

Abstract
Background
Notch may behave as an oncogene or a tumor suppressor gene in lung cancer cells. Notch receptor undergoes cleavage by enzymes, including γ-secretase, generating the active Notch intracellular domain (NICD). The aim of the present study was to investigate the effect of DAPT, a γ-secretase inhibitor, in non-small cell lung cancer (NSCLC) cells, as well as the impact of epidermal growth factor (EGF) that is over-expressed by NSCLC cells, on Notch signaling. H23, A549, H661 and HCC827 human NSCLC cell lines were used, expressing various NICD and EGF receptor (EGFR) protein levels.
Results
DAPT decreased the number of H661 cells in a concentration-dependent manner, while it had a small effect on H23 and A549 cells and no effect on HCC827 cells that carry mutated EGFR. Notch inhibition did not affect the stimulatory effect of EGF on cell proliferation, while EGF prevented DAPT-induced NICD decrease in H23 and H661 cells. The type of cell death induced by DAPT seems to depend on the cell type.
Conclusions
Our data indicate that inhibition of Notch cleavage may not affect cell number in the presence of EGFR mutations and that EGFR may affect Notch signalling suggesting that a dual inhibition of these pathways might be promising in NSCLC.
Electronic supplementary material
The online version of this article (doi:10.1186/s12929-015-0196-1) contains supplementary material, which is available to authorized users.
Keywords: Notch intracellular domain, Epidermal growth factor receptor, Non-small cell lung cancer cells, Apoptosis, Autophagy, Cell cycle arrest
Background
Lung cancer is the leading cause of cancer death. Its incidence and mortality is 52.5/100,000 and 48.7/100,000 per year, respectively [1]. Non-small cell lung cancer (NSCLC) is the most common subtype, accounting for approximately 90 % of lung cancer cases [1]. Most patients are presented with advanced stage disease and since patient outcomes are largely dependent on stage, 5 year survival rate for lung cancer remains low, at about 15 % [2]. Until recently, chemotherapy with cytotoxic agents was the only available treatment for lung cancer [1]. Advances in the research field have led to the elucidation of unique pathogenic mechanisms, critical pathways and molecules involved in cancer development, which have resulted in the introduction of targeted therapies as new treatment options in cancer. A typical example is the epidermal growth factor receptor (EGFR) activity that has been found to be overexpressed in about 50–80 % of NSCLC [3]. Anti-angiogenic agent bevacizumab and EGFR tyrosine kinase inhibitors erlotinib and gefitinib have been approved for NSCLC treatment [4, 5]. Moreover, continuous ongoing clinical trials investigate the efficacy and safety of anti-angiogenic and anti-EGFR agents combinations, as well as other agents such as multi-targeted anti-angiogenic tyrosine kinase inhibitors and anti-HER2 agents [6–8].
Despite the existence of many therapeutic options, the prognosis of patients with metastatic disease remains poor, with a median survival of about 12 months with the newer regimens. This shows how important the development of new strategies remains. Although targeted therapies are a step forward, it is essential to clarify the biology of lung cancer cells [9].
Notch is a transmembrane heterodimeric receptor with four distinct members (Notch1 to Notch4) in humans and rodents. Notch signaling pathway is initiated upon ligand binding, where the receptor subjects into two proteolytic cleavages. The γ-secretase, a complex enzyme, regulates the second cleavage of Notch, through which the Notch intracellular domain (NICD) is liberated to the cytoplasm and then enters the nucleus in order to activate the transcription of Notch targeted genes [10]. Notch is known for playing a key role in embryogenesis and organogenesis by regulating cell proliferation and differentiation [11]. In addition, Notch seems to be involved in carcinogenesis and a cell-type dependent profile has been observed with Notch to act as oncogene or tumor suppressor gene [12–14]. Both behaviors of Notch have been described in lung cancer. Notch 1 and 2 proteins are frequently expressed in NSCLC. Notch 1 is rarely expressed in small cell lung cancer (SCLC), whereas a subset of SCLC exhibit Notch 2 expression. It is suspected that Notch has a growth promoting function in NSCLC, whereas in SCLC it exerts an inhibitory effect [11, 12]. Research also shows potential cross talk between Notch pathway and others, such as EGFR and Wnt [15–19]. Taking those findings together, Notch pathway is currently under investigation, with various agents being tested as potential new therapeutic options for patients with NSCLC [20].
The cross-talk between the Notch and EGFR signaling has been previously described in genetic model systems, where these pathways can function in either an antagonistic or synergistic fashion, depending on tissue and developmental context [21]. However, the mechanism through which this interplay occurs, remains unknown [19]. In breast cancer cells, Notch pathway is used by cancer cells to compensate for EGFR targeted inhibition [22]. The case of breast cancer along with previous data from lung cancers cells demonstrating that Notch 1 might have a role in acquired resistance to gefitinib [23], prompted us to investigate the inhibition of Notch in NSCLC cell lines after cell treatment with γ-secretase inhibitor; N- [N- (3, 5- difluorophenacetyl)- 1- alanyl]- S- phenylglycine t- butyl ester (DAPT), as well as the impact of epidermal growth factor (EGF) in Notch signaling. Our data indicated that Notch inhibition was not effective in all NSCLC cells and this effect was dependent on EGFR protein levels and mutations.
Methods
Cell culture and reagents
NSCLC cell lines H23, A549, H661 and HCC827 were purchased from the American Type Culture Collection (ATCC) and cultured as manufacturer recommends. According to ATCC, H23, A549 and H661 cells express wild type EGFR, while HCC827 cells express mutated EGFR bearing E746-A750 deletion (https://www.lgcstandards-atcc.org/) as shown in Additional file 1: Table S1. Cells were cultured in RPMI 1640 medium with 2 mM L-glutamine and supplemented with 1 mM sodium pyruvate, 4.5 g/L glucose, 1.5 g/L sodium bicarbonate, 100 μg/ml penicillin G/streptomycin, 2.5 μg/ml amphotericin B, 50 μg/ml gentamycin and 10 % foetal bovine serum (FBS). Cells were cultured at 37 °C, 5 % CO2 and 100 % humidity.
