

Abstract
Background
Tobacco smoke contains numerous agonists of the aryl-hydrocarbon receptor (AhR) pathway, and activation of the AhR pathway was shown to promote atherosclerosis in mice. Intriguingly, cigarette smoking is most strongly and robustly associated with DNA modifications to an AhR pathway gene, the aryl-hydrocarbon receptor repressor (AHRR). We hypothesized that altered AHRR methylation in monocytes, a cell type sensitive to cigarette smoking and involved in atherogenesis, may be a part of the biological link between cigarette smoking and atherosclerosis.
Methods and Results
DNA methylation profiles of AHRR in monocytes (542 CpG sites ± 150kb of AHRR, using Illumina 450K array) were integrated with smoking habits and ultrasound-measured carotid plaque scores from 1,256 participants of the Multi-Ethnic Study of Atherosclerosis (MESA). Methylation of cg05575921 significantly associated (p = 6.1×10−134) with smoking status (current vs. never). Novel associations between cg05575921 methylation and carotid plaque scores (p = 3.1×10−10) were identified, which remained significant in current and former smokers even after adjusting for self-reported smoking habits, urinary cotinine, and well-known CVD risk factors. This association replicated in an independent cohort using hepatic DNA (n = 141). Functionally, cg05575921 was located in a predicted gene expression regulatory element (enhancer), and had methylation correlated with AHRR mRNA profiles (p = 1.4×10−17) obtained from RNA sequencing conducted on a subset (n = 373) of the samples.
Conclusions
These findings suggest AHRR methylation may be functionally related to AHRR expression in monocytes, and represents a potential biomarker of subclinical atherosclerosis in smokers.
Keywords: smoking, atherosclerosis, gene expression/regulation, epidemiology, epigenetics, DNA methylation
Background
Smoking is a major preventable risk factor for many human diseases including cardiovascular diseases1, 2 (CVD), which cause a substantial health and economic burden worldwide. Decoding the molecular mechanisms by which smoking drives the development of CVD may provide both preclinical diagnostic biomarkers and drug targets for therapeutics of CVD.
Epigenetic modifications such as DNA methylation of cytosine-(phosphate)-guanine (CpG) dinucleotides are functional biochemical alterations to DNA that are implicated in regulation of gene expression and disease development2, 3. Cigarette smoke contains thousands of chemicals4, including many known carcinogens such as polycyclic aromatic hydrocarbons (PAHs)5, which may lead to both mutagenic and epigenetic alterations to the genomes of smokers. Recent genome-wide association studies have shown that smoking is most significantly and robustly associated with DNA methylation alterations of the Aryl Hydrocarbon Receptor Repressor (AHRR)6–9, which appear to be at least partially reversible upon smoking cessation9, and associated with higher mRNA expression levels of AHRR in human lung10, and lymphoblasts11.
Previous studies indicate that AHRR, a basic helix–loop–helix/Per-ARNT-Sim (bHLH/PAS) family transcription factor, is transcriptionally regulated through activation of the Ah Receptor (AhR) pathway12, 13. Tobacco smoke contains numerous agonists of AhR signaling14 and persistent activation of the AhR signaling pathway has been hypothesized to contribute to atherogenesis15–18. While there is currently no known role for AHRR in atherogenesis, recent studies implicate up-regulation of AHRR expression in monocyte-to-macrophage differentiation19 and suppression of anti-inflammation13. We hypothesized that altered AHRR methylation levels in CD14+ monocytes, a cell type sensitive to cigarette smoking20 and involved in atherogenesis21, 22, might provide a biological link between smoking and atherosclerotic plaque formation. Therefore, we integrated methylation profiles of CpG sites within 150kb of AHRR (measured using the Illumina 450K array) in primary CD14+ monocytes with smoking status and ultrasound-measured carotid plaque scores from 1,256 participants of the Multi-Ethnic Study of Atherosclerosis (MESA)23, and sought replication in the Pathobiological Determinants of Atherosclerosis in Youth Study (PDAY, n = 141)24. To look for potential functional effects of smoking-associated AHRR methylation, we incorporated histone modification data in monocytes from the ENCODE25 and BLUEPRINT26 projects, and gene expression measured in monocytes from a subset of the population (n = 373) using RNA-sequencing. Methylation and gene expression results were validated in independent monocyte samples using reduced representation bisulfite sequencing (RRBS, n = 4) and real-time PCR (n = 10).
Methods
Study Population
The Multi-Ethnic Study of Atherosclerosis (MESA) was designed to investigate the prevalence, correlates, and progression of subclinical cardiovascular disease in a population cohort of 6,814 participants. Since its inception in 2000, five clinic visits collected extensive clinical, socio-demographic, lifestyle, behavior, laboratory, nutrition, and medication data23. The present analysis is primarily based on analyses of purified monocyte samples from the April 2010 – February 2012 examination (exam 5) of 1,264 randomly selected MESA participants from four MESA field centers (Baltimore, MD; Forsyth County, NC; New York, NY; and St. Paul, MN). The study protocol was approved by the Institutional Review Board at each site. All participants signed informed consent.
Data collection
Smoking status, average number of cigarettes smoked per day, age of smoking cessation, and pack-years of cigarette smoking were assessed via standard American Thoracic Society questionnaire items27. Participants who reported smoking fewer than 100 cigarettes in their lifetimes were classified as never smokers. Among participants who reported smoking greater than 100 cigarettes in their lifetime, those who reported smoking during the last 30 days were classified as current smokers and those who did not were classified as former smokers. Pack-years of cigarette smoking were calculated from age of starting to quitting (or current age among current smokers) × (cigarettes per day/20). Urinary cotinine levels were measured via immunoassay (Immulite 2000 Nicotine Metabolite Assay; Diagnostic Products Corp., Los Angeles, CA)28.
To obtain carotid artery plaque scores, readers adjudicated carotid plaque presence or absence, defined as a focal abnormal wall thickness (carotid IMT >1.5 mm) or a focal thickening of >50% of the surrounding IMT, as reported previously29, 30. Presence or absence of plaque acoustic shadowing was recorded. A total plaque score (range 0–12) was calculated to describe carotid plaque burden. One point per plaque was allocated for the near and far walls of each segment (CCA, bulb, ICA) of each carotid artery that was interrogated. For carotid plaque presence, intra-reader reproducibility was excellent (per reader Kappa = 0.82–1.0, overall Kappa = 0.83, 95% confidence interval [CI] 0.70–0.96), as was inter-reader reproducibility (Kappa = 0.89; 95% CI 0.72 – 1.00)31.
