

Abstract
Malformations of cortical development constitute a variety of pathological brain abnormalities that commonly cause severe, medically-refractory epilepsy, including focal lesions, such as focal cortical dysplasia, hetereotopias, and tubers of tuberous sclerosis complex, and diffuse malformations, such as lissencephaly. Although some cortical malformations result from environmental insults during cortical development in utero, genetic factors are increasingly recognized as primary pathogenic factors across the entire spectrum of malformations. Genes implicated in causing different cortical malformations are involved in a variety of physiological functions, but many are focused on regulation of cell proliferation, differentiation, and neuronal migration. Advances in molecular genetic methods have allowed the engineering of increasingly sophisticated animal models of cortical malformations and associated epilepsy. These animal models have identified some common mechanistic themes shared by a number of different cortical malformations, but also revealed the diversity and complexity of cellular and molecular mechanisms that lead to the development of the pathological lesions and resulting epileptogenesis.
Keywords: seizure, mice, focal cortical dysplasia, heterotopia, lissencephaly, tuberous sclerosis
1. Introduction
Malformations of cortical development represent a spectrum of pathological abnormalities of the brain, which are commonly associated with epilepsy (Aronica et al., 2012; Guerrini and Dobyns, 2014). Depending on the type of cortical malformation, epilepsy is typically severe, often with onset of seizures early in childhood or infancy. In addition, epilepsy associated with malformation of cortical development is frequently intractable or resistant to available anti-seizure medications. Patients with localized, isolated cortical malformations, such as focal cortical dysplasia, may be candidates for epilepsy surgery (Hauptman and Mathern, 2012). However, many malformations of cortical development, such as lissencephaly, have extensive, bilateral involvement of the cortex and generate generalized or multifocal seizures, making epilepsy surgery difficult or impossible. Furthermore, even in patients with localized malformations, the actual region of cortical disorganization may extend microscopically beyond the gross anatomical lesion apparent on imaging tests, often leading to failure of epilepsy surgery. Thus, understanding the pathogenesis of malformations of cortical development and the associated mechanisms of epileptogenesis is critical for developing more effective therapies for epilepsy related to these cortical malformations.
Although some malformations of cortical development are caused by environmental insults that occur during cortical development in utero, genetic factors also play a critical role in the pathogenesis of many cortical malformations (Guerrini and Dobyns, 2014). With the explosion in diagnostic technologies in genetics, specific genetic mutations are increasingly identified as causes of different malformations of cortical development. Recognized gene defects may disrupt a variety of different cellular and molecular processes during cortical development. However, many of these genes are involved in relatively selective functions, such as regulation of cell proliferation and survival, cell cycle, and neuronal migration.
The impact of a genetic mutation in causing a particular type of cortical malformation also depends on the stage of cortical development that is most directly affected. Cortical development is divided into at least three, overlapping stages: cellular proliferation and differentiation, neuronal migration, and postmigrational cortical organization. The standard classification scheme for cortical malformations is based on the stage at which cortical development is first disrupted (Barkovich et al., 2012; Barkovich et al., 2009). Although there are numerous types of malformations of cortical development, only a few categories are most commonly utilized in clinical practice. For example, most types of focal cortical dysplasia and dysplastic megalencephaly are typically considered defects in cellular proliferation and differentiation. The spectrum of lissencephalies and heterotopias likely results from disorders of neuronal migration. Polymicrogyria may primarily relate to abnormalities in postmigrational cortical organization. Despite this logical mechanistic-based classification scheme, it is increasingly recognized that these categorizations are overly simplified. There is considerable overlap between different types of malformations of cortical development, and some may result from defects in multiple stages of cortical development. To a large extent, advances in genetics, and the associated biological pathways involved, will help refine the classification system and provide insights into the true mechanistic relationships between different types of cortical malformations.
Animal models, particularly transgenic mice, represent powerful tools for investigating genetic mechanisms of cortical malformations and epilepsy. Many genetic mutations causing human cortical malformations and epilepsy have been engineered in mice and other model systems. Although there are significant limitations to the degree and fidelity to which animal models may mimic human pathology and physiology, animal models have begun to reveal important, translationally-relevant insights into the pathogenesis of cortical malformations and associated mechanisms of epileptogenesis. In this paper, we will review selected examples of animal models of malformations of cortical development and epilepsy, representing different types within the mechanistic classification scheme, and discuss general limitations and future directions for these models.
2. Animal Models of Different Types of Malformations of Cortical Development
2.1 Focal Cortical Dysplasia
Focal cortical dysplasia is a common cause of drug-resistant focal epilepsy and often identified in pathological specimens resected from patients receiving epilepsy surgery. Pathologically, focal cortical dysplasia is primarily characterized by discrete localized regions of disorganized cortical lamination and morphologically abnormal cells. A standardized classification system for focal cortical dysplasia has been proposed, including three main tiers (Blumcke et al., 2011; Marin-Valencia et al., 2014). Type I primarily involves localized areas of abnormal cortical layering. In addition to cortical dyslamination, Type II is characterized by dysmorphic neurons, with the subtype IIb also containing stereotypic, undifferentiated balloon cells. Finally, Type III is associated with other dual pathologies, such as hippocampal sclerosis, developmental tumors, and vascular malformations. Many cases of focal cortical dysplasia Type I and Type III may result from late postmigrational cortical defects or injuries. In contrast, focal cortical dysplasia Type II represents the prototypical malformation of cortical development presumed to result from primary defects in cell proliferation and differentiation and has been strongly linked to genetic mutations in pathways controlling these functions, such as the mammalian target of rapamycin (mTOR) pathway.
2.1.1 Irradiation Model in Rats
In utero irradiation of rats serves as a model of Type Ib human focal cortical dysplasia. Although it has been included in this review article for comparison, it is not a genetic model of epilepsy. It is an injury-based model. Exposing fetal rats in utero to about 200 cGy of external radiation on embryonic day 17 (E17) results in offspring with diffuse (not focal) cortical abnormalities that include microcephaly, thinning of the cortical mantle, loss of lamination of the neocortex, and spatial disorientation of the neurons within the neocortex (Cowan and Geller, 1960; Roper et al. 1995). In addition, the animals show nodular periventricular and subcortical heterotopia. They also show focal areas of ectopic neurons in the CA1 region of the hippocampus, something that is not commonly seen in human malformations of cortical development. The mechanism of creating the dysplasia involves the selective killing of a large number of cells in the fetal cerebral hemispheres followed by an attempt by the surviving cells to create the cortex in an altered environment. Immature, migrating neuroblasts are particularly sensitive to the radiation (Altman et al., 1968). Radial glia in the developing cortex are also severely damaged (Roper et al. 1997). Since the radial glia serve as the guidance structures for many of the neurons that migrate to the cortical plate, it is not surprising that many of the abnormalities seen in this model may reflect disordered neuronal migration. The progenitor cells in the ventricular and subventricular zones are relatively spared by the radiation-induced cell death as are the more mature neurons that already reside in the cortical plate by E17. Although the effects of the radiation produce a diffuse cortical dysplasia, there are some gradations in terms of severity of the histological abnormalities. The dysplasia is much more pronounced near the midline and gradually diminishes laterally near the rhinal sulcus. It is also more pronounced as one goes from anterior to posterior in the cerebral hemispheres.
Irradiated rats have spontaneous seizures as documented in both EEG and video EEG studies (Kellinghaus et al., 2004; Kondo et al., 2001). However the frequency and severity of the seizures in this model appear to be mild relative to many other model of epilepsy; also the percentage of animals that demonstrate spontaneous seizures may be lower than in most models of epilepsy. Irradiated rats also show increased susceptibility to seizures in response to pilocarpine (Setkowicz et al., 2003). Irradiated rats show learning and memory impairment relative to both hippocampal and neocortical dysfunction (Zhou et al., 2011) and long-term potentiation is impaired in the CA1 region of the hippocampus in irradiated rats (Zhou and Roper, 2012). This means that this model recapitulates one of the most common and debilitating co-morbidities seen in humans with intractable epilepsy.
Although a number of histological and physiological abnormalities have been found in irradiated rats, the biggest change in these animals involves damage to the inhibitory interneurons of the neocortex with subsequent loss of inhibition in the cortical circuitry. GABAergic, inhibitory interneurons of the neocortex can be classified based on protein markers and intrinsic firing patterns (Kawaguchi and Kubota, 1997). Most of the cortical interneurons are comprised of 3 types: parvalbumin-immunoreactive (PV-ir), somatostatin-immunoreactive (SS-ir) and calretinin-immunoreactive (CR-ir). Mature irradiated rats have about a 50% loss of PV-ir and SS-ir interneurons based on density (relative to total neuron counts) as well as absolute numbers based on stereological cell counting studies (Akakin et al., 2013; Deukmedjian et al., 2004). This is seen in physiological recordings as a loss of spontaneous and miniature inhibitory postsynaptic current (IPSCs) in the excitatory pyramidal cells of the central “layers” of the dysplastic cortex (Zhu and Roper, 2000). This loss of inhibition results in an imbalance of excitation and inhibition in the dysplastic cortex and likely predisposes to seizures and an increased propensity for epileptiform activity. In addition, the surviving PV-ir and SS-ir interneurons in dysplastic cortex have a relative loss of excitatory post-synaptic currents (Zhou et al. 2009; Zhou and Roper 2011). This correlated with a reduced resting firing rate in those interneurons as well (Zhou and Roper, 2011). These abnormalities would cause further impairment of inhibition in the dysplastic cortex. Finally, at the level of paired whole cell recordings from nearby neurons in the neocortex, irradiated rats show decreased strength of inhibitory synaptic connections between PV-ir interneurons and pyramidal cells (Zhou and Roper, 2014). Taken together, these studies demonstrate that a single in utero insult can produce a permanent injury that results in a profound and multifactorial impairment in the inhibitory system of the neocortex. We feel that these findings may have direct implications for human Type Ib focal cortical dysplasia as well as other malformations of cortical development that may result from in utero and perinatal injury.
