

Abstract
Background
Next-generation sequencing provides a powerful means of molecular characterization. However, methods such as single-nucleotide polymorphism detection or whole-chromosome sequence analysis are computationally expensive, prone to errors, and are still less accessible than traditional typing methods. Here, we present the Listeria monocytogenes core-genome sequence typing method for molecular characterization. This method uses a high-confidence core (HCC) genome, calculated to ensure accurate identification of orthologs. We also developed an evolutionarily relevant nomenclature based upon phylogenetic analysis of HCC genomes. Finally, we created a pipeline (LmCGST; https://sourceforge.net/projects/lmcgst/files/) that takes in raw next-generation sequencing reads, calculates a subject HCC profile, compares it to an expandable database, assigns a sequence type, and performs a phylogenetic analysis.
Results
We analyzed 29 high-quality, closed Listeria monocytogenes chromosome sequences and identified loci that are reliable targets for automated molecular characterization methods. We identified 1013 open-reading frames that comprise our high-confidence core (HCC) genome. We then populated a database with HCC profiles from 114 taxa. We sequenced 84 randomly selected isolates from the Listeriosis Reference Service for Canada’s collection and analysed them with the LmCGST pipeline. In addition, we generated pulsed-field gel electrophoresis, ribotyping, and in silico multi-locus sequence typing (MLST) data for the 84 isolates and compared the results to those obtained using the CGST method. We found that all of the methods yielded results that are generally congruent. However, due to the increased numbers of categories, the CGST method provides much greater discriminatory power than the other methods tested here.
Conclusions
We show that the CGST method provides increased discriminatory power relative to typing methods such as pulsed-field gel electrophoresis, ribotyping, and multi-locus sequence typing while it addresses several shortcomings of other methods of molecular characterization with next-generation sequence data. It uses discrete, well-defined groupings (types) of organisms that are phylogenetically relevant and easily interpreted. In addition, the CGST scheme can be expanded to include additional loci and HCC profiles in the future. In total, the CGST method provides an approach to the molecular characterization of Listeria monocytogenes with next-generation sequence data that is highly reproducible, easily standardized, portable, and accessible.
Electronic supplementary material
The online version of this article (doi:10.1186/s12866-015-0526-1) contains supplementary material, which is available to authorized users.
Keywords: Listeria monocytogenes, Typing, Molecular characterization, Next-generation sequencing, Pulsed-field gel electrophoresis, PFGE, Multi-locus sequence typing, MLST, wgMLST, cgMLST
Background
Listeria monocytogenes is a facultatively anaerobic, Gram-positive bacterium that occurs naturally in plant, soil, and surface water environments [1]. However, L. monocytogenes may also be isolated from domestic cattle, sheep, goats, and poultry [2] and can make its way into the food supply, causing sporadic and outbreak cases of foodborne listeriosis [3]. Listeriosis is commonly associated with life-threatening meningitis and septicemia in adults and may lead to miscarriages in pregnant women [4]. Thus, molecular characterization (typing) of L. monocytogenes is important to clinical microbiology and to the epidemiological analysis of listeriosis [5].
Molecular typing methods such as pulsed-field gel electrophoresis [6], ribotyping [7], and multi-locus sequence typing (MLST) [8] have provided greatly improved resolving power relative to previously used phenotypic methods, such as serotyping, phage typing, biotyping, antibiotic susceptibility testing, and bacteriocin typing [9]. Recent advances in DNA sequencing technologies and reduced costs have made high-quality whole-genome sequence (WGS) data readily available [10]. Comprehensive sequencing and analysis of bacterial genomes has been shown to be a valuable tool for epidemiological studies [11, 12]. In particular, WGS data are commonly used to identify nucleotide differences, so-called single-nucleotide polymorphisms (SNPs), between bacterial chromosomes [13]. However, studies have shown that the use of either de-novo or reference-guided assemblies for identifying SNP differences between subjects and references can lead to errors that make interpretation difficult [14–16]. Alternatively, WGS data may be assembled de-novo into chromosome sequence data that can be aligned and phylogenetically analyzed in order to compare them directly. However, we show that this method can generate misleading results, presumably due to differences between sequencing runs and/or the computational challenges of aligning short-read sequence data without a reference. Most importantly, the manner in which these methods are currently used ultimately relies upon the accurate interpretation of phylogenetic trees, a requirement that can make the assignment of discrete groupings (types) difficult and can make these approaches less accessible than the typing methods they are intended to replace.
Whole-genome multi-locus sequence typing (wgMLST) has been shown to be a powerful alternative to SNP or whole-chromosome analyses [17] that is amenable to the assignment of discrete groupings in addition to phylogenetic analysis. In concept, this method is nearly identical to the MLST approach, during which 7 or so loci are studied, with the exception that all the genes in a genome are examined. This scheme takes advantage of the discriminating power of WGS data while providing the basis for the grouping of organisms into sequence types. wgMLST also makes the interpretation of data easier and more accessible. However, the accurate identification of orthologous sequences is computationally expensive and there is no current consensus on how this should be accomplished, making wgMLST methods difficult to standardize and distribute. Researchers have shown that using the loci that comprise an organisms core-genome (cgMLST) provides a powerful means of analyzing WGS data that can be standardized [18, 19]. While cgMLST is an improvement over wgMLST, both methods, like standard MLST, rely upon allele numbering systems that often provide scant information regarding the evolutionary relationships of organisms.
Here, we present a cgMLST-style typing method for the molecular characterization of L. monocytogenes (core-genome sequence typing; CGST) that has several advantages over other proposed methods. In order to ensure that orthologs are properly identified, we calculated a high-confidence core (HCC) of genes that is useful for reliable and efficient large-scale typing. Furthermore, we developed a nomenclature that is based upon nucleotide distances between HCC profiles, providing phylogenetically meaningful groupings. Finally, we wrote a bioinformatic pipeline (LmCGST available at https://sourceforge.net/projects/lmcgst/files/) that can: i) analyze raw reads, contiguous sequences, or closed chromosome sequences; ii) identify an organism’s HCC profile; iii) compare that profile to an expandable database; iv) provide an evolutionarily meaningful sequence type assignment; and v) generate a phylogenetic analysis that illustrates the evolutionary relationship of the subject to the members of the database. In total, the CGST method provides an approach that is highly reproducible, easily standardized, portable, and accessible.