Weight was measured with a Detecto Platform Balance Scale to the nearest 0.5 kg. Height was measured with a stadiometer (Accu-Hite Measure Device with level bubble) to the nearest 0.1 cm. BMI was defined as weight in kilograms divided by square of height in meters. Type 2 Diabetes (T2D) was defined as fasting glucose ≥ 6.99 mmol/L (≥ 126 mg/dL) or use of hypoglycemic medication. Fasting serum glucose was measured by rate reflectance spectrophotometry using thin-film adaptation of the glucose oxidase method on the Vitros analyzer (Johnson & Johnson Clinical Diagnostics, Rochester, NY). To achieve consistency of the serum glucose assay over examinations, 200 samples were reanalyzed and then recalibrated the original observations. LDL-cholesterol was calculated using the Friedewald equation. Resting blood pressure was measured three times in the seated position using a Dinamap model Pro 100 automated oscillometric sphygmomanometer (Critikon, Tampa, FL). The average of the last two measurements was used in analysis. Hypertension was assessed using the JNC VI (1997) criteria (systolic pressure greater than or equal to 140 mm Hg, diastolic pressure greater than or equal to 90 mm Hg or current use of anti-hypertensive medication).
Purification of Monocytes
Monocytes were isolated with anti-CD14 monoclonal antibody coated magnetic beads, using AutoMACs automated magnetic separation unit (Miltenyi Biotec, Bergisch Gladbach, Germany). Initially flow cytometry analysis of 18 monocyte samples collected from all four MESA field centers was performed to assess the cell purification quality across the labs and technicians. The purity was > 90% for all samples (see Data Supplement for details).
DNA/RNA Extraction
DNA and RNA were isolated from samples simultaneously using the AllPrep DNA/RNA Mini Kit (Qiagen, Inc., Hilden, Germany; see Data Supplement for details).
Methylation Quantification
Illumina HumanMethylation450 BeadChips and HiScan reader were used to perform the epigenome-wide methylation analysis (see Data Supplement for details). This methylation data has been deposited in the NCBI Gene Expression Omnibus and is accessible through GEO Series accession number (GSE56046).
Quality Control and Pre-Processing of Microarray Data
Data pre-processing and quality control (QC) analyses were performed in R (http://www.r-project.org/) using Bioconductor (http://www.bioconductor.org/) packages (see Data Supplement for detailed methods). The Illumina HumanMethylation450 BeadChip included probes for 485K CpG sites. Of these CpG sites, 484,817 passed the QC elimination criteria including: ‘detected’ methylation levels <90% of MESA samples using a detection p-value cut-off of 0.05 or overlap with a repetitive element or region.
To estimate residual sample contamination for data analysis, we generated separate enrichment scores for neutrophils, B cells, T cells, monocytes, and natural killer cells. We implemented a Gene Set Enrichment Analysis32 as previously described33 to calculate the enrichment scores using the gene signature of each blood cell type from previously defined lists34.
To remove technical error in methylation levels associated with batch effects across the multiple chips, positional effects of the sample on the chip, and residual sample contamination with non-monocyte cell types, we adjusted methylation M-values for chip, sample position on the chip, and estimated residual sample contamination with neutrophils, B cells, T cells, monocytes, and natural killer cells.
mRNA quantification using RNA seq
A subset of 374 samples was randomly selected from the 1,264 MESA monocyte samples for RNA sequencing of mRNA (see Data Supplement for details).
Association Analyses
The overall goal of the association analysis was to characterize associations between smoking and AHRR methylation, and AHRR methylation and carotid plaque scores. Analyses were performed using the linear model (lm) function of the Stats package and the stepAIC function of the MASS package in R. Reported correlations (r) represent the partial Pearson product-moment correlation coefficient, after adjusting for covariates.
To identify methylation sites associated with smoking, we fit separate linear regression models with smoking status as a predictor of the M-value for each CpG site genome-wide that passed QC. Covariates were age, sex, race/ethnicity, and study site. P-values were adjusted for multiple testing using the q-value FDR method35. The FDR was calculated at the genome-wide level, including all 484,817 CpGs passing QC, although only results from CpG sites within 150 kb of AHRR (542 CpGs) are presented.
To identify potential bimodal distributions, we calculated the kurtosis and skewness of the M-value distributions of the 542 probes investigated. Six probes (1.1%) were found to have strong negative kurtosis (kurtosis < −.75) and some skewness (skewness < −.3 or skewness > .3), suggesting bimodal distributions. Although linear regression estimates are robust to the normality assumption in large samples, we next performed permutation testing with the 542 CpGs (H0: beta = 0, i.e. no difference in current smoking vs. never smoking) to ensure the validity of the linear regression results. The number of permutations was set to be 1,000 for all CpGs, and for CpGs with p-value < .10, we also ran 100,000 permutations. We found very consistent permutation p-values compared to the normal p-values from the linear regression analysis for all probes, including the six probes with bimodal distributions. The correlation between the normal and permutation p-values is 0.999, the correlation between the ranks based on the normal and permutation p-values is also 0.999, and the sum of the absolute differences between the normal and permutation p-values divided by the sum of the normal p-values is 2% (0.02).
To characterize the association between AHRR methylation and carotid plaque levels, we fit separate linear regression models with the M-value for the 542 CpGs within 150 kb of AHRR as a predictor of carotid plaque scores [natural log (carotid plaque score + 1)]. Initial analysis covariates were age, sex, race/ethnicity, and study site. Secondary analyses adjusted for smoking status, the number of cigarettes per day, pack-years, traditional CVD risk factors (BMI, LDL cholesterol, hypertension, diabetes mellitus) and statin use.
Methylation of the most significantly differentially methylated CpG associated with smoking status and with carotid plaques scores (i.e. cg05575921) was also investigated for association with nearby gene expression levels and genetic variants. We arbitrarily chose to investigate a large window (± 1Mb) surrounding the CpG to avoid missing any potential effects. For gene expression analysis, we fit separate linear regression models with the M-value as a predictor of mRNA expression for any autosomal gene within 1 Mb of the CpG. Covariates were age, sex, and race/ethnicity, and study site. For analyses of nearby genetic variants, we fit separate linear regression models with single nucleotide polymorphisms (SNPs) located within 1 Mb as a predictor of the M-value in the MESA samples from Caucasian participants, including SNPs with a minor allele frequency > 0.05 in the MESA Caucasian population. P-values were adjusted for multiple testing using the q-value FDR method35.
Mediation analysis
We performed mediation analysis to investigate the hypothesis that smoking may have an effect on atherosclerosis mediated through methylation alteration. We used Structural Equation Modeling (SEM) with bootstrapping as implemented in the R package lavaan36 to estimate the indirect effects (mediated through methylation) of current smoking (cigarettes per day) and cumulative exposure (pack-years) on carotid plaque score in current and former smokers for the most significantly differentially methylated CpG associated with smoking status and with carotid plaques score.
