

ABSTRACT
We determined whether there is turnover of the peptidoglycan (PG) cell wall of the ovococcus bacterial pathogen Streptococcus pneumoniae (pneumococcus). Pulse-chase experiments on serotype 2 strain D39 radiolabeled with N-acetylglucosamine revealed little turnover and release of PG breakdown products during growth compared to published reports of PG turnover in Bacillus subtilis. PG dynamics were visualized directly by long-pulse–chase–new-labeling experiments using two colors of fluorescent d-amino acid (FDAA) probes to microscopically detect regions of new PG synthesis. Consistent with minimal PG turnover, hemispherical regions of stable “old” PG persisted in D39 and TIGR4 (serotype 4) cells grown in rich brain heart infusion broth, in D39 cells grown in chemically defined medium containing glucose or galactose as the carbon source, and in D39 cells grown as biofilms on a layer of fixed human epithelial cells. In contrast, B. subtilis exhibited rapid sidewall PG turnover in similar FDAA-labeling experiments. High-performance liquid chromatography (HPLC) analysis of biochemically released peptides from S. pneumoniae PG validated that FDAAs incorporated at low levels into pentamer PG peptides and did not change the overall composition of PG peptides. PG dynamics were also visualized in mutants lacking PG hydrolases that mediate PG remodeling, cell separation, or autolysis and in cells lacking the MapZ and DivIVA division regulators. In all cases, hemispheres of stable old PG were maintained. In PG hydrolase mutants exhibiting aberrant division plane placement, FDAA labeling revealed patches of inert PG at turns and bulge points. We conclude that growing S. pneumoniae cells exhibit minimal PG turnover compared to the PG turnover in rod-shaped cells.
IMPORTANCE PG cell walls are unique to eubacteria, and many bacterial species turn over and recycle their PG during growth, stress, colonization, and virulence. Consequently, PG breakdown products serve as signals for bacteria to induce antibiotic resistance and as activators of innate immune responses. S. pneumoniae is a commensal bacterium that colonizes the human nasopharynx and opportunistically causes serious respiratory and invasive diseases. The results presented here demonstrate a distinct demarcation between regions of old PG and regions of new PG synthesis and minimal turnover of PG in S. pneumoniae cells growing in culture or in host-relevant biofilms. These findings suggest that S. pneumoniae minimizes the release of PG breakdown products by turnover, which may contribute to evasion of the innate immune system.
INTRODUCTION
Peptidoglycan (PG) biosynthesis and placement are dynamic processes that determine the shapes, sizes, chaining, and resistance to turgor of bacterial cells (1–6). In Gram-positive bacteria, PG also serves as the scaffolding for covalent attachment of surface wall teichoic acid, capsule, and sortase-attached proteins (7–9). The seminal work of Park and Uehara demonstrated that PG is rapidly turned over and the breakdown components recycled in some Gram-negative bacteria, such as Escherichia coli (10). The turnover and recycling pathways are mediated by specific sets of genes that encode PG cleavage enzymes that break down PG, transporters to take up and recover PG breakdown products, and additional enzymes that convert PG breakdown products into metabolic intermediates (10–13). Turnover and release of PG fragments to culture medium in certain Gram-positive bacteria, such as Bacillus subtilis, has been known for some time. However, Gram-positive PG recycling was only discovered recently, because genes that encode PG recycling enzymes are subject to catabolite repression and most previous work was done in glucose-containing media (13, 14).
Because PG is unique to eubacteria, PG breakdown products serve important roles in signaling both to bacteria and to eukaryotic host cells. In E. coli, the PG recycling pathway was recently implicated in a futile cycle that increases the efficacy of β-lactam antibiotics, which inhibit the transpeptidase activity of penicillin binding proteins (PBPs) (15). In some Gram-negative bacteria, such as Citrobacter freundii and Enterobacter cloacae, recycled anhydro-murotripeptide acts as an inducer for the expression of β-lactamase gene expression, thereby providing antibiotic resistance (12, 16). An analogous signaling system functions in certain Gram-positive bacteria, such as Bacillus licheniformis, where recycled PG peptides (e.g., γ-d-Glu-m-DAP [DAP is diaminopimelic acid]) relieve the expression of a β-lactamase gene (12). Other Gram-positive species, such as Staphylococcus aureus, encode integral membrane proteins with extracellular β-lactam-binding domains connected to cytoplasmic domains that function as a repressor-specific protease to induce the expression of β-lactamases and β-lactam-resistant penicillin-binding protein 2a (PBP2a) (12). Finally, besides acting in antibiotic stress and resistance responses, PG fragments are signals in bacterial spore germination pathways (17, 18).
PG fragments function in two diametrically opposed ways with regard to eukaryotic host cells. Some PG fragments, such as the anhydro-muramic-tripeptides of Neisseria and Bordetella species, act as potent toxins of ciliated epithelial cells by inducing inflammatory cytokine production (19). These PG fragments are produced by bacterial PG hydrolases and lytic transglycosylases and play important roles in pathogenesis. In contrast, PG fragments produced by bacterial autolysis mechanisms, PG turnover pathways, or host PG lysozymes and other PG hydrolases are major signals of infection to host innate immune systems (11, 17, 20, 21). Extracellular PG fragments interact with Toll-like receptors and PG recognition proteins to stimulate innate immune responses (11, 17, 22). PG fragments produced intracellularly by lysozyme digestion in phagocytes stimulate Nod receptors (20, 21). For some phagocytosed extracellular pathogens, such as Streptococcus pneumoniae (pneumococcus), lysozyme digestion concomitantly produces PG fragments and releases a pore-forming toxin that damages the phagosome membrane (23). This damage enables the release of PG fragments into the cytosol, where they can interact with Nod2 receptors that induce proinflammatory signaling, leading to the recruitment of additional phagocytic cells to infection sites (23).
Because PG fragments act as potent signals to activate host immunity, planktonic bacteria likely minimize the shedding of PG fragments. One mechanism to reduce PG shedding is to synchronize the activities of PG turnover and recycling pathways, as occurs in E. coli (10). Another mechanism, which operates in Staphylococcus aureus, is to remodel the PG to reduce recognition by PG recognition proteins of the innate immune system (24). A third mechanism to reduce PG fragment shedding would be to not carry out PG turnover and recycling at all. Certain Gram-positive species, such as S. pneumoniae, Staphylococcus aureus, and Enterococcus faecalis, do not encode obvious homologues of certain key enzymes that are required in the PG recycling pathways of other Gram-positive bacteria (13, 25), although as noted above, S. aureus does encode surface sensor proteins that covalently bind and respond to β-lactam antibiotics (12). However, it remains possible that these bacteria use previously unidentified enzymes or activities to complete PG recycling pathways.
S. pneumoniae is a human commensal bacterium that colonizes the nasopharynx as a biofilm and that can become an opportunistic pathogen in individuals recovering from influenza or with compromised immune systems, causing a number of serious respiratory and invasive diseases, such as pneumonia (26–28). Alterations in PG biosynthesis have strong effects on the efficiency of pneumococcal colonization and infection. Mutations in numerous genes implicated in PG synthesis and remodeling that do not ostensibly affect growth in culture strongly attenuate colonization and lung infection in transposon-sequencing (Tn-Seq) screens (29). Moreover, cell chaining, which is especially sensitive to the function of PG hydrolases, favors colonization in the nasopharynx but makes cells vulnerable to phagocytosis in the lung (30, 31). In this paper, we used fluorescent d-amino acid (FDAA) probes (32, 33) to visualize PG dynamics in capsulated and unencapsulated mutants of S. pneumoniae growing in culture and in host-relevant biofilms in vitro. We also used FDAA probes to visualize defects in PG morphology caused by PG hydrolases and key cell division mutants. Our combined results indicate minimal turnover of pneumococcal PG, consistent with the lack of an apparent complete PG recycling pathway. Minimal PG turnover may be a mechanism that allows S. pneumoniae cells to grow planktonically without releasing PG breakdown fragments that would alert the human innate immune system.
MATERIALS AND METHODS
Pneumococcal strains and growth conditions.
Most mutant strains used in this study were derived from strains IU1824 and IU1945, unencapsulated derivatives of serotype 2 S. pneumoniae strain D39 (IU1690) (see Table S1 in the supplemental material). Strains containing antibiotic markers were constructed by transforming linear DNA amplicons synthesized by overlapping fusion PCR into competent pneumococcal cells as described previously (34–36). Primers used for the generation of amplicons are listed in Table S2. All constructs were confirmed by DNA sequencing of chromosomal regions corresponding to the amplicon region used for transformation. Genes encoding PG hydrolases LytA and LytB, d,d-carboxypeptidase DacA, putative lytic transglycosylase Pmp23, and class A Pbp1a are in single-gene operons or are at the ends of operons; consequently, replacement of these reading frames with antibiotic cassettes causes minimal polarity, as discussed previously and confirmed by construction of markerless deletions (37; data not shown). The murMN gene (PG peptide cross-bridge formation), pgdA gene (GlcNAc deacetylase), and adr gene (O-acetylated MurNAc) may also be in single-gene operons based on transcriptome analyses (data not shown). As expected from previous reports (38, 39), replacement of these reading frames with antibiotic resistance cassettes did not change cell morphology and growth. High sensitivity of the ΔpgdA mutant to lysozyme (39) was confirmed (data not shown). PcsB expression was decreased by a promoter replacement as reported previously (40). The cell morphology phenotypes of mutants with reading frame replacements in dacB (l,d-carboxypeptidase) or spd_0703 (WalRK regulon gene of unknown function) were confirmed previously by complementation (37). Markerless deletions of the division genes, mapZ (alternative name, locZ) and divIVA, fused short (20-amino acid) stretches from the beginning and end of the genes to maintain possible translational coupling (see Tables S1 and S2). The phenotypes of the markerless ΔmapZ and ΔdivIVA mutants were similar to those reported previously for other mapZ and divIVA mutants (41–43).
Bacteria were grown on petri plates containing Trypticase soy agar II (modified; Becton-Dickinson) and 5% (vol/vol) defibrinated sheep blood (TSAII-BA). Plates were incubated at 37°C in an atmosphere of 5% CO2. For antibiotic selection, TSAII BA plates contained 250 μg kanamycin ml−1, 150 μg spectinomycin ml−1, 0.3 μg erythromycin ml−1, 250 μg streptomycin ml−1, or 2.5 μg chloramphenicol ml−1. In most experiments, strains were cultured statically in Becton-Dickinson brain heart infusion (BHI) broth at 37°C in an atmosphere of 5% CO2, and growth was monitored by the optical density at 620 nm (OD620) as described previously (34–36). Where indicated, bacteria were grown in a chemically defined medium (CDM) containing 1.0% (wt/vol) glucose or 1.0% (wt/vol) galactose as the carbon source (44). Bacteria were inoculated into BHI broth from frozen cultures or colonies, serially diluted into the same medium, and propagated overnight. For growth experiments, overnight cultures that were still in exponential phase (OD620 of 0.1 to 0.4) were diluted back to an OD620 of ≈0.003 to start the final cultures, which lacked antibiotics. For inoculation of CDM cultures, 1-ml amounts of overnight cultures of bacteria grown in BHI were centrifuged at 16,000 × g for 5 min at 4°C, and pellets were washed once in cold 1× phosphate-buffered saline (PBS). Following centrifugation, pellets were resuspended in 2 ml of warm CDM, which was further diluted into warm CDM cultures.
Long-pulse–chase experiments to determine release of [14C]GlcNAc from pneumococcal PG sacculi.