Results and discussion
Calculation of the Listeria monocytogenes core-genome
In order to calculate the pan- and core-genomes of Listeria monocytogenes, we used a semi-automated approach in which protein sequence translations of open reading frames (ORFs) predicted from a set of 29 high-quality, phylogenetically diverse chromosome sequences obtained from the National Center for Biotechnology Information (NCBI) were analyzed (Additional file 1, red labels and Additional file 2). A pan-genome is defined as the total pool of ORFs present within the genomes examined (i.e., the union of ORFs) and a core-genome is defined as the subset of ORFs that are present within every genome (i.e., the intersection of ORFs) [20–22]. Based upon pairwise comparisons with the Basic Local Alignment Search Tool (BLAST) [23], using a protein sequence similarity cut-off of 60 % and a minimum coverage of 80 % [24], we determined that the L. monocytogenes pan- and core-genomes consist of 4766 and 2114 ORFs, respectively (Additional file 3). Our results are consistent with the analysis of 17 L. monocytogenes genomes performed by Kuenne, et al. (Additional file 3, diamond) [24].
Calculation of a high-confidence core
During the course of calculating the L. monocytogenes pan- and core-genomes, we identified conditions that confound pairwise homology searches, such as loci that retrieve multiple hits (i.e., multi-copy genes), the use of different ORF prediction software, and the presence of low complexity regions within ORFs. Therefore, we further curated the set of 2114 core ORFs as described below in order to develop a robust database, ensuring that all orthologs used for downstream comparisons are properly identified. We found and removed 439 predicted multi-copy ORFs. In addition, we annotated the 29 high-quality, closed genome sequences obtained from NCBI with GeneMark [25], Glimmer [26, 27], and Prokka v1.10 [28]. We then used these datasets to estimate three different single-copy core-genomes that are composed of 1772, 1438, and 1826 ORFs, respectively. Differences in the numbers of ORFs predicted may occur as programs use a variety of methods to identify start/stop codons and overlapping ORFs [29]. We performed an all versus all bidirectional protein sequence BLAST to identify 1061 ORFs that are shared in all four datasets. Then, we performed an all versus all bidirectional nucleotide BLAST with 114 taxa in order to identify ORFs that fail to generate hits (likely due to the presence of low complexity regions) or generate ambiguous results. Any ORF that did not generate hits to all other taxa when used as a query were removed from the analysis. This resulted in the removal of an additional 48 ORFs. In total, 1013 ORFs were identified (Additional file 4) that are reliable targets of nucleotide and protein sequence BLAST searches and, so, comprise hour high-confidence core (HCC) of predicted genes.
In order to characterize the HCC, we assigned Clusters of Orthologous Groups (COGs) categories to the full set of strain 08–5578 protein sequences [GenBank: NC_013766] and compared it to the suite of predicted proteins in the HCC (Additional file 5). The distributions of both the full-set of proteins and the HCC-set within COGs categories are similar. This result indicates that the HCC provides a good approximation of the diversity of functions present in the full complement of L. monocytogenes genes. We also mapped the strain 08–5578 HCC genes and the HCC loci appear to be evenly distributed throughout the chromosome (Additional file 6).
Core-genome sequence typing of Listeria monocytogenes
We developed a bioinformatic pipeline using the Perl programming language (LmCGST.pl, available at https://sourceforge.net/projects/lmcgst/files/) for identifying HCC loci in genome sequence data and comparing a subject HCC to a database that has been seeded with HCC profiles calculated from 114 unique, high-quality chromosome sequences (Additional file 7). The script can take raw short-read sequence data (i.e., fastq files), contiguous sequences, closed chromosome sequences, or fully annotated ORFs as input. The software compares the subject HCC to the database and identifies the HCC profile with the fewest nucleotide differences. A file is also generated that reports on the numbers of perfect ORF matches, partial ORF matches, missing ORFs, and the numbers of SNPs, along with the identities of SNP-containing ORFs and the positions of the SNPs.
A phylogenetically relevant typing scheme was developed by grouping HCC profiles by evolutionary lineages (Fig. 1). Then we calculated the pairwise nucleotide distances between HCC profiles of each member of the database. We identified and grouped those HCC profiles with less than or equal to 100 base-pair differences and those with less than or equal to 10 base-pair differences. Finally, unique HCC profiles that differ by fewer than 10 nucleotides were numbered in the order that they were processed. A core-genome sequence type is, therefore, defined as a unique set of HCC sequences and subjects must be 100 % identical at the nucleotide level with no missing or partial ORFs to be considered a match to any HCC profile in the database. This typing scheme allows for the assignment of unique identifiers that specify the evolutionary relationships of subjects to members of the database within the L. monocytogenes phylogeny (Additional file 8). Finally, a phylogenetic analyses of the HCC profiles of the subject and the database are generated in order to visualize their evolutionary relationships (Additional file 9).
Fig. 1.
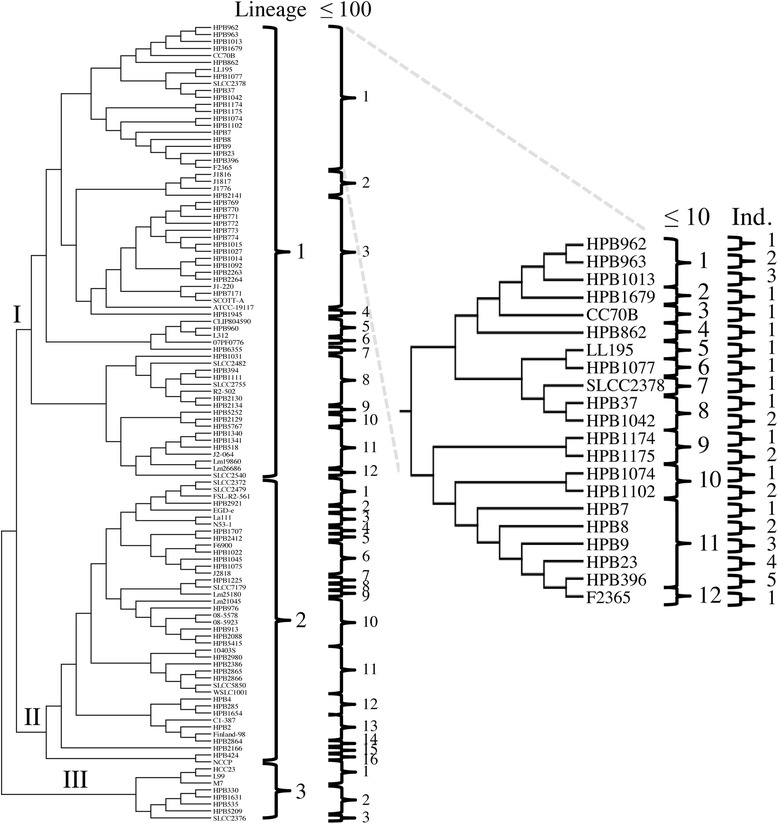
Listeria monocytogenes high-confidence core profiles grouped by nucleotide distances. A cladogram was calculated by aligning and concatenating 1013 loci that comprise the L. monocytogenes high-confidence core (HCC) genomes of 114 taxa and analyzing the resulting alignment of 1,067,173 nucleotide positions with the Randomized Axelerated Maximum Likelihood tool (GTRCATI + 25γ). The best of 100 bootstrap replicates is shown. Nucleotide distances were measured with PHYLIP. Taxa were grouped by evolutionary lineage (I, II, or III) and those that have 100 and 10 or fewer nucleotide differences, while unique HCC profiles that differ by no more than 10 nucleotides were numbered in the order that they were processed
In order to establish the levels of genome sequence coverage necessary for accurate core-genome sequence typing, we tested the pipeline with a set of 12 L. monocytogenes strain 08–5578 Illumina short-read sequence datasets obtained from sequencing runs of varying qualities (Additional file 10 and Additional file 11). We found that de-novo sequence assemblies of at least 66-fold coverage provide reliable results, with no false SNPs, missing ORFs, or partial ORFs.