Replication study
Replication analysis for the most significantly differentially methylated CpG associated with smoking status and with carotid plaques score included samples from 141 males from the Pathobiological Determinants of Atherosclerosis in Youth Study (PDAY)24 (see Data Supplement for details). Tests for association between methylation measured in hepatic samples and the extent of fatty streaks observed in the right coronary arteries and the left halves of the aortas were performed using linear regression, including the following risk factors: age, race, lipids (HDL, non-HDL), BMI, and glucose levels.
Reduced Representation Bisulfite Sequencing (RRBS) methods and DMR Identification
To investigate the relationship between smoking and CpG methylation not captured by microarray, RRBS libraries of CD14+ monocytes from two smokers and two non-smokers were generated as previously described37 with minor modifications (see Data Supplement).
To identify genomic regions that are differentially methylated regions (DMRs) between smokers and non-smokers, we used the method described in Ziller et al.38. Briefly, this method first uses the methylated and unmethylated counts to identify a set of dynamic CpGs, which are CpGs that are significantly differentially methylated between groups, using a model based on the beta difference distribution. Next, it merges any dynamic CpGs whose genomic locations are within 500 bp into a set of CpG clusters. Finally, all clusters that are significantly differentially methylated between groups are retained as the final set of DMRs. We defined the DMR location as the region between the outermost CpGs in the cluster. We excluded any DMRs that had fewer than five CpGs with nonzero methylation counts, and we also excluded any DMRs that had an overall methylation difference of less than 10%.
Functional characterization
Histone modifications in monocytes and monocyte-derived macrophages from BLUEPRINT19 were downloaded from GEO (Series GSE58310) including H3K27ac and H3K4me1. Other histone modifications in monocytes from ENCODE39 were downloaded through the UCSC browser25, including H3K4me1 (GEO accession GSM1003535), H3K4me3 (GEO accession GSM1003536), and H3K27me3 (GEO accession GSM1003564), which were produced by the Bernstein lab at the BROAD Institute.
RT-qPCR
Reverse Transcription Quantitative Polymerase Chain Reaction (RTqPCR) was used for replication of gene expression in CD14+ monocytes from nonsmokers and smokers collected at the NIEHS Clinical Research Unit (see Data Supplement).
Results
The study population (n = 1,256; Table 1) was composed of 47% Caucasians, 32% Hispanics, and 21% African Americans that were recruited from four different study sites across the US, as previously described33.
Table 1.
Characteristics of the MESA participants
| Cigarette Smoking Status
|
||||||
|---|---|---|---|---|---|---|
| Overall (n = 1,256) |
Never (n = 502) |
Former (n = 640) |
Current (n = 114) |
|||
| Proportion, Mean (SD), or Median (IQR) | Proportion, Mean (SD), or Median (IQR) | Proportion, Mean (SD), or Median (IQR) | P-val* | Proportion, Mean (SD), or Median (IQR) | P-val* | |
| Demographic | ||||||
| Mean age (years) | 70 (9) | 70 (10) | 70 (9) | ns | 65 (8) | <0.001 |
| Gender (% female) | 51 | 62 | 44 | <0.001 | 45 | <0.001 |
| Race | ||||||
| Caucasian (%) | 47 | 43 | 50 | 0.03 | 45 | ns |
| Hispanic (%) | 32 | 34 | 31 | ns | 25 | ns |
| African American (%) | 21 | 23 | 19 | ns | 31 | ns |
| Smoking exposure | ||||||
| Mean pack-years | 16 (20) | 36 (27) | ||||
| Mean cigarettes per day | 12 (8) | |||||
| Median urine cotinine (ng/ml)† | 7 (0) | 7 (0) | 7 (0) | <0.001 | 3,708 (3,772) | <0.001 |
| Clinical and laboratory | ||||||
| Mean BMI | 30 (6) | 30 (6) | 30 (5) | ns | 28 (6) | 0.05 |
| Mean LDL cholesterol (mg/dl) | 105 (32) | 108 (32) | 102 (33) | 0.005 | 105 (32) | ns |
| Diabetes (%) | 20 | 19 | 21 | ns | 26 | ns |
| Hypertension (%) | 61 | 60 | 63 | ns | 54 | ns |
| Median carotid plaque score† | 2 (4) | 1 (3) | 2 (4) | <0.001 | 2 (4) | 0.003 |
SD, standard deviation; IQR, interquartile range (IQR); ns, P-val > 0.05
compared to never smokers; t-test to compare means, Wilcoxon rank sum test to compare medians, and chi-square test to compare proportions
measured in a subset of samples: carotid plaque (n = 1,176) urine cotinine (n = 928)
AHRR Methylation and Smoking
We identified 33 of 542 CpGs within 150 kb of AHRR whose degree of methylation was significantly associated with cigarette smoking at a genome-wide FDR threshold of 5% (p ranging 6.1×10−134 – 2.5×10−4, Figure 1) in monocytes from 114 current smokers and 502 never smokers. Methylation of cg05575921 (chr5:373,378; hg19) within the AHRR gene body represented the most significantly (p = 6.07×10−134, FDR = 2.67×10−128) differentially methylated CpG.
Figure 1.
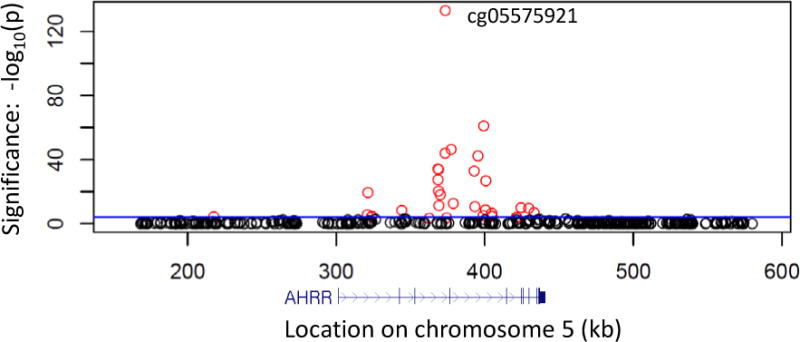
Cigarette smoking status is strongly associated with DNA methylation profiles of AHRR in monocytes from current smokers and never smokers. Regional association plot shows the significance (y-axis: −log10 p-value; FDRgenome-wide < 0.05 colored red; FDRgenome-wide ≥ 0.05 colored black) of the association between smoking status (114 current vs. 502 never smokers) and methylation in CD14+ monocytes (542 CpGs within 150 kb of AHRR, measured using the Illumina 450K microarray). The blue horizontal line represents a Bonferroni corrected significance threshold (α = 0.05) for the 542 CpGs examined. The genomic position (hg19) of AHRR relative to the CpGs is shown below the association plot (in blue). Regression covariates included age, sex, race, and study site.