2.1.2 Pten
In contrast to the irradiation model, other animal models of focal cortical dysplasia have been developed that involve specific genetic mutations linked to human focal cortical dysplasia. Although there may be a variety of genetic mechanisms associated with focal cortical dysplasia, a recurring theme that has emerged in recent years is a common link to the mechanistic/mammalian target of rapamycin (mTOR pathway), a relatively ubiquitous cell signaling pathway that controls cell growth and proliferation. Genetic mutations in signaling molecules that are upstream modulators of the mTOR pathway leads to abnormal, hyperactivation of the mTOR pathway, which then drives increased cell growth and proliferation, causing many of the pathological features of focal cortical dysplasia, as well as other related malformations of cortical development, such as hemimegalencephaly (Figure 1). One such gene, DEPDC5, has recently been found to cause autosomal dominant familial focal epilepsy with variable foci, including cases of isolated focal cortical dysplasia (Scheffer et al., 2014). Another gene that has been particularly well-studied in animal models with epilepsy is PTEN, the phosphatase and tensin homolog gene.
Figure 1.
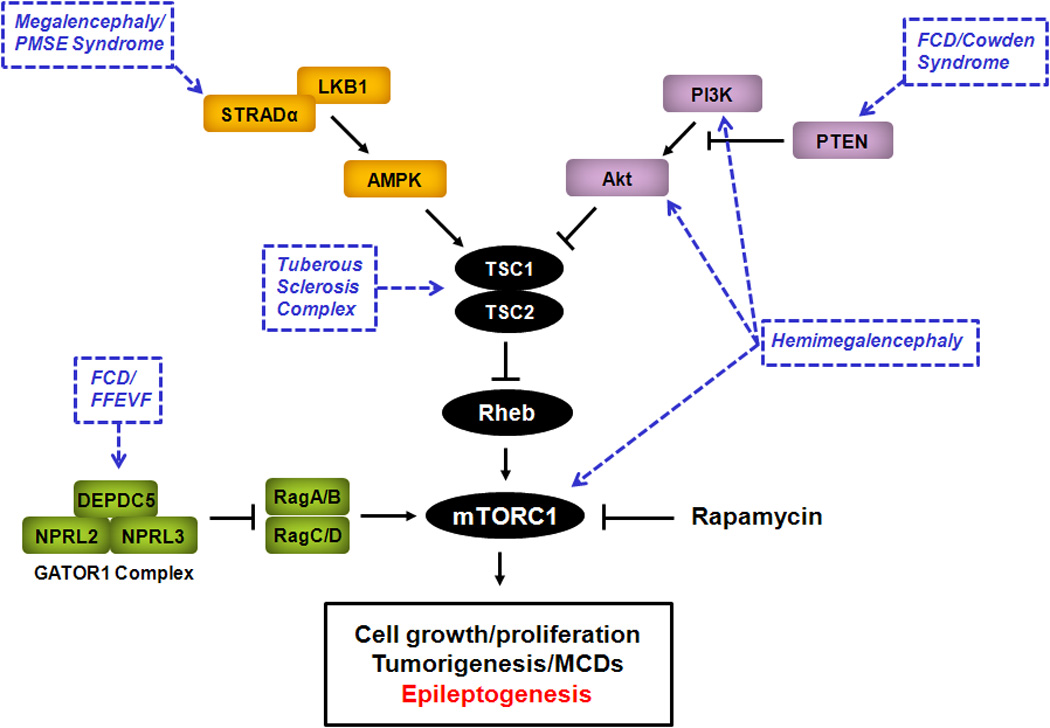
The mTOR pathway as a central signaling pathway causing malformations of cortical development and epilepsy. The mTOR pathway involves two complexes, the rapamycin-sensitive, mTORC1, and the relatively rapamycin-insensitive, mTORC2 (not shown), is regulated by a number of upstream signaling pathways, and controls many important downstream functions, including cell growth and proliferation. Genetic mutations in different components of the mTOR pathway have been identified that cause abnormal activation of mTORC1 and may lead to various malformations of cortical development and epilepsy syndromes involving increased cell growth and proliferation (blue). Inhibition of mTORC1 by rapamycin or other mTORC1 inhibitors may represent a rational therapy for epilepsy and malformations of cortical development due to abnormal mTOR pathway activation. AMPK - 5' adenosine monophosphate-activated protein kinase; DEPDC5 - Dishevelled, Egl-10 and Pleckstrin domain-containing protein 5; FCD - focal cortical dysplasia; MCD - malformation of cortical development; FFEVF - familial focal epilepsy with variable foci; GATOR1 - GTPase-activating protein activity towards Rags; mTORC1 - mammalian target of rapamycin complex 1; NPRL2 - nitrogen permease regulator 2-like protein; NPRL3 - nitrogen permease regulator 3-like protein; PI3K - phosphoinositide-3 kinase; PMSE - polyhydramnios, megalencephaly, symptomatic epilepsy; PTEN - phosphatase and tensin homolog on chromosome 10; Rheb - Ras homolog enriched in brain; STRADα - STE20-related kinase adapter alpha; TSC1 - tuberous sclerosis complex 1 protein; TSC2 - tuberous sclerosis complex 2 protein.
PTEN mutations have been linked to several human disorders, some of which involve cortical dysplasia and epilepsy, such as Cowden syndrome and isolated focal cortical dysplasia (Cheung et al., 2014; O'Rourke et al., 2012). Several Pten knock-out mouse models have been developed involving inactivation of Pten in neurons or astrocytes in the brain, using cell promoter-targeted Cre-Lox mediated recombination of the Pten gene (Backman et al., 2001; Kwon et al., 2003; Ljungberg et al., 2009; Sunnen et al., 2011; Zhou et al., 2009). Pathologically, the primary features of these models are megalencephaly, hypertrophic neurons, and cellular proliferation, which at least partially mimic cellular features of focal cortical dysplasia. However, one significant limitation of these mouse models are that the cellular abnormalities are diffuse, leading to megalencephaly of the entire brain; these models to not appear to recapitulate the localized cortical lesions of focal cortical dysplasia.
The different Pten knock-out mice are consistent in featuring severe epilepsy. The specific mechanisms by which Pten gene inactivation and the associated cellular abnormalities in growth and proliferation actually cause epileptogenesis are not well-understood. However, consistent with the predicted regulation of the mTOR pathway by PTEN, epilepsy in Pten mice is dependent on mTOR activation, as seizures can be suppressed by rapamycin, an mTOR inhibitor, in these mice (Kwon et al., 2003; Ljungberg et al., 2009; Sunnen et al., 2011; Zhou et al., 2009). As also seen in other syndromes involving malformations of cortical development described below, the mTOR pathway may hold the key to understanding the downstream mechanisms of epileptogenesis in focal cortical dysplasia and other cortical malformations. Most importantly, from a therapeutic standpoint, mTOR inhibitors represent a novel mechanistic class of drugs that may not only be effective for treating seizures, but could have anti-epileptogenic properties for preventing epilepsy related to malformations of cortical development (Wong, 2013, 2010).
2.2 Tubers/Tuberous Sclerosis Complex
Tuberous sclerosis complex (TSC) is an autosomal dominant genetic disorder characterized by hamartoma and tumor formation in multiple organs due to mutation of either the TSC1 or TSC2 gene (Crino et al., 2006; Orlova and Crino, 2010). TSC represents one of the most common genetic causes of epilepsy, with up to 90% of patients with TSC having epilepsy, and two-thirds of those having drug-resistant epilepsy (Chu-Shore et al., 2010). Epilepsy in TSC is typically related to cortical tubers, as surgical removal of tubers can sometimes eliminate seizures in some TSC patients, although the specific cellular and molecular mechanisms of epileptogenesis in TSC are still incompletely understood and are not necessarily dependent on tubers in all cases (Wong, 2008). Pathologically, tubers closely resemble focal cortical dysplasia Type IIB, including the presence of undifferentiated giant cells, analogous to balloon cells of focal cortical dysplasia Type IIB. Similar to focal cortical dysplasia Type IIB, tubers are hypothesized to represent a primary defect in neuroglial proliferation and differentiation during early cortical development. As the TSC genes are negative regulators of mTOR, hyperactivation of the mTOR pathway due to TSC gene mutations provides a rational mechanistic basis for abnormal cell growth and proliferation causing tumors and other developmental lesions in TSC. mTOR inhibitors are already a proven, approved treatment for brain and kidney tumors in TSC (Bissler et al., 2013; Franz et al., 2013; Krueger et al., 2010).