DAPT (N- [N- (3, 5- difluorophenacetyl)- 1- alanyl]- S- phenylglycine t- butyl ester (DAPT, LY374973) and EGF were purchased from Sigma (Sigma-Aldrich Chemie GmbH, Germany). All experiments were performed according to the following conditions: After reaching 80 % confluence, serum starvation followed for 24 h. Cells were then treated with DAPT at the concentrations of 0.01, 0.1, 1, 10 and 20 μM or/and EGF at the concentration of 1 μg/ml. In case of DAPT and EGF co-treatment, EGF was added 30 min prior to DAPT. The duration of the treatment was determined by the assay that followed.
Cell proliferation assay
To determine whether DAPT alone or in combination with EGF, affects the proliferation of cell proliferation, the 3-[4,5-dimethylthiazol-2-yl]-2,5-dimethyltetrazolium bromide (MTT) assay was used, as previously described [24]. Briefly, cells were seeded at a density of 1,5 ×104 cells/well in 24-well plates and cells treated as described above. Forty-eight hours after agents’ application, MTT solution (5 mg/ml in PBS) was added at a volume equal to 1/10, to each well and incubated for 2 h, at 37 °C. Medium was removed and 100 μl acidified isopropanol (0.33 ml HCl in 100 ml isopropanol) was added in each well in order to solubilize the dark blue formazan crystals. The solution was transferred to 96-well plates and was immediately read in a microplate reader (Tecan, Sunrise, Magellan 2) at a wavelength of 570 nm using reference wavelength 620 nm.
Apoptosis assay
All cell lines were plated at 1 × 105 cells per well in 6-well plates. DAPT was added as previously described. At the end of a 24 h incubation, cells were washed twice with PBS, trypsinized for 7 min and centrifuged for 4 min at 166 g. Cells were resuspended in 200 μl 1X binding buffer (10 mM HEPES pH 7.4, 140 mM NaCl, 2.5 mM CaCl2). The cell suspension was incubated with 5 μl Annexin V-FITC in the dark at 25 °C, for 10 min. Then, 10 μl of the 20 μg/ml propidium iodide stock solution was added, followed by 200 μl of binding buffer and the cells were immediately analysed by flow cytometry [25] (EPICS-XL of Coulter) according to the manufacturer’s instructions (rh Annexin V/FITC kit, Bender MedSystems, Germany). The application of Annexin-V (An) along with propidium iodide (PI) distinguishes 4 populations; the viable (An−/PI−), the early apoptotic (An+/PI−), the late apoptotic (An+/PI+) and the necrotic (An−/PI+) cells. The sum of early and late apoptotic cells calculated as apoptotic cells.
Cell cycle analysis
All cell lines were plated at 1 × 106 cells per Petri dish and DAPT was added as earlier described. At the end of the 24 or 48 h incubation, cells were washed twice with PBS, trypsinized for 7 min and centrifuged for 4 min at 166 g. Cell cycle analysis was performed using the Muse™ Cell Cycle kit according to the manufacturer’s instruction (Merck-Millipore, Germany). Briefly, cells were washed once with PBS and centrifuged for 5 min at 300 g. The supernatants were discarded leaving approximately 50 μl of PBS. The pellet of cells was resuspended in the residual PBS was added drop-wise into a tube containing 1 ml of ice cold 70 % ethanol while vortexing at medium speed. The samples were kept at −20 °C for at least 3 h. Then, 200 μl of fixed cells were centrifuged at 300 g, for 5 min. The pellet of cells was resuspended in 200 μl of Muse™ Cell Cycle Reagent and cells were incubated for 30 min, protected from light. After the incubation, cells were analysed by Muse™ Cell Analyzer, according to the manufacturer’s instructions (Muse™ software, Merck-Millipore, Germany).
Western blot analysis
Cells were plated at Petri dishes. After reaching 80 % confluence, cells were treated as described above. At the indicated time point after agents’ application, cells were collected with scrapper and lysed using lysis buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 5 mM EDTA, 1 % Triton, 10 % glycerol, 1 mM phenylmethyl-sulphonyl-fluoride, 2 mM Na-orthovanadate and 10 mM leupeptin). Protein concentration was determined by Bradford assay. Samples were analysed by immunoblotting as previously noted [24]. For Beclin-1, an autophagy indicator, cells were collected 24 h after appropriate treatment and a goat anti-Beclin-1 (dilution 1:500, Santa Cruz, CA, USA) was used. For Notch-1 intracelluler domain (NICD), time course experiments were performed and a sheep anti-Notch-1 ICD (dilution 1:1000, R&D Systems, Germany) was used. For EGFR detection, time course experiments were performed and a rabbit anti-EGFR (dilution 1:6000, Millipore,Temecula, CA, USA) was used. Actin was used as internal control for protein quantification and a monoclonal anti-actin antibody (dilution 1:1000, Chemicon, Millipore, Temecula, CA, USA) was used.
Detection of the immunoreactive proteins was performed by chemiluminescence using horseradish peroxidase substrate SuperSignal (Pierce, Rockford, IIL, USA), according to the manufacturer’s instructions.
Transmission electron microscopy (TEM)
H661 cells were plated at 1 × 105 cells per well in 6-well plates. DAPT was added as previously described. At the end of a 24 h incubation cells were washed with PBS once, fixed with 2.5 % glutaraldehyde at 4 °C for 2 h and then washed twice with PBS. Cells were dehydrated in a graded series of ethanol (50, 70, 80, 90 and 100 %), transferred in to 100 % acetone for 15 min and embedded in SPURR resin. Ultrathin sections were stained with uranyl acetate and lead citrate and examined and photographed with a JEOL 100S transmission electron microscope equipped with an Olympus MegaView G2 digital camera.
Statistical analysis
Differences between groups and controls were tested by one-way ANOVA. Each experiment included at least triplicate measurements. All results are expressed as mean ± SEM from at least three independent experiments.