In order to predict the amount of time required to run the LmCGST pipeline as the size of the database increases, we documented the amount of time necessary for the pipeline to complete the genome assembly, annotation, HCC comparisons, and phylogenetic analyses for databases containing 25, 50, and 100 randomly selected HCC profiles (Additional file 12). In addition, we estimated the amount of time required to assemble and annotate a single genome. Genome assembly with SPAdes takes approximately 2.69 h, while it takes approximately 0.09 h to perform annotation with Prokka. Although these times may change with different genomes and short-read sequence datasets, they did not increase with the size of the database (Additional file 13). For the HCC comparison and optional tree-building steps, processing times did increase with the size of the database. We fitted the amount of time necessary to perform the HCC comparisons and phylogenetic analyses to a polynomial regression model (y = 0.0015x2 + 2.027x-6.4733 and y = 0.0631x2 + 9.973x + 33.137, respectively). With 100 taxa occupying the database, the comparison step took approximately 0.059 h and the tree-building step took approximately 0.46 h. When we predicted processing times with 500 taxa present in the database, we estimated that it will take approximately 0.38 h for the comparison step and 5.77 h to estimate the phylogeny. The total amount of time that is predicted to be necessary for the LmCGST pipeline to run with a database of 500 HCC profiles is 8.93 h, with the assembly, annotation, and comparison steps accounting for approximately 3.16 h.
Comparison of molecular typing methods
In order to compare the core-genome sequence typing (CGST) method to currently used molecular typing methods, we randomly selected 84 strains from the collection at the Listeriosis Reference Service for Canada. Then, we performed pulsed-field gel electrophoresis (PFGE) [30] with both ApaI and AscI restriction enzymes and ribotyping [7]. In addition, we generated whole-genome sequence data with an Illumina MiSeq benchtop sequencer and performed in silico multi-locus sequence typing (MLST) [8] with abcZ, bglA, cat, dapE, dat, ldh, and lhkA loci [31] and CGST (Fig. 2). Previous studies have shown that in silico MLST analyses of next-generation sequence data allow for high levels of allelic identification and are highly-concordant with published and publicly available sequence types [32]. We analyzed the congruence and discriminating power of each typing method individually and in two combinations in which we analyzed ApaI and AscI PFGE together (labelled PFGE) and we analyzed ApaI, AscI, and ribotyping together (labelled PFGE + Ribo). We began by calculating the Simpson’s index of diversity [33] with 95 % confidence intervals (Additional file 14). The values for all categories range from 0.889 (Ribotyping) to 0.995 (CGST) and the data indicate that CGST has higher discriminating power than the other typing methods, either individually or combined. In addition, the data indicate that the strains randomly selected for this study are sufficiently diverse for purposes of comparing these different typing methods.
Fig. 2.
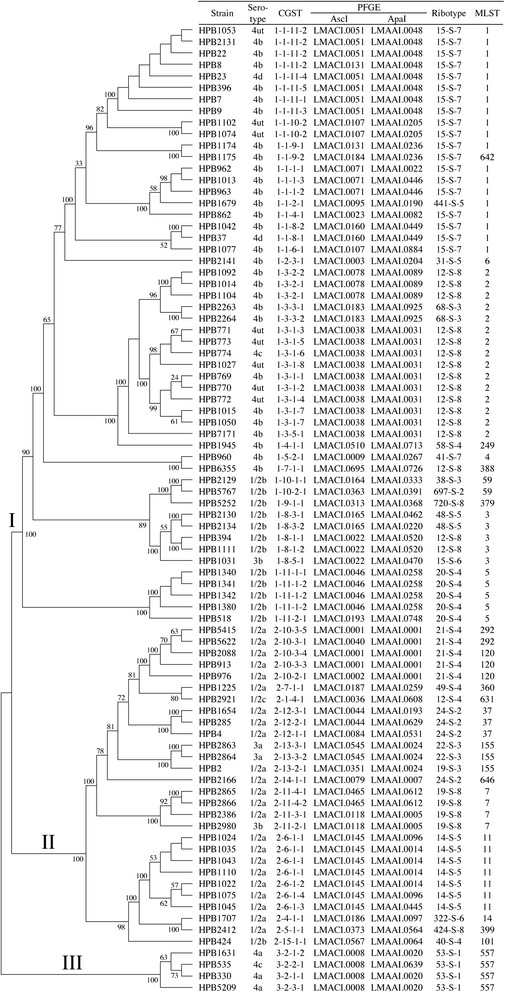
Typing data derived from 84 Listeria monocytogenes strains. Strains were selected randomly from the collection stored at the Listeriosis Reference Service for Canada. Standard typing assays, such as serotyping, AscI and ApaI pulsed-field gel electrophoresis (PFGE), and ribotyping were performed. In addition, whole-genome sequence data were generated and analyzed with in silico multi-locus sequence typing (MLST) and core-genome sequence typing (CGST)
We then calculated the adjusted Wallace coefficients [34, 35] with 95 % confidence intervals [36] for the typing datasets (Additional file 15 and Table 1). The data indicate that the typing methods tested here are fairly congruent. For example, if two strains are identified as belonging to the same group using the CGST method, there is approximately a 65.7 % chance that they will be grouped together with PFGE or PFGE + Ribo and a 100.0 % chance they will be grouped together with ribotyping or MLST methods (Table 1, first row). The data also indicate that, due to the increased number of categories, the CGST method has greater discriminatory power than the other methods tested here. That is, if two strains are identified as belonging to the same group with either PFGE or PFGE + Ribo, there is a 12.0 % chance that they will be grouped together with the CGST method, while ribotyping and MLST methods yielded values of 4.6 and 4.8 %, respectively (Table 1, first column).
Table 1.