Numerous animal models of TSC have been developed involving spontaneous or induced inactivation of the Tsc1 or Tsc2 gene, with varying degrees of pathological brain abnormalities and evidence of hyperexcitability or seizures (Table 1). The classic Eker rat, with a spontaneous heterozygous Tsc2 mutation, has a very mild neurological phenotype, with very rare cerebral hamartomas that resemble tubers and slightly increased kindling of chemical-induced seizures, but no spontaneous epilepsy (Waltereit et al., 2006). Similarly, adult mice with heterozygyous Tsc1 or Tsc2 mutations have not been reported to have pathological brain abnormalities or seizures, although they do have learning deficits (Ehninger et al., 2008; Goorden et al., 2007). Recently, however, young Tsc1+/− heterozygous mice have been reported to have seizures during a restricted developmental time period (Lozovaya et al., 2014). While conventional homozygous Tsc KO mice are embryonic lethal, viable mice with severe neurological manifestations, including pathological abnormalities and epilepsy, have consistently been generated with homozygous inactivation of Tsc1 or Tsc2 targeted specifically to subsets of brain cell types, such as neurons (Fu et al., 2012; Meikle et al., 2007; Normand et al., 2013), astrocytes (Uhlmann et al., 2002; Zeng et al., 2011) or neuroglial progenitor cells (Carson et al., 2012; Goto et al., 2011; Kassai et al., 2014; Magri et al., 2011; Magri et al., 2013; Way et al., 2009). Phenotypically, many of these brain-targeted Cre-mediated knock-out mice feature progressive epilepsy, and some have also been documented to have impaired learning and potential autistic/social deficits. The more severe phenotypes of homozygous knock-out mice compared with heterozygous mutants suggest that a “second hit” or loss of heterozygosity may be required for neurological manifestations of TSC. Similar to the Pten knock-out mice, many of the TSC models share pathological features of megalencephaly, cellular hypertrophy, and proliferation, which can be directly attributed to mTOR pathway hyperactivation. Using specific cell-targeting, some models not surprisingly find that Tsc1 gene inactivation in neurons is sufficient to cause epilepsy, but other models also implicate non-neuronal cells, such as astrocytes. In addition, on a molecular level, defects in neurotransmitter receptors, transporters, and ion channels are found in some of these models, which may directly affect neuronal excitability and seizure susceptibility (Jansen et al., 2005; Lozovaya et al., 2014; Wong et al., 2003; Zeng et al., 2007).
Table 1.
Animal Models of Tuberous Sclerosis Complex
| Brain Pathological Abnormalities | Neurological Phenotype | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Animal Model /Genotype (Cell type targeted) |
Focal (F)/ Diffuse (D) |
Megalencephaly | Cytomegaly | Other | Epilepsy | Cognitive deficits |
Other | Reversal by rapamycin |
References |
| Eker rat (Tsc2+/−) | F/− | − | + | rare cerebral hamartomas | − | − | increased PTZ kindling | n/a | Waltereit et al. 2006 |
| Adult Tsc1+/− mice | − | − | − | no pathological abnormalities | − | + | impaired social behavior | n/a | Goorden et al. 2007 |
| Adult Tsc2+/− mice | − | − | − | no pathological abnormalities | − | + | + | Ehninger et al. 2008 | |
| Infant/juvenile Tsc1+/− mice | − | − | − | no pathological abnormalities | + | n/a | seizures at P9-P18 only | n/a | Lozovaya et al. 2014 |
| Synapsin-Tsc1 CKO mice (neurons) |
D | − | + | enlarged dysplastic neurons decreased myelination |
+ | n/a | + |
Meikle et al. 2007 Meikle et al. 2008 |
|
| CamKII-Tsc1 CKO mice (neurons) |
D | + | + | neuronal hypertrophy, astrogliosis |
n/a | n/a | + | Ehninger et al. 2008 | |
| Dlx5/6-Tsc1 CKO mice (GABAergic interneurons) |
D | − | + | decreased GABAergic interneurons, cluster of ectopic interneurons |
− | n/a | decreased seizure threshold to fluorythyl |
n/a | Fu et al. 2012 |
| Inducible Gbx2-Tsc1 CKO mice (thalamic neurons) |
F | − | + | thalamic neuron cytomegaly, altered thalamocortical projections |
+ | n/a | n/a | Normand et al. 2013 | |
| L7-Tsc1 CKO mice (cerebellar purkinje neurons) |
F | − | + | reduced number, increased size of cerebellar purkinje neurons |
n/a | n/a | impaired social behavior, repetitive behaviors |
+ | Tsai et al. 2012 |
| Gfap-Tsc1 CKO mice (primarily astrocytes) |
D | + | + | astrocyte proliferation, neuronal dispersion, abnormal Glt1, Kir expression |
+ | + | + |
Uhlmann et al. 2002 Wong et al. 2003 Jansen et al. 2004 Zeng et al. 2007, 2008 |
|
| Gfap-Tsc2 CKO mice (primarily astrocytes) |
D | + | + | astrocyte proliferation, neuronal dispersion |
+ | n/a | + | Zeng et al. 2011 | |
| Gfap2-Tsc1 CKO mice (radial glial progenitor cells) |
D | + | + | abnormal cortical lamination, astrogliosis, decreased myelination |
+ | n/a | + | Magri et al. 2013 | |
| Gfap2-Tsc2 CKO mice (radial glial progenitor cells) |
D | + | + | abnormal cortical lamination, astrogliosis, decreased myelination |
+ | n/a | + |
Way et al. 2009 Way et al. 2012 |
|
| Emx1-Tsc1 CKO mice (neural progenitor cells) |
D | + | + | abnormal cortical lamination astrogliosis, decreased myelination |
+ | n/a | + |
Magri et al. 2011 Carson et al. 2012 |
|
| Inducible nestin-Tsc1 CKO mice (neuroprogenitor cells) |
D | + | + | giant cells, dendritic abnormalities increased ER stress |
+ | n/a | + | Goto et al. 2011 | |
| In utero electroporation Tsc1 Mice (neuroprogenitor cells) |
F | − | + | Heterotopic nodules with | − | n/a | decreased seizure threshold to PTZ |
n/a | Feliciano et al. 2011 |
| Inducible Cag-Tsc1 CKO adult mice (all cells) |
− | − | − | rare giant cells, but otherwise no pathological abnormalities |
+ | n/a | increased neuronal excitability and LTP |
+ | Abs et al. 2013 |
n/a – not applicable or not reported
While the mechanisms of epileptogenesis in these mouse models of TSC may be multifactorial, abnormal mTOR activity may be a central trigger for the range of pathological and molecular defects, given mTOR’s involvement in regulating cell growth, proliferation, and protein synthesis. mTOR inhibitors can reverse most of these molecular and pathological abnormalities, and can also prevent epilepsy, consistent with an antiepileptogenic effect (Carson et al., 2012; Goto et al., 2011; Meikle et al., 2008; Way et al., 2012; Zeng et al., 2011; Zeng et al., 2008). These preclinical studies have started to be translated into TSC patients, as mTOR inhibitors are now being tested in clinical trials for epilepsy in TSC (Krueger et al., 2010; Krueger et al., 2013).
Despite the large number and variety of animal models of TSC available, one significant limitation to most of these models is the failure to recapitulate focal lesions of tubers and related focal cortical dysplasia. This limitation may be related to a couple of factors. First, since rodent cortex is normally lissencephalic, the natural lack of gyri and sulci may not provide the necessary substrate to allow localized lesions to be generated. More importantly, the typical experimental approaches for inducing gene inactivation may not have sufficient spatial specificity. Conventional knock-out mice involve gene inactivation typically affecting large populations of cells throughout extensive regions of the cortex and brain in general. In contrast, more advanced, spatially-targeted methods for localized gene inactivation may allow for the development of focal lesions. For example, in utero electroporation to inactivate Tsc1 in selected neuronal populations leads to discrete focal lesions containing cytomegalic neurons in cortex, which may be a more realistic model of tubers in mice (Feliciano et al., 2011). However, although there is evidence of lower seizure threshold, spontaneous seizures have not been reported in these mice, indicating that these lesions may not be sufficient to cause epilepsy in this model.
On the other hand, another novel animal TSC-related model suggests that gross pathological lesions are not necessary to produce seizures. Temporal control of gene inactivation utilizing a tamoxifen-estrogen receptor-Cre system allows for knock-out of Tsc1 gene in a normal adult brain (Abs et al., 2013). This leads to development of seizures within a few days, in the absence of overt pathological changes, indicating that gross pathological lesions are not necessary to cause epilepsy and instead implicating non-structural cellular and molecular defects in promoting epileptogenesis in TSC.