Results
Profile of NSCLC cells according to NICD and EGFR protein levels
Initially, NSCLC cell lines H23, A549, H661 and HCC827 (Fig. 1a and b) were screened for NICD and EGFR protein levels. Among four cell lines, H23 expressed the highest, H661 and HCC827 intermediate and A549 the lowest NICD protein levels (Fig. 1a). Concerning EGFR, H23 expressed the highest, A549 and HCC827 intermediate and H661 the lowest, almost undetectable, protein levels (Fig. 1b). According to ATCC, H23, A549 and H661 cells have wild type (wt) EGFR and HCC827 cells have mutated (mt) EGFR (https://www.lgcstandards-atcc.org/) (see Additional file 1: Table S1).
Fig. 1.
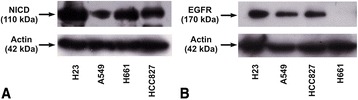
Screening of NSCLC cell lines for a) NICD and b) EGFR protein levels. The images of western blot analysis are representative of three independent experiments
The effect of Notch inhibition in NSCLC cells’ proliferation
Since all cell lines express activated Notch, we studied the effect of DAPT on the number of unstimulated cells. The number of H661 cells was significantly decreased in a concentration-dependent manner 48 h after DAPT addition. DAPT had only a minor effect on H23 and A549 cells, while HCC827 cells were resistant to DAPT at any tested concentration (Fig. 2a). Based on these data, the inhibitory effect of DAPT on the number of cells initiated at 10 μM and thus this concentration was used for the following experiments.
Fig. 2.
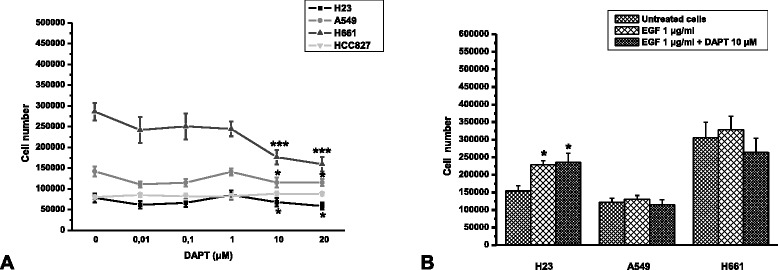
The effect of DAPT a) alone and b) after stimulation of NSCLC cells with EGF in cell proliferation, 48 h after DAPT application. Each value represents the means ± SEM from at least three independent experiments. The asterisks denote statistically significant difference between experimental groups and untreated cells. C *P < 0,05 and ***P < 0,0001
To investigate a possible implication of Notch pathway with EGFR signalling, we stimulated cells with EGF 1 μg/ml for 30 min prior to addition of DAPT. HCC827 cells were not used in these assays, since they carry active mutated EGFR (see Additional file 1: Table S1) and do not further respond to EGF [26, 27]. EGF alone significantly increased only the number of H23 cells, while it had no effect on A549 and H661 cells. DAPT did not reverse the stimulatory effect of EGF in H23 cells. On the other hand, in all cells, the presence of EGF prevented the inhibitory effect of DAPT (Fig. 2a and b).
The effect of DAPT in NICD and EGFR protein levels
In order to correlate the effect of DAPT on cell proliferation with inhibition of the Notch pathway, we checked the effect of DAPT on NICD protein levels. DAPT decreased NICD protein levels in both H23 and H661 cells, reaching a maximum effect at 6 h (Fig. 3). Interestingly, DAPT was more effective in H661 cells, in line with its effect on the number of cells. We next studied the effect of EGF alone or in combination with DAPT. EGF alone did not cause any statistical significant effect on NICD protein levels either in H23 (Fig. 4a and b) or in H661 (Fig. 4c and d) cells. When EGF was applied 30 min prior to DAPT, it prevented NICD decrease caused by DAPT, in both H23 and H661 cells (Fig. 4e, f, g and h), in line with the corresponding effects on the number of cells. In case of H23 cells, this prevention was observed up to 4 h after cells’ treatment, while at 6 h there was an attenuation effect of EGF to DAPT-induced NICD protein decrease (Fig. 4e and f). In H661 cells there was a full prevention at all-time points tested (Fig. 4g and h).
Fig. 3.
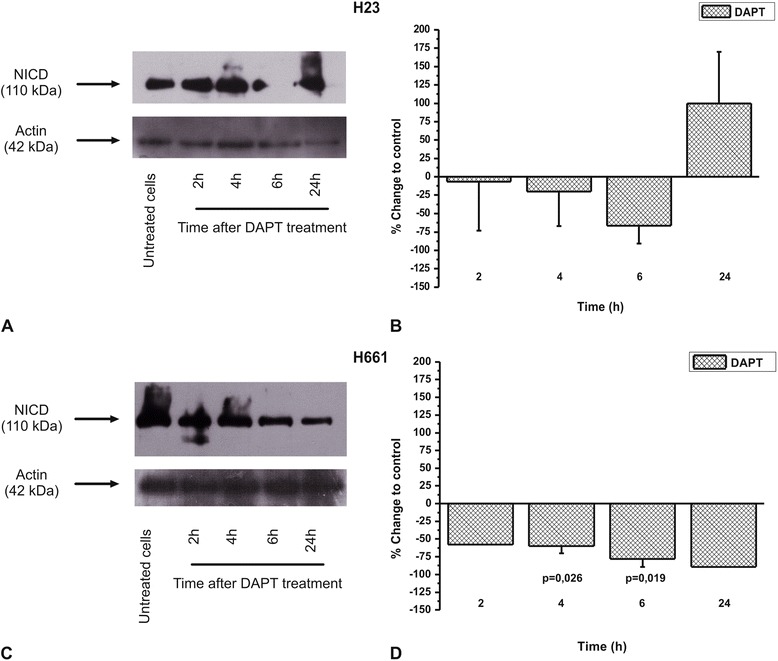
The DAPT effect on NICD protein levels in a) H23 and c) H661 cells in a time dependent manner. The images are representative of three independent experiments. Western blots were semi-quantified using appropriate software. The results in b) and d) are expressed as % change ± SEM compared to untreated cells from at least three independent experiments
Fig. 4.