To explore the smoking-associated cg05575921 methylation in more detail, we examined the relationship between smoking history/status, the dosage of exposure, the level of urinary cotinine, and cg05575921 methylation. In agreement with the proposed reversibility of smoking-responsive cg05575921 methylation9, the average methylation measured in former smokers (mean beta = 0.79) was more similar to never smokers (mean beta = 0.83, Figure 2a) than current smokers (mean beta = 0.59). Furthermore, we found that cg05575921 methylation was positively correlated with time since quitting in former smokers (partial correlation: r = 0.35, p < 2×10−16, Figure 2b), and was negatively correlated with recent exposure measurements in current smokers such as the average number of cigarettes smoked per day (r = −0.39, p = 6.1×10−5, Figure 2c) and urinary cotinine levels (r = −0.43, p < 9.09×10−4, Figure 2d), after adjusting for age, sex, race, and study site. Consistent with these findings, cg05575921 methylation was also significantly negatively correlated with cumulative exposure measured by pack-years in current smokers (r = −0.34, p = 3.97×10−4) and former smokers (r = −0.35, p < 2×10−16; Data Supplement Figure 1).
Figure 2.
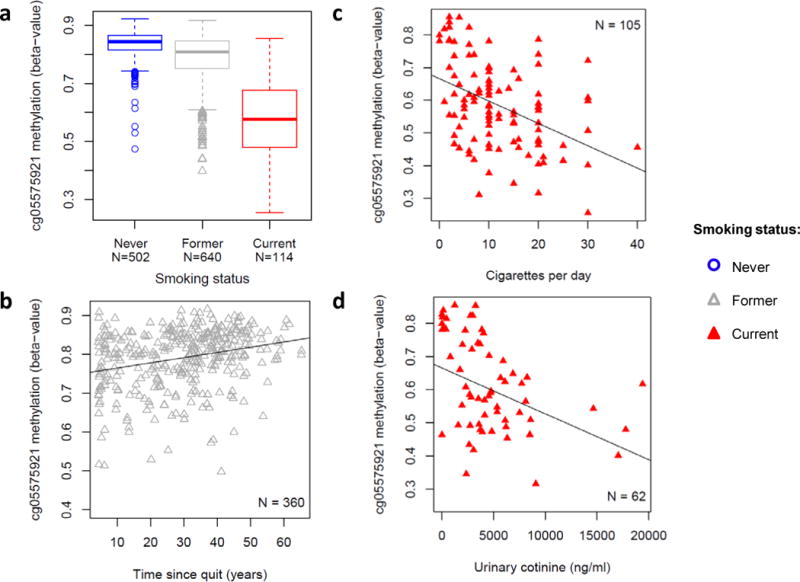
Most significant smoking-associated methylation profile in monocytes strongly reflects smoking exposure. (a) Box plots showing distributions of AHRR cg05575921 methylation (y-axis: beta-value) by smoking status (b–d) cg05575921 methylation (y axis: beta-value) was (b) positively correlated with years since quitting smoking in former smokers (r = 0.35, 95% CI: 0.26 – 0.44, p < 2×10−16) and negatively correlated with (c) the number of cigarettes smoked per day (r = −0.39, 95% CI: −0.54 – −0.22, p = 6.1×10−5) and (d) urinary cotinine levels (ng/ml) (r = −0.43, 95% CI: −0.62 – −0.21, p = 9.09×10−4) in current smokers. Regression covariates included age, sex, race, and study site.
AHRR Methylation and Carotid Plaque Scores
Among all investigated CpGs within 150 kb of AHRR, methylation profiles of two CpGs were associated with higher carotid plaque scores (cg05575921, p = 3.08×10−10; cg21161138, p = 7.73×10−8; Figure 3a), both of which were also associated with smoking. Including both CpG methylation profiles in the same model did not find independent effects for these CpGs on carotid plaque score (methylation correlation = 0.37).
Figure 3.
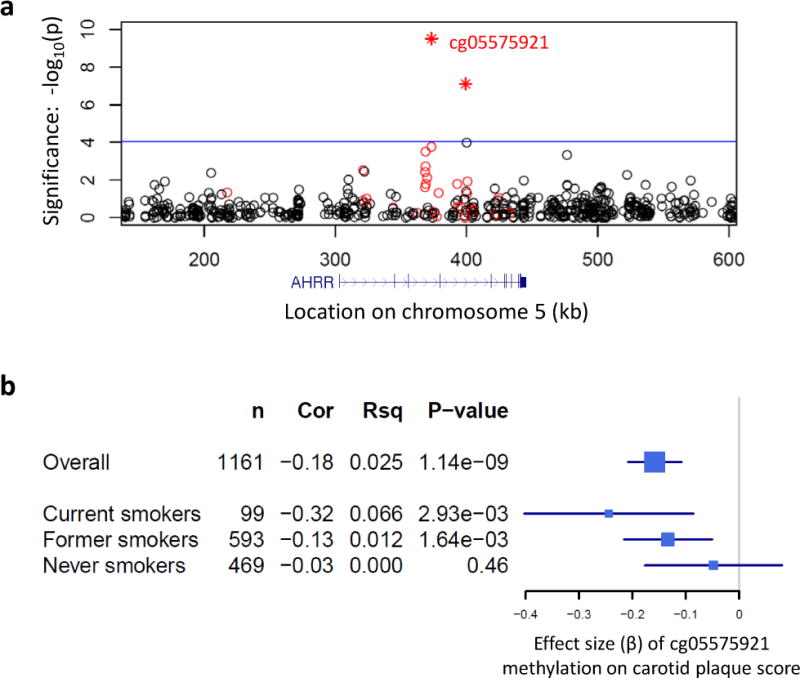
AHRR methylation is associated with carotid plaque score. (a) Regional association plot shows the significance (y-axis: log10 P-value) of the association between methylation (542 CpGs within 150 kb of AHRR measured in 1,176 CD14+ monocyte samples) and ultrasound-measured carotid plaque score [ln(plaque score + 1)]. Two methylation profiles (cg05575921 and cg21161138, r = 0.37) significantly associated with carotid plaque score (indicated as stars), based on a Bonferroni adjusted significance threshold for 542 CpGs (α = 0.05; indicated by the blue horizontal line). CpGs with methylation associated with smoking (FDRgenome-wide<0.05) are colored red. The genomic position (hg19) of AHRR relative to the CpGs is shown below the association plot (in blue). Regression covariates included age, sex, race, and study site. (b) Forest plot shows the estimated effect size (x-axis: beta and 95% CI) of cg05575921 methylation on carotid plaque score (overall and stratified by smoking status). Also shown are the sample size (n), the partial Pearson’s correlation coefficient (Cor) between methylation and carotid plaque score, the unique variance (Rsq) of carotid plaque score explained by cg05575921 methylation, and the significance (P-value). Regression covariates included age, sex, race, study site, BMI, LDL cholesterol, diabetes, hypertension, and statin use.