Overall, the variety of animal models of TSC demonstrate the complexity and diversity of mechanisms that may be involved in generating tubers and other pathological abnormalities in TSC, as well as in promoting epileptogenesis. There are likely both shared (e.g., cell growth and proliferation) and distinct (e.g., cell-specific channels) mechanisms contributing to epileptogenesis in different knockout mice targeting different cell types. On the molecular level, the mTOR pathway is central to generating a range of pathological, cellular, and molecular abnormalities that may contribute to lesion formation and associated epilepsy. However, while different animal models have implicated a range of cellular and molecular players, an overall mechanistic scheme accounting for tuber formation and epileptogeneisis remains elusive.
2.3 Megalencephaly
While focal cortical dysplasia Type IIb and cortical tubers of TSC are localized, focal malformations of cortical development featuring defects in cell proliferation and differentiation, a spectrum of more extensive cortical malformations involving megalencephaly also share similar pathological features and may involve common mechanisms of dysregulated brain growth and proliferation (Crino, 2007; Mirzaa and Poduri, 2014). In particular, hemimegalencephaly is characterized by disordered cortical lamination and enlarged balloon cells, similar to focal cortical dysplasia Type IIb and tubers. The mTOR pathway has again been identified as a common pathogenic mechanism for these related malformations of cortical development (Figure 1), as somatic mutations in mTOR or upstream regulators of mTOR, such as PI3 kinase or AKT, have been found in brain specimens of patients with hemimegalencephaly and intractable epilepsy (Lee et al., 2012; Poduri et al., 2012). In addition, a rare, newly-described autosomal recessive disorder, polyhydramnios, megalencephaly, and symptomatic epilepsy syndrome (PMSE) or pretzel syndrome, also features megalencephaly and other cortical malformations and is caused by mutations in the LYK5/STRADA gene, which leads to mTOR hyperactivation (Figure 1) (Orlova et al., 2010). Treatment with rapamycin led to a reduction in seizures in several patients with PMSE (Parker et al., 2013).
Animal models of hemimegalencephaly involving identified human mutations in the mTOR pathway have not been specifically developed. However, both Pten and Tsc1 KO mice should have similar molecular effects in causing dysregulated mTOR signaling via inactivation of upstream regulators of mTOR. In fact, as mentioned above, most Pten and Tsc1 KO mice do not exhibit focal abnormalities resembling focal cortical dysplasia or tubers, but rather show diffuse cellular hypertrophy, proliferation, and increased cortical size (Goto et al., 2011; Kwon et al., 2003; Ljungberg et al., 2009; Uhlmann et al., 2002; Zeng et al., 2011); thus, these models are probably better models of megalencephaly. In addition, an animal model of PMSE syndrome has been developed by knocking down STRADA in mouse cortex, which results in aberrant cortical lamination (Orlova et al., 2010; Parker et al., 2013).
2.4 Heterotopias
While abnormalities in cellular proliferation and differentiation during early cortical development may underlie a spectrum of focal and more diffuse malformations of cortical development, including focal cortical dysplasia Type II, cortical tubers, and hemimegalencephaly, heterotopias likely result from defects of cortical migration. During cortical development, most neurons are generated in the germinal ventricular zone and migrate to their final position in the cortex in an “inside-out” fashion, with the later neurons migrating past earlier neurons to form outer layers of cortex. Heterotopias appear to result from an abnormal arrest of migration, so that collections of neurons end up in an inappropriate location, such as in subcortical white matter. Similar to the spectrum between focal cortical dysplasia and megalencephaly, heterotopias range from nodular, with a small focal collection of misplaced neurons, to band heterotopias, which may consist of a thin zone or layer of dense heterotopic neurons in white matter running parallel to the cortex (“double cortex”). Although some genes for nodular heterotopia have been identified, many cases of isolated nodular heterotopia may be related to environmental insults. However, a genetic basis for band heterotopia has been strongly established and may overlap with other, more extensive malformations of cortical development, such as lissencephaly. In particular, doublecortin was one of the first genes implicated in causing malformations of cortical development, including both subcortical band heterotopia and lissencephaly (Gleeson et al., 1998). The doublecortin (DCX) gene is located on the X-chromosome, and mutations cause severe X-linked lissencephaly in boys and milder subcortical band heterotopia in girls. Consistent with a potential role in cell motility and migration, DCX has been found to function as a microtubule-binding and stabilizing protein.
A couple of rodent models of DCX-related heterotopias have been developed. Dcx KO mice surprisingly show no obvious lamination defects in cortex, but the hippocampus shows a double layer of pyramidal neurons, which in some respects mimic the abnormal migration of heterotopic neurons (Nosten-Bertrand et al., 2008). Furthermore, these Dcx and other related KO mice have spontaneous seizures arising from hippocampus (Kerjan et al., 2009; Nosten-Bertrand et al., 2008). Other experimental approaches have attempted to model the cortical lesions of heterotopia. In utero electroporation of RNAi against Dcx in rats leads to formation of subcortical band heterotopias (Ramos et al., 2006). Although spontaneous seizures have not been reported in this model, rats with subcortical band heterotopia do have a decreased seizure threshold to pentylenetetrazole (Manent et al., 2009). Besides the pathological abnormalities, the specific cellular and molecular mechanisms leading to epileptogenesis in these models are not well-defined.
2.5 Lissencephaly
Lissencephaly represents one of the most severe malformations of cortical development and is almost uniformly associated with drug-resistant epilepsy and intellectual disability, often including infantile spasms. With absent or greatly simplified gyri, lissencephaly is the prototypical cortical malformation attributed to defects in neuronal migration and all cases are presumed to be genetic. A number of genes have been identified as causing different types of lissencephaly. Not surprisingly, the function of many of these genes relates to cell motility, in particular microtubule-dependent molecular motors. Several subtypes of lissencephaly have been recognized based on pathological features, as distinguished by the number of layers of cortex affected and differences in an anterior versus posterior predominance. While X-linked lissencephaly due to DCX mutations tend to have anterior predominance with four-layered cortex, many other forms of lisscephaly have posterior predominance, such as caused by LIS1 or TUBA1A mutations (Lo Nigro et al., 1997; Poirier et al., 2007). Another X-linked lissencephaly caused by ARX mutations is characterized by three-layered cortex and agenesis of the corpus callosum (Kitamura et al., 2002).
Several animal models of lissencephaly have been created based on genetic mutations causing human lissencephaly. As the normal rodent cortex is already lissencephalic, development of an accurate gross pathological model of lissencephaly in rodents is essentially impossible. However, cellular and molecular abnormalities due to the implicated gene mutations can be investigated. For example, insertion of a heterozygous human Lis1 mutation into mice causes heterotopic hippocampal pyramidal neurons, similar to Dcx KO mice, and increased neuronal excitability and seizures (Fleck et al., 2000; Greenwood et al., 2009). Several mouse models incorporating Arx mutations have been generated. These models show aberrant cortical lamination or defects in interneurons (Kitamura et al., 2009; Marsh et al., 2009; Price et al., 2009). Spontaneous seizures occur in some of these models, including spasm-like seizures in pups (Price et al., 2009). From a translational standpoint, treatment with estrogen during an early developmental time window decreased the emergence of spasms and later seizures in adulthood, while restoring the loss of inhibitory interneurons thought to be related to impaired migration (Olivetti et al., 2014). Although very early, prenatal intervention would be required to prevent lissencephaly in people, these animal studies at least provide proof of principle that epileptogenesis related to even the most severe malformations of cortical development could potentially be modulated therapeutically.
2.6 Polymicrogyria
Polymicrogyria, or excessive small gyri, is perhaps the most heterogeneous and poorly-delineated among the more common malformations of cortical development (Stutterd and Leventer, 2014). Like other types of cortical malformations, there is a range of severity of polymicrogyria, from very focal, unilateral forms to extensive bilateral involvement. Furthermore, polymicrogyria may occur concurrently with other malformations of cortical development, such as schizencephaly and heterotopia. Traditionally, many cases of polymicrogyria were thought to result from environmental insults during later stages of cortical development, typically during cortical organization following neuronal migration. However, some genetic causes have recently been identified. First, there is overlap and phenotypic heterogeneity in genes causing lissencephaly, particularly those involving tubulin proteins that constitute microtubules (TUBA1A, TUBB2B). While some patients with TUBA1A or TUBB2B mutations may have a classic lissencephaly phenotype, others with the same mutations may exhibit a polymicrogyria pattern in appearance. Recently, a specific syndrome, bilateral frontoparietal polymicrogyria, which features a characteristic cobblestone pattern of the cortex, has been attributed to the GPR56 gene, a large G-protein coupled receptor (Piao et al., 2004). Only a few mouse models incorporating mutations in polymicrogyria-associated genes have been reported. The gross pathological features of these models do not necessarily resemble human polymicrogyria. Furthermore, epilepsy has not been investigated or reported in these models. Mutations in the Tubb2b gene in mice leads to cortical thinning, ventriculomegaly, and perinatal lethality associated with increased apoptosis of cortical neurons (Stottmann et al., 2013). GPR56 knock-out mice exhibit ectopic cortical neurons due to overmigration of neurons through defective pial membranes (Li et al., 2008). But again, the pathophysiological consequences of these abnormalities related to epilepsy have not been reported.