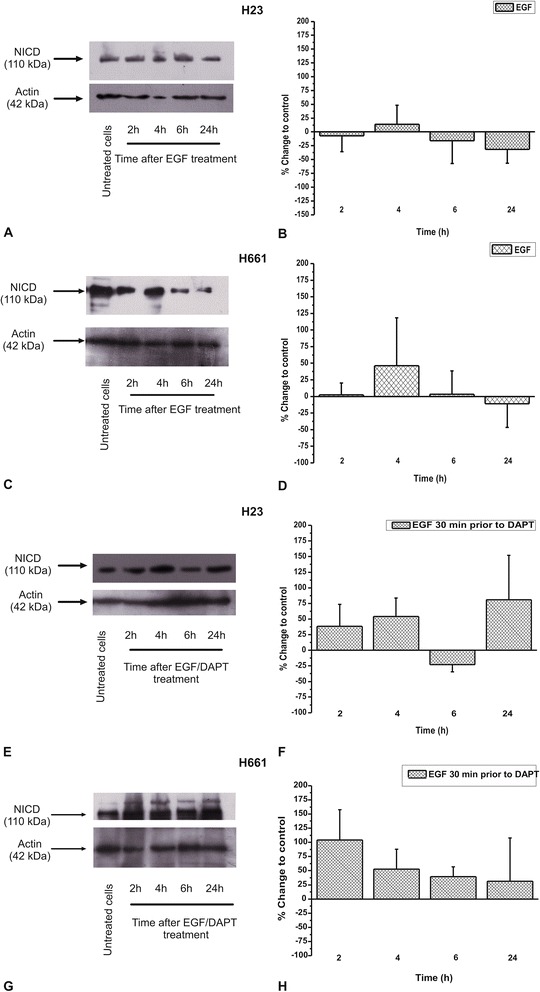
The EGF effect on NICD protein levels, alone in a) H23 and c) H661 cells or prior to DAPT in e) H23 and g) H661 cells in a time dependent manner. The images are representative of three independent experiments. Western blots were semi-quantified using appropriate software. The results in b), d), f) and h) are expressed as % change ± SEM compared to untreated cells from at least three independent experiments
We then tested whether DAPT affected EGFR protein levels in lung cancer cells. DAPT increased the protein levels of EGFR in H23 (Fig. 5a) but not H661 cells (Fig. 5b).
Fig. 5.
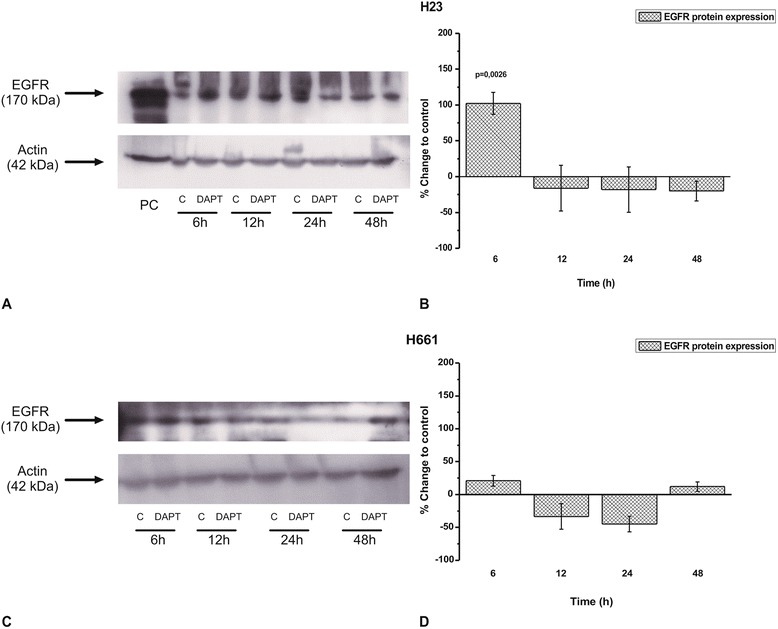
The effect of DAPT in EGFR protein levels in several time points in a) H23 and c) H661 cells. The images are representative of three independent experiments. Western blots were semi-quantified using appropriate software. The results in b) and d) are expressed as % change ± SEM compared to untreated cells from at least three independent experiments
Notch inhibition in NSCLC cells’ death
We estimated whether the reduction in cell number after DAPT application in lung cancer cells was due to stimulation of any of the two types of programmed cell death, apoptosis and autophagy. DAPT did not affect apoptosis in H23 and HCC827 cells, but increased cell apoptosis in A549 and H661 cells in a statistically significant manner (Fig. 6a and Additional file 2: Figure S1). Regarding autophagy, we found that DAPT increased the protein levels of Beclin-1, an indicator of autophagy [28, 29], only in H661 cells. This increase was 88 % ± 23 and was observed 24 h after DAPT addition to cells (Fig. 6b and c). Indeed, autophagosomes were detected in H661 cells 24 h after their treatment with DAPT, using TEM (Fig. 6d). Although the untreated cells appeared to have granular cytoplasm, no sign of autophagy was detected compared to cells after treatment with DAPT.
Fig. 6.