Adjusted Wallace coefficient and 95 % confidence intervals
| CGST | PFGE + Ribo | PFGE | ApaI | AscI | Ribotype | MLST | |
|---|---|---|---|---|---|---|---|
| CGST | 0.657 (0.400–0.914) | 0.657 (0.400–0.914) | 0.828 (0.645–1.000) | 0.827 (0.644–1.000) | 1.000 (1.000–1.000) | 1.000 (1.000–1.000) | |
| PFGE + Ribo | 0.120 (0.019–0.222) | 1.000 (1.000–1.000) | 1.000 (1.000–1.000) | 1.000 (1.000–1.000) | 1.000 (1.000–1.000) | 0.977 (0.955–0.999) | |
| PFGE | 0.120 (0.019–0.222) | 1.000 (1.000–1.000) | 1.000 (1.000–1.000) | 1.000 (1.000–1.000) | 1.000 (1.000–1.000) | 0.977 (0.955–0.999) | |
| ApaI | 0.128 (0.024–0.233) | 0.845 (0.743–0.947) | 0.845 (0.743–0.947) | 0.844 (0.741–0.947) | 0.980 (0.962–0.999) | 0.931 (0.896–0.967) | |
| AscI | 0.118 (0.017–0.220) | 0.781 (0.711–0.851) | 0.781 (0.711–0.851) | 0.780 (0.709–0.850) | 0.982 (0.965–0.999) | 0.982 (0.965–0.999) | |
| Ribotype | 0.046 (0.000–0.095) | 0.251 (0.137–0.365) | 0.251 (0.137–0.365) | 0.291 (0.171–0.412) | 0.316 (0.207–0.425) | 0.787 (0.641–0.932) | |
| MLST | 0.048 (0.000–0.099) | 0.256 (0.144–0.368) | 0.256 (0.144–0.368) | 0.289 (0.168–0.410) | 0.329 (0.223–0.436) | 0.820 (0.683–0.957) |
The Wallace coefficient measures agreement between groupings made with different typing methods. Row headers indicate methods from which two random samples were drawn and column headers identify the methods that were compared. The probabilities that two samples grouped together with one method (rows) will also be grouped together with another method (columns) are shown along with 95 % confidence intervals (parentheses)
ApaI and AscI data were combined to generate the PFGE category and ApaI, AscI, and Ribotype data were combined to generate the PFGE + Ribo category
Conclusions
Here, we have calculated a Listeria monocytogenes high-confidence core (HCC) genome which serves as the basis for an extended multi-locus sequence typing regime called core-genome sequence typing (CGST). We have shown that analysis of next-generation sequence data with CGST provides significantly increased power to distinguish isolates of L. monocytogenes relative to currently used methods of molecular characterization. Furthermore, CGST provides several advantages over other typing methods that utilize next-generation sequence data, such as the analysis of single-nucleotide polymorphisms (SNPs) or phylogenetic analysis of whole-chromosome sequence data. Recent studies have shown that SNP analyses can be problematic and can result in phylogenetic artifacts that may obscure the true relationships between isolates, whether a de-novo or reference-guided approach is used [14–16]. Theoretically, simply performing phylogenetic analysis of whole-chromosome sequence data should provide the highest levels of resolution. However, differences between next-generation sequencing runs may introduce errors that make chromosome sequences appear more evolutionarily distant than they really are (Additional file 16 and Additional file 17). That is, phylogenetic analyses may yield branches separating taxa, due to differences between sequencing runs, when no differences between chromosomes actually exist. Although, it is possible that high sequence coverage (e.g., 155.48-fold) may reduce the numbers of differences, the CGST method is capable of accurately resolving relationships with much lower levels of sequence coverage (Additional file 10). Furthermore, accurate alignment and phylogenetic analysis of large numbers of whole-chromosomes is computationally expensive and accessible methods are currently lacking. Finally, both SNP and whole-chromosome analyses ultimately rely upon the interpretation of phylogenetic trees while keeping all of these considerations in mind. While methods that utilize next-generation sequence data can deliver increased resolution, they may lack discrete, well-defined types and may be less accessible than the typing methods they are meant to replace.
The aim of developing the extended MLST scheme presented here is to remedy the shortcomings of other methods of molecular characterization that utilize next-generation sequence data by providing discrete, well-defined groupings (types) of organisms that are phylogenetically relevant and easily interpreted. In addition, because we target 1013 HCC loci retrieved from de-novo genome assemblies, only the most reliable portions of chromosome sequence assemblies are used; regions that are common sources of error, such as gaps and repetitive regions, are avoided [37]. An additional, significant benefit of the CGST scheme is that it can be expanded in the future to include multi-copy and accessory genes, as necessary or desired, and studies correlating nucleotide differences between loci with important phenotypes can be incorporated. Furthermore, the database can be continually improved with the addition of novel HCC profiles. Thus, the CGST provides the best of next-generation sequence data analysis while avoiding several sources of error.
Methods
Calculation of the Listeria monocytogenes core-genome
We downloaded chromosome, gene, and protein sequence data for 29 high-quality, closed Listeria monocytogenes chromosome sequences from the National Center for Biotechnology Information (Additional file 2) [38–48]. Then, we used the methods of Kuenne et al. [24] to calculate the L. monocytogenes pan- and core-genomes. Briefly, we used the Basic Local Alignment Search Tool (BLAST) [23] to establish orthology by performing pairwise protein sequence alignments (i.e., all versus all BLASTp) with a minimum coverage threshold of 80 % and an identity cut-off of 60 % [24]. Sequences encoding proteins present in all 29 datasets were counted as members of the core-genome and the entire collection of sequences constitutes the pan-genome. Pan- and core-genome distributions were calculated by randomly selecting between 2 and 28 taxa randomly, with replacement and calculating 1000 times (Additional file 3).
Calculation of a high-confidence core
The high-confidence core (HCC) was calculated by identifying and removing open-reading frames from the calculated Listeria monocytogenes core-genome whose products yielded multiple hits with the all versus all pairwise alignment analysis of the 29 datasets. We then re-annotated the chromosome sequences three times with GeneMarkS v2.8 [25], Glimmer v3.02 [26, 27], and Prokka v1.10 [28]. Using BLASTp with the 80 % coverage, 60 % identity cut-offs, we identified sequences present in all four datasets (NCBI, GeneMark, Glimmer, and Prokka). Finally, we performed an all versus all BLASTn analysis in order to identify genes that, when used as a query, reliably retrieve (“hit”) homologous nucleotide sequences. The composition of HCC loci were compared to the total complement of genes present in the L. monocytogenes strain 08–5578 genome the by using the Clusters of Orthologous Genes database (“2003 COGs, original format”) available at http://www.ncbi.nlm.nih.gov/COG/ [49, 50] to analyze the calculated 08–5578 HCC and the NCBI annotation.