3. Discussion/Conclusions
A number of animal models of malformations of cortical development exist that almost match the range and diversity of human cortical malformations. With advances in genetic technology, an expanding number of genetic etiologies have been identified in causing various malformations of cortical development. Transgenic mice incorporating these human genetic mutations represent appropriate models of the genetic basis of these malformations of cortical development. Some of these models also recapitulate the pathological, cellular, and molecular abnormalities and clinical features (e.g., epilepsy) observed in the corresponding human disorders. In a couple instances, this has led to identification of potential therapeutic targets for epileptogenesis related to malformations of cortical development, such as in the mTOR pathway. However, there are significant limitations to most of these animal models in mimicking the full range of pathological and clinical features of human MCDs, and there is an obvious need to develop better experimental approaches and models of MCDs for the future.
3.1 Limitations of Animal Models of Malformations of Cortical Development
Transgenic mice can accurately reproduce the precise genetic defects causing human malformations of cortical development. However, the subsequent phenotypes resulting from these genetic manipulations in animals are more variable in their ability to precisely model the specific clinical and pathological features of human malformations of cortical development. In particular, rodent models of most malformations of cortical development are limited in the degree to which the gross pathological lesions of malformations of cortical development can be recapitulated. For example, while cellular features of cytomegaly are reliably produced in most genetic models of malformations of cortical development related to abnormal proliferation and differentiation, most animal models of focal cortical dysplasia and TSC-related tubers show diffuse pathological abnormalities and do not exhibit focal lesions seen in the human disorders. Similarly rodent models incorporating gene mutations implicated in polymicrogyria do not necessarily exhibit the gross pathological features of numerous small gyri. Perhaps the most fidelity has been obtained in modeling heterotopias, especially the double cortical layers of band heterotopias, but less progress has been made in recapitulating nodular heterotopias in animals. The obvious differences in gross structure between normal rodent and human cortex, particularly the normally lissencephalic cortex of rodents, suggest that the rodent brain may lack the necessary structural substrate to fully recapitulate many, if not all, human malformations of cortical development. Attempting to model lissencephaly effectively faces the opposite problem – as rodent cortex normally lacks gyri at baseline, the effect of genetic mutations in inducing lissencephaly in a brain that normally has gyri cannot be fully assessed in rodent models.
Despite these limitations in mimicking some of the gross pathological features of human malformations of cortical development, rodent models still offer many advantages in investigating cellular and molecular features of human cortical malformations. A prototypic example is the establishment of the role of the mTOR pathway in mouse models of TSC, Pten, and now several other malformations of cortical development. Although most of the relevant animal models do not recapitulate the focal pathological lesions seen in these malformations of cortical development, the basic finding of cellular and molecular dysregulation of the mTOR pathway in these models has definite clinical relevance to these disorders, as well as potential therapeutic applications in the use of mTOR inhibitors.
Even if the pathological lesions of human malformations of cortical development can be accurately mimicked in animal models, there are additional limitations in assessing mechanisms of epileptogenesis in these models. Lesional epileptogenesis may involve at least two mechanistic components or stages. First are the mechanisms disrupted during cortical development that cause the formation of the lesion, and secondly are the mechanisms that actually produce neuronal hyperexcitability and seizures. Of course, these mechanisms may be overlapping, but they may also be distinct or sequential, especially given that all malformations of cortical development are presumably formed by birth, but many affected patients do not present with seizures until after the neonatal period. Thus, studies of animal models of cortical malformation-related epilepsy should attempt to distinguish those mechanisms involved in lesion formation versus hyperexcitability.
Related to this mechanistic distinction, it is often debated in cases of focal malformations of cortical development, such as focal cortical dysplasia, tubers, and heterotopias, whether seizures start within the lesions themselves or in the perilesional regions (Wong, 2008). If the perilesional hypothesis is correct, the specific pathological and cellular features of the lesion itself may not be critical. Rather, a more non-specific disruption of normal networks adjacent to the lesion may account for seizure generation. Furthermore, alterations in the biochemical or molecular properties in the perilesional region may be key. Thus, complete studies of epileptogenesis in models of malformations of cortical development should include both lesional and non-lesional/peri-lesional mechanisms.
3.2 Future Directions
Given the limitations of rodent models of malformations of cortical development, there are a number of future directions to advance the field. Continued efforts to more precisely mimic the pathological lesions of human malformations of cortical development are needed in different animal models. This will likely involve refinement of temporally and spatially-controlled gene inactivation techniques, including in utero electroporation and inducible gene knockout of specific cell types in selected brain regions during critical periods of brain development. Given the intrinsic limitations of lissencephalic cortex in rodents, this might also extend to using more advanced vertebrate species. Beyond the pathological lesions, careful characterization of perilesional regions and their contribution to epileptogenesis should be investigated more thoroughly. In particular, direct comparisons between any microscopic cellular, molecular, and electrophysiological abnormalities of the perilesional regions with the gross pathological lesions should shed insight on the interactions and relative importance of the lesion and perilesional areas in epilepsy. Furthermore, it will be important to understand the role of other genetic and environmental modifying factors in contributing to phenotypical heterogeneity with different malformations of cortical development resulting from mutations in the same gene. Finally, more detailed characterization of the behavioral phenotypes of animal models of malformations of cortical development, which, in the absence of overt, documented epilepsy, may be relatively subtle and require more sensitive behavioral assays.
The ultimate translational application of animal models of disease is to facilitate development of novel or improved treatments. Again animal models have already provided some insights into potential biochemical targets for treating epilepsy associated with some malformations of cortical development, particularly with mTOR inhibitors. The variety of genetic mutations linked to different malformations of cortical development provides additional opportunities for animal models to investigate other signaling pathways and molecular mechanisms that may have therapeutic applications. Besides identifying other potential drug targets, genetic and molecular analysis of animal models may provide insights into the mechanisms of drug resistance that commonly occurs with epilepsy due to many malformations of cortical development. Finally, even surgical approaches to epilepsy and malformations of cortical development can be improved with a better understanding of the extent of the true lesion, gross and microscopic, that needs to be resected to maximize seizure freedom following epilepsy surgery. The continued development and refinement of genetic models of malformations of cortical development should lead to significant advances in the treatment of cortical malformation-related epilepsy.
Highlights.
Genetic animal models mimic the spectrum of human malformations of cortical development.
Advanced genetic methods have engineered increasingly sophisticated animal models of cortical malformations.
Distinct and overlapping mechanisms of epileptogenesis occur across different animal models of cortical malformations.
Animal models have identified novel therapeutic targets for epilepsy, such as the mTOR pathway.