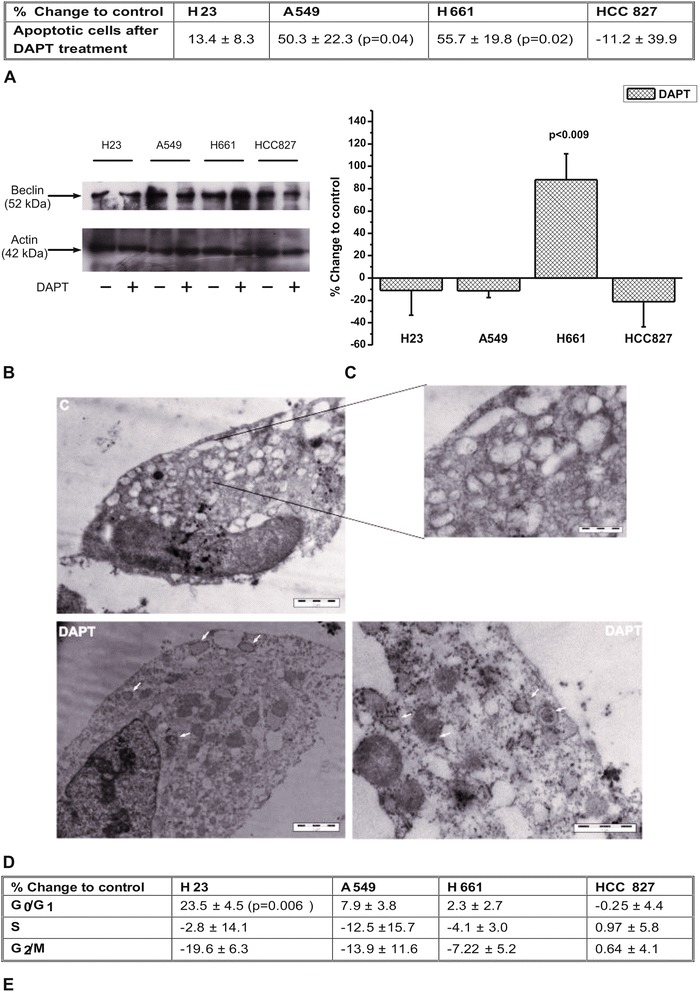
The effect of DAPT in NSCLC cell death. a Apoptosis is expressed as the % change ± SEM compared to untreated cells from at least three independent experiments. b Autophagy is estimated according to Beclin-1 protein levels. The image is a representative of three independent experiments. c The images from western blot analysis were semi-quantified using appropriate software. d Autophagosomes were detected in H661 cells after their treatment with DAPT under observation of samples using TEM. e Cell cycle arrest is expressed as % change ± SEM compared to untreated cells from at least three independent experiments
We then studied the effect of DAPT in cell necrosis and no significant differences were found in any cell line tested (data not shown).
Finally, we tested whether DAPT affected cell cycle distribution 24 h after DAPT application to cells and we found no effect (data not shown). However, DAPT increased to a small extent the percentage of H23 cells in G0/G1 phase at 48 h. A slight increase was also found in A549 cells, without being statistically significant. No significant effect was observed in H661 and HCC827 cells (Fig. 6e and Additional file 3: Figure S2).
Discussion
The EGFR pathway plays a vital role in the pathogenesis and progression of NSCLC. Although the contribution of EGFR signalling in lung cancer development is well established, the importance of Notch pathway in lung cancer is under investigation. The existing data for the role of Notch in lung cancer are conflicting and not limited between NSCLC and SCLC groups, but differences are also demonstrated among NSCLC groups, as well as among the members of Notch family. Chen et al. reported that Notch 1 was down-regulated in NSCLC cell lines, while constitutive expression of active Notch 1 in NSCLC cells caused cell death depending on oxygen concentration [30]. Another study showed that Notch 3 is active in NSCLC and treatment of cells with a γ-secretase inhibitor caused a cell proliferation reduction and increase in apoptosis [17]. Yin et al. suggested that the controversial effects of Notch signaling are highly context-dependent [31]. In addition, it has been found that Notch effect might be dose-dependent in mammary epithelial MCF-10A cells, whereas high Notch activity caused inhibition of cell proliferation and low Notch activity stimulated a strong hyperproliferative response [32]. All these conflicting data reveal a significant but complicated role of Notch in cancer development and progression.
In the current study, we selected four NSCLC cell lines expressing different levels of NICD and EGFR protein levels. We found that the cell lines exhibited different response to the γ-secretase inhibitor DAPT and surprisingly, this behavior seems to be related to EGFR status. DAPT was effective in proliferation of cells expressing wtEGFR, while it did not affect HCC827 cells expressing mtEGFR. In addition, differences were observed among the cells with wtEGFR. We found that although H23 and A549 cells exerted a similar small response to DAPT regarding cell proliferation, the decrease in cell number was possibly due to cell cycle arrest for H23 cells and increase in apoptosis for A549 cells. In H661 cells that were more sensitive to DAPT, the decrease in cell number was due to an increase of both apoptosis and autophagy. Our results verify that the impact of Notch inhibition may vary depending on cell context, since different types of cell death occurred in different cell lines. Although in the literature both cell cycle arrest [33, 34] and apoptosis stimulation [17, 35] have been described to be induced in cancer cells by Notch inhibition, there is no previous evidence that Notch inhibition triggers autophagy in cancer cells. However, it is known that apoptosis and autophagy are two mechanisms of programmed cell death that may co-exist and act synergistically [36]. A link between Notch pathway and autophagy was presented in a recent paper where the authors observed that loss of autophagy leads to precocious Notch activation during Drosophila oogenesis [37]. We might assume that H661 cells were more sensitive to DAPT because of the dual induction of apoptosis and autophagy compared with H23 and A549 cells, where only one type of cell death was activated. The sensitivity of H661 cells to DAPT might be correlated with the EGFR protein levels, since H661 cells expressed the lowest EGFR levels compared with H23 and A549 cells. The low EGFR protein levels may render H661 cells more sensitive to EGFR-independent signaling pathways regarding cell proliferation. This hypothesis could be supported by the failure of EGF alone to stimulate H661 cell proliferation. Although, EGF failed to stimulate A549 cell proliferation too, this may be explained by the existence of Kras mutation in these cells [38].