DNA extraction, library construction, and DNA sequencing
Listeria monocytogenes isolates frozen in glycerol were streaked on pre-warmed Tryptose Agar plates and incubated at 37 °C over-night. Single colonies were picked and used to inoculate 5 ml pre-warmed Brain Heart Infusion (BHI) broth and incubated over-night at 37 °C with shaking (200 rpm). Then, 200 μl of the cultures were transferred to 50 ml pre-warmed BHI and incubated at 37 °C with shaking for 6 h to achieve mid-logarithmic growth phase [51, 52]. Approximately 25 ml of the cultures were decanted into 50 ml falcon tubes and centrifuged at 3800 RCF for 5 min. The pellets were completely dissolved in 500 μl Tris-ethylenediaminetetraacetic acid by vortexing. We added 500 μl phenol-chloroform (1:1), 30 μl sodium acetate (3 M, pH 5.2), and 30 μl sodium dodecyl sulfate and mixed vigorously by shaking. The mixtures were then pipetted into 2 ml screw-cap tubes filled with approximately 0.5 ml glass beads (0.1 mm). The tubes were shaken in a Mini-Beadbeater machine (BioSpec products, Bartlesville, Oklahoma) for 45 s using the “Homogenizer” setting and placed on ice for 45 s. Shaking was repeated an additional four times. Approximately 300 μl of the mixtures were then added to Maxwell 16 Cell DNA Purification Kit cartridges and samples were run using the standard DNA Blood/Cells protocol on a Maxwell 16 machine (Promega, Madison, Wisconsin) with elution in 300 μl nuclease-free water. RNA contamination was removed by adding 2 μl RNase A (Qiagen Sciences, Maryland) and incubating the samples for 10 min at 37 °C. Single phenol-chloroform-isoamyl alcohol (25:24:1) extractions followed by two ethanol precipitations were done. Samples were indexed with Nextera XT DNA Sample Preparation Kits (Illumina, San Diego, California) according to the standard protocol and sequenced (2 × 250 bp reads) on a MiSeq benchtop sequencer (Illumina).
For the L. monocytogenes strain 08–5578 test dataset, the sample was split into four subsamples. Each subsample was indexed and sequenced as described above three separate times for a total of twelve sets of short-read sequences. These data have been deposited to the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) under accession numbers SRR1342176, SRR1342220, SRR1373524, SRR1373525, SRR1373527, SRR1373529, SRR1373530, SRR1373531, SRR1373534, SRR1373535, SRR1507228, and SRR1508282.
Core-genome sequence typing of Listeria monocytogenes
A Perl script was developed (LmCGST) that takes raw short-read (fastq), contiguous, or chromosome sequence files, identifies the high-confidence core (HCC) genome of a subject, compares the subject HCC profile to the database of HCC profiles, and generates a phylogenetic tree to illustrate the relationships of the subject to members of the database. If fastq files are provided, LmCGST assembles the reads de-novo using SPAdes v3.0.0 [53] and the BayesHammer error correction tool [54]. The resulting contiguous sequences are then annotated with Prokka v1.10 and the HCC loci are identified with bidirectional BLAST. Assemblies yielding partial (i.e., less than 60 %) or missing ORFs will generate a warning as sequence quality may be insufficient for genome-scale typing. Phylogenetic analyses are calculated with PHYLIP by first using the “dnadist” module to calculate distances (F84 model and default settings) and then by using the “neighbor” module (Neighbor-joining with default settings) to generate the tree.
We used this script and a database originally seeded with HCC profiles calculated from 29 high-quality, closed genome sequences (Additional file 2) to analyze additional sequence data obtained from NCBI as well as data whole-genome sequence data generated as described above. A total of 14 completely unique HCC profiles calculated from the data were added to the database (Additional file 18). We also calculated HCC profiles using sequence data from 84 strains from the Listeriosis Reference Service for Canada's collection; [55] 71 strains were identified as having unique HCC profiles and were added subsequently added to the database (Additional file 18), while HCC profiles calculated from the remaining 13 datasets matched an HCC profile already populating the database (Additional file 18, asterisks).
The pipeline was run on a desktop computer with an AMD Phenom II X6 1090 T processor and 16GB of DDR3 RAM using 5 cores.
Comparison of molecular typing methods
Serotyping of Listeria monocytogenes strains was performed using commercial O-antigen Listeria antisera (Denka Seiken, Tokyo, Japan), according to the manufacturer’s recommendations. Consistent with PulseNet standardized protocols, isolates were subtyped using pulsed-field gel electrophoresis with AscI and ApaI restriction endonucleases, as previously described [56]. Automated ribotyping was performed with the restriction enzyme EcoRI and the RiboPrinter Microbial Characterization System (Qualicon Inc., Wilmington, Delaware), according to the manufacturer’s manual [57]. Multi-locus sequence typing was performed in silico using allele sequences and profiles downloaded from the Pasteur Institute (http://bigsdb.web.pasteur.fr/), and a custom Perl script. Finally, we used the Comparing Partitions on-line tool (http://darwin.phyloviz.net/ComparingPartitions/index.php?link=Tool) to calculate Simpson’s index of diversity [33, 35] and Adjusted Wallace [34] with 95 % confidence intervals [36].
Whole-chromosome phylogenetic analysis
We assembled the 4 largest short-read sequence datasets generated during the 12 L. monocytogenes strain 08–5578 sequencing runs described above (SRR1373534, SRR1508282, SRR1507228, and SRR1373535). The datasets were assembled de-novo with SPAdes v3.0.0. Contiguous sequences were aligned with Mauve v2.3.1 and 3,138,152 nucleotide positions were phylogenetically analyzed with RAxML v8.1.1 [58] (GTRCATI + 25γ) for 100 bootstrap replicates.
Availability of supporting data
Genome sequence data supporting the results of this article are available in the National Center for Biotechnology Information Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra) under accession numbers SRR1342176, SRR1342220, SRR1373524, SRR1373525, SRR1373527, SRR1373529, SRR1373530, SRR1373531, SRR1373534, SRR1373535, SRR1507228, and SRR1508282.
Acknowledgements
This work was funded by Genomics R&D Initiative (GRDI) grant #4500834, awarded to FP. The authors are grateful to Karine Hébert and Kevin Tyler for access to their serotyping, pulsed-field gel electrophoresis, and ribotyping database. We thank Robyn Kenwell for running the Illumina MiSeq sequencer. We also thank two anonymous reviewers whose insightful comments greatly improved the quality of this manuscript.