Footnotes
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Abs E, Goorden SM, Schreiber J, Overwater IE, Hoogeveen-Westerveld M, Bruinsma CF, Aganovic E, Borgesius NZ, Nellist M, Elgersma Y. TORC1-dependent epilepsy caused by acute biallelic Tsc1 deletion in adult mice. Ann Neurol. 2013;74:569–579. doi: 10.1002/ana.23943. [DOI] [PubMed] [Google Scholar]
- 2.Akakin D, Martinez-Diaz H, Chen HX, Roper SN. Reduced densities of parvalbumin-and somatostatin-expressing interneurons in experimental cortical dysplasia and heterotopia in early postnatal development. Epilepsy Res. 2013;104:226–233. doi: 10.1016/j.eplepsyres.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 3.Altman J, Anderson WJ, Wright KA. Differential radiosensitivity of stationary and migratory primitive cells in the brains of infant rats. Exp Neurol. 1968;22:52–74. doi: 10.1016/0014-4886(68)90019-8. [DOI] [PubMed] [Google Scholar]
- 4.Aronica E, Becker AJ, Spreafico R. Malformations of cortical development. Brain pathology (Zurich, Switzerland) 2012;22:380–401. doi: 10.1111/j.1750-3639.2012.00581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Backman SA, Stambolic V, Suzuki A, Haight J, Elia A, Pretorius J, Tsao MS, Shannon P, Bolon B, Ivy GO, Mak TW. Deletion of Pten in mouse brain causes seizures, ataxia and defects in soma size resembling Lhermitte-Duclos disease. Nature genetics. 2001;29:396–403. doi: 10.1038/ng782. [DOI] [PubMed] [Google Scholar]
- 6.Barkovich AJ, Guerrini R, Kuzniecky RI, Jackson GD, Dobyns WB. A developmental and genetic classification for malformations of cortical development: update 2012. Brain. 2012;135:1348–1369. doi: 10.1093/brain/aws019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barkovich AJ, Millen KJ, Dobyns WB. A developmental and genetic classification for midbrain-hindbrain malformations. Brain. 2009;132:3199–3230. doi: 10.1093/brain/awp247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bissler JJ, Kingswood JC, Radzikowska E, Zonnenberg BA, Frost M, Belousova E, Sauter M, Nonomura N, Brakemeier S, de Vries PJ, Whittemore VH, Chen D, Sahmoud T, Shah G, Lincy J, Lebwohl D, Budde K. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet. 2013;381:817–824. doi: 10.1016/S0140-6736(12)61767-X. [DOI] [PubMed] [Google Scholar]
- 9.Blumcke I, Thom M, Aronica E, Armstrong DD, Vinters HV, Palmini A, Jacques TS, Avanzini G, Barkovich AJ, Battaglia G, Becker A, Cepeda C, Cendes F, Colombo N, Crino P, Cross JH, Delalande O, Dubeau F, Duncan J, Guerrini R, Kahane P, Mathern G, Najm I, Ozkara C, Raybaud C, Represa A, Roper SN, Salamon N, Schulze-Bonhage A, Tassi L, Vezzani A, Spreafico R. The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc Task Force of the ILAE Diagnostic Methods Commission. Epilepsia. 2011;52:158–174. doi: 10.1111/j.1528-1167.2010.02777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carson RP, Van Nielen DL, Winzenburger PA, Ess KC. Neuronal and glia abnormalities in Tsc1-deficient forebrain and partial rescue by rapamycin. Neurobiol Dis. 2012;45:369–380. doi: 10.1016/j.nbd.2011.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheung KM, Lam CW, Chan YK, Siu WK, Yong L. Atypical focal cortical dysplasia in a patient with Cowden syndrome. Hong Kong medical journal = Xianggang yi xue za zhi / Hong Kong Academy of Medicine. 2014;20:165–167. doi: 10.12809/hkmj133863. [DOI] [PubMed] [Google Scholar]
- 12.Chu-Shore CJ, Major P, Camposano S, Muzykewicz D, Thiele EA. The natural history of epilepsy in tuberous sclerosis complex. Epilepsia. 2010;51:1236–1241. doi: 10.1111/j.1528-1167.2009.02474.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crino PB. Focal brain malformations: a spectrum of disorders along the mTOR cascade. Novartis Foundation symposium. 2007;288:260–272. doi: 10.1002/9780470994030.ch18. discussion 72–81. [DOI] [PubMed] [Google Scholar]
- 14.Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. The New England Journal of Medicine. 2006;355:1345–1356. doi: 10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]
- 15.Cowan D, Geller LM. Long-term pathological effects of prenatal X-irradiation on the central nervous system of the rat. J Neuropathol Exp Neurol. 1960;19:488–527. doi: 10.1097/00005072-196010000-00002. [DOI] [PubMed] [Google Scholar]
- 16.Deukmedjain AJ, King MA, Cuda C, Roper SN. The GABAergic system of the developing neocortex has a reduced capacity to recover from in utero injury in experimental cortical dysplasia. J Neuropathol Exp Neurol. 2004;63:1265–1273. doi: 10.1093/jnen/63.12.1265. [DOI] [PubMed] [Google Scholar]
- 17.Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, Kwiatkowski DJ, Ramesh V, Silva AJ. Reversal of learning deficits in a Tsc2+/− mouse model of tuberous sclerosis. Nature medicine. 2008;14:843–848. doi: 10.1038/nm1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feliciano DM, Su T, Lopez J, Platel JC, Bordey A. Single-cell Tsc1 knockout during corticogenesis generates tuber-like lesions and reduces seizure threshold in mice. The Journal of clinical investigation. 2011;121:1596–1607. doi: 10.1172/JCI44909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fleck MW, Hirotsune S, Gambello MJ, Phillips-Tansey E, Suares G, Mervis RF, Wynshaw-Boris A, McBain CJ. Hippocampal abnormalities and enhanced excitability in a murine model of human lissencephaly. J Neurosci. 2000;20:2439–2450. doi: 10.1523/JNEUROSCI.20-07-02439.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Franz DN, Belousova E, Sparagana S, Bebin EM, Frost M, Kuperman R, Witt O, Kohrman MH, Flamini JR, Wu JY, Curatolo P, de Vries PJ, Whittemore VH, Thiele EA, Ford JP, Shah G, Cauwel H, Lebwohl D, Sahmoud T, Jozwiak S. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. 2013;381:125–132. doi: 10.1016/S0140-6736(12)61134-9. [DOI] [PubMed] [Google Scholar]
- 21.Fu C, Cawthon B, Clinkscales W, Bruce A, Winzenburger P, Ess KC. GABAergic interneuron development and function is modulated by the Tsc1 gene. Cerebral cortex (New York, N.Y.: 1991) 2012;22:2111–2119. doi: 10.1093/cercor/bhr300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gleeson JG, Allen KM, Fox JW, Lamperti ED, Berkovic S, Scheffer I, Cooper EC, Dobyns WB, Minnerath SR, Ross ME, Walsh CA. Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell. 1998;92:63–72. doi: 10.1016/s0092-8674(00)80899-5. [DOI] [PubMed] [Google Scholar]
- 23.Goorden SM, van Woerden GM, van der Weerd L, Cheadle JP, Elgersma Y. Cognitive deficits in Tsc1+/− mice in the absence of cerebral lesions and seizures. Ann Neurol. 2007;62:648–655. doi: 10.1002/ana.21317. [DOI] [PubMed] [Google Scholar]
- 24.Goto J, Talos DM, Klein P, Qin W, Chekaluk YI, Anderl S, Malinowska IA, Di Nardo A, Bronson RT, Chan JA, Vinters HV, Kernie SG, Jensen FE, Sahin M, Kwiatkowski DJ. Regulable neural progenitor-specific Tsc1 loss yields giant cells with organellar dysfunction in a model of tuberous sclerosis complex. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:E1070–E1079. doi: 10.1073/pnas.1106454108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Greenwood JS, Wang Y, Estrada RC, Ackerman L, Ohara PT, Baraban SC. Seizures, enhanced excitation, and increased vesicle number in Lis1 mutant mice. Ann Neurol. 2009;66:644–653. doi: 10.1002/ana.21775. [DOI] [PubMed] [Google Scholar]
- 26.Guerrini R, Dobyns WB. Malformations of cortical development: clinical features and genetic causes. Lancet Neurol. 2014;13:710–726. doi: 10.1016/S1474-4422(14)70040-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hauptman JS, Mathern GW. Surgical treatment of epilepsy associated with cortical dysplasia: 2012 update. Epilepsia. 2012;53(Suppl 4):98–104. doi: 10.1111/j.1528-1167.2012.03619.x. [DOI] [PubMed] [Google Scholar]
- 28.Jansen LA, Uhlmann EJ, Crino PB, Gutmann DH, Wong M. Epileptogenesis and reduced inward rectifier potassium current in tuberous sclerosis complex-1-deficient astrocytes. Epilepsia. 2005;46:1871–1880. doi: 10.1111/j.1528-1167.2005.00289.x. [DOI] [PubMed] [Google Scholar]
- 29.Kassai H, Sugaya Y, Noda S, Nakao K, Maeda T, Kano M, Aiba A. Selective activation of mTORC1 signaling recapitulates microcephaly, tuberous sclerosis, and neurodegenerative diseases. Cell reports. 2014;7:1626–1239. doi: 10.1016/j.celrep.2014.04.048. [DOI] [PubMed] [Google Scholar]
- 30.Kawaguchi Y, Kubota Y. GABAergic cell subtypes and their synaptic connections in rat frontal cortex. Cereb Cortex. 1997;7:476–486. doi: 10.1093/cercor/7.6.476. [DOI] [PubMed] [Google Scholar]