Since our data indicated that the effect of γ-secretase inhibitors might be affected by the EGFR status, the three cell lines expressing wtEGFR were stimulated with EGF prior to DAPT addition. The stimulation of all cells with EGF fully prevented the inhibition of cell proliferation by DAPT. This is in agreement with the lack of effect of DAPT in HCC827 cells bearing active mtEGFR [26, 27]. Although our data are not conclusive, this is the first indication in the literature that EGFR activation may affect Notch signalling in NSCLC cells. A similar effect regarding NICD protein levels was observed when cells were treated with EGF prior to DAPT application. EGF attenuated the reduction of NICD levels caused by DAPT. Nevertheless, since there is no direct evidence for the impact of EGF to NICD protein, we might assume that ERK1/2 phosphorylation induced by EGF activates Notch pathway [39]. Regarding a bidirectional effect between Notch and EGFR pathway, to our knowledge, there is evidence from breast cancer cells where Notch over-expression caused EGFR up-regulation [19]. A similar effect has been described in gliomas through p53 regulation [40]. Αn opposite effect has been described in breast cancer cells where HER-2 inhibition using transtuzumab caused increased Notch 1 activity [41]. In this study, inhibition of Notch activation by DAPT increased EGFR protein levels in H23 cells without affecting the EGFR levels in H661 cells. We might speculate that this increase offered a relative resistance in H23 cells compared to H661 cells in cell proliferation after DAPT treatment.
This study presents indications that EGFR and Notch signalling pathways crosstalk in human lung cancer cell lines. In more recent research, it has been shown that in hypoxia, ADAM12 could be the linker between the two pathways since it mediates the release of heparin-binding EGF-like growth factor after Notch activation that leads to EGFR activation and cell invasion promotion. These experiments performed in head and neck, lung and pancreatic cancer cells [42].
Conclusions
In conclusion, the interaction of Notch with EGFR revealed from our data might imply that a dual inhibition of these pathways might be promising in NSCLC cells that express high EGFR protein levels. This indicates an attractive new avenue of combination approaches for cancer therapy that may enhance the potency of EGFR inhibitory agents on tumours.
Acknowledgements
We would like to thank EOGE Oncological Research Fund for financial support.
Additional files
Classification of cells according to ATCC-LGC Promochem instructions (https://www.lgcstandards-atcc.org/), regarding tissue type of origin and EGFR mutation status. (PDF 26 kb)
Representative plots for apoptosis and necrosis detection in H23, A549, H661 and HCC827 cells as described in “Materials and Methods” section. Control: untreated cells and DAPT: cells treated with DAPT. Four populations are distinguished: the viable (An-/PI-) [region 3], the early apoptotic (An+/PI-) [region 4], the late apoptotic (An+/PI+) [region 2] and the necrotic (An-/PI+) [region 1] cells. (PDF 399 kb)
Representative plots for cell cycle analysis in H23, A549, H661 and HCC827 cells as described in “Materials and Methods” section. Control: untreated cells, DAPT: cells treated with DAPT. The G0/G1, S and G2/M phases are indicated with blue, red and green colour, respectively. (PDF 113 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
EG has made substantial contribution to conception and design of the project, participated in acquisition and analysis of data and was involved in drafting the manuscript, AN participated in acquisition of data, as well as data analysis and was involved in drafting the manuscript, DK and AL participated in acquisition of data, SR participated in acquisition of data regarding TEM images, EP and ADT were involved in drafting the manuscript and revised it critically for important intellectual content, TM has made substantial contribution to conception and design of the project and was involved in drafting the manuscript, KF participated in acquisition of data regarding TEM images, HPK has made substantial contribution to conception and design of the project, was involved in drafting the manuscript and revised it critically for important intellectual content. All authors have given final approval of the version to be published.
Authors’ information
EG is researcher at the Clinical Oncology Laboratory at the Medical School of University of Patras and she is very experienced in the methods described in this manuscript as well as in targeting EGFR in cancer cells. AN is MD and PhD student at the Clinical Oncology Laboratory at the Medical School of University of Patras. His thesis is about targeting EGFR in NSCLC. He is also interested to gain full training in Medical Oncology. DK is Pharmacist and her MSc is related to Notch inhibition in NSCLC. AL is Pharmacist and PhD student and her thesis is related to dual inhibition of Notch and EGFR in NSCLC. SR is undergraduate student at Electron Microscopy Laboratory, Faculty of Crop Production, Agricultural University of Athens and she is training in electron microscopy technology. EP is Pharmacist and Professor of Molecular Pharmacology, at the Department of Pharmacy of the University of Patras. She is very experienced in issues and techniques in angiogenesis and tumor growth in vivo and in vitro as well as in pharmacological studies on the mechanisms involved in cancer. ADT is Chemist and Associate Professor of Biochemistry and Molecular Biology in the University of Patras. He is very experienced in issues regarding Pathobiology of diseases such as expression and biological role of extracellular matrix in disease development, in cell growth and functions and molecular targeted therapies. TM is Medical Oncologist and Assistant Professor of Medical Oncology in Division of Oncology, at Medical School at the University of Patras. He is very experienced Oncologist and his research interests include the translational research for the disclosure of reliable, sensitive and responsive prognostic biomarkers to predict cancer behaviour, response to therapy and outcome of the disease. Also, he is interested for the role of HER family in cancer as well as its interaction with other pathways such as Notch and IGFR. KF is Biologist, Professor at Faculty of Crop Production and he is Head of Electron Microscopy Laboratory at Agricultural University of Athens. KF is very experienced at Electron Microscopy technique with many relevant publications. HPK is MD, Professor of Medical Oncology and Head of Division of Oncology at University of Patras. HPK exerts a significant clinical work through his participation in more than 150 clinical trials and subsequent publications and as a coordinator of the Clinical Oncology Laboratory attempts to transfer knowledge, skills and ideas from the clinical to the practical bench and vice versa to improve the management of cancer patients. The main goals of his research include the elucidation of the pathogenetic mechanism of action for new agents to treat various types of cancer and the identification of reliable, sensitive and specific predictive and prognostic biomarkers to predict the response of patients to antineoplastic therapies and to estimate the outcome of the disease.