Cells from an overnight culture were diluted to an OD of ≈0.002 in 4.0 ml of warmed BHI broth containing [14C]GlcNAc (specific activity, 55 mCi per mmol; GE Healthcare) to a final concentration of 7.3 μM. Following growth of the culture to an OD620 of 0.25, cells were centrifuged at 16,000 × g for 10 min at 4°C and resuspended in 1.0 ml of cold 1× PBS buffer. Cells were collected and washed two more times, and 400-μl aliquots from each of the three 1× PBS washes were scintillation counted to verify that unincorporated [14C]GlcNAc was washed away. Cells were resuspended to an OD620 of 0.1 in 10 ml of BHI broth containing 1.0 mM unlabeled GlcNAc and incubated at 37°C. Four-hundred-microliter aliquots were taken at 0, 10, 20, and 30 min and every 30 min thereafter for 360 min. Aliquots were placed on ice for 2 min and then centrifuged at 16,000 × g for 10 min in the cold. Supernatants were collected, added to 4.0 ml of scintillation fluid, and counted using a Packard scintillation counter. Pellets were resuspended in 400 μl of boiling 5% (wt/vol) SDS for 30 min. PG sacculi were pelleted by centrifugation at 130,000 × g for 20 min at room temperature. Supernatants were discarded, and sacculus pellets were resuspended in 150 μl H2O, which was added to 4.0 ml scintillation fluid and counted.
Long pulse-chase-new labeling with FDAA probes of pneumococcal cells in culture imaged by two-dimensional (2-D) epifluorescence microscopy.
FDAAs HADA (7-hydroxycoumarin-3-carboxylic acid 3-amino–d-alanine), NADA (4-chloro-7-nitrobenzofurazan 3-amino–d-alanine), and TADA (tetramethylrhodamine 3-amino–d-alanine) were synthesized as reported previously (32, 45), with the following change: TADA was synthesized as reported for TDL [5(6)-carboxytetramethylrhodamine-d-lysine; TAMRA-d-lysine], except that Boc-d-DAP-OH (N-alpha-t-butyloxycarbonyl-d-2,3-diaminopropionic acid) was used in place of Boc-d-Lys-OH (N-alpha-t-butyloxycarbonyl-d-lysine). Cells from overnight cultures were diluted to an OD620 of 0.01 in 2.0 ml of warmed BHI broth. At an OD620 of 0.02, 1.0 μl of 500 mM HADA (in dimethyl sulfoxide [DMSO]) was added to a final concentration of 250 μM. Cultures were grown to an OD620 of 0.20 to 0.25, after which they were transferred to 2.0-ml centrifuge tubes, which were placed in an ice bath for 1 min to halt labeling and centrifuged for 5 min at 16,000 × g in the cold. Supernatants were discarded, and pellets were resuspended in 1.0 ml of cold 1× PBS. Cells were centrifuged (2.5 min at 16,000 × g) and washed with cold 1× PBS two more times. After the third spin, cell pellets were diluted to an OD620 of 0.05 in 2.0 ml of warmed BHI to which 1.0 μl of 500 mM NADA or TADA (in DMSO) had already been added. Cultures were placed back in an incubator and grown at 37°C, and 200-μl aliquots were taken at 0, 20, 40, 60, 80, and 100 min after addition of NADA or TADA. These aliquots were centrifuged at 16,000 × g for 2.5 min at 4°C, supernatants were discarded, and pellets were resuspended in 50 μl of cold 1× PBS. An amount of 1.5 μl of labeled live cells (no fixation) was placed on a slide, covered with a glass coverslip, and imaged and processed as described previously using a Nikon E-400 epifluorescence phase-contrast microscope and NIS-Elements AR imaging software (Nikon) (33, 46).
For examination of stationary-phase cells, the following changes were made: a 4.0-ml cell culture was labeled with FDAAs, and samples were taken at 0, 10, 20, and 30 min after the second labeling; additional samples were taken every 0.5 h until 270 min. For examination of cells labeled with HADA for 30 min and TADA for 5 min, the following changes were made: cells were grown without FDAAs to an OD620 of 0.1, after which 1.0 μl of 500 mM HADA (in DMSO) was added to a final concentration of 250 μM, cultures were placed back in the 37°C incubator for 30 min (approximately one generation), washed as described above, labeled with TADA for 5 min, washed again, fixed, and examined microscopically as described previously (33, 46).
Examination of cells prelabeled with fluorescent antibiotics before HADA labeling was performed as follows. Cells from overnight cultures grown without FDAAs were diluted to an OD620 of 0.2, at which point several 500-μl aliquots of cultures were taken and centrifuged at room temperature in microcentrifuge tubes. Pellets were resuspended in 250 μl of BHI broth containing either 10 μg fluorescent cephalosporin/ml (47), 5 μg fluorescent bocillin/ml (48), 2 μg fluorescent vancomycin/ml (33), or no antibiotic. Cultures containing fluorescent bocillin or fluorescent vancomycin were incubated for 10 min at room temperature in the dark, whereas cultures containing fluorescent cephalosporin were incubated for 30 min at room temperature in the dark. HADA was then added to a final concentration of 250 μM, and cultures were incubated for 5 min at room temperature. Cells were centrifuged in the cold and washed twice with cold 1× PBS and were finally resuspended in 50 μl of cold 1× PBS. Cells were viewed without fixation as described above.
Long pulse-chase-new labeling with FDAA probes of B. subtilis cells in culture imaged by 2-D epifluorescence microscopy.
B. subtilis laboratory strain P479 (from Dan Kearns, Indiana University—Bloomington) was streaked onto LB plates, which were incubated overnight at 37°C in air. Single colonies were inoculated into fresh LB broth, and cultures were grown at 37°C with aeration and shaking at 400 rpm. After 2 h, exponential cultures were diluted in fresh LB broth to an OD620 of 0.02, and long-pulse labeling of B. subtilis cells with HADA was performed as described above for S. pneumoniae, except that B. subtilis cultures were shaken in air. At an OD620 of 0.20 to 0.25, HADA was washed away (chase) and TADA (new labeling) was added as described above for new labeling of S. pneumoniae with NADA. Exponential cultures were shaken at 37°C and sampled every 20 min, at which times live cells were washed to remove unincorporated TADA and observed by epifluorescence microscopy as described above for S. pneumoniae cells.
2-D time-lapse microscopy of single pneumococcal cells labeled with TADA.
Cells from an overnight culture were diluted to an OD620 of 0.01 in 2.0 ml of warmed BHI broth. An amount of 1.0 μl of 500 mM TADA (in DMSO) was added to a final concentration of 250 μM. Cultures were grown to an OD620 of 0.1 to 0.15, when they were transferred to a 2-ml centrifuge tube, placed in an ice bath for 1 min to stop the labeling, and centrifuged for 5 min at 16,000 × g in the cold. Supernatants were discarded, and pellets were resuspended in 1 ml of cold 1× PBS. Cells were centrifuged (2.5 min at 16,000 × g) and washed with cold 1× PBS two more times. After the second wash, supernatants were discarded, and pellets were resuspended in 1 ml of warmed BHI broth lacking FDAAs. An amount of 5 μl of culture was pipetted onto a large coverslip on the microscope stage of a Nikon Eclipse Ti inverted microscope equipped with NIS-Elements AR imaging software. A section of BHI–1% (wt/vol) agar cut out of a plate was placed on the sample, which was surrounded by a chamber covered by a second coverslip. The chamber wall was warmed to 32°C by a heating fan. Different individual cells were selected, and their growth into microcolonies was imaged every 2 min for 17 h.
High-performance liquid chromatography (HPLC) analysis of the composition of PG peptides isolated from pneumococcal cells labeled with HADA.
PG peptides were purified by a procedure adapted from reference 49. Cells from overnight cultures were diluted into 200 ml (for total PG peptide profiles) or 25 ml (for fluorescent PG peptide profiles) of warmed BHI broth to an OD620 of 0.01 and incubated at 37°C. At an OD620 of 0.02, HADA (in DMSO) was added to a final concentration of 250 μM, and cultures were grown to an OD620 of 0.5. Cells were centrifuged at 16,000 × g for 10 min at 4°C, and pellets were resuspended in 10 ml of cold 1× PBS. Cells were centrifuged and washed twice more, and after the last spin, cells were resuspended in 1 ml of cold 50 mM Tris-HCl, pH 7.0. Cell suspensions were added dropwise to 5 ml of boiling 5% (wt/vol) SDS and boiled for 30 min. Sacculi were collected by centrifugation at 18,500 × g for 20 min at room temperature and resuspended in 1.0 M NaCl. Centrifugation and washing with 1.0 M NaCl was done one more time. Pellets were then centrifuged, washed three more times with cold H2O, and finally resuspended in 6 ml of H2O. Samples were added to a 15-ml tube containing lysing matrix B (MP Biomedicals), and sacculi were further disrupted in a FastPrep 24 homogenizer in the cold using 4 pulses of 40 s each at a speed of 6 m/s. Tubes were centrifuged at 9,300 × g for 1 min at 4°C, and supernatants were centrifuged at 130,000 × g for 45 min at 4°C (these conditions were used for all subsequent spins in this procedure). Pellets were resuspended in 100 mM Tris-HCl, pH 7.5, 20 mM MgSO4. DNase A was added to 10 μg/ml and RNase A was added to 50 μg/ml, and samples were incubated at 37°C for 1 h with shaking at 400 rpm. CaCl2 and trypsin were added to 10 mM and 10 μg/ml, respectively, and samples were incubated at 37°C with shaking at 400 rpm for approximately 18 h. SDS was added to a concentration of 1% (wt/vol), and samples were incubated at 80°C for 15 min. Following centrifugation, sacculus pellets were resuspended in 1 ml of 8 M LiCl and incubated at 37°C for 15 min. Following centrifugation, sacculus pellets were resuspended in 10 mM EDTA for 15 min at 37°C. Samples were centrifuged and washed 3× with H2O, and pellets were resuspended in 1 ml of 50 mM Tris-HCl, pH 7.0.
An amount of 1.0 μl of pneumococcal LytA amidase (7.0 mg/ml) was added, and samples were incubated at 37°C in the dark with shaking at 400 rpm for 24 h. LytA was purified as described previously (50). Two additional digestions were performed by adding 1.0 μl more of LytA and incubating for 8 h and then 16 h. The final digested samples were filtered through a 3K Amicon spin filter, and the flowthrough containing released PG peptides was collected. Samples were placed in a vacuum centrifuge at 37°C until all solvent had evaporated. The final residue was resuspended in 200 μl of 0.05% (vol/vol) trifluoroacetic acid. Peptides were resolved on a C18 column (Vydac 218 TP54) attached to a Shimadzu 10A HPLC system at a flow rate of 0.5 ml per min. Total PG peptide profiles from 200 ml of culture were resolved using the gradient described in reference 51, with UV detection at A210 and fluorescence detection (excitation, 405 nm; emission, 450 nm) (see Fig. S2 in the supplemental material). For unlabeled cells, the A210 chromatogram of these D39 strains was similar to that reported previously (51). Fluorescent peptide profiles from 25 ml of culture were resolved using the gradient described in reference 37, with fluorescence detection (excitation, 405 nm; emission, 450 nm) (see Fig. 4).
FIG 4.

Rapid turnover of sidewall PG in exponentially growing cells of B. subtilis detected by FDAA long-pulse–chase–new-labeling experiments. B. subtilis wild-type laboratory strain P479 was grown exponentially in LB broth (±1% [wt/vol] glucose; results without glucose are shown), labeled with HADA (pseudocolored green; old PG) for about 3 generations, washed, and then chased in the presence of a second color of FDAA, TADA (pseudocolored red; new PG synthesis) as described in Materials and Methods. Live cells were imaged by epifluorescence phase-contrast microscopy at the indicated time points after the removal of HADA (chase) and the addition of TADA. Minimal variation in labeling patterns was observed for ≈200 individual cells examined in microscopic fields at different time points after the start of the chase/TADA labeling, and representative images are shown. Labeling was done twice (once with added glucose and once without) with similar results. Scale bar = 5 μm. P, phase-contrast image; H, HADA labeling (old PG); T, TADA labeling (new PG synthesis); O, overlay of H and T images. The arrow at 40 min of chase indicates faint residual old (green) sidewall PG remaining. Arrows at 60 and 80 min indicate faint inert PG that likely resulted from partial synthesis. See the text for additional details.