Abbreviations
- MLST
Multi-locus sequence typing
- SNP
Single-nucleotide polymorphism
- WGS
Whole-genome sequence
- wgMLST
Whole-genome multi-locus sequence typing
- cgMLST
Core-genome multi-locus sequence typing
- CGST
Core-genome sequence typing
- HCC
High-confidence core
- ORF
Open-reading frame
- NCBI
National center for biotechnology information
- BLAST
Basic local alignment search tool
Additional files
Phylogenetic analysis of 68 aligned Listeria monocytogenes genomes. Evolutionary relationships of 68 L. monocytogenes strains, including 29 that were used to calculate pan- and core-genomes during this study. (PDF 121 kb)
Sources and general features of closed chromosomes compared for calculation of pan- and core-genomes. Serotypes, NCBI accession numbers, sources, years collected, countries of origin, chromosome length, GC content, numbers of proteins, and references for 29 L. monocytogenes. (PDF 97 kb)
Pan- and core-genome sizes of 29 closed Listeria monocytogenes chromosome sequences. Estimated pan- and core-genome sizes calculated from 2–29 randomly selected chromosome sequences. (PDF 251 kb)
List of 1013 open-reading frames that comprise the high-confidence core genome with NCBI numbers for Listeria monocytogenes strain 08–5578. (PDF 403 kb)
Comparisons of the frequencies of proteins within Clusters of Orthologous Groups categories. Frequencies of different functional classes of proteins encoded by open-reading frames from a complete Listeria monocytogenes chromosome (08–5578) and the high-confidence core. (PDF 117 kb)
Distribution of 1013 genes encoding ORFs present in the calculated high-confidence core-genome of L. monocytogenes strain 08–5578. ORFs mapped onto a chromosome map. (PDF 389 kb)
Diagram illustrating tasks performed by the Listeria monocytogenes Core-Genome Sequence Typer. (PDF 117 kb)
Listeria monocytogenes high-confidence core profiles grouped by nucleotide distances. Proposed nomenclature in which Listeria monocytogenes high-confidence core profiles are grouped by nucleotide distances. (PDF 144 kb)
Phylogenetic analysis of 115 high-confidence core profiles. Phylogenetic relationships of 114 HCC profiles that comprise the CGST database and a single randomly selected test subject. (PDF 112 kb)
Comparison of 12 sets of Illumina MiSeq data of varying qualities. Numbers of: a) SNPs, b) missing ORFs, and c) partial ORFs observed when genome sequence datasets of low to high quality are analyzed using the LmCGST pipeline. (PDF 112 kb)
Summary statistics describing data obtained from Illumina sequencing runs of Listeria monocytogenes strain 08–5578 genomic DNA samples. (PDF 97 kb)
Processing times for steps of the LmCGST pipeline with different database sizes. (PDF 86 kb)
Predicted LmCGST processing times with increasing database sizes. Amounts of time predicted to be required for the assembly, annotation, HCC comparison, and tree-building steps with database sizes from 25 to 500 HCC profiles. (PDF 108 kb)
Simpson’s index of diversity and 95 % confidence intervals. Measurements of the probabilities that two strains sampled randomly from a population will be assigned to two different types with the CGST, PFGE, ribotyping, and MLST methods. (PDF 85 kb)
Diagram of adjusted Wallace coefficients calculated from pulsed-field gel electrophoresis (PFGE), ribotyping, and core-genome sequence typing (CGST) data. (PDF 124 kb)
Phylogenetic analysis of de-novo assemblies calculated from short-read sequence data obtained from four sequencing runs of a single Listeria monocytogenes strain 08–5578 DNA extraction. (PDF 117 kb)
Distance analysis of de-novo assemblies calculated from short-read sequence data obtained from four sequencing runs of a single Listeria monocytogenes strain 08–5578 DNA extraction. (PDF 96 kb)
Data used to calculate unique Listeria monocytogenes high-confidence core genomes. (PDF 109 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AWP and FP conceived the project. AWP designed the experiment and analyzed the data. AWP and NP developed the software. FP provided the ribotyping and pulsed-field gel electrophoresis data. AWP and FP contributed to the writing of the manuscript. All authors read and approved the final manuscript.
Contributor Information
Arthur W. Pightling, Email: Arthur.Pightling@fda.hhs.gov
Nicholas Petronella, Email: Nicholas.Petronella@hc-sc.gc.ca.
Franco Pagotto, Email: Franco.Pagotto@hc-sc.gc.ca.
References
- 1.Weis J, Seeliger HP. Incidence of Listeria monocytogenes in nature. Appl Microbiol. 1975;30:29–32. doi: 10.1128/am.30.1.29-32.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gray ML, Killinger AH. Listeria monocytogenes and listeric infections. Bacteriol Rev. 1966;30:309–82. doi: 10.1128/br.30.2.309-382.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wehr HM. Listeria monocytogenes - a current dilemma. J Assoc Off Anal Chem. 1987;70:769–72. [PubMed] [Google Scholar]
- 4.Farber JM, Losos JZ. Listeria monocytogenes: a foodborne pathogen. CMAJ. 1988;138:413–8. [PMC free article] [PubMed] [Google Scholar]
- 5.Pagotto F, Corneau N, Scherf C, Leopold P, Clark C, Farber JM. Molecular typing and differentiation of foodborne bacterial pathogens. In: Fratamico PM, Bhunia AK, Smith JL, editors. Foodborne pathogens. Norfolk, UK: Caister Academic Press; 2005. pp. 51–75. [Google Scholar]
- 6.Goering RV. Pulsed field gel electrophoresis: a review of application and interpretation in the molecular epidemiology of infectious disease. Infect Genet Evol. 2010;10:866–75. doi: 10.1016/j.meegid.2010.07.023. [DOI] [PubMed] [Google Scholar]
- 7.Gottlieb P, Rudner R. Restriction site polymorphism of ribosomal ribonucleic acid gene sets in members of the genus Bacillus. Int J Syst Bacteriol. 1985;35:244–52. doi: 10.1099/00207713-35-3-244. [DOI] [Google Scholar]
- 8.Maiden MC, Bygraves JA, Feil E, Morelli G, Russell JE, Urwin R, Zhang Q, Zhou J, Zurth K, Caugant DA, Feavers IM, Achtman M, Spratt BG. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci U S A. 1998;95:3140–5. doi: 10.1073/pnas.95.6.3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Farber JM. An introduction to the hows and whys of molecular typing. J Food Prot. 1996;59:1091–101. doi: 10.4315/0362-028X-59.10.1091. [DOI] [PubMed] [Google Scholar]
- 10.Loman NJ, Constantinidou C, Chan JZM, Halachev M, Sergeant M, Penn CW, Robinson ER, Pallen MJ. High-throughput bacterial genome sequencing: an embarrassment of choice, a world of opportunity. Nat Rev Microbiol. 2012;10:599–606. doi: 10.1038/nrmicro2850. [DOI] [PubMed] [Google Scholar]