References
- 1.D’Addario G, Felip E. Non-small-cell lung cancer: ESMO clinical recommendations for diagnosis, treatment and follow-up. Ann Oncol. 2009;20(Suppl 4):68–70. doi: 10.1093/annonc/mdp132. [DOI] [PubMed] [Google Scholar]
- 2.Hammerschmidt S, Wirtz H. Lung cancer: current diagnosis and treatment. Dtsch Arztebl Int. 2009;106(49):809–18. doi: 10.3238/arztebl.2009.0809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reade CA, Ganti AK. EGFR targeted therapy in non-small cell lung cancer: potential role of cetuximab. Biogeosciences. 2009;3:215–24. [PMC free article] [PubMed] [Google Scholar]
- 4.Aita M, Fasola G, Defferrari C, Brianti A, Bello MG, Follador A, et al. Targeting the VEGF pathway: antiangiogenic strategies in the treatment of non-small cell lung cancer. Crit Rev Oncol Hematol. 2008;68(3):183–96. doi: 10.1016/j.critrevonc.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 5.Adamo V, Franchina T, Adamo B, Denaro N, Gambadauro P, Chiofalo G, et al. Gefitinib in lung cancer therapy: clinical results, predictive markers of response and future perspectives. Cancer Biol Ther. 2009;8(3):206–12. doi: 10.4161/cbt.8.3.7465. [DOI] [PubMed] [Google Scholar]
- 6.Scagliotti G, Govindan R. Targeting angiogenesis with multitargeted tyrosine kinase inhibitors in the treatment of non-small cell lung cancer. Oncologist. 2010;15(5):436–46. doi: 10.1634/theoncologist.2009-0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pennell NA, Lynch TJ., Jr Combined inhibition of the VEGFR and EGFR signaling pathways in the treatment of NSCLC. Oncologist. 2009;14(4):399–411. doi: 10.1634/theoncologist.2008-0276. [DOI] [PubMed] [Google Scholar]
- 8.Andre F, Le Chevalier T, Soria JC. Her2-neu: a target in lung cancer? Ann Oncol. 2004;15(1):3–4. doi: 10.1093/annonc/mdh033. [DOI] [PubMed] [Google Scholar]
- 9.Mahalingam D, Mita A, Mita MM, Nawrocki ST, Giles FJ. Targeted therapy for advanced non-small cell lung cancers: historical perspective, current practices, and future development. Curr Probl Cancer. 2009;33(2):73–111. doi: 10.1016/j.currproblcancer.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 10.Miele L, Golde T, Osborne B. Notch signaling in cancer. Curr Mol Med. 2006;6(8):905–18. doi: 10.2174/156652406779010830. [DOI] [PubMed] [Google Scholar]
- 11.Collins BJ, Kleeberger W, Ball DW. Notch in lung development and lung cancer. Semin Cancer Biol. 2004;14(5):357–64. doi: 10.1016/j.semcancer.2004.04.015. [DOI] [PubMed] [Google Scholar]
- 12.Westhoff B, Colaluca IN, D’Ario G, Donzelli M, Tosoni D, Volorio S, et al. Alterations of the Notch pathway in lung cancer. Proc Natl Acad Sci U S A. 2009;106(52):22293–8. doi: 10.1073/pnas.0907781106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Axelson H. Notch signaling and cancer: emerging complexity. Semin Cancer Biol. 2004;14(5):317–9. doi: 10.1016/j.semcancer.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 14.Wang NJ, Sanborn Z, Arnett KL, Bayston LJ, Liao W, Proby CM, et al. Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proc Natl Acad Sci U S A. 2011;108(43):17761–6. doi: 10.1073/pnas.1114669108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li C, Zhang Y, Lu Y, Cui Z, Yu M, Zhang S, et al. Evidence of the cross talk between Wnt and Notch signaling pathways in non-small-cell lung cancer (NSCLC): Notch3-siRNA weakens the effect of LiCl on the cell cycle of NSCLC cell lines. J Cancer Res Clin Oncol. 2011;137(5):771–8. doi: 10.1007/s00432-010-0934-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baumgart A, Seidl S, Vlachou P, Michel L, Mitova N, Schatz N, et al. ADAM17 regulates epidermal growth factor receptor expression through the activation of Notch1 in non-small cell lung cancer. Cancer Res. 2010;70(13):5368–78. doi: 10.1158/0008-5472.CAN-09-3763. [DOI] [PubMed] [Google Scholar]
- 17.Konishi J, Yi F, Chen X, Vo H, Carbone DP, Dang TP. Notch3 cooperates with the EGFR pathway to modulate apoptosis through the induction of bim. Oncogene. 2010;29(4):589–96. doi: 10.1038/onc.2009.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Y, Burns JA, Cheney CA, Zhang N, Vitelli S, Wang F, et al. Distinct expression profiles of Notch-1 protein in human solid tumors: Implications for development of targeted therapeutic monoclonal antibodies. Biogeosciences. 2010;4:163–71. doi: 10.2147/btt.s11021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dai J, Ma D, Zang S, Guo D, Qu X, Ye J, et al. Cross-talk between Notch and EGFR signaling in human breast cancer cells. Cancer Invest. 2009;27(5):533–40. doi: 10.1080/07357900802563036. [DOI] [PubMed] [Google Scholar]
- 20.Wang Z, Li Y, Ahmad A, Azmi AS, Banerjee S, Kong D, et al. Targeting Notch signaling pathway to overcome drug resistance for cancer therapy. Biochim Biophys Acta. 2010;1806(2):258–67. doi: 10.1016/j.bbcan.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doroquez DB, Rebay I. Signal integration during development: mechanisms of EGFR and Notch pathway function and cross-talk. Crit Rev Biochem Mol Biol. 2006;41(6):339–85. doi: 10.1080/10409230600914344. [DOI] [PubMed] [Google Scholar]