Long pulse-chase-new labeling with FDAA probes of pneumococcal cells in host-relevant biofilms imaged by 3-D–SIM.
Pneumococcal biofilms were formed on fixed human lung epithelial cells by a method adapted from references 52 and 53. Briefly, bacteria from BHI overnight cultures were diluted into 2.0 ml CDM to an OD620 of 0.01, and CDM cultures were grown at 37°C to an OD620 of 0.1. Bacterial cultures were then diluted to 2 × 104 CFU per 500 μl of CDM, which was added to round coverslips containing a confluent monolayer of H292 human lung epithelial cells that were previously fixed with 4% (vol/vol) paraformaldehyde as described in reference 53. The coverslips were contained in 24-well culture plates, which were incubated at 34°C for 48 h in an atmosphere of 5% CO2, with CDM changes every 12 h. At 48 h, adherent bacteria in biofilms were labeled with TADA (final concentration, 250 μM) in 250 μl of fresh CDM, and cultures were incubated at 34°C for 2 h. Wells were gently washed twice with 250 μl of cold 1× PBS, and HADA (final concentration, 250 μM) in 250 μl CDM was added to each well. Plates were incubated at 34°C for 2 h, after which concanavalin A-Alexa Fluor 488 was added to a final concentration of 50 μg per ml, and incubation was continued for 15 min. Wells were washed gently twice with 250 μl of cold 1× PBS, and 500 μl of 4% (vol/vol) paraformaldehyde was added to each well. Adherent bacteria in biofilms were fixed for 1 h at 34°C before 500 μl of 1.0 M glycine was added to quench the cross-linking reaction. Wells were washed twice with 250 μl of cold 1× PBS, and coverslips were removed from the wells and allowed to air dry. An amount of 3.0 μl of SlowFade gold antifade reagent (Invitrogen) was added to each coverslip, which was placed on a slide and covered by a second coverslip. Images were obtained by three-dimensional structured-illumination microscopy (3-D–SIM), which was performed with the OMX 3-D–SIM superresolution system located in the Indiana University—Bloomington Light Microscopy Imaging Center (http://www.indiana.edu/~lmic/microscopes/OMX.html) as described previously (33, 46). For examination of 4′,6-diamidino-2-phenylindole (DAPI)-labeled biofilms, bacterial biofilms were formed in BHI broth, instead of CDM, on a monolayer of fixed H292 human lung cells as described above. After 2 h of TADA labeling, wells were gently washed twice with 250 μl of cold 1× PBS, and samples were fixed with 4% (vol/vol) paraformaldehyde as described above. An amount of 3.0 μl of SlowFade gold antifade reagent plus DAPI reagent (Invitrogen) was added to each coverslip, and coverslips were observed by 3-D–SIM as described above.
RESULTS AND DISCUSSION
Classical long-pulse–chase radiolabeling demonstrates a low rate of release of [14C]GlcNAc from PG sacculi of growing pneumococcal cells.
In a study focused on the coordination of the timing of PG and teichoic acid synthesis in S. pneumoniae, Tomasz and coworkers performed pulse-chase experiments to determine the release of incorporated radiolabeled Lys or Glu from PG peptides of cells of S. pneumoniae strain R6 growing exponentially in chemically defined medium (54). Based on a limited number of timed samples, they reported negligible release of PG peptides and concluded that there is likely minimal turnover of pneumococcal PG from cells. We reprised this experiment using a variation of a pulse-chase protocol that showed substantial release of PG fragments from B. subtilis, indicative of high PG turnover (55, 56). An unencapsulated mutant of serotype 2 strain D39 was labeled for approximately 7 generations with 14C-radiolabeled N-acetylglucosamine ([14C]GlcNAc) in BHI broth (see Materials and Methods). Cells were washed and chased with excess cold GlcNAc, and radioactivity was determined in PG sacculi and in the culture medium from samples taken at different stages of growth (Fig. 1). In exponentially growing cells (to 90 min; doubling time of 35 min) (Fig. 1) before the start of stationary-phase autolysis, there was a low rate of release of [14C]GlcNAc into the growth medium from the PG sacculi of the pulse-labeled pneumococcal cells (5.6% ± 1.6% [mean ± standard error of the mean {SEM}; n = 2] per generation). In contrast, there was ≈27% release per generation of [14C]GlcNAc into the growth medium from the PG sacculi of pulse-labeled, exponentially growing B. subtilis cells (55). The release of [14C]GlcNAc from the pneumococcal PG sacculi was approximately matched by the recovery of radioactivity from the spent medium when values were corrected for quenching differences.
FIG 1.

Low rate of release of [14C]GlcNAc from sacculi into spent medium by S. pneumoniae strain IU1945 (D39 Δcps) during exponential growth and early stationary phase. Bacteria growing exponentially in BHI broth were labeled with [14C]GlcNAc for about 7 generations, washed, and resuspended in BHI containing a chase of unlabeled GlcNAc (time zero) as described in Materials and Methods. At the times indicated, medium and cells were collected. Cells were converted to sacculi by boiling in SDS, and samples of medium and sacculi were scintillation counted. The experiment was performed twice with similar results, and a representative curve from one experiment is shown. Quenching of 14C counting was approximately 1.4-fold greater for spent medium than for sacculus samples. See the text for additional details.
The interpretation of such a low rate of release of [14C]GlcNAc from pneumococcal sacculi is complicated by a low level of cell death (≈5%) and residual autolysis that occurs in exponential cultures of S. pneumoniae cells in BHI broth (L.-T. Sham, unpublished data). This low-level cell death is dependent on the medium and can be largely suppressed by the addition of Mn2+ to BHI broth (Sham, unpublished). Whether low-level cell death and residual autolysis occur for planktonically growing S. pneumoniae in host niches is unknown. Autolysis, like PG turnover, can release PG breakdown products from bacterial cells; however, PG turnover is a controlled process in normal PG synthesis of live cells (10, 12, 13), whereas autolysis results in cell death. Fratricidal PG lysis of noncompetent pneumococcal cells is pronounced in biofilms, which were studied and are discussed below.
Within the limitations of these pulse-chase experiments, the low rate of release of [14C]GlcNAc from pneumococcal sacculi is consistent with the earlier results of Tomasz and coworkers (54) and contrasts with the high turnover observed for growing B. subtilis cells (55, 56). Similar long-pulse–chase experiments have revealed that other ovococcus-shaped bacteria, such as Enterococcus faecalis and Streptococcus mutans, exhibit low rates of PG fragment release from sacculi (56, 57). One interpretation of these combined results is that there is minimal PG turnover and PG fragment release in exponentially growing S. pneumoniae and, likely, in other Gram-positive ovococcus species compared to the PG turnover and fragment release in B. subtilis. An alternative interpretation is that Gram-positive ovococci exhibit high rates of PG turnover and recycling, similar to E. coli, which limits its net release of PG breakdown fragments into growth media from exponentially growing cells to ≈6% (10).
Long-pulse–chase–new-labeling experiments with FDAA probes support minimal turnover of pneumococcal PG.
To distinguish between the hypotheses of minimal PG turnover versus high PG turnover coupled to recycling, we visualized PG dynamics directly by long-pulse–chase–new-labeling experiments using FDAA probes (32, 33, 45). FDAAs consist of a d-Ala-like molecule linked to differently colored fluorophores that are covalently incorporated into bacterial PG in regions of active PBP transpeptidase activity (32, 33, 45). We labeled the PG of a D39 Δcps strain uniformly with the blue FDAA HADA (Fig. 2, left, pseudocolored green) for approximately 3 generations of exponential growth in BHI broth (see Materials and Methods). Cells were washed, rinsed, and resuspended in a chase of fresh BHI broth containing the green FDAA NADA (Fig. 2, left, pseudocolored red). Cells were collected at different times during exponential growth and early stationary phase before the onset of autolysis, and live cells were viewed by epifluorescence microscopy. In this labeling scheme, regions of old PG appear green in the images, and regions of newly synthesized PG appear red (Fig. 2, left).
FIG 2.
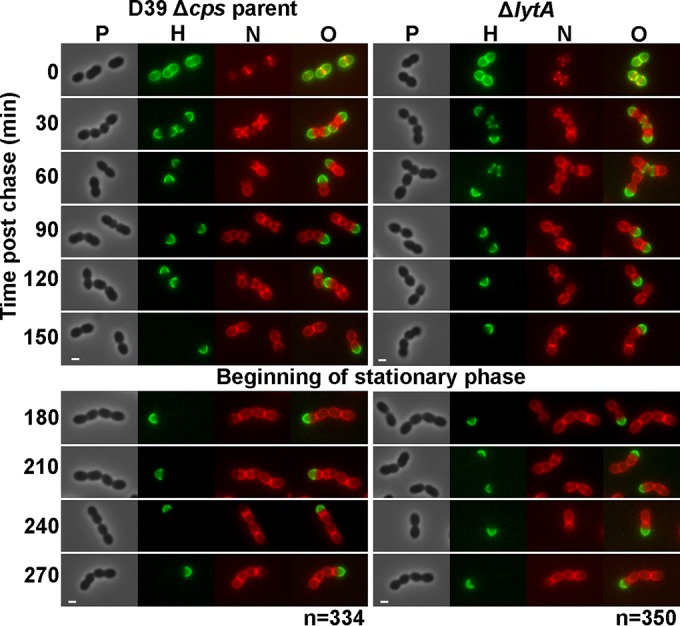
Persistence of hemispheres of stable old PG in exponentially growing and early-stationary-phase cells of S. pneumoniae detected by FDAA long-pulse–chase–new-labeling experiments. Parent strain IU1945 (D39 Δcps) and an isogenic lytA mutant (IU3900) were grown exponentially in BHI broth, labeled with HADA (pseudocolored green; old PG) for about 3 generations, washed, and then chased in the presence of a second color of FDAA, NADA (pseudocolored red; new PG synthesis) as described in Materials and Methods. Live cells were imaged by epifluorescence phase-contrast microscopy at the indicated time points after the removal of HADA (chase) and the addition of NADA. Minimal variation in labeling patterns was observed for >330 individual cells in diplococci or short chains of each strain examined in microscopic fields at different time points after the start of the chase/NADA labeling, and representative images are shown. Labeling of the parent strain was done numerous independent times, and labeling of the lytA mutant was done twice with similar results. Scale bar = 1 μm. P, phase-contrast image; H, HADA labeling (old PG); N, NADA labeling (new PG synthesis); O, overlay of H and N images. Quantitation indicating hemispherical labeling is in the text.
Consistent with minimal PG turnover, the old PG remained as extensive regions of hemispherical labeling that persisted indefinitely as cells entered stationary phase (Fig. 2, left). The demarcation between regions of old and newly labeled PG was distinct, with no regions of overlap in parent cells (Fig. 2, left, see single-color images). The sharp demarcation between old and new PG was further visualized by reducing the time of HADA labeling to 30 min, followed by a 5-min pulse of TADA labeling (see Fig. S1A in the supplemental material). In this labeling scheme, old PG (green) appeared as separate hemispheres banded by rings of newly synthesized PG (red) at active septa (see Fig. S1A, arrows). The other hemispheres of the cells were unlabeled, resulting in flat tops. In addition, we determined the extent of old PG labeling by measuring the lengths of green-labeled regions relative to the total lengths of newly divided and late-divisional cells in exponentially growing cultures (Fig. 2, left, 0 to 150 min). The old PG comprised 48% ± 1%, 49% ± 1%, 48% ± 1%, 50% ± 1%, and 48% ± 1% (mean ± SEM; n = 15) of the surface area of cells chased for 30 min, 60 min, 90 min, 120 min, and 150 min, respectively. Thus, old PG remained as complete hemispheres, supporting the hypothesis of minimal PG turnover in growing pneumococcal cells.