Importantly, even after adjusting for statin use and traditional CVD risk factors including body mass index (BMI), low-density lipoprotein (LDL) cholesterol, hypertension, and diabetes mellitus, the CpG with methylation most strongly associated with carotid plaque score remained significant (cg05575921, r = −0.18, p = 1.14×10−9, Figure 3b). After additionally adjusting for smoking status, the effect of cg05575921 methylation on carotid plaque score remained significant (r = −0.13, p = 2.2×10−5). We then stratified by smoking status and found the estimated effect of cg05575921 methylation was strongest among current smokers (r = −0.32, p = 2.93×10−3) and was attenuated in former smokers (r = −0.13, p = 1.64×10−3). No significant relationship was detected between AHRR methylation and carotid plaque score in never smokers (r = −0.03; p = 0.46), likely due to the predominantly hypermethylated state of cg05575921 in never smokers, and the associated reduced variability of methylation in never smokers compared with current and former smokers.
In current and former smokers, the effect of cg05575921 methylation on carotid plaque score was further attenuated, but remained significant (p < 0.05) after including self-reported pack-years and urinary cotinine levels in the model (Data Supplement Table 1). Standardized effect estimates of cg05575921 methylation (beta = −0.13, p = 0.01) and pack-years (beta = 0.13, p = 0.007) on carotid plaque score were similar in magnitude, each uniquely explaining ~1% of the variance of carotid plaque score. The standardized effect size of cg05575921 methylation on carotid plaque score was similar in magnitude to the effect of LDL cholesterol on carotid plaque score (beta = 0.16, p = 0.001), as shown in the full model results presented in the Data Supplement Table 1.
Using mediation analysis to explore if methylation was intermediate in the association between smoking (pack-years and cigarettes per day) and higher carotid plaque score, we found cg05575921 methylation significantly mediated 37% of the total effect of self-reported pack-years on carotid plaque score (indirect effect: β ± SE: 0.25 ± 0.06; p = 3.1×10−4) in current and former smokers, and 60% of the total effect of cigarettes per day on carotid plaque score (indirect effect: β ± SE: 0.011 ± 0.004; p = 4.3×10−3) in current smokers.
We replicated both the associations of cg05575921 methylation (in hepatic cells) with smoking (p = 0.002), and with subclinical atherosclerosis (extent of fatty streaks in the right coronary arteries and left halves of the aortas; p = 0.002) measured at autopsy in biopsies from 141 young males (< 35 years) from the PDAY study.
Functional genomic characterization of the cg05575921 locus
Using reduced representation bisulfite sequencing (RRBS) to measure CpG methylation in non-MESA monocyte samples from two smokers and two non-smokers, we recapitulated the association between smoking and methylation of cg05575921. Additionally, RRBS revealed seven adjacent (within 180 bp) CpGs not included on the Illumina 450K microarray which displayed similar directional changes in methylation with smoking status, composing a smoking-associated differentially methylated region (DMR) (Figure 4 and Data Supplement Figure 2).
Figure 4.
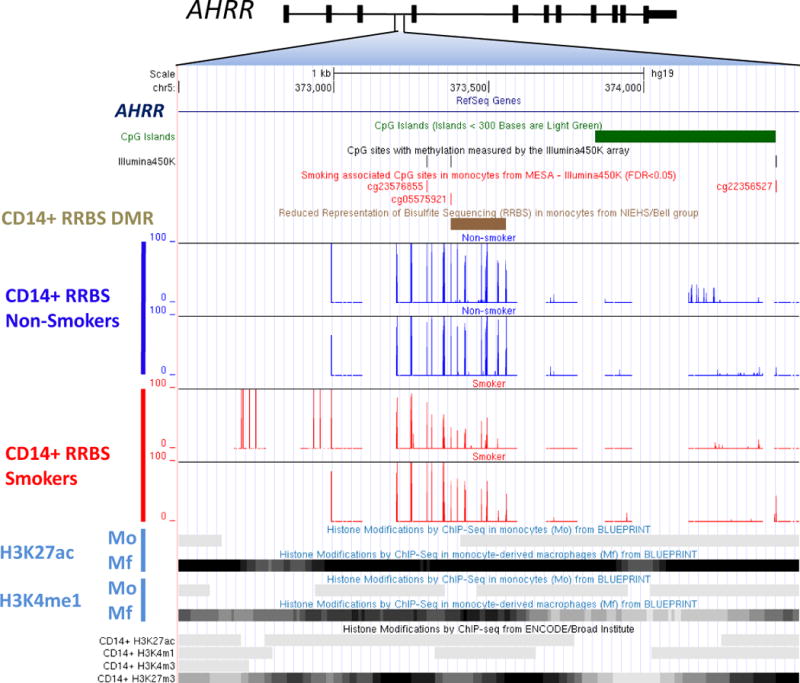
Functional genomic characterization of the smoking-and carotid plaque score-associated locus within AHRR. Cg05575921 is located on chromosome 5p15.33 in an AHRR intron and a CpG island ‘shore’. Within 500 bp of cg05575921, the Illumina450 microarray captured two additional CpGs, both of which were associated with smoking in MESA (FDRgenome-wide < 0.05). Fine mapping using reduced representation of bisulfite sequencing (RRBS) revealed a statistically significant (p = 2.39×10−5) differentially methylation region (DMR, brown bar, chr5:373,378 – 373,556 hg19) by smoking status, including cg05575921 and seven other CpGs within 200 bp that were not included on the Illumina 450K microarray. RRBS was performed in non-MESA monocyte samples from two non-smokers (shown in dark blue) and two current smokers (shown in red), with the resulting percent methylation detected for nearby CpGs shown on the y-axis. This DMR overlaps potential functional features including histone modifications indicative of an active enhancer (H3K27ac and H3K4me1) in monocyte-derived macrophages (Mf; from BLUEPRINT) and a poised enhancer (active: H3K4m1; repressed: H3K27m3) in a CD14+ monocyte sample (from ENCODE).
The AHRR DMR (chr5:373,378–373,556 hg19) is located in a potential poised enhancer region, based on the presence of both activating (H3K27ac/H3K4me1) and repressive (H3K27me3) histone modifications in a monocyte sample (from ENCODE25). This enhancer region appears to be strongly activated (based on the enrichment of histone marks H3K27ac/H3K4me1) upon monocyte differentiation to macrophage (from BLUEPRINT19, 26; Figure 4).
DNA methylation of cg05575921 and cis-gene expression
In a subset of the MESA monocyte samples with both RNA-sequencing and methylation profiles (n=373, including 29 current smokers and 152 never smokers), cg05575921 methylation significantly (FDR < 0.05) associated with mRNA expression of only one gene within 1 MB, AHRR (r = −0.42, p = 1.4×10−17, FDR = 4.32×10−15; Figure 5a). Analysis stratified by smoking status revealed that AHRR expression was strongly up-regulated in current smokers (p = 3.29×10−21, Data Supplement Figure 3a), which was replicated using real-time PCR in a set of independent monocyte samples (p = 0.003; Data Supplement Figure 3b).