- 22.Baker AT, Zlobin A, Osipo C. Notch-EGFR/HER2 Bidirectional Crosstalk in Breast Cancer. Front in oncol. 2014;4:360. doi: 10.3389/fonc.2014.00360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xie M, He CS, Wei SH, Zhang L. Notch-1 contributes to epidermal growth factor receptor tyrosine kinase inhibitor acquired resistance in non-small cell lung cancer in vitro and in vivo. Eur J Cancer. 2013;49(16):3559–72. doi: 10.1016/j.ejca.2013.07.007. [DOI] [PubMed] [Google Scholar]
- 24.Koutras A, Giannopoulou E, Kritikou I, Antonacopoulou A, Evans TR, Papavassiliou AG, et al. Antiproliferative effect of exemestane in lung cancer cells. Mol Cancer. 2009;8:109. doi: 10.1186/1476-4598-8-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuwada SK, Scaife CL, Kuang J, Li X, Wong RF, Florell SR, et al. Effects of trastuzumab on epidermal growth factor receptor-dependent and -independent human colon cancer cells. Int J Cancer. 2004;109(2):291–301. doi: 10.1002/ijc.11686. [DOI] [PubMed] [Google Scholar]
- 26.Furugaki K, Iwai T, Moriya Y, Harada N, Fujimoto-Ouchi K. Loss of an EGFR-amplified chromosome 7 as a novel mechanism of acquired resistance to EGFR-TKIs in EGFR-mutated NSCLC cells. Lung Cancer. 2014;83(1):44–50. doi: 10.1016/j.lungcan.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 27.Cragg MS, Kuroda J, Puthalakath H, Huang DC, Strasser A. Gefitinib-induced killing of NSCLC cell lines expressing mutant EGFR requires BIM and can be enhanced by BH3 mimetics. PLoS Med. 2007;4(10):1681–9. doi: 10.1371/journal.pmed.0040316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.He Y, Mo Z, Xue Z, Fang Y. Establish a flow cytometric method for quantitative detection of Beclin-1 expression. Cytotechnology. 2013;65(4):481–9. doi: 10.1007/s10616-012-9503-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Giannopoulou E, Antonacopoulou A, Matsouka P, Kalofonos HP. Autophagy: novel action of panitumumab in colon cancer. Anticancer Res. 2009;29(12):5077–82. [PubMed] [Google Scholar]
- 30.Chen Y, De Marco MA, Graziani I, Gazdar AF, Strack PR, Miele L, et al. Oxygen concentration determines the biological effects of NOTCH-1 signaling in adenocarcinoma of the lung. Cancer Res. 2007;67(17):7954–9. doi: 10.1158/0008-5472.CAN-07-1229. [DOI] [PubMed] [Google Scholar]
- 31.Yin L, Velazquez OC, Liu ZJ. Notch signaling: emerging molecular targets for cancer therapy. Biochem Pharmacol. 2010;80(5):690–701. doi: 10.1016/j.bcp.2010.03.026. [DOI] [PubMed] [Google Scholar]
- 32.Mazzone M, Selfors LM, Albeck J, Overholtzer M, Sale S, Carroll DL, et al. Dose-dependent induction of distinct phenotypic responses to Notch pathway activation in mammary epithelial cells. Proc Natl Acad Sci U S A. 2010;107(11):5012–7. doi: 10.1073/pnas.1000896107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu J, Zhu X, Lu Q. Antiproliferative effects of gamma-secretase inhibitor, a Notch signalling inhibitor, in multiple myeloma cells and its molecular mechanism of action. J Int Med Res. 2013;41(4):1017–26. doi: 10.1177/0300060513485912. [DOI] [PubMed] [Google Scholar]
- 34.Zang S, Ji C, Qu X, Dong X, Ma D, Ye J, et al. A study on Notch signaling in human breast cancer. Neoplasma. 2007;54(4):304–10. [PubMed] [Google Scholar]
- 35.Cao H, Hu Y, Wang P, Zhou J, Deng Z, Wen J. Down-regulation of Notch receptor signaling pathway induces caspase-dependent and caspase-independent apoptosis in lung squamous cell carcinoma cells. APMIS. 2012;120(6):441–50. doi: 10.1111/j.1600-0463.2011.02825.x. [DOI] [PubMed] [Google Scholar]
- 36.Liu JJ, Lin M, Yu JY, Liu B, Bao JK. Targeting apoptotic and autophagic pathways for cancer therapeutics. Cancer Lett. 2011;300(2):105–14. doi: 10.1016/j.canlet.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 37.Barth JM, Hafen E, Kohler K. The lack of autophagy triggers precocious activation of Notch signaling during Drosophila oogenesis. BMC Dev Biol. 2012;12:35. doi: 10.1186/1471-213X-12-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Furugaki K, Iwai T, Shirane M, Kondoh K, Moriya Y, Mori K. Schedule-dependent antitumor activity of the combination with erlotinib and docetaxel in human non-small cell lung cancer cells with EGFR mutation, KRAS mutation or both wild-type EGFR and KRAS. Oncol Rep. 2010;24(5):1141–6. doi: 10.3892/or_00000965. [DOI] [PubMed] [Google Scholar]
- 39.Kang JH, Lee EH, Park SW, Chung IY. MUC5AC expression through bidirectional communication of Notch and epidermal growth factor receptor pathways. J Immunol. 2011;187(1):222–9. doi: 10.4049/jimmunol.1003606. [DOI] [PubMed] [Google Scholar]
- 40.Purow BW, Sundaresan TK, Burdick MJ, Kefas BA, Comeau LD, Hawkinson MP, et al. Notch-1 regulates transcription of the epidermal growth factor receptor through p53. Carcinogenesis. 2008;29(5):918–25. doi: 10.1093/carcin/bgn079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Osipo C, Patel P, Rizzo P, Clementz AG, Hao L, Golde TE, et al. ErbB-2 inhibition activates Notch-1 and sensitizes breast cancer cells to a gamma-secretase inhibitor. Oncogene. 2008;27(37):5019–32. doi: 10.1038/onc.2008.149. [DOI] [PubMed] [Google Scholar]
- 42.Diaz B, Yuen A, Iizuka S, Higashiyama S, Courtneidge SA. Notch increases the shedding of HB-EGF by ADAM12 to potentiate invadopodia formation in hypoxia. J Cell Biol. 2013;201(2):279–92. doi: 10.1083/jcb.201209151. [DOI] [PMC free article] [PubMed] [Google Scholar]