We used single-cell time-lapse microscopy to confirm the formation of hemispherical regions of stable pneumococcal PG that appeared not to turn over during multiple generations of division (Fig. 3; see Materials and Methods). In this experiment, single cells labeled uniformly in BHI broth with the photostable red FDAA TADA (Fig. 3, pseudocolored orange) were deposited onto a BHI-agar layer. Individual cells were repeatedly photographed over time as single cells grew into microcolonies, where regions of new PG synthesis lacked labeling (Fig. 3). Remarkably, four hemispheres of old PG (Fig. 3, orange), corresponding to the four hemispheres in the starting diplococcus cells, persisted without evident change in signal intensity as the microcolony enlarged into hundreds of cells in 5 h. Despite likely photobleaching, the average total intensities of the four labeled hemispheres did indeed remain nearly constant for the 5 images from 0 to 112 min (99.4 ± 10.3, 104.2 ± 7.5, 104.2 ± 10.5, and 93.1 ± 7.2 [mean ± standard deviation {SD}; n = 5] in arbitrary units), after which time cells in the microcolony started to overlap (Fig. 3). This result is consistent with retention of [14C]GlcNAc in the PG of labeled cells per sample volume, despite the growth of unlabeled cells (Fig. 1). Together, these FDAA long-pulse–chase experiments reveal minimal turnover of PG in growing S. pneumoniae cells, in agreement with the low rate of PG fragment release in radiolabeling experiments (Fig. 1) (54).
FIG 3.
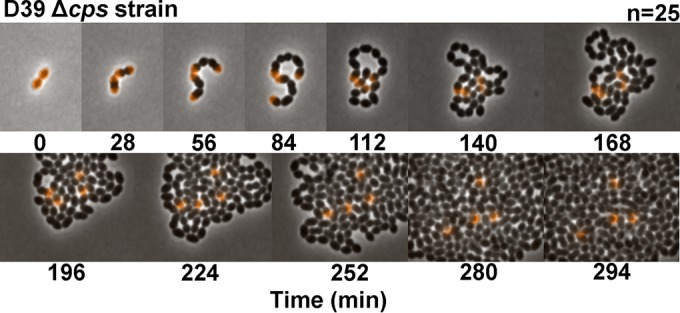
Retention of hemispheres of stable TADA-labeled PG in single pneumococcal cells viewed by time-lapse microscopy. Cells of strain IU1945 (D39 Δcps) growing exponentially in BHI broth were labeled with TADA (pseudocolored orange) for abut 4 generations, washed, resuspended in BHI broth lacking FDAA, deposited onto a large coverslip, and covered with a pad of BHI agar as described in Materials and Methods. Division of single cells into microcolonies was imaged every 2 min at 32°C using a 2-D epifluorescence phase-contrast microscope. Fluorescence intensities of hemispheres were nearly equal and constant with time (see the text). The image series shown is representative of 25 individual starting cells that were recorded.
To further validate the FDAA pulse-chase approach, we visualized the rapid turnover of the PG sidewall of exponentially growing B. subtilis cells (Fig. 4). B. subtilis laboratory strain P479 was grown exponentially in LB or LB plus 1% (wt/vol) glucose with aeration, and long-pulse (HADA)–chase–new-labeling (TADA) experiments were performed exactly as described for S. pneumoniae (Fig. 2, left), except for the aeration (Fig. 4) (see Materials and Methods). In contrast to the results for S. pneumoniae labeling (Fig. 2), old sidewall PG (Fig. 4, green) was barely visible at 40 min after the start of the chase (Fig. 4, arrow) and was not detected in 65% and 95% of cells chased for 60 and 80 min, respectively. Inert PG persisted at cell poles at all time points but covered <9% of cell bodies (Fig. 4, arrows). The weak intensity of some inert PG labeling (Fig. 4, 60 and 80 min, arrows) likely reflects partial labeling rather than turnover of inert PG. Unlike the distinct demarcation of regions of old and newly synthesized PG in S. pneumoniae (Fig. 2, left; see also Fig. S1 in the supplemental material), regions of old and newly synthesized PG overlapped in sidewalls and even in inert PG (Fig. 4, last row) of growing B. subtilis cells. The nearly complete disappearance of sidewall PG by 40 min (approximately 2 doublings) is consistent with the PG turnover rate of ≈50% per generation reported previously for B. subtilis (13, 14) and was not changed whether PG recycling was presumably active (Fig. 4, no glucose) or repressed (with glucose; data not shown). These results indicate that PG turnover can readily be detected by the FDAA pulse-chase protocol used in these experiments. Moreover, the labeling pattern characteristic of rapid turnover of B. subtilis sidewall PG is completely different from that observed for S. pneumoniae cells, in which hemispheres of stable PG persist for long periods of time (Fig. 2 and 3). This difference in labeling patterns further supports the hypothesis of minimal PG turnover in growing pneumococcal cells.
To reliably interpret the labeling patterns of S. pneumoniae cells, we determined the positions of FDAA labeling in PG. FDAA probes are thought to indicate regions of active transpeptidase activity by acting as a mimic of d-alanine that reverses a covalent acyl-intermediate formed between PBPs and PG tetrapeptides (33, 58). This side reaction of PBP transpeptidases restores PG pentapeptides that contain fluorescent d-Ala at their ends. We confirmed that the FDAA probes label PG pentapeptides at a low level and do not perceptibly alter the overall distribution of PG peptides in S. pneumoniae D39 strains (Fig. 5; see also Fig. S2 in the supplemental material). Fluorescent PG peptides from D39 Δcps cells labeled for about 5 generations with an FDAA (HADA) correspond exactly to PG peptides whose amounts are increased ≈10× in a ΔdacA mutant lacking a d,d-carboxypeptidase that accumulates pentapeptides in monomeric and cross-linked PG peptide species (Fig. 5) (37). No fluorescent signal was detected in unlabeled cells (Fig. 5). This exact correspondence is only consistent with PG pentapeptide labeling by FDAAs in the parent cells. Nonfluorescent PG peptides from the unlabeled and HADA-labeled D39 Δcps parent and an isogenic ΔdacA mutant were detected by measuring the A210 using a different HPLC gradient reported previously for D39 strains (see Fig. S2) (51). With the exception of additional A210-absorbing peaks in the HADA-labeled samples that eluted at ≈100 min (see Fig. S2A, arrow), the wild-type amounts and distributions of PG peptides were not significantly perturbed by HADA labeling of the parent strain (see Fig. S2A). The relatively small peak detected in the HADA-labeled parent strain (see Fig. S2A, arrow) is consistent with a small amount of fluorescent PG pentapeptide, given that fluorophore-labeled PG pentapeptides likely have greater absorbance at A210 than the amide bonds of unlabeled PG peptides.
FIG 5.

The same PG peptides were labeled by HADA in parent strain IU1945 (D39 Δcps) (A) and isogenic mutant IU7204 (D39 ΔdacA) (B), which lacks the d,d-carboxypeptidase that converts PG pentapeptides to PG tetrapeptides. Bacteria were grown in BHI broth and labeled with HADA, PG was purified, and PG peptides released with purified LytA amidase as described in Materials and Methods. HPLC was used to resolve PG peptides using the gradient conditions described in reference 37, with fluorescence detection (excitation, 405 nm; emission, 450 nm). The fluorescence intensity scale (y axis; expressed in arbitrary millivolt [mv] units) is about 8-fold greater for panel B than for panel A. Baseline and chromatograms of PG peptides from unlabeled cells are indicated. The experiment was performed independently twice with similar results.
We confirmed the dependence of FDAA labeling on active PBP transpeptidase activity by prelabeling D39 Δcps cells with fluorescent antibiotics (Fig. 6, pseudocolored red) that inhibit transpeptidase activity before pulsing with HADA for 5 min (Fig. 6, pseudocolored green). A fluorescent cephalosporin (Ceph C-T) characterized previously specifically labels only PBP1b and PBP3 (DacA) in S. pneumoniae but not other PBPs (47). PBP1b and PBP3 (DacA) localize to the septum of dividing cells or over the surface, respectively, as indicated by Ceph C-T labeling (Fig. 6) and previous localization studies of DacA (37). Prelabeling with Ceph C-T did not occlude HADA labeling of equatorial and septal regions, where the transpeptidase activities of other PBPs are active. In contrast, prelabeling with fluorescent vancomycin or fluorescent bocillin showed the expected labeling at cell equators and division septa (Fig. 6). However, prelabeling with these general PBP transpeptidase inhibitors blocked specific labeling by HADA, which was now mostly distributed nonspecifically in cells. Thus, specific labeling by FDAAs requires PBP transpeptidase activity. We did not distinguish further whether the nonspecific distribution of HADA reflected uptake into or surface deposition onto the antibiotic-stressed cells.
FIG 6.
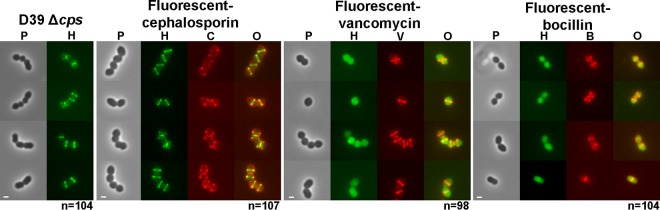
Prelabeling pneumococcal cells with fluorescent vancomycin or fluorescent bocillin but not fluorescent cephalosporin prevents localized labeling with the FDAA HADA. Strain IU1945 (D39 Δcps) cells were grown exponentially in BHI broth, pretreated with the indicated antibiotics (pseudocolored red), washed, and labeled with HADA for 5 min (pseudocolored green), washed, and imaged by epifluorescence microscopy as described in Materials and Methods. There was minimal variation in labeling patterns for >90 individual cells in diplococci or short chains for each treatment examined in microscopic fields in two biological replicates, and representative cells are shown. Fluorescent cephalosporin is specific to a limited number of PBPs (47), whereas fluorescent bocillin and fluorescent vancomycin label all PBPs and PG pentapeptides, respectively (see references 33 and 48). See the text for additional details. Scale bar = 1. P, phase-contrast image; H, HADA labeling; C, V, and B, fluorescent-cephalosporin, vancomycin, and bocillin, respectively; O, overlay of green and red fluorescence images.
We performed several additional experiments that support the general conclusion of minimal turnover of S. pneumoniae PG detected in FDAA long-pulse–chase–new-labeling experiments. We observed similar persistence of hemispherical regions of PG in the wild-type encapsulated serotype 2 D39 strain and the serotype 4 TIGR4 strain (see Fig. S3 in the supplemental material). The D39 and TIGR4 strains produce different exopolysaccharide capsules and have ≈10% overall differences in their total gene complements (59). Therefore, minimal PG turnover is not confined to D39 strains, which were used for the remainder of the experiments in this study. Growing the D39 Δcps strain in chemically defined medium containing glucose or galactose instead of glucose-containing rich BHI broth (Fig. 2, left) did not change the persistence of stable PG hemispheres (see Fig. S4). Thus, minimal PG turnover was not dependent on the medium, nor did turnover increase when catabolite repression was relieved with galactose as the carbon source.
We checked whether the degree of (Ser/Ala-Ala) cross-bridge formation in PG peptides influenced PG turnover. The MurMN enzymes catalyze cross-bridge formation in D39 and other pneumococcal strains (60–63). We showed previously that laboratory strain R6 contains substantially more cross-bridges than its D39 progenitor strain (40). Both strain R6 and D39 Δcps ΔmurMN showed the same persistence of PG hemispheres as the D39 Δcps parent strain (see Fig. S5 in the supplemental material). Last, we examined whether the minimal PG turnover was altered by ΔpgdA or Δadr mutation, each of which alters PG glycan chain modification and, thus, could potentially serve as a turnover signal. PgdA deacetylates GlcNAc in the PG glycan chains of wild-type cells, thereby imparting resistance to host lysozyme produced by the innate immune response (23, 39, 64). Adr O-acetylates the 6 position of MurNAc in PG glycan chains, which imparts lysozyme and penicillin resistance to strains of some serotypes (38, 64). The ΔpgdA and Δadr mutants were indistinguishable from their wild-type parent strain and contained hemispheres of stable PG (data not shown), indicating that these glycan chain modifications did not detectably alter the minimal turnover of PG in growing cells.