Figure 5.
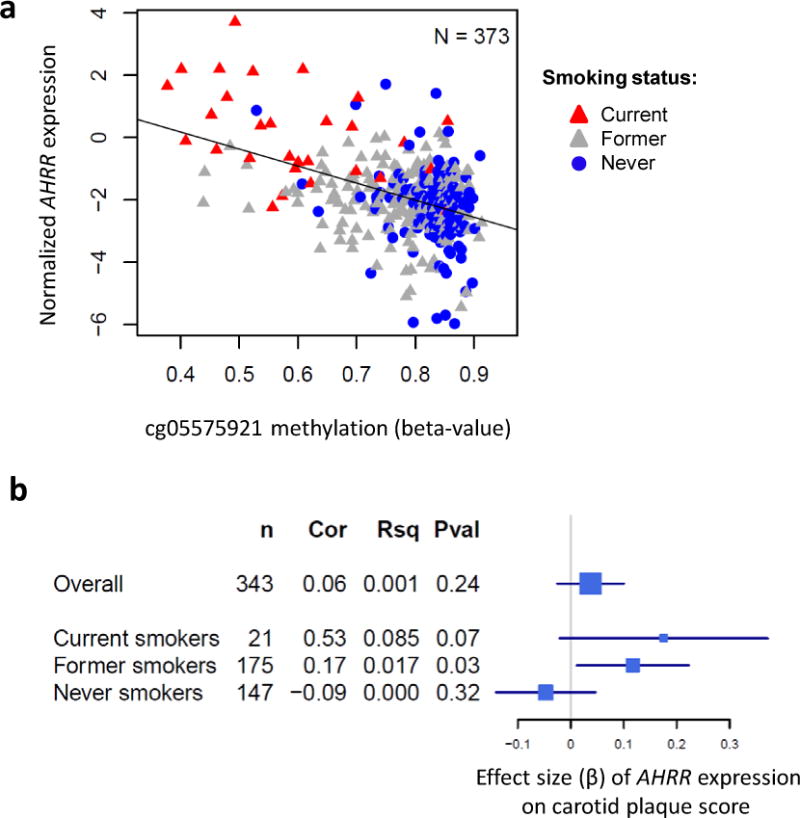
AHRR expression associations with AHRR methylation and carotid plaque score. (a) mRNA expression profiles of AHRR measured using RNA sequencing (y-axis, normalized values) negatively correlate (r = −0.42, p = 1.36×10−17) with methylation of AHRR cg05575921 (x-axis, beta-value) measured using the Illumina 450K microarray. Regression covariates included age, sex, race, and study site. (b) Forest plot shows the estimated effect size (x-axis: beta and 95% CI) of AHRR mRNA expression on carotid plaque score (overall and stratified by smoking status). Also shown are the sample size (n), the partial Pearson’s correlation coefficient (Cor) between AHRR expression and carotid plaque score, the unique variance of carotid plaque score explained by AHRR expression (Rsq), and the significance (Pval). Regression covariates included age, sex, race, BMI, LDL cholesterol, diabetes, and hypertension.
Next, we tested for associations between AHRR expression and carotid plaque score and found suggestive evidence, even in our small sample size of current smokers with RNA-sequencing measurements, for a positive association between AHRR expression and carotid plaque score (r = 0.53, p = 0.07, n = 21; Figure 5b). Carotid plaque score was also positively associated with AHRR expression in former smokers (r = 0.17, p = 0.03, n = 175), but not in never smokers (p > 0.05; n = 147).
Nearby genetic variants were also investigated for association with cg05575921 methylation; however, no significant (FDR < 0.05) associations were observed with common genetic variants.
Discussion
Extending previous findings in other tissues, we found decreased methylation levels in human monocytes at the AHRR CpG cg05575921 is a significant biomarker of smoking. This finding is in line with several previous reports in other cell types6, 7, 11. Cg05575921 methylation appeared to be dosage-dependent and at least partially reversible upon smoking cessation. These results, along with other published reports7, indicate that methylation levels at cg05575921 represent a biomarker of “effect” that integrates both smoking dose and duration.
Importantly, we uncovered novel associations between smoking-responsive methylation of AHRR in monocytes and subclinical atherosclerosis in a large, multi-ethnic human cohort, and replicated the association between subclinical atherosclerosis and AHRR methylation in hepatic cells from an independent cohort. These results suggest that smoking-altered methylation of AHRR in multiple cell types may represent a biomarker for the risk of atherosclerosis associated with smoking.
Our results also indicate that AHRR methylation could be a useful biomarker of atherosclerosis in addition to self-reported smoking exposure and other known CVD risk factors. For instance, in current and former smokers, AHRR methylation remained significantly associated with subclinical atherosclerosis even after adjustment for self-reported smoking exposure, urinary cotinine, and many other known CVD risk factors. However, longitudinal studies are necessary to confirm AHRR methylation as a predictor of atherosclerosis.
Genome-wide association studies have suggested that differential DNA methylation is often associated with expression changes of nearby genes33. Functionally, we found reduced methylation levels at cg05575921, measured by microarray, were associated with up-regulated AHRR mRNA expression in monocytes from 373 participants, measured using RNA-sequencing, and in an independent sample using RT-PCR. Combined with a recent report in lung tissue from five smokers and five non-smokers that found smoking-associated methylation of AHRR was linked with increased AHRR expression (p=0.0047)10, these results provide evidence that altered AHRR methylation profiles may mediate increased transcription of AHRR in smokers.
Previous studies indicate that AHRR is transcriptionally up-regulated by activation of the Ah Receptor pathway12, 13. Interestingly, RRBS showed that the AHRR cg05575921 locus is located in a smoking-associated DMR, which appears to overlap an intragenic enhancer that is activated, accompanied by up-regulation of AHRR expression, when monocytes differentiate into macrophages19. Taken together with the ability of DNA methylation profiles to predict enhancer activity40, 41, we hypothesize that smoking-induced Ah Receptor pathway activation drives methylation changes in the AHRR gene accompanied by histone modifications and enhancer activation, resulting in up-regulation of AHRR expression in monocytes. Residual levels of altered methylation and enhancer activity at the cg05575921 DMR in former smokers could partially explain why AHRR gene expression was previously reported to be up-regulated in former smokers compared with never smokers even decades after smoking cessation42.