- 11.Laksanalamai P, Joseph LA, Silk BJ, Burall LS, Tarr LC, Gerner-Smidt P, Datta AR. Genomic characterization of Listeria monocytogenes strains involved in a multistate listeriosis outbreak associated with cantaloupe in US. PLoS One. 2012;7:e42448. doi: 10.1371/journal.pone.0042448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gilmour M, Graham M, Van Domselaar G, Tyler S, Kent H, Trout-Yakel KM, Larios O, Allen V, Lee B, Nadon C. High-throughput genome sequencing of two Listeria monocytogenes clinical isolates during a large foodborne outbreak - 1471-2164-11-120.pdf. BMC Genomics. 2010;11:120. doi: 10.1186/1471-2164-11-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nielsen R, Paul JS, Albrechtsen A, Song YS. Genotype and SNP calling from next-generation sequencing data. Nat Rev Genet. 2011;12:443–51. doi: 10.1038/nrg2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pightling AW, Petronella N, Pagotto F. Choice of reference sequence and assembler for alignment of Listeria monocytogenes short-read sequence data greatly influences rates of error in SNP analyses. PLoS One. 2014;9:e104579. doi: 10.1371/journal.pone.0104579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bertels F, Silander OK, Pachkov M, Rainey PB, Van Nimwegen E. Automated reconstruction of whole-genome phylogenies from short-sequence reads. Mol Biol Evol. 2014;31:1077–88. doi: 10.1093/molbev/msu088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pettengill JB, Luo Y, Davis S, Chen Y, Gonzalez-Escalona N, Ottesen A, Rand H, Allard MW, Strain E. An evaluation of alternative methods for constructing phylogenies from whole genome sequence data: a case study with Salmonella. PeerJ. 2014;2:e620. doi: 10.7717/peerj.620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cody AJ, McCarthy ND, Van Rensburg MJ, Isinkaye T, Bentley SD, Parkhill J, Dingle KE, Bowler ICJW, Jolley KA, Maiden MCJ. Real-time genomic epidemiological evaluation of human Campylobacter isolates by use of whole-genome multilocus sequence typing. J Clin Microbiol. 2013;51:2526–34. doi: 10.1128/JCM.00066-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kohl TA, Diel R, Harmsen D, Rothgänger J, Meywald Walter K, Merker M, Weniger T, Niemann S. Whole-genome-based mycobacterium tuberculosis surveillance: a standardized, portable, and expandable approach. J Clin Microbiol. 2014;52:2479–86. doi: 10.1128/JCM.00567-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bennett JS, Jolley KA, Maiden MCJ. Genome sequence analyses show that Neisseria oralis is the same species as “Neisseria mucosa var. Heidelbergensis.”. Int J Syst Evol Microbiol. 2013;63:3920–6. doi: 10.1099/ijs.0.052431-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D, Ward NL, Angiuoli SV, Crabtree J, Jones AL, Durkin AS, Deboy RT, Davidsen TM, Mora M, Scarselli M, Margarit y Ros I, Peterson JD, Hauser CR, Sundaram JP, Nelson WC, Madupu R, Brinkac LM, Dodson RJ, Rosovitz MJ, Sullivan SA, Daugherty SC, Haft DH, Selengut J, Gwinn ML, Zhou L, Zafar N, et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc Natl Acad Sci U S A. 2005;102:13950–5. doi: 10.1073/pnas.0506758102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lefébure T, Stanhope MJ. Evolution of the core and pan-genome of Streptococcus: positive selection, recombination, and genome composition. Genome Biol. 2007;8:R71. doi: 10.1186/gb-2007-8-5-r71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hogg JS, Hu FZ, Janto B, Boissy R, Hayes J, Keefe R, Post JC, Ehrlich GD. Characterization and modeling of the Haemophilus influenzae core and supragenomes based on the complete genomic sequences of Rd and 12 clinical nontypeable strains. Genome Biol. 2007;8:R103. doi: 10.1186/gb-2007-8-6-r103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuenne C, Billion A, Mraheil MA, Strittmatter A, Daniel R, Goesmann A, Barbuddhe S, Hain T, Chakraborty T. Reassessment of the Listeria monocytogenes pan-genome reveals dynamic integration hotspots and mobile genetic elements as major components of the accessory genome. BMC Genomics. 2013;14:47. doi: 10.1186/1471-2164-14-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Besemer J, Lomsadze A, Borodovsky M. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001;29:2607–18. doi: 10.1093/nar/29.12.2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Delcher AL, Harmon D, Kasif S, White O, Salzberg SL. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999;27:4636–41. doi: 10.1093/nar/27.23.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Delcher AL, Bratke KA, Powers EC, Salzberg SL. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics. 2007;23:673–9. doi: 10.1093/bioinformatics/btm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seemann T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–9. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 29.Koonin EV, Galperin MY. Sequence - evolution - function: computational approaches in comparative genomics. Boston: Kluwer; 2003. [PubMed] [Google Scholar]
- 30.Schwartz DC, Cantor CR. Separation of yeast chromosome-sized DNAs by pulsed field gradient gel electrophoresis. Cell. 1984;37:67–75. doi: 10.1016/0092-8674(84)90301-5. [DOI] [PubMed] [Google Scholar]
- 31.Salcedo C, Arreaza L, Alcalá B, De la Fuente L, Vázquez JA. Development of a multilocus sequence typing method for analysis of Listeria monocytogenes clones. J Clin Microbiol. 2003;41:757–62. doi: 10.1128/JCM.41.2.757-762.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carrillo CD, Kruczkiewicz P, Mutschall S, Tudor A, Clark C, Taboada EN. A framework for assessing the concordance of molecular typing methods and the true strain phylogeny of campylobacter jejuni and C. Coli using draft genome sequence data. Frontiers Cellular Infection Microbiol. 2012;2:57. doi: 10.3389/fcimb.2012.00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Simpson EH. Measurement of Diversity. Nature. 1949;163:688. doi: 10.1038/163688a0. [DOI] [Google Scholar]