Minimal turnover of PG in S. pneumoniae cells growing in host-relevant biofilms.
We wanted to confirm the persistence of stable PG hemispheres in S. pneumoniae cells growing in a host-relevant biofilm model of colonization (Fig. 7). Hakansson and coworkers reported that encapsulated strains of S. pneumoniae, which do not form robust biofilms on abiotic surfaces, form physiologically relevant biofilms when in contact with fixed human respiratory tract epithelial cells (53, 65). Pneumococcal cells from these biofilms can be used to seed long-term, growing biofilms on live epithelial cells (52). Furthermore, bacteria in the biofilms growing on the fixed or live epithelial cells transition to virulent planktonic cells when the biofilms are exposed to stimuli produced during infections (52, 65). We repeated FDAA long-pulse–chase–new-labeling experiments with unencapsulated and encapsulated D39 strains growing in biofilms on fixed human epithelial cells (Fig. 7; see Materials and Methods). In the images from these experiments (Fig. 7), PG from the TADA long pulse (i.e., old PG) or with HADA new labeling (i.e., new PG) is pseudocolored red and blue, respectively, and the layer of fixed human epithelial cells is labeled with fluorescent concanavalin A (pseudocolored green). The images were generated by high-resolution 3-D structured-illumination microscopy (3-D–SIM) as described in Materials and Methods. Some of each biofilm was washed away during the multiple washes required to remove unincorporated TADA and HADA. The remaining biofilms clearly show stable hemispheres of old PG (now red) in the unencapsulated and encapsulated D39 strains (Fig. 7, arrows). We conclude that S. pneumoniae growing in host-relevant biofilms shows the same minimal turnover of PG as bacteria growing planktonically in BHI broth or CDM (Fig. 2, left; see also Fig. S3 and S4 in the supplemental material).
FIG 7.

Persistence of hemispheres of stable old PG that are indicative of minimal PG turnover in S. pneumoniae cells growing in host-relevant biofilms. Bacterial strains were grown as biofilms in CDM on a stratum of fixed H292 human lung epithelial cells, labeled with TADA for 2 h (pseudocolored red; old PG), washed, and chased in the presence of HADA for 2 h (pseudocolored blue; newly synthesized PG) as described in Materials and Methods. The H292 human cells were stained with concanavalin A-Alexa Fluor 488 (pseudocolored green), the bacterial biofilms were fixed with paraformaldehyde, and the biofilms and host cell strata were imaged by high-resolution 3-D–SIM. (A) Strain IU1945 (D39 Δcps). (B) Rotation (90°) of image in panel A. (C) Close up of bacterial cells outlined by white rectangle in panel A. (D) Strain IU1690 (D39). (E) Rotation (90°) of image in panel D. (F) Close up of bacterial cells outlined by white rectangle in panel D. The labeling patterns were similar for >200 cells of each strain examined in microscopic fields in two biological replicates, and representative images are shown. Hemispheres of stable PG (red) are indicated by arrows.
Previous characterization of S. pneumoniae cells in biofilms on human epithelial cells indicated considerable release of DNA, likely by the induction of fratricide as part of increased competence of pneumococci in this host-relevant biofilm model (65–67). The extracellular DNA released is thought to act as a major matrix component in the biofilms (65, 68). Consistent with this expectation, pneumococcal cells in biofilms formed on fixed epithelial cells were surrounded by a haze of DAPI-stained DNA outside the host cell nuclei (data not shown). Despite the release of bacterial DNA and PG peptide fragments by competence-induced fratricide, pneumococcal cells in biofilms formed on living epithelial cells induce minimal innate immune responses (reviewed in reference 69). This observation has important implications. Our results show minimal PG turnover during growth, implying that S. pneumoniae may remain hidden from the innate immune system when growing planktonically or even in biofilms (Fig. 2 and 7; see also Fig. S3 in the supplemental material). Once in a biofilm, PG lysis of noncompetent cells by the fratricidal PG hydrolases, CbpD endopeptidase, LytC lysozyme, and LytA amidase (67, 70–72), releases PG fragments and substantial bacterial DNA. However, these potent innate immune signals must be largely confined to the biofilm matrix, thus preventing interaction with host cells and strong induction of innate and secondary adaptive immune responses.
Mutations in PG-remodeling hydrolase genes lead to placement of inert PG at abnormal division vertices.
PG hydrolases are required for PG turnover in E. coli and B. subtilis (10, 12, 13). In S. pneumoniae, PG hydrolases carry out three broad functions: remodeling during PG synthesis (e.g., PcsB:FtsEX, LytB, Pmp23, DacA, and DacB) (see references 37, 43, and 50), autolysis in late stationary phase and during antibiotic stress responses (LytA) (71, 73), and fratricide during competence (LytA, CbpD, and LytC) (67, 70–72). PG hydrolases involved in PG remodeling are mostly attached to the outer surface of the cell membrane, and deficiency of these PG hydrolases causes cell morphology defects, including cell rounding, nonparallel division, and chaining (Fig. 8; see also Fig. S6 in the supplemental material). We used the long-pulse–chase–new-labeling protocol described above (Fig. 2, left) to survey exponentially growing mutants deficient in PG-remodeling or -autolytic hydrolases (37) for altered persistence of stable PG compared to that in wild-type cells or for regions of inert peptidoglycan deposition in mutants that exhibit aberrant cell morphologies (Fig. 2 [left], 3, and 8; see also Fig. S3 and S6).
FIG 8.
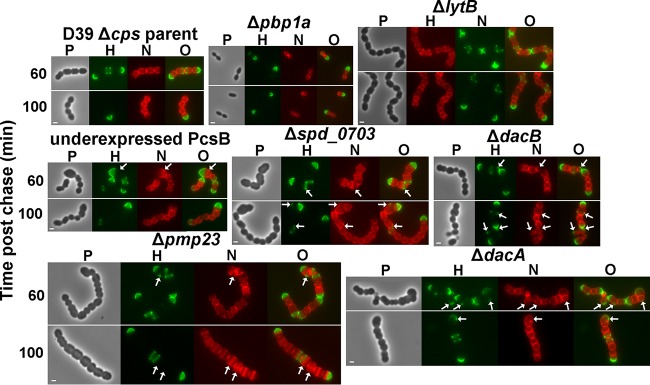
Mutants deficient in PG hydrolases retain hemispheres of stable PG and show regions of inert PG at aberrant division points. Strains IU1945 (D39 Δcps parent), IU6647 (Δpbp1a), IU3877 (ΔlytB), IU2336 (PcsB underexpression [40]), IU3881 (Δspd_0703), IU3880 (ΔdacB), IU3875 (Δpmp23), and IU7204 (ΔdacA) (see Table S1 in the supplemental material) were grown exponentially in BHI broth, labeled with HADA (pseudocolored green; old PG), washed, and chased in the presence of NADA (pseudocolored red; newly synthesized PG) as described in the legend to Fig. 2 and in Materials and Methods. Live cells were imaged by epifluorescence phase-contrast microscopy at the indicated time points after the removal of HADA (chase) and the addition of NADA. Additional time points for each strain are shown in Fig. S6. Minimal variation in labeling patterns was observed for >190 individual cells in diplococci or chains examined for each strain in microscopic fields at different time points after the start of the chase/NADA labeling in two biological replicates, and representative images are shown. Scale bar = 1 μm. P, phase-contrast image; H, HADA labeling (old PG); N, NADA labeling (new PG synthesis); O, overlay of H and N images. Arrows indicate regions in kinked chains bounded by green spots of inert peptidoglycan, by red spots or regions of newly synthesized PG trapped between regions of inert green PG, or by closely spaced red regions of newly synthesized PG. See the text for additional details.
Absence of the major LytA autolysin did not alter the persistence of hemispheres of stable PG in exponentially growing or early-stationary-phase cells (Fig. 2, right), and cells of the lytA mutant and D39 Δcps lytA+ parent were indistinguishable under these culture conditions (Fig. 2). In some lytA mutant cells, regions of PG synthesis at intermediate stages were labeled with HADA (Fig. 2, right, green) at the time of washing and the start of the NADA chase (Fig. 2, right, red). These persistent regions of green labelling appear between nonoverlapping regions of red labelling in dividing cells (Fig. 2, right, arrows) and were also observed for some parent cells (data not shown). These results reiterate the previous conclusion that in the D39 genetic background, the function of the LytA amidase is confined to stress and fratricide and it plays a minimal role in remodeling during normal cell division (37). Likewise, the absence of the LytB N-acetylglucosaminidase, which catalyzes the final separation of divided cells (74, 75), led to the formation of long chains of cells with the same persistence of stable PG hemispheres as observed in the parent strain (Fig. 8, top row). Finally, we examined the PG stability in a Δpbp1a mutant lacking the major class A PBP1a, which likely functions in both septal and peripheral (i.e., midcell sidewall) PG synthesis (43, 76). Δpbp1a mutants have the distinctive phenotype of forming considerably smaller but normally shaped cells compared to their pbp1a+ parent strain (Fig. 8, top row) (76). The persistence of stable PG hemispheres was not altered by the Δpbp1a mutation or the decrease in cell size and volume.
A deficiency of PG hydrolases involved in PG remodeling during cell division led to the formation of cells in chains (Fig. 8, middle and bottom rows; see also Fig. S6 in the supplemental material). However, unlike the parent strain and lytA, pbp1a, and lytB mutants, which contain well-spaced, parallel division planes perpendicular to the long axes of cells (Fig. 2 and 8, top row), the cells of PG-remodeling hydrolase mutants were often larger than normal, misshapen, and in chains containing kinks, indicative of aberrant cell division in nonparallel planes (Fig. 8, middle and bottom rows, arrows; see also Fig. S6). PcsB is an essential PG hydrolase regulated by interactions with FtsEX that functions as a PG peptide amidase or endopeptidase in PG remodeling during cell division (50, 77, 78). Cells lacking Pmp23, a putative lytic transglycosylase (79) or Spd_0703, a WalRK regulon member of unknown function (37), formed chains of kinked, blocky, unseparated cells, sometimes containing closely spaced regions of PG transpeptidase activity (Fig. 8, middle and bottom rows, red; see also Fig. S6). Although they were located in kinked chains, cells lacking the DacA d,d-carboxypeptidase or DacB l,d-carboxypeptidase, which sequentially trim PG peptides to tetramers and then trimers, respectively (37, 40, 80, 81), were more spherical than cells of the parent strain or other mutants. Notably, each PG hydrolase mutant retained hemispheres of stable old PG in long-pulse–chase–new-labeling experiments (Fig. 8, green; see also Fig. S6), supporting a role for these PG hydrolases in division rather than in PG turnover, which again appeared minimal. In addition, long pulse-chase-new labeling revealed that regions of chain kinking were often bounded or marked by green spots of inert peptidoglycan, by red spots or regions of newly synthesized PG trapped between regions of inert green PG, or by closely spaced red regions of newly synthesized PG (Fig. 8, middle and bottom rows, arrows). This result supports the interpretation from E. coli that regions of inert PG can block and limit the placement of FtsZ division planes, resulting in directional changes in cell shape and contour (82, 83). We conclude that the defects in PG hydrolysis in these mutants ultimately leads to aberrant PG placement that distorts the direction of future PG synthesis, resulting in cell shape and morphology defects. The spatiotemporal function of these PG hydrolases in remodeling during PG synthesis and PG cleavage during cell separation remains largely unknown, and this knowledge is required to understand the mechanisms underlying these cell shape and morphology defects.