- 31.Kellinghaus C, Kunieda T, Ying Z, Pan A, Lüders HO, Najm IM. Severity of histopathologic abnormalities and in vivo epileptogenicity in the in utero radiation model of rats is dose dependent. Epilepsia. 2004;45:583–591. doi: 10.1111/j.0013-9580.2004.41103.x. [DOI] [PubMed] [Google Scholar]
- 32.Kerjan G, Koizumi H, Han EB, Dube CM, Djakovic SN, Patrick GN, Baram TZ, Heinemann SF, Gleeson JG. Mice lacking doublecortin and doublecortin-like kinase 2 display altered hippocampal neuronal maturation and spontaneous seizures. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:6766–6771. doi: 10.1073/pnas.0812687106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kitamura K, Itou Y, Yanazawa M, Ohsawa M, Suzuki-Migishima R, Umeki Y, Hohjoh H, Yanagawa Y, Shinba T, Itoh M, Nakamura K, Goto Y. Three human ARX mutations cause the lissencephaly-like and mental retardation with epilepsy-like pleiotropic phenotypes in mice. Human Molecular Genetics. 2009;18:3708–3724. doi: 10.1093/hmg/ddp318. [DOI] [PubMed] [Google Scholar]
- 34.Kitamura K, Yanazawa M, Sugiyama N, Miura H, Iizuka-Kogo A, Kusaka M, Omichi K, Suzuki R, Kato-Fukui Y, Kamiirisa K, Matsuo M, Kamijo S, Kasahara M, Yoshioka H, Ogata T, Fukuda T, Kondo I, Kato M, Dobyns WB, Yokoyama M, Morohashi K. Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nature genetics. 2002;32:359–369. doi: 10.1038/ng1009. [DOI] [PubMed] [Google Scholar]
- 35.Kondo S, Najm I, Kunieda T, Perryman S, Yacubova K, Lüders HO. Electroencephalographic characterization of an adult rat model of radiation-induced cortical dysplasia. Epilepsia. 2001;42:1221–1227. doi: 10.1046/j.1528-1157.2001.38300.x. [DOI] [PubMed] [Google Scholar]
- 36.Krueger DA, Care MM, Holland K, Agricola K, Tudor C, Mangeshkar P, Wilson KA, Byars A, Sahmoud T, Franz DN. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. The New England journal of medicine. 2010;363:1801–1811. doi: 10.1056/NEJMoa1001671. [DOI] [PubMed] [Google Scholar]
- 37.Krueger DA, Wilfong AA, Holland-Bouley K, Anderson AE, Agricola K, Tudor C, Mays M, Lopez CM, Kim MO, Franz DN. Everolimus treatment of refractory epilepsy in tuberous sclerosis complex. Ann Neurol. 2013;74:679–687. doi: 10.1002/ana.23960. [DOI] [PubMed] [Google Scholar]
- 38.Kwon CH, Zhu X, Zhang J, Baker SJ. mTor is required for hypertrophy of Pten-deficient neuronal soma in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:12923–12928. doi: 10.1073/pnas.2132711100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee JH, Huynh M, Silhavy JL, Kim S, Dixon-Salazar T, Heiberg A, Scott E, Bafna V, Hill KJ, Collazo A, Funari V, Russ C, Gabriel SB, Mathern GW, Gleeson JG. De novo somatic mutations in components of the PI3K–AKT3-mTOR pathway cause hemimegalencephaly. Nature Genetics. 2012;44:941–945. doi: 10.1038/ng.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li S, Jin Z, Koirala S, Bu L, Xu L, Hynes RO, Walsh CA, Corfas G, Piao X. GPR56 regulates pial basement membrane integrity and cortical lamination. J Neurosci. 2008;28:5817–5826. doi: 10.1523/JNEUROSCI.0853-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ljungberg MC, Sunnen CN, Lugo JN, Anderson AE, D'Arcangelo G. Rapamycin suppresses seizures and neuronal hypertrophy in a mouse model of cortical dysplasia. Disease models & mechanisms. 2009;2:389–398. doi: 10.1242/dmm.002386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lo Nigro C, Chong CS, Smith AC, Dobyns WB, Carrozzo R, Ledbetter DH. Point mutations and an intragenic deletion in LIS1, the lissencephaly causative gene in isolated lissencephaly sequence and Miller-Dieker syndrome. Human molecular genetics. 1997;6:157–164. doi: 10.1093/hmg/6.2.157. [DOI] [PubMed] [Google Scholar]
- 43.Lozovaya N, Gataullina S, Tsintsadze T, Tsintsadze V, Pallesi-Pocachard E, Minlebaev M, Goriounova NA, Buhler E, Watrin F, Shityakov S, Becker AJ, Bordey A, Milh M, Scavarda D, Bulteau C, Dorfmuller G, Delalande O, Represa A, Cardoso C, Dulac O, Ben-Ari Y, Burnashev N. Selective suppression of excessive GluN2C expression rescues early epilepsy in a tuberous sclerosis murine model. Nature communications. 2014;5:4563. doi: 10.1038/ncomms5563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Magri L, Cambiaghi M, Cominelli M, Alfaro-Cervello C, Cursi M, Pala M, Bulfone A, Garcia-Verdugo JM, Leocani L, Minicucci F, Poliani PL, Galli R. Sustained activation of mTOR pathway in embryonic neural stem cells leads to development of tuberous sclerosis complex-associated lesions. Cell stem cell. 2011;9:447–462. doi: 10.1016/j.stem.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 45.Magri L, Cominelli M, Cambiaghi M, Cursi M, Leocani L, Minicucci F, Poliani PL, Galli R. Timing of mTOR activation affects tuberous sclerosis complex neuropathology in mouse models. Disease models & mechanisms. 2013;6:1185–1197. doi: 10.1242/dmm.012096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Manent JB, Wang Y, Chang Y, Paramasivam M, LoTurco JJ. Dcx reexpression reduces subcortical band heterotopia and seizure threshold in an animal model of neuronal migration disorder. Nature medicine. 2009;15:84–90. doi: 10.1038/nm.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marin-Valencia I, Guerrini R, Gleeson JG. Pathogenetic mechanisms of focal cortical dysplasia. Epilepsia. 2014;55:970–978. doi: 10.1111/epi.12650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marsh E, Fulp C, Gomez E, Nasrallah I, Minarcik J, Sudi J, Christian SL, Mancini G, Labosky P, Dobyns W, Brooks-Kayal A, Golden JA. Targeted loss of Arx results in a developmental epilepsy mouse model and recapitulates the human phenotype in heterozygous females. Brain. 2009;132:1563–1576. doi: 10.1093/brain/awp107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meikle L, Pollizzi K, Egnor A, Kramvis I, Lane H, Sahin M, Kwiatkowski DJ. Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. J Neurosci. 2008;28:5422–5432. doi: 10.1523/JNEUROSCI.0955-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Meikle L, Talos DM, Onda H, Pollizzi K, Rotenberg A, Sahin M, Jensen FE, Kwiatkowski DJ. A mouse model of tuberous sclerosis: neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced myelination, seizure activity, and limited survival. J Neurosci. 2007;27:5546–5558. doi: 10.1523/JNEUROSCI.5540-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mirzaa GM, Poduri A. Megalencephaly and hemimegalencephaly: breakthroughs in molecular etiology. American journal of medical genetics. Part C, Seminars in medical genetics. 2014;166c:156–172. doi: 10.1002/ajmg.c.31401. [DOI] [PubMed] [Google Scholar]
- 52.Normand EA, Crandall SR, Thorn CA, Murphy EM, Voelcker B, Browning C, Machan JT, Moore CI, Connors BW, Zervas M. Temporal and mosaic Tsc1 deletion in the developing thalamus disrupts thalamocortical circuitry, neural function, and behavior. Neuron. 2013;78:895–909. doi: 10.1016/j.neuron.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nosten-Bertrand M, Kappeler C, Dinocourt C, Denis C, Germain J, Phan Dinh Tuy F, Verstraeten S, Alvarez C, Metin C, Chelly J, Giros B, Miles R, Depaulis A, Francis F. Epilepsy in Dcx knockout mice associated with discrete lamination defects and enhanced excitability in the hippocampus. PloS one. 2008;3:e2473. doi: 10.1371/journal.pone.0002473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.O'Rourke DJ, Twomey E, Lynch SA, King MD. Cortical dysplasia associated with the PTEN mutation in Bannayan Riley Ruvalcaba syndrome: a rare finding. Clinical dysmorphology. 2012;21:91–92. doi: 10.1097/MCD.0b013e328351639d. [DOI] [PubMed] [Google Scholar]
- 55.Olivetti PR, Maheshwari A, Noebels JL. Neonatal estradiol stimulation prevents epilepsy in Arx model of X-linked infantile spasms syndrome. Science translational medicine. 2014;6:220ra12. doi: 10.1126/scitranslmed.3007231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Orlova KA, Crino PB. The tuberous sclerosis complex. Annals of the New York Academy of Sciences. 2010;1184:87–105. doi: 10.1111/j.1749-6632.2009.05117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Orlova KA, Parker WE, Heuer GG, Tsai V, Yoon J, Baybis M, Fenning RS, Strauss K, Crino PB. STRADalpha deficiency results in aberrant mTORC1 signaling during corticogenesis in humans and mice. The Journal of clinical investigation. 2010;120:1591–1602. doi: 10.1172/JCI41592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Parker WE, Orlova KA, Parker WH, Birnbaum JF, Krymskaya VP, Goncharov DA, Baybis M, Helfferich J, Okochi K, Strauss KA, Crino PB. Rapamycin prevents seizures after depletion of STRADA in a rare neurodevelopmental disorder. Science translational medicine. 2013;5:182ra53. doi: 10.1126/scitranslmed.3005271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Piao X, Hill RS, Bodell A, Chang BS, Basel-Vanagaite L, Straussberg R, Dobyns WB, Qasrawi B, Winter RM, Innes AM, Voit T, Ross ME, Michaud JL, Descarie JC, Barkovich AJ, Walsh CA. G protein-coupled receptor-dependent development of human frontal cortex. Science (New York, N.Y.) 2004;303:2033–2036. doi: 10.1126/science.1092780. [DOI] [PubMed] [Google Scholar]
