995;6:271–8. [PubMed] [Google Scholar]
- 6.Kepp O, Semeraro M, Bravo-San Pedro JM, Bloy N, Buqué A, Huang X, et al. eIF2α phosphorylation as a biomarker of immunogenic cell death. Semin Cancer Biol. 2015 doi: 10.1016/j.semcancer.2015.02.004. pii: S1044–579X. [DOI] [PubMed] [Google Scholar]
- 7.Mu Y, Zang G, Engström U, Busch C, Landström M. TGFβ-induced phosphorylation of Par6 promotes migration and invasion in prostate cancer cells. Br J Cancer. 2015 doi: 10.1038/bjc.2015.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Colaluca IN, Tosoni D, Nuciforo P, Senic-Matuglia F, Galimberti V, Viale G, et al. NUMB controls p53 tumour suppressor activity. Nature. 2008;451:76–80. doi: 10.1038/nature06412. [DOI] [PubMed] [Google Scholar]
- 9.Carter S, Vousden KH. A role for Numb in p53 stabilization. Genome Biol. 2008;9:221. doi: 10.1186/gb-2008-9-5-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dhami GK, Liu H, Galka M, Voss C, Wei R, Muranko K, et al. Dynamic methylation of Numb by Set8 regulates its binding to p53 and apoptosis. Mol Cell. 2013;50:565–76. doi: 10.1016/j.molcel.2013.04.028. [DOI] [PubMed] [Google Scholar]
- 11.Aparicio S, Eaves CJ. p53: a new kingpin in the stem cell arena. Cell. 2009;138:1060–1062. doi: 10.1016/j.cell.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 12.Kawamura T, Suzuki J, Wang YV, Menendez S, Morera LB, Raya A, et al. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature. 2009;460:1140–1144. doi: 10.1038/nature08311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marión RM, Strati K, Li H, Murga M, Blanco R, Ortega S, et al. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature. 2009;460:1149–1153. doi: 10.1038/nature08287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Y, Feng H, Gu H, Lewis DW, Yuan Y, Zhang L, et al. The p53-PUMA axis suppresses iPSC generation. Nat Commun. 2013;4:2174. doi: 10.1038/ncomms3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bric A, Miething C, Bialucha CU, Scuoppo C, Zender L, Krasnitz A, et al. Functional identification of tumor-suppressor genes through an in vivo RNA interference screen in a mouse lymphoma model. Cancer Cell. 2009;16:324–335. doi: 10.1016/j.ccr.2009.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.March HN, Rust AG, Wright NA, ten Hoeve J, de Ridder J, Eldridge M, et al. Insertional mutagenesis identifies multiple networks of cooperating genes driving intestinal tumorigenesis. Nat Genet. 2011;43:1202–1209. doi: 10.1038/ng.990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Siddique HR, Saleem M. Role of BMI1, a stem cell factor, in cancer recurrence and chemoresistance: preclinical and clinical evidences. Stem Cells. 2012;30:372–8. doi: 10.1002/stem.1035. [DOI] [PubMed] [Google Scholar]
- 18.Visvader JE. Cells of origin in cancer. Nature. 2011;469:314–322. doi: 10.1038/nature09781. [DOI] [PubMed] [Google Scholar]
- 19.Amson R, Pece S, Lespagnol A, Vyas R, Mazzarol G, Tosoni D, et al. Reciprocal repression between P53 and TCTP. Nat Med. 2012;18:91–99. doi: 10.1038/nm.2546. [DOI] [PubMed] [Google Scholar]
- 20.Chen CL, Tsukamoto H, Liu JC, Kashiwabara C, Feldman D, Sher L, et al. Reciprocal regulation by TLR4 and TGF-β in tumor-initiating stem-like cells. J Clin Invest. 2013;123:2832–2849. doi: 10.1172/JCI65859. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Carlsson G, Gullberg B, Hafström L. Estimation of liver tumor volume using different formulas - an experimental study in rats. J Cancer Res Clin Oncol. 1983;105:20–23. doi: 10.1007/BF00391826. [DOI] [PubMed] [Google Scholar]
- 22.StataCorp. Stata Statistical Software: Release 11. College Station, TX: StataCorp LP; 2009. [Google Scholar]
- 23.Nishimura T, Kaibuchi K. Numb controls integrin endocytosis for directional cell migration with aPKC and PAR-3. Dev Cell. 2007;13:15–28. doi: 10.1016/j.devcel.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 24.Machida K, Feldman DE, Tsukamoto H. TLR4-Dependent Tumor-Initiating Stem Cell-Like Cells (TICs) in Alcohol-Associated Hepatocellular Carcinogenesis. Adv Exp Med Biol. 2015;815:131–144. doi: 10.1007/978-3-319-09614-8_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guntur VP, Waldrep JC, Guo JJ, Selting K, Dhand R. Increasing p53 protein sensitizes non-small cell lung cancer to paclitaxel and cisplatin in vitro. Anticancer Res. 2010;30:3557–3564. [PubMed] [Google Scholar]
- 26.Ibrahim EE, Babaei-Jadidi R, Saadeddin A, Spencer-Dene B, Hossaini S, Abuzinadah M, et al. Embryonic NANOG activity defines colorectal cancer stem cells and modulates through AP1- and TCF-dependent mechanisms. Stem Cells. 2012;30:2076–2087. doi: 10.1002/stem.1182. [DOI] [PubMed] [Google Scholar]
- 27.Wirtz-Peitz F, Nishimura T, Knoblich JA. Linking cell cycle to asymmetric division: Aurora-A phosphorylates the Par complex to regulate Numb localization. Cell. 2008;135:161–173. doi: 10.1016/j.cell.2008.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brooks CL, Gu W. p53 regulation by ubiquitin. FEBS Lett. 2011;585:2803–2809. doi: 10.1016/j.febslet.2011.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.