The consequences of AHRR up-regulation in monocytes is unclear, but given the potential role of AHRR in macrophage biology19 and suppression of anti-inflammation13, up-regulation of AHRR could plausibly alter the function of monocytes and macrophages in smokers. While there is currently no known role for AHRR in atherogenesis, a number of studies have shown that activation of the Ah Receptor pathway promotes atherosclerosis. For instance, using in vitro and in vivo models in mice, Wu et al18 demonstrated that Ah Receptor pathway activation with treatments such as cigarette smoke extract or 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD), increased expression of inflammatory markers in macrophages, and were associated with the accumulation of lipids in macrophages and plaque formation. Vogel et al.43 showed that Ah Receptor pathway activation increased cytokine production and foam cell formation. A limitation of our study is it remains unclear if smoking-associated AHRR expression is causally related to atherogenesis, or occurs in parallel to the Ah Receptor activation-related effects on plaque formation noted above. Mediation analysis results do not distinguish confounding from mediating effects; therefore, although we observe cg05575921 methylation significantly mediating the effect of pack-years on carotid plaque score, these results do not confirm biological mediation and should be interpreted with caution.
In conclusion, we identified smoking-responsive methylation profiles at the cg05575921 locus which are associated with up-regulated AHRR expression in monocytes, and with subclinical atherosclerosis in a human population. Future functional studies are necessary to understand the potential role of AHRR in response to cigarette smoke and in atherogenesis.
Supplementary Material
Acknowledgments
The authors thank the other investigators, the staff, and the participants of the MESA study for their valuable contributions. A full list of participating MESA investigators and institutions can be found at http://www.mesa-nhlbi.org. The authors would also like to thank Drs. Michael Ziller and Alexander Meissner of the Broad Institute of MIT and Harvard for their generosity in providing the R code to identify differentially methylated regions.
Funding Sources: This research was supported by contracts N01-HC-from the National Heart, Lung and Blood Institute, by grants UL1-RR-024156 and UL1-RR-025005 from the National Center for Research Resources, and support from the Intramural Research Program (projects: ZO1-ES100475 and Z01 ES046008) of the National Institute of Environmental Health Sciences. The MESA Epigenomics & Transcriptomics Study was funded by NHLBI grant R01HL101250 to Wake Forest University Health Sciences. Urinary Cotinine measurements are available to the MESA study courtesy of MESA Lung contract R01-HL077612. The research described in this publication was funded in part by the U.S Environmental Protection Agency through RD831697 to the University of Washington (MESA Air); it has not been subjected to the Agency’s required peer and policy review and therefore does not necessarily reflect the views of the Agency and no official endorsement should be inferred.
Footnotes
Conflict of Interest Disclosures: None.
Journal Subject Terms: Epidemiology; Functional Genomics; Gene Expression and Regulation
References
- 1.Csiszar A, Podlutsky A, Wolin MS, Losonczy G, Pacher P, Ungvari Z. Oxidative stress and accelerated vascular aging: implications for cigarette smoking. Front Biosci (Landmark Ed) 2009;14:3128–3144. doi: 10.2741/3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Breitling LP. Current genetics and epigenetics of smoking/tobacco-related cardiovascular disease. Arterioscler Thromb Vasc Biol. 2013;33:1468–1472. doi: 10.1161/ATVBAHA.112.300157. [DOI] [PubMed] [Google Scholar]
- 3.Petronis A. Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature. 2010;465:721–727. doi: 10.1038/nature09230. [DOI] [PubMed] [Google Scholar]
- 4.Talhout R, Schulz T, Florek E, van BJ, Wester P, Opperhuizen A. Hazardous compounds in tobacco smoke. Int J Environ Res Public Health. 2011;8:613–628. doi: 10.3390/ijerph8020613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muto H, Takizawa Y. Dioxins in cigarette smoke. Arch Environ Health. 1989;44:171–174. doi: 10.1080/00039896.1989.9935882. [DOI] [PubMed] [Google Scholar]
- 6.Joubert BR, Haberg SE, Nilsen RM, Wang X, Vollset SE, Murphy SK, et al. 450K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environ Health Perspect. 2012;120:1425–1431. doi: 10.1289/ehp.1205412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Philibert RA, Beach SR, Lei MK, Brody GH. Changes in DNA methylation at the aryl hydrocarbon receptor repressor may be a new biomarker for smoking. Clin Epigenetics. 2013;5:19. doi: 10.1186/1868-7083-5-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dogan MV, Shields B, Cutrona C, Gao L, Gibbons FX, Simons R, et al. The effect of smoking on DNA methylation of peripheral blood mononuclear cells from African American women. BMC Genomics. 2014;15:151. doi: 10.1186/1471-2164-15-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zeilinger S, Kuhnel B, Klopp N, Baurecht H, Kleinschmidt A, Gieger C, et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS One. 2013;8:e63812. doi: 10.1371/journal.pone.0063812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shenker NS, Polidoro S, van Veldhoven K, Sacerdote C, Ricceri F, Birrell MA, et al. Epigenome-wide association study in the European Prospective Investigation into Cancer and Nutrition (EPIC-Turin) identifies novel genetic loci associated with smoking. Hum Mol Genet. 2013;22:843–851. doi: 10.1093/hmg/dds488. [DOI] [PubMed] [Google Scholar]
- 11.Monick MM, Beach SR, Plume J, Sears R, Gerrard M, Brody GH, et al. Coordinated changes in AHRR methylation in lymphoblasts and pulmonary macrophages from smokers. Am J Med Genet B Neuropsychiatr Genet. 2012;159B:141–151. doi: 10.1002/ajmg.b.32021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kazantseva MG, Highton J, Stamp LK, Hessian PA. Dendritic cells provide a potential link between smoking and inflammation in rheumatoid arthritis. Arthritis Res Ther. 2012;14:R208. doi: 10.1186/ar4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Awji EG, Chand H, Bruse S, Smith KR, Colby JK, Mebratu Y, et al. Wood Smoke Enhances Cigarette Smoke-Induced Inflammation by Inducing the Aryl Hydrocarbon Receptor Repressor in Airway Epithelial Cells. Am J Respir Cell Mol Biol. 2015;52:377–386. doi: 10.1165/rcmb.2014-0142OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dertinger SD, Silverstone AE, Gasiewicz TA. Influence of aromatic hydrocarbon receptor-mediated events on the genotoxicity of cigarette smoke condensate. Carcinogenesis. 1998;19:2037–2042. doi: 10.1093/carcin/19.11.2037. [DOI] [PubMed] [Google Scholar]