- 60.Poduri A, Evrony GD, Cai X, Elhosary PC, Beroukhim R, Lehtinen MK, Hills LB, Heinzen EL, Hill A, Hill RS, Barry BJ, Bourgeois BF, Riviello JJ, Barkovich AJ, Black PM, Ligon KL, Walsh CA. Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron. 2012;74:41–48. doi: 10.1016/j.neuron.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Poirier K, Keays DA, Francis F, Saillour Y, Bahi N, Manouvrier S, Fallet-Bianco C, Pasquier L, Toutain A, Tuy FP, Bienvenu T, Joriot S, Odent S, Ville D, Desguerre I, Goldenberg A, Moutard ML, Fryns JP, van Esch H, Harvey RJ, Siebold C, Flint J, Beldjord C, Chelly J. Large spectrum of lissencephaly and pachygyria phenotypes resulting from de novo missense mutations in tubulin alpha 1A (TUBA1A) Human mutation. 2007;28:1055–1064. doi: 10.1002/humu.20572. [DOI] [PubMed] [Google Scholar]
- 62.Price MG, Yoo JW, Burgess DL, Deng F, Hrachovy RA, Frost JD, Jr, Noebels JL. A triplet repeat expansion genetic mouse model of infantile spasms syndrome, Arx(GCG)10+7, with interneuronopathy, spasms in infancy, persistent seizures, and adult cognitive and behavioral impairment. J Neurosci. 2009;29:8752–8763. doi: 10.1523/JNEUROSCI.0915-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ramos RL, Bai J, LoTurco JJ. Heterotopia formation in rat but not mouse neocortex after RNA interference knockdown of DCX. Cerebral cortex (New York, N.Y.: 1991) 2006;16:1323–1331. doi: 10.1093/cercor/bhj074. [DOI] [PubMed] [Google Scholar]
- 64.Roper SN, Abraham LA, Streit WJ. Exposure to in utero irradiation produces disruption of radial glia in rats. Dev Neurosci. 1997;19:521–528. doi: 10.1159/000111249. [DOI] [PubMed] [Google Scholar]
- 65.Roper SN, Gilmore RL, Houser CR. Experimentally induced disorders of neuronal migration produce an increased propensity for electrographic seizures in rats. Epilepsy Res. 1995;21:205–219. doi: 10.1016/0920-1211(95)00027-8. [DOI] [PubMed] [Google Scholar]
- 66.Scheffer IE, Heron SE, Regan BM, Mandelstam S, Crompton DE, Hodgson BL, Licchetta L, Provini F, Bisulli F, Vadlamudi L, Gecz J, Connelly A, Tinuper P, Ricos MG, Berkovic SF, Dibbens LM. Mutations in mammalian target of rapamycin regulator DEPDC5 cause focal epilepsy with brain malformations. Ann Neurol. 2014;75:782–787. doi: 10.1002/ana.24126. [DOI] [PubMed] [Google Scholar]
- 67.Setkowicz Z, Klak K, Janeczko K. Long-term changes in postnatal susceptibility to pilocarpine-induced seizures in rats exposed to gamma radiation at different stages of prenatal development. Epilepsia. 2003;44:1267–1273. doi: 10.1046/j.1528-1157.2003.08203.x. [DOI] [PubMed] [Google Scholar]
- 68.Stottmann RW, Donlin M, Hafner A, Bernard A, Sinclair DA, Beier DR. A mutation in Tubb2b, a human polymicrogyria gene, leads to lethality and abnormal cortical development in the mouse. Human molecular genetics. 2013;22:4053–4063. doi: 10.1093/hmg/ddt255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stutterd CA, Leventer RJ. Polymicrogyria: a common and heterogeneous malformation of cortical development. American journal of medical genetics. Part C, Seminars in medical genetics. 2014;166c:227–239. doi: 10.1002/ajmg.c.31399. [DOI] [PubMed] [Google Scholar]
- 70.Sunnen CN, Brewster AL, Lugo JN, Vanegas F, Turcios E, Mukhi S, Parghi D, D'Arcangelo G, Anderson AE. Inhibition of the mammalian target of rapamycin blocks epilepsy progression in NS-Pten conditional knockout mice. Epilepsia. 2011;52:2065–2075. doi: 10.1111/j.1528-1167.2011.03280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Uhlmann EJ, Wong M, Baldwin RL, Bajenaru ML, Onda H, Kwiatkowski DJ, Yamada K, Gutmann DH. Astrocyte-specific TSC1 conditional knockout mice exhibit abnormal neuronal organization and seizures. Ann Neurol. 2002;52:285–296. doi: 10.1002/ana.10283. [DOI] [PubMed] [Google Scholar]
- 72.Waltereit R, Welzl H, Dichgans J, Lipp HP, Schmidt WJ, Weller M. Enhanced episodic-like memory and kindling epilepsy in a rat model of tuberous sclerosis. Journal of neurochemistry. 2006;96:407–413. doi: 10.1111/j.1471-4159.2005.03538.x. [DOI] [PubMed] [Google Scholar]
- 73.Way SW, McKenna J, 3rd, Mietzsch U, Reith RM, Wu HC, Gambello MJ. Loss of Tsc2 in radial glia models the brain pathology of tuberous sclerosis complex in the mouse. Human molecular genetics. 2009;18:1252–1265. doi: 10.1093/hmg/ddp025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Way SW, Rozas NS, Wu HC, McKenna J, 3rd, Reith RM, Hashmi SS, Dash PK, Gambello MJ. The differential effects of prenatal and/or postnatal rapamycin on neurodevelopmental defects and cognition in a neuroglial mouse model of tuberous sclerosis complex. Human molecular genetics. 2012;21:3226–3236. doi: 10.1093/hmg/dds156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wong M. A critical review of mTOR inhibitors and epilepsy: from basic science to clinical trials. Expert review of neurotherapeutics. 2013;13:657–669. doi: 10.1586/ern.13.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wong M. Mammalian target of rapamycin (mTOR) inhibition as a potential antiepileptogenic therapy: From tuberous sclerosis to common acquired epilepsies. Epilepsia. 2010;51:27–36. doi: 10.1111/j.1528-1167.2009.02341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wong M. Mechanisms of epileptogenesis in tuberous sclerosis complex and related malformations of cortical development with abnormal glioneuronal proliferation. Epilepsia. 2008;49:8–21. doi: 10.1111/j.1528-1167.2007.01270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wong M, Ess KC, Uhlmann EJ, Jansen LA, Li W, Crino PB, Mennerick S, Yamada KA, Gutmann DH. Impaired glial glutamate transport in a mouse tuberous sclerosis epilepsy model. Ann Neurol. 2003;54:251–256. doi: 10.1002/ana.10648. [DOI] [PubMed] [Google Scholar]
- 79.Zeng LH, Ouyang Y, Gazit V, Cirrito JR, Jansen LA, Ess KC, Yamada KA, Wozniak DF, Holtzman DM, Gutmann DH, Wong M. Abnormal glutamate homeostasis and impaired synaptic plasticity and learning in a mouse model of tuberous sclerosis complex. Neurobiol Dis. 2007;28:184–196. doi: 10.1016/j.nbd.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zeng LH, Rensing NR, Zhang B, Gutmann DH, Gambello MJ, Wong M. Tsc2 gene inactivation causes a more severe epilepsy phenotype than Tsc1 inactivation in a mouse model of tuberous sclerosis complex. Human Molecular Genetics. 2011;20:445–454. doi: 10.1093/hmg/ddq491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zeng LH, Xu L, Gutmann DH, Wong M. Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann Neurol. 2008;63:444–453. doi: 10.1002/ana.21331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhou FW, Chen HX, Roper SN. Balance of inhibitory and excitatory synaptic activity is altered in fast-spiking interneurons in experimental cortical dysplasia. J Neurophysiol. 2009;102:2514–2525. doi: 10.1152/jn.00557.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhou FW, Rani A, Martinez-Diaz H, Foster TC, Roper SN. Altered behavior in experimental cortical dysplasia. Epilepsia. 2011;52:2293–2303. doi: 10.1111/j.1528-1167.2011.03267.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhou FX, Roper SN. Altered firing rates and patterns in interneurons in experimental cortical dysplasia. Cereb Cortex. 2011;21:1645–1658. doi: 10.1093/cercor/bhq234. [DOI] [PubMed] [Google Scholar]
- 85.Zhou FW, Roper SN. Impaired hippocampal memory function and synaptic plasticity in experimental cortical dysplasia. Epilepsia. 2012;53:850–859. doi: 10.1111/j.1528-1167.2012.03431.x. [DOI] [PubMed] [Google Scholar]
- 86.Zhou FW, Roper SN. Reduced chemical and electrical connections of fast-spiking interneurons in experimental cortical dysplasia. J Neurophysiol. 2014;112:1277–1290. doi: 10.1152/jn.00126.2014. [DOI] [PubMed] [Google Scholar]
- 87.Zhou J, Blundell J, Ogawa S, Kwon CH, Zhang W, Sinton C, Powell CM, Parada LF. Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knock-out mice. J Neurosci. 2009;29:1773–1783. doi: 10.1523/JNEUROSCI.5685-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhu WJ, Roper SN. Reduced inhibition in an animal model of cortical dysplasia. J Neurosci. 2000;20:8925–8931. doi: 10.1523/JNEUROSCI.20-23-08925.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]