Hemispheres of stable PG are maintained in ΔmapZ and ΔdivIVA cell division mutants.
Recently, it was reported that proteins that bind to regions of cell curvature, such as MreB, can alter PG synthesis and eliminate inert PG placement at the poles of E. coli cells under the specific experimental conditions tested (84). Consequently, we examined whether the MapZ and DivIVA cell division regulators of S. pneumoniae alter the location and persistence of stable PG observed in the wild-type parent cells (Fig. 9). MapZ (also called LocZ) is a recently reported protein that has been hypothesized to direct FtsZ to the equators of future dividing daughter cells (42, 85, 86). Using FtsZ-green fluorescent protein (GFP) fusions, FtsZ was reported to be greatly mislocalized in ΔmapZ mutants, which showed severe morphology defects (42, 86). We examined ΔmapZ cell morphologies in D39 strains labeled with FDAAs. Similar to the results reported in reference 42, ΔmapZ D39 mutant cells showed occasional minor distortions in shape but were similar in overall morphology to wild-type cells under these growth conditions (Fig. 9, left). This result was somewhat unexpected, given that MapZ has been postulated to be required for FtsZ positioning (42, 85, 86). Consistent with their relatively normal cell morphologies, long pulse-chase-new labeling of these cells revealed only occasional misplaced PG synthesis, which is a surrogate indicator of FtsZ location (33), in the ΔmapZ mutant compared to that in the parent strain (Fig. 9, left). Important to this study, the absence of MapZ did not have any obvious effects on the hemispherical persistence of stable PG, indicating that MapZ does not modulate PG turnover. The nonessentiality of mapZ, the moderate cell morphology defects in the absence of FtsZ-GFP fusions (A. Perez, unpublished data), and the frequent midcell placement of PG synthesis in the ΔmapZ mutant under these culture conditions suggest that S. pneumoniae D39 likely has redundant systems for FtsZ positioning.
FIG 9.
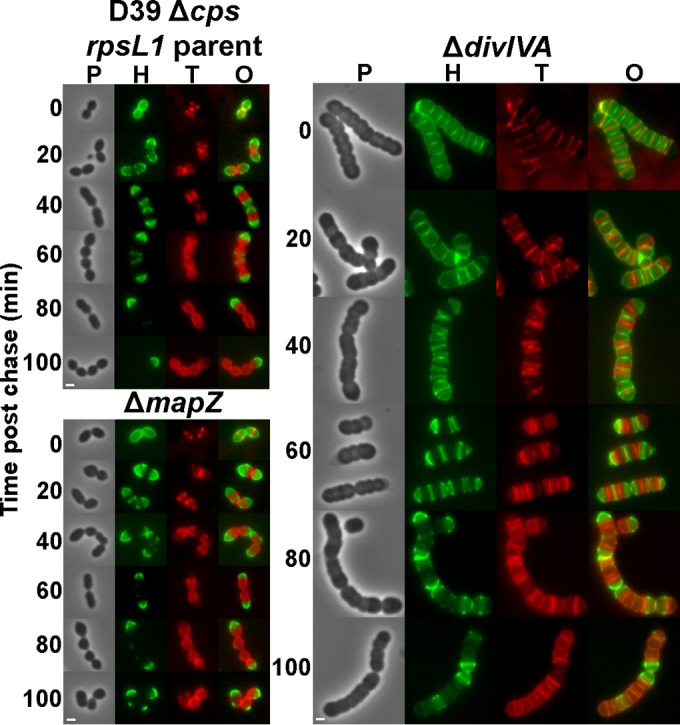
Hemispheres of stable PG are maintained in ΔmapZ and ΔdivIVA cell division mutants, but ΔdivIVA mutants exhibit alternating patterns of new and old PG along the lengths of cells. Strains IU1824 (D39 Δcps rpsL1 parent), IU5661 (ΔdivIVA), and IU9175 (ΔmapZ) (see Table S1 in the supplemental material) were grown exponentially in BHI broth, labeled with HADA (pseudocolored green; old PG), washed, and chased in the presence of TADA (pseudocolored red; newly synthesized PG) as described in the legend to Fig. 2 and in Materials and Methods. Live cells were imaged by epifluorescence phase-contrast microscopy at the indicated time points after the removal of HADA (chase) and the addition of TADA. Similar labeling patterns were observed for >200 individual cells in diplococci or chains examined for each strain in microscopic fields at different time points after the start of the chase/TADA labeling in two biological replicates, and representative images are shown. Scale bar = 1 μm. P, phase-contrast image; H, HADA labeling (old PG); T, TADA labeling (new PG synthesis); O, overlay of H and T images.
Based on results from B. subtilis, DivIVA binds to regions of negative curvature in Gram-positive bacterial cells (87, 88). In S. pneumoniae, ΔdivIVA mutant cells form long chains of significantly enlarged, compressed-sphere-shaped cells (Fig. 9, right), as reported first by Massidda and coworkers (41, 43). At earlier times in long-pulse–chase–new-labeling experiments, regions of new PG synthesis were visible at the equators of ΔdivIVA cells. However, at intermediate times (20 to 60 min of chase), complicated labeling patterns emerged that showed alternating regions of old and new PG along the length of the chains (Fig. 9, right). Unlike the wild-type strains and the other mutants examined here, where regions of old PG were distinctly demarcated from regions of new PG synthesis (Fig. 2 and 3; see also Fig. S3 in the supplemental material), regions of stable PG partially overlapped regions of continuing PG synthesis in the ΔdivIVA mutant (Fig. 9, right). At later times (80 to 100 min of chase), closely spaced, irregular band patterns of newly synthesized PG (red) were still visible over faint regions of old PG (green). Thus, the absence of DivIVA caused a partial loss of boundaries between regions of old PG and new PG synthesis not observed in other mutants examined here. Relevant to this study, despite the complicated PG synthesis patterns of the ΔdivIVA mutant, hemispherical regions of old PG persisted. Thus, the lack of the DivIVA curvature recognition protein did not eliminate caps of stable PG in S. pneumoniae cells. The functions of DivIVA in pneumococcal cell division are not well understood (43), and the results presented here underscore the importance of DivIVA in organizing the placement of FtsZ-directed PG synthesis in dividing S. pneumoniae cells.
Conclusions.
It has sometimes been assumed that S. pneumoniae, like B. subtilis and E. coli, may turn over its PG during growth (e.g., see reference 89). Two lines of evidence presented in this paper argue against this assumption and support minimal PG turnover in growing pneumococcal cells. Classical pulse-chase experiments tracking radiolabeled amino acids or a glycan sugar showed very low rates of release of PG fragments from sacculi of growing S. pneumoniae cells into the medium (Fig. 1) (54). However, radiolabel release experiments alone cannot distinguish between minimal PG turnover versus rapid turnover and recycling. Long-pulse–chase–new-labeling experiments with FDAAs strongly supported the hypothesis of minimal PG turnover instead of rapid PG turnover and recycling. In pneumococcal cells growing planktonically in culture or in host-relevant biofilms, hemispheres of old PG persisted, and there was no overlap between regions of old and new PG synthesis (Fig. 2 and 7; see also Fig. S1 in the supplemental material). FDAA pulse-chase of single cells followed by time-lapse microscopy confirmed the persistence of hemispheres of stable PG without significant loss of signal intensity in microcolonies (Fig. 3). In contrast, FDAA long-pulse–chase–new-labeling experiments revealed the expected rapid turnover of sidewall PG in growing B. subtilis cells, whether recycling was repressed or induced (Fig. 4) (13, 14, 55). The labeling pattern characteristic of rapid turnover of B. subtilis sidewall PG (Fig. 4) is completely different from that observed for S. pneumoniae cells (Fig. 2 and 3; see also Fig. S1), and this difference further supports the hypothesis of minimal PG turnover in growing pneumococcal cells.
Minimal PG turnover is consistent with the apparent absence of certain genes in S. pneumoniae that are required for PG recycling in other bacteria (13, 25). In addition, results in this paper indicate the same pattern of minimal PG turnover in encapsulated serotype 2 and 4 stains (see Fig. S3 in the supplemental material) and in an unencapsulated mutant of a serotype 2 strain growing in rich medium or CDM with different carbon sources (Fig. 2; see also Fig. S4). Thus, minimal turnover is a general property of the PG of growing S. pneumoniae cells. There may also be minimal PG turnover in other ovococcus species, based on an apparent absence of recycling enzymes and results from previous radiolabeling experiments showing low rates of PG fragment release from sacculi (13, 56, 57).
Minimal turnover of pneumococcal PG has several mechanistic and physiological implications. In particular, PG remodeling, which is the trimming of PG by endopeptidases and lytic transglycosylases required for the insertion of new glycan strands during PG biosynthesis, does not release substantial amounts of fragments from the PG of growing S. pneumoniae cells. Indeed, pneumococcal mutants deficient in key PG hydrolases and lytic transglycosylases implicated in PG remodeling still exhibited minimal PG turnover and accumulated inert PG at vertices of abnormal growth and division (Fig. 8; see also Fig. S6 in the supplemental material). Likewise, pneumococcal mutants lacking the major cell division regulators MapZ (LocZ) and DivIVA maintained minimal PG turnover, although the separation of old and new PG synthesis was distorted in the divIVA mutant (Fig. 9). Mutants deficient in glycan chain modification or peptide cross-bridge formation also did not deviate from the fundamental pattern of minimal PG turnover and persistence of hemispheres of old PG that is characteristic of the parent strain (Fig. 2; see also Fig. S5).
The function of the DacA (PBP3) d,d-carboxypeptidase is particularly interesting with regard to the persistence of old PG labeled by FDAAs in these experiments (Fig. 2 and 3; see also Fig. S1 in the supplemental material). PG peptide composition determinations showed that FDAA-labeled PG pentapeptides are clearly substrates of DacA, because they accumulated to high levels in a ΔdacA mutant (Fig. 5). Nevertheless, FDAA-labeled PG pentapeptides persisted for long periods of time in hemispheres of stable PG in S. pneumoniae cells (Fig. 2 and 3; see also Fig. S1), despite DacA's localization over the pneumococcal cell surface (37). DacA and its homologue in E. coli, PBP5, are bound closely to the outer surface of the cell membrane by an amphipathic helix domain (90). The persistence of FDAA-labeled PG pentapeptides in old PG implies that surface-bound DacA cannot reach FDAA-labeled pentapeptides, which are synthesized by PBP transpeptidase activities tethered further out from DacA in the cell membrane (90).
Another implication of minimal PG turnover of planktonically growing pneumococcal cells is the possible diminution of detection by the human host innate immune system. Unlike many bacterial species that announce their presence by releasing significant amounts of PG fragments (see the introduction), planktonically growing pneumococcus may remain hidden from detection. In this regard, biofilms present a paradox that needs to be studied further. On the one hand, S. pneumoniae cells growing in biofilms retain minimal PG turnover (Fig. 7). On the other hand, competence and accompanying fratricide increase in biofilms, releasing PG fragments and DNA fragments that form part of the biofilm matrix (65). Yet, despite the formation of these potential signals, host cells in contact with biofilms do not exhibit strong innate immune responses (69), implying sequestration of released DNA and PG fragments. We conclude that fratricidal PG destruction in biofilms provides a mode of de facto PG turnover without recycling for S. pneumoniae cells and that studies of the structure and composition of the pneumococcal biofilm matrix are needed to understand the sequestration of PG fragments and DNA from host cell strata.
Supplementary Material
ACKNOWLEDGMENTS
We thank Amilcar Perez, Tiffany Tsui, Jiaqi Zheng, and Britta Rued for helpful discussions and comments about this work. We thank Lok-To Chris Sham for permission to present unpublished results, Erin Carlson for Ceph C-T fluorescent cephalosporin, and Dan Kearns for a B. subtilis laboratory strain.