- 15.Kerley-Hamilton JS, Trask HW, Ridley CJ, Dufour E, Lesseur C, Ringelberg CS, et al. Inherent and benzo[a]pyrene-induced differential aryl hydrocarbon receptor signaling greatly affects life span, atherosclerosis, cardiac gene expression, and body and heart growth in mice. Toxicol Sci. 2012;126:391–404. doi: 10.1093/toxsci/kfs002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Humblet O, Birnbaum L, Rimm E, Mittleman MA, Hauser R. Dioxins and cardiovascular disease mortality. Environ Health Perspect. 2008;116:1443–1448. doi: 10.1289/ehp.11579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Savouret JF, Berdeaux A, Casper RF. The aryl hydrocarbon receptor and its xenobiotic ligands: a fundamental trigger for cardiovascular diseases. Nutr Metab Cardiovasc Dis. 2003;13:104–113. doi: 10.1016/s0939-4753(03)80026-1. [DOI] [PubMed] [Google Scholar]
- 18.Wu D, Nishimura N, Kuo V, Fiehn O, Shahbaz S, Van WL, et al. Activation of aryl hydrocarbon receptor induces vascular inflammation and promotes atherosclerosis in apolipoprotein E−/− mice. Arterioscler Thromb Vasc Biol. 2011;31:1260–1267. doi: 10.1161/ATVBAHA.110.220202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saeed S, Quintin J, Kerstens HH, Rao NA, Aghajanirefah A, Matarese F, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. 2014;345:1251086. doi: 10.1126/science.1251086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bergmann S, Siekmeier R, Mix C, Jaross W. Even moderate cigarette smoking influences the pattern of circulating monocytes and the concentration of sICAM-1. Respir Physiol. 1998;114:269–275. doi: 10.1016/s0034-5687(98)00098-x. [DOI] [PubMed] [Google Scholar]
- 21.Ley K, Miller YI, Hedrick CC. Monocyte and macrophage dynamics during atherogenesis. Arterioscler Thromb Vasc Biol. 2011;31:1506–1516. doi: 10.1161/ATVBAHA.110.221127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lessner SM, Prado HL, Waller EK, Galis ZS. Atherosclerotic lesions grow through recruitment and proliferation of circulating monocytes in a murine model. Am J Pathol. 2002;160:2145–2155. doi: 10.1016/S0002-9440(10)61163-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bild DE, Bluemke DA, Burke GL, Detrano R, Diez Roux AV, Folsom AR, et al. Multi-ethnic study of atherosclerosis: objectives and design. Am J Epidemiol. 2002;156:871–881. doi: 10.1093/aje/kwf113. [DOI] [PubMed] [Google Scholar]
- 24.Wissler RW. USA Multicenter Study of the pathobiology of atherosclerosis in youth. Ann N Y Acad Sci. 1991;623:26–39. doi: 10.1111/j.1749-6632.1991.tb43716.x. [DOI] [PubMed] [Google Scholar]
- 25.Rosenbloom KR, Sloan CA, Malladi VS, Dreszer TR, Learned K, Kirkup VM, et al. ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res. 2013;41:D56–D63. doi: 10.1093/nar/gks1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adams D, Altucci L, Antonarakis SE, Ballesteros J, Beck S, Bird A, et al. BLUEPRINT to decode the epigenetic signature written in blood. Nat Biotechnol. 2012;30:224–226. doi: 10.1038/nbt.2153. [DOI] [PubMed] [Google Scholar]
- 27.Ferris BG. Epidemiology Standardization Project (American Thoracic Society) Am Rev Respir Dis. 1978;118:1–120. [PubMed] [Google Scholar]
- 28.Rodriguez J, Jiang R, Johnson WC, MacKenzie BA, Smith LJ, Barr RG. The association of pipe and cigar use with cotinine levels, lung function, and airflow obstruction: a cross-sectional study. Ann Intern Med. 2010;152:201–210. doi: 10.1059/0003-4819-152-4-201002160-00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stein JH, Korcarz CE, Hurst RT, Lonn E, Kendall CB, Mohler ER, et al. Use of carotid ultrasound to identify subclinical vascular disease and evaluate cardiovascular disease risk: a consensus statement from the American Society of Echocardiography Carotid Intima-Media Thickness Task Force. Endorsed by the Society for Vascular Medicine. J Am Soc Echocardiogr. 2008;21:93–111. doi: 10.1016/j.echo.2007.11.011. [DOI] [PubMed] [Google Scholar]
- 30.Touboul PJ, Hennerici MG, Meairs S, Adams H, Amarenco P, Bornstein N, et al. Mannheim carotid intima-media thickness consensus (2004–2006). An update on behalf of the Advisory Board of the 3rd and 4th Watching the Risk Symposium, 13th and 15th European Stroke Conferences, Mannheim, Germany, 2004, and Brussels, Belgium, 2006. Cerebrovasc Dis. 2007;23:75–80. doi: 10.1159/000097034. [DOI] [PubMed] [Google Scholar]
- 31.Tattersall MC, Gassett A, Korcarz CE, Gepner AD, Kaufman JD, Liu KJ, et al. Predictors of carotid thickness and plaque progression during a decade: the multi-ethnic study of atherosclerosis. Stroke. 2014;45:3257–3262. doi: 10.1161/STROKEAHA.114.005669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Y, Ding J, Reynolds LM, Lohman K, Register TC, de la Fuente A, et al. Methylomics of gene expression in human monocytes. Hum Mol Genet. 2013;22:5065–5074. doi: 10.1093/hmg/ddt356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abbas AR, Baldwin D, Ma Y, Ouyang W, Gurney A, Martin F, et al. Immune response in silico (IRIS): immune-specific genes identified from a compendium of microarray expression data. Genes Immun. 2005;6:319–331. doi: 10.1038/sj.gene.6364173. [DOI] [PubMed] [Google Scholar]
- 35.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rosseel Y. lavaan: An R Package for Structural Equation Modeling. J Stat Softw. 2012;48:1–36. [Google Scholar]
- 37.Boyle P, Clement K, Gu H, Smith ZD, Ziller M, Fostel JL, et al. Gel-free multiplexed reduced representation bisulfite sequencing for large-scale DNA methylation profiling. Genome Biol. 2012;13:R92. doi: 10.1186/gb-2012-13-10-r92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ziller MJ, Gu H, Muller F, Donaghey J, Tsai LT, Kohlbacher O, et al. Charting a dynamic DNA methylation landscape of the human genome. Nature. 2013;500:477–481. doi: 10.1038/nature12433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmidl C, Klug M, Boeld TJ, Andreesen R, Hoffmann P, Edinger M, et al. Lineage-specific DNA methylation in T cells correlates with histone methylation and enhancer activity. Genome Res. 2009;19:1165–1174. doi: 10.1101/gr.091470.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wiench M, John S, Baek S, Johnson TA, Sung MH, Escobar T, et al. DNA methylation status predicts cell type-specific enhancer activity. EMBO J. 2011;30:3028–3039. doi: 10.1038/emboj.2011.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bosse Y, Postma DS, Sin DD, Lamontagne M, Couture C, Gaudreault N, et al. Molecular signature of smoking in human lung tissues. Cancer Res. 2012;72:3753–3763. doi: 10.1158/0008-5472.CAN-12-1160. [DOI] [PubMed] [Google Scholar]
- 43.Vogel CF, Sciullo E, Matsumura F. Activation of inflammatory mediators and potential role of ah-receptor ligands in foam cell formation. Cardiovasc Toxicol. 2004;4:363–373. doi: 10.1385/ct:4:4:363. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.