- 34.Severiano A, Pinto FR, Ramirez M, Carriço JA. Adjusted Wallace coefficient as a measure of congruence between typing methods. J Clin Microbiol. 2011;49:3997–4000. doi: 10.1128/JCM.00624-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hunter PR, Gaston MA. Numerical index of the discriminatory ability of typing systems: an application of Simpson’s index of diversity. J Clin Microbiol. 1988;26:2465–6. doi: 10.1128/jcm.26.11.2465-2466.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pinto FR, Melo-Cristino J, Ramirez M. A confidence interval for the wallace coefficient of concordance and its application to microbial typing methods. PLoS One. 2008;3:e3696. doi: 10.1371/journal.pone.0003696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsai IJ, Otto TD, Berriman M. Improving draft assemblies by iterative mapping and assembly of short reads to eliminate gaps. Genome Biol. 2010;11:R41. doi: 10.1186/gb-2010-11-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Edman DC, Pollock MB, Hall ER. Listeria monocytogenes L forms. I. Induction maintenance, and biological characteristics. J Bacteriol. 1968;96:352–7. doi: 10.1128/jb.96.2.352-357.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bécavin C, Bouchier C, Lechat P, Archambaud C, Creno S, Gouin E, Wu Z, Kühbacher A, Brisse S, Graciela Pucciarelli M, García-del Portillo F, Hain T, Portnoy DA, Chakraborty T, Lecuit M, Pizarro-Cerdá J, Moszer I, Bierne H, Cossart P. Comparison of widely used Listeria monocytogenes strains EGD, 10403S, and EGD-e highlights genomic differences underlying variations in pathogenicity. MBio. 2014;5:e00969. doi: 10.1128/mBio.00969-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Olsen SJ, Patrick M, Hunter SB, Reddy V, Kornstein L, MacKenzie WR, Lane K, Bidol S, Stoltman GA, Frye DM, Lee I, Hurd S, Jones TF, LaPorte TN, Dewitt W, Graves L, Wiedmann M, Schoonmaker-Bopp DJ, Huang AJ, Vincent C, Bugenhagen A, Corby J, Carloni ER, Holcomb ME, Woron RF, Zansky SM, Dowdle G, Smith F, Ahrabi-Fard S, Ong AR, et al. Multistate outbreak of Listeria monocytogenes infection linked to delicatessen turkey meat. Clin Infect Dis. 2005;40:962–7. doi: 10.1086/428575. [DOI] [PubMed] [Google Scholar]
- 41.Holch A, Webb K, Lukjancenko O, Ussery D, Rosenthal BM, Gram L. Genome sequencing identifies two nearly unchanged strains of persistent Listeria monocytogenes isolated at two different fish processing plants sampled 6 years apart. Appl Environ Microbiol. 2013;79:2944–51. doi: 10.1128/AEM.03715-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Steele CL, Donaldson JR, Paul D, Banes MM, Arick T, Bridges SM, Lawrence ML. Genome sequence of lineage III Listeria monocytogenes strain HCC23. J Bacteriol. 2011;193:3679–80. doi: 10.1128/JB.05236-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hain T, Ghai R, Billion A, Kuenne C, Steinweg C, Izar B, Mohamed W, Mraheil M, Domann E, Schaffrath S, Kärst U, Goesmann A, Oehm S, Pühler A, Merkl R, Vorwerk S, Glaser P, Garrido P, Rusniok C, Buchrieser C, Goebel W, Chakraborty T. Comparative genomics and transcriptomics of lineages I, II, and III strains of Listeria monocytogenes. BMC Genomics. 2012;13:144. doi: 10.1186/1471-2164-13-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen J, Xia Y, Cheng C, Fang C, Shan Y, Jin G, Fang W. Genome sequence of the nonpathogenic Listeria monocytogenes serovar 4a strain M7. J Bacteriol. 2011;193:5019–20. doi: 10.1128/JB.05501-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.David McMullen P, Gillaspy AF, Gipson J, Bobo LD, Skiest DJ, Freitag NE. Genome sequence of Listeria monocytogenes 07PF0776, a Cardiotropic serovar 4b strain. J Bacteriol. 2012;194:3552. doi: 10.1128/JB.00616-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nelson KE, Fouts DE, Mongodin EF, Ravel J, DeBoy RT, Kolonay JF, Rasko DA, Angiuoli SV, Gill SR, Paulsen IT, Peterson J, White O, Nelson WC, Nierman W, Beanan MJ, Brinkac LM, Daugherty SC, Dodson RJ, Durkin AS, Madupu R, Haft DH, Selengut J, Van Aken S, Khouri H, Fedorova N, Forberger H, Tran B, Kathariou S, Wonderling LD, Uhlich GA, et al. Whole genome comparisons of serotype 4b and 1/2a strains of the food-borne pathogen Listeria monocytogenes reveal new insights into the core genome components of this species. Nucleic Acids Res. 2004;32:2386–95. doi: 10.1093/nar/gkh562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen Y, Strain EA, Allard M, Brown EW. Genome sequences of Listeria monocytogenes strains J1816 and J1-220, associated with human outbreaks. J Bacteriol. 2011;193:3424–5. doi: 10.1128/JB.05048-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weinmaier T, Riesing M, Rattei T, Bille J, Arguedas-Villa C, Stephan R, Tasara T. Complete Genome Sequence of Listeria monocytogenes LL195, a Serotype 4b Strain from the 1983–1987 Listeriosis Epidemic in Switzerland. Genome Announc. 2013;1:8–9. doi: 10.1128/genomeA.00152-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, Rao BS, Smirnov S, Sverdlov AV, Vasudevan S, Wolf YI, Yin JJ, Natale DA. The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mulkidjanian AY, Koonin EV, Makarova KS, Mekhedov SL, Sorokin A, Wolf YI, Dufresne A, Partensky F, Burd H, Kaznadzey D, Haselkorn R, Galperin MY. The cyanobacterial genome core and the origin of photosynthesis. Proc Natl Acad Sci U S A. 2006;103:13126–31. doi: 10.1073/pnas.0605709103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Toledo-Arana A, Dussurget O, Nikitas G, Sesto N, Guet-Revillet H, Balestrino D, Loh E, Gripenland J, Tiensuu T, Vaitkevicius K, Barthelemy M, Vergassola M, Nahori M-A, Soubigou G, Régnault B, Coppée J-Y, Lecuit M, Johansson J, Cossart P. The Listeria transcriptional landscape from saprophytism to virulence. Nature. 2009;459:950–6. doi: 10.1038/nature08080. [DOI] [PubMed] [Google Scholar]
- 52.Oliver HF, Orsi RH, Ponnala L, Keich U, Wang W, Sun Q, Cartinhour SW, Filiatrault MJ, Wiedmann M, Boor KJ. Deep RNA sequencing of L. monocytogenes reveals overlapping and extensive stationary phase and sigma B-dependent transcriptomes, including multiple highly transcribed noncoding RNAs. BMC Genomics. 2009;10:641. doi: 10.1186/1471-2164-10-641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J Comput Biol. 2012;19:455–77. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nikolenko SI, Korobeynikov AI, Alekseyev MA. BayesHammer: Bayesian clustering for error correction in single-cell sequencing. BMC Genomics. 2013;14(Suppl 1):S7. doi: 10.1186/1471-2164-14-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pightling AW, Lin M, Pagotto F. Draft Genome Sequence of Listeria monocytogenes Strain LI0521 (syn. HPB7171), Isolated in 1983 during an Outbreak in Massachusetts Caused by Contaminated Cheese. Genome Announc. 2014;2:2013–4. doi: 10.1128/genomeA.00729-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Graves LM, Swaminathan B. PulseNet standardized protocol for subtyping Listeria monocytogenes by macrorestriction and pulsed-field gel electrophoresis. Int J Food Microbiol. 2001;65:55–62. doi: 10.1016/S0168-1605(00)00501-8. [DOI] [PubMed] [Google Scholar]
- 57.Bruce JL, Hubner RJ, Cole EM, McDowell CI, Webster JA. Sets of EcoRI fragments containing ribosomal RNA sequences are conserved among different strains of Listeria monocytogenes. Proc Natl Acad Sci U S A. 1995;92:5229–33. doi: 10.1073/pnas.92.11.5229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stamatakis A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]