This work was supported by MPI grant 1R01GM113172 (to Y.V.B., M.S.V., and M.E.W).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00541-15.
REFERENCES
- 1.Cava F, de Pedro MA. 2014. Peptidoglycan plasticity in bacteria: emerging variability of the murein sacculus and their associated biological functions. Curr Opin Microbiol 18:46–53. doi: 10.1016/j.mib.2014.01.004. [DOI] [PubMed] [Google Scholar]
- 2.Egan AJ, Vollmer W. 2013. The physiology of bacterial cell division. Ann N Y Acad Sci 1277:8–28. doi: 10.1111/j.1749-6632.2012.06818.x. [DOI] [PubMed] [Google Scholar]
- 3.Frirdich E, Gaynor EC. 2013. Peptidoglycan hydrolases, bacterial shape, and pathogenesis. Curr Opin Microbiol 16:767–778. doi: 10.1016/j.mib.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 4.Lovering AL, Safadi SS, Strynadka NC. 2012. Structural perspective of peptidoglycan biosynthesis and assembly. Annu Rev Biochem 81:451–478. doi: 10.1146/annurev-biochem-061809-112742. [DOI] [PubMed] [Google Scholar]
- 5.Randich AM, Brun YV. 2015. Molecular mechanisms for the evolution of bacterial morphologies and growth modes. Front Microbiol 6:580. doi: 10.3389/fmicb.2015.00580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Typas A, Banzhaf M, Gross CA, Vollmer W. 2011. From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat Rev Microbiol 10:123–136. doi: 10.1038/nrmicro2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Denapaite D, Bruckner R, Hakenbeck R, Vollmer W. 2012. Biosynthesis of teichoic acids in Streptococcus pneumoniae and closely related species: lessons from genomes. Microb Drug Resist 18:344–358. doi: 10.1089/mdr.2012.0026. [DOI] [PubMed] [Google Scholar]
- 8.Eberhardt A, Hoyland CN, Vollmer D, Bisle S, Cleverley RM, Johnsborg O, Havarstein LS, Lewis RJ, Vollmer W. 2012. Attachment of capsular polysaccharide to the cell wall in Streptococcus pneumoniae. Microb Drug Resist 18:240–255. doi: 10.1089/mdr.2011.0232. [DOI] [PubMed] [Google Scholar]
- 9.Schneewind O, Missiakas D. 2014. Sec-secretion and sortase-mediated anchoring of proteins in Gram-positive bacteria. Biochim Biophys Acta 1843:1687–1697. doi: 10.1016/j.bbamcr.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park JT, Uehara T. 2008. How bacteria consume their own exoskeletons (turnover and recycling of cell wall peptidoglycan). Microbiol Mol Biol Rev 72:211–227. doi: 10.1128/MMBR.00027-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boudreau MA, Fisher JF, Mobashery S. 2012. Messenger functions of the bacterial cell wall-derived muropeptides. Biochemistry 51:2974–2990. doi: 10.1021/bi300174x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Johnson JW, Fisher JF, Mobashery S. 2013. Bacterial cell-wall recycling. Ann N Y Acad Sci 1277:54–75. doi: 10.1111/j.1749-6632.2012.06813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reith J, Mayer C. 2011. Peptidoglycan turnover and recycling in Gram-positive bacteria. Appl Microbiol Biotechnol 92:1–11. doi: 10.1007/s00253-011-3486-x. [DOI] [PubMed] [Google Scholar]
- 14.Litzinger S, Duckworth A, Nitzsche K, Risinger C, Wittmann V, Mayer C. 2010. Muropeptide rescue in Bacillus subtilis involves sequential hydrolysis by β-N-acetylglucosaminidase and N-acetylmuramyl-l-alanine amidase. J Bacteriol 192:3132–3143. doi: 10.1128/JB.01256-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cho H, Uehara T, Bernhardt TG. 2014. Beta-lactam antibiotics induce a lethal malfunctioning of the bacterial cell wall synthesis machinery. Cell 159:1300–1311. doi: 10.1016/j.cell.2014.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fisher JF, Mobashery S. 2014. The sentinel role of peptidoglycan recycling in the beta-lactam resistance of the Gram-negative Enterobacteriaceae and Pseudomonas aeruginosa. Bioorg Chem 56:41–48. doi: 10.1016/j.bioorg.2014.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bertsche U, Mayer C, Gotz F, Gust AA. 2015. Peptidoglycan perception—sensing bacteria by their common envelope structure. Int J Med Microbiol 305:217 doi: 10.1016/j.ijmm.2014.12.019. [DOI] [PubMed] [Google Scholar]
- 18.Shah IM, Laaberki MH, Popham DL, Dworkin J. 2008. A eukaryotic-like Ser/Thr kinase signals bacteria to exit dormancy in response to peptidoglycan fragments. Cell 135:486–496. doi: 10.1016/j.cell.2008.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan YA, Hackett KT, Dillard JP. 2012. The lytic transglycosylases of Neisseria gonorrhoeae. Microb Drug Resist 18:271–279. doi: 10.1089/mdr.2012.0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clarke TB, Weiser JN. 2011. Intracellular sensors of extracellular bacteria. Immunol Rev 243:9–25. doi: 10.1111/j.1600-065X.2011.01039.x. [DOI] [PubMed] [Google Scholar]
- 21.Wheeler R, Chevalier G, Eberl G, Gomperts Boneca I. 2014. The biology of bacterial peptidoglycans and their impact on host immunity and physiology. Cell Microbiol 16:1014–1023. doi: 10.1111/cmi.12304. [DOI] [PubMed] [Google Scholar]
- 22.Royet J, Gupta D, Dziarski R. 2011. Peptidoglycan recognition proteins: modulators of the microbiome and inflammation. Nat Rev Immunol 11:837–851. doi: 10.1038/nri3089. [DOI] [PubMed] [Google Scholar]
- 23.Lemon JK, Weiser JN. 2015. Degradation products of the extracellular pathogen Streptococcus pneumoniae access the cytosol via its pore-forming toxin. mBio 6(1):e02110-14. doi: 10.1128/mBio.02110-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Atilano ML, Pereira PM, Vaz F, Catalao MJ, Reed P, Grilo IR, Sobral RG, Ligoxygakis P, Pinho MG, Filipe SR. 2014. Bacterial autolysins trim cell surface peptidoglycan to prevent detection by the Drosophila innate immune system. eLife 3:e02277. doi: 10.7554/eLife.02277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lanie JA, Ng WL, Kazmierczak KM, Andrzejewski TM, Davidsen TM, Wayne KJ, Tettelin H, Glass JI, Winkler ME. 2007. Genome sequence of Avery's virulent serotype 2 strain D39 of Streptococcus pneumoniae and comparison with that of unencapsulated laboratory strain R6. J Bacteriol 189:38–51. doi: 10.1128/JB.01148-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Donkor ES. 2013. Understanding the pneumococcus: transmission and evolution. Front Cell Infect Microbiol 3:7. doi: 10.3389/fcimb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Henriques-Normark B, Tuomanen EI. 2013. The pneumococcus: epidemiology, microbiology, and pathogenesis. Cold Spring Harb Perspect Med 3(7):a010215. doi: 10.1101/cshperspect.a010215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vernatter J, Pirofski LA. 2013. Current concepts in host-microbe interaction leading to pneumococcal pneumonia. Curr Opin Infect Dis 26:277–283. doi: 10.1097/QCO.0b013e3283608419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Opijnen T, Camilli A. 2012. A fine scale phenotype-genotype virulence map of a bacterial pathogen. Genome Res 22:2541–2551. doi: 10.1101/gr.137430.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodriguez JL, Dalia AB, Weiser JN. 2012. Increased chain length promotes pneumococcal adherence and colonization. Infect Immun 80:3454–3459. doi: 10.1128/IAI.00587-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weiser JN. 2013. The battle with the host over microbial size. Curr Opin Microbiol 16:59–62. doi: 10.1016/j.mib.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuru E, Hughes HV, Brown PJ, Hall E, Tekkam S, Cava F, de Pedro MA, Brun YV, VanNieuwenhze MS. 2012. In situ probing of newly synthesized peptidoglycan in live bacteria with fluorescent d-amino acids. Angew Chem Int Ed Engl 51:12519–12523. doi: 10.1002/anie.201206749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tsui HC, Boersma MJ, Vella SA, Kocaoglu O, Kuru E, Peceny JK, Carlson EE, VanNieuwenhze MS, Brun YV, Shaw SL, Winkler ME. 2014. Pbp2x localizes separately from Pbp2b and other peptidoglycan synthesis proteins during later stages of cell division of Streptococcus pneumoniae D39. Mol Microbiol 94:21–40. doi: 10.1111/mmi.12745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramos-Montanez S, Tsui HC, Wayne KJ, Morris JL, Peters LE, Zhang F, Kazmierczak KM, Sham LT, Winkler ME. 2008. Polymorphism and regulation of the spxB (pyruvate oxidase) virulence factor gene by a CBS-HotDog domain protein (SpxR) in serotype 2 Streptococcus pneumoniae. Mol Microbiol 67:729–746. [DOI] [PubMed] [Google Scholar]
- 35.Tsui HC, Keen SK, Sham LT, Wayne KJ, Winkler ME. 2011. Dynamic distribution of the SecA and SecY translocase subunits and septal localization of the HtrA surface chaperone/protease during Streptococcus pneumoniae D39 cell division. mBio 2(5):e00202-11. doi: 10.1128/mBio.00202-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsui HC, Mukherjee D, Ray VA, Sham LT, Feig AL, Winkler ME. 2010. Identification and characterization of noncoding small RNAs in Streptococcus pneumoniae serotype 2 strain D39. J Bacteriol 192:264–279. doi: 10.1128/JB.01204-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barendt SM, Sham LT, Winkler ME. 2011. Characterization of mutants deficient in the l,d-carboxypeptidase (DacB) and WalRK (VicRK) regulon, involved in peptidoglycan maturation of Streptococcus pneumoniae serotype 2 strain D39. J Bacteriol 193:2290–2300. doi: 10.1128/JB.01555-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Crisostomo MI, Vollmer W, Kharat AS, Inhulsen S, Gehre F, Buckenmaier S, Tomasz A. 2006. Attenuation of penicillin resistance in a peptidoglycan O-acetyl transferase mutant of Streptococcus pneumoniae. Mol Microbiol 61:1497–1509. doi: 10.1111/j.1365-2958.2006.05340.x. [DOI] [PubMed] [Google Scholar]
- 39.Vollmer W, Tomasz A. 2000. The pgdA gene encodes for a peptidoglycan N-acetylglucosamine deacetylase in Streptococcus pneumoniae. J Biol Chem 275:20496–20501. doi: 10.1074/jbc.M910189199. [DOI] [PubMed] [Google Scholar]
- 40.Barendt SM, Land AD, Sham LT, Ng WL, Tsui HC, Arnold RJ, Winkler ME. 2009. Influences of capsule on cell shape and chain formation of wild-type and pcsB mutants of serotype 2 Streptococcus pneumoniae. J Bacteriol 191:3024–3040. doi: 10.1128/JB.01505-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fadda D, Santona A, D'Ulisse V, Ghelardini P, Ennas MG, Whalen MB, Massidda O. 2007. Streptococcus pneumoniae DivIVA: localization and interactions in a MinCD-free context. J Bacteriol 189:1288–1298. doi: 10.1128/JB.01168-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Holeckova N, Doubravova L, Massidda O, Molle V, Buriankova K, Benada O, Kofronova O, Ulrych A, Branny P. 2015. LocZ is a new cell division protein involved in proper septum placement in Streptococcus pneumoniae. mBio 6(1):e01700-14. doi: 10.1128/mBio.01700-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Massidda O, Novakova L, Vollmer W. 2013. From models to pathogens: how much have we learned about Streptococcus pneumoniae cell division? Environ Microbiol 15:3133–3157. doi: 10.1111/1462-2920.12189. [DOI] [PubMed] [Google Scholar]
- 44.van de Rijn I, Kessler RE. 1980. Growth characteristics of groupA Streptococci in a new chemically defined medium. Infect Immun 27:444–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuru E, Tekkam S, Hall E, Brun YV, Van Nieuwenhze MS. 2015. Synthesis of fluorescent d-amino acids and their use for probing peptidoglycan synthesis and bacterial growth in situ. Nat Protoc 10:33–52. doi: 10.1038/nprot.2014.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Land AD, Tsui HC, Kocaoglu O, Vella SA, Shaw SL, Keen SK, Sham LT, Carlson EE, Winkler ME. 2013. Requirement of essential Pbp2x and GpsB for septal ring closure in Streptococcus pneumoniae D39. Mol Microbiol 90:939–955. doi: 10.1111/mmi.12408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kocaoglu O, Calvo RA, Sham LT, Cozy LM, Lanning BR, Francis S, Winkler ME, Kearns DB, Carlson EE. 2012. Selective penicillin-binding protein imaging probes reveal substructure in bacterial cell division. ACS Chem Biol 7:1746–1753. doi: 10.1021/cb300329r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kocaoglu O, Tsui HC, Winkler ME, Carlson EE. 2015. Profiling of beta-lactam selectivity for penicillin-binding proteins in Streptococcus pneumoniae D39. Antimicrob Agents Chemother 59:3548–3555. doi: 10.1128/AAC.05142-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bui NK, Eberhardt A, Vollmer D, Kern T, Bougault C, Tomasz A, Simorre JP, Vollmer W. 2012. Isolation and analysis of cell wall components from Streptococcus pneumoniae. Anal Biochem 421:657–666. doi: 10.1016/j.ab.2011.11.026. [DOI] [PubMed] [Google Scholar]
- 50.Sham LT, Barendt SM, Kopecky KE, Winkler ME. 2011. Essential PcsB putative peptidoglycan hydrolase interacts with the essential FtsXSpn cell division protein in Streptococcus pneumoniae D39. Proc Natl Acad Sci U S A 108:E1061–E1069. doi: 10.1073/pnas.1108323108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garcia-Bustos J, Tomasz A. 1990. A biological price of antibiotic resistance: major changes in the peptidoglycan structure of penicillin-resistant pneumococci. Proc Natl Acad Sci U S A 87:5415–5419. doi: 10.1073/pnas.87.14.5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marks LR, Davidson BA, Knight PR, Hakansson AP. 2013. Interkingdom signaling induces Streptococcus pneumoniae biofilm dispersion and transition from asymptomatic colonization to disease. mBio 4(4):e00438–13. doi: 10.1128/mBio.00438-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marks LR, Parameswaran GI, Hakansson AP. 2012. Pneumococcal interactions with epithelial cells are crucial for optimal biofilm formation and colonization in vitro and in vivo. Infect Immun 80:2744–2760. doi: 10.1128/IAI.00488-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tomasz A, McDonnell M, Westphal M, Zanati E. 1975. Coordinated incorporation of nascent peptidoglycan and teichoic acid into pneumococcal cell walls and conservation of peptidoglycan during growth. J Biol Chem 250:337–341. [PubMed] [Google Scholar]
- 55.Bisicchia P, Noone D, Lioliou E, Howell A, Quigley S, Jensen T, Jarmer H, Devine KM. 2007. The essential YycFG two-component system controls cell wall metabolism in Bacillus subtilis. Mol Microbiol 65:180–200. doi: 10.1111/j.1365-2958.2007.05782.x. [DOI] [PubMed] [Google Scholar]
- 56.Doyle RJ, Chaloupka J, Vinter V. 1988. Turnover of cell walls in microorganisms. Microbiol Rev 52:554–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mesnage S, Chau F, Dubost L, Arthur M. 2008. Role of N-acetylglucosaminidase and N-acetylmuramidase activities in Enterococcus faecalis peptidoglycan metabolism. J Biol Chem 283:19845–19853. doi: 10.1074/jbc.M802323200. [DOI] [PubMed] [Google Scholar]
- 58.Lupoli TJ, Tsukamoto H, Doud EH, Wang TS, Walker S, Kahne D. 2011. Transpeptidase-mediated incorporation of d-amino acids into bacterial peptidoglycan. J Am Chem Soc 133:10748–10751. doi: 10.1021/ja2040656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tettelin H, Hollingshead SK. 2004. Comparative genomics of Streptococcus pneumoniae intrastrain diversity and genome plasticity, p 15–29. In Tuomanen EI, Mitchell TJ, Morrison DA, Spratt BG (ed), The pneumococcus. ASM Press, Washington, DC. [Google Scholar]
- 60.De Pascale G, Lloyd AJ, Schouten JA, Gilbey AM, Roper DI, Dowson CG, Bugg TD. 2008. Kinetic characterization of lipid II-Ala:alanyl-tRNA ligase (MurN) from Streptococcus pneumoniae using semisynthetic aminoacyl-lipid II substrates. J Biol Chem 283:34571–34579. doi: 10.1074/jbc.M805807200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Filipe SR, Pinho MG, Tomasz A. 2000. Characterization of the murMN operon involved in the synthesis of branched peptidoglycan peptides in Streptococcus pneumoniae. J Biol Chem 275:27768–27774. [DOI] [PubMed] [Google Scholar]
- 62.Greene NG, Narciso AR, Filipe SR, Camilli A. 2015. Peptidoglycan branched stem peptides contribute to Streptococcus pneumoniae virulence by inhibiting pneumolysin release. PLoS Pathog 11(6):e1004996. doi: 10.1371/journal.ppat.1004996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lloyd AJ, Gilbey AM, Blewett AM, De Pascale G, El Zoeiby A, Levesque RC, Catherwood AC, Tomasz A, Bugg TD, Roper DI, Dowson CG. 2008. Characterization of tRNA-dependent peptide bond formation by MurM in the synthesis of Streptococcus pneumoniae peptidoglycan. J Biol Chem 283:6402–6417. doi: 10.1074/jbc.M708105200. [DOI] [PubMed] [Google Scholar]
- 64.Davis KM, Weiser JN. 2011. Modifications to the peptidoglycan backbone help bacteria to establish infection. Infect Immun 79:562–570. doi: 10.1128/IAI.00651-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chao Y, Marks LR, Pettigrew MM, Hakansson AP. 2014. Streptococcus pneumoniae biofilm formation and dispersion during colonization and disease. Front Cell Infect Microbiol 4:194. doi: 10.3389/fcimb.2014.00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Marks LR, Reddinger RM, Hakansson AP. 2012. High levels of genetic recombination during nasopharyngeal carriage and biofilm formation in Streptococcus pneumoniae. mBio 3(5):e00200-12. doi: 10.1128/mBio.00200-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wei H, Havarstein LS. 2012. Fratricide is essential for efficient gene transfer between pneumococci in biofilms. Appl Environ Microbiol 78:5897–5905. doi: 10.1128/AEM.01343-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Domenech M, Garcia E, Prieto A, Moscoso M. 2013. Insight into the composition of the intercellular matrix of Streptococcus pneumoniae biofilms. Environ Microbiol 15:502–516. doi: 10.1111/j.1462-2920.2012.02853.x. [DOI] [PubMed] [Google Scholar]
- 69.Gilley RP, Orihuela CJ. 2014. Pneumococci in biofilms are non-invasive: implications on nasopharyngeal colonization. Front Cell Infect Microbiol 4:163. doi: 10.3389/fcimb.2014.00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Eldholm V, Johnsborg O, Straume D, Ohnstad HS, Berg KH, Hermoso JA, Havarstein LS. 2010. Pneumococcal CbpD is a murein hydrolase that requires a dual cell envelope binding specificity to kill target cells during fratricide. Mol Microbiol 76:905–917. doi: 10.1111/j.1365-2958.2010.07143.x. [DOI] [PubMed] [Google Scholar]
- 71.Mellroth P, Daniels R, Eberhardt A, Ronnlund D, Blom H, Widengren J, Normark S, Henriques-Normark B. 2012. LytA, major autolysin of Streptococcus pneumoniae, requires access to nascent peptidoglycan. J Biol Chem 287:11018–11029. doi: 10.1074/jbc.M111.318584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Perez-Dorado I, Gonzalez A, Morales M, Sanles R, Striker W, Vollmer W, Mobashery S, Garcia JL, Martinez-Ripoll M, Garcia P, Hermoso JA. 2010. Insights into pneumococcal fratricide from the crystal structures of the modular killing factor LytC. Nat Struct Mol Biol 17:576–582. doi: 10.1038/nsmb.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mellroth P, Sandalova T, Kikhney A, Vilaplana F, Hesek D, Lee M, Mobashery S, Normark S, Svergun D, Henriques-Normark B, Achour A. 2014. Structural and functional insights into peptidoglycan access for the lytic amidase LytA of Streptococcus pneumoniae. mBio 5(1):e01120-13. doi: 10.1128/mBio.01120-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Garcia P, Gonzalez MP, Garcia E, Lopez R, Garcia JL. 1999. LytB, a novel pneumococcal murein hydrolase essential for cell separation. Mol Microbiol 31:1275–1281. doi: 10.1046/j.1365-2958.1999.01238.x. [DOI] [PubMed] [Google Scholar]
- 75.De Las Rivas B, Garcia JL, Lopez R, Garcia P. 2002. Purification and polar localization of pneumococcal LytB, a putative endo-beta-N-acetylglucosaminidase: the chain-dispersing murein hydrolase. J Bacteriol 184:4988–5000. doi: 10.1128/JB.184.18.4988-5000.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Land AD, Winkler ME. 2011. The requirement for pneumococcal MreC and MreD is relieved by inactivation of the gene encoding PBP1a. J Bacteriol 193:4166–4179. doi: 10.1128/JB.05245-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sham LT, Jensen KR, Bruce KE, Winkler ME. 2013. Involvement of FtsE ATPase and FtsX extracellular loops 1 and 2 in FtsEX-PcsB complex function in cell division of Streptococcus pneumoniae D39. mBio 4(4):e00431-13. doi: 10.1128/mBio.00431-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bartual SG, Straume D, Stamsas GA, Munoz IG, Alfonso C, Martinez-Ripoll M, Havarstein LS, Hermoso JA. 2014. Structural basis of PcsB-mediated cell separation in Streptococcus pneumoniae. Nat Commun 5:3842. doi: 10.1038/ncomms4842. [DOI] [PubMed] [Google Scholar]
- 79.Pagliero E, Dublet B, Frehel C, Dideberg O, Vernet T, DiGuilmi AM. 2008. The inactivation of a new peptidoglycan hydrolase Pmp23 leads to abnormal septum formation in Streptococcus pneumoniae. Open Microbiol J 2:107–114. doi: 10.2174/1874285800802010107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Abdullah MR, Gutierrez-Fernandez J, Pribyl T, Gisch N, Saleh M, Rohde M, Petruschka L, Burchhardt G, Schwudke D, Hermoso JA, Hammerschmidt S. 2014. Structure of the pneumococcal L,D-carboxypeptidase DacB and pathophysiological effects of disabled cell wall hydrolases DacA and DacB. Mol Microbiol 93:1183–1206. doi: 10.1111/mmi.12729. [DOI] [PubMed] [Google Scholar]
- 81.Hoyland CN, Aldridge C, Cleverley RM, Duchene MC, Minasov G, Onopriyenko O, Sidiq K, Stogios PJ, Anderson WF, Daniel RA, Savchenko A, Vollmer W, Lewis RJ. 2014. Structure of the LdcB LD-carboxypeptidase reveals the molecular basis of peptidoglycan recognition. Structure 22:949–960. doi: 10.1016/j.str.2014.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Potluri LP, de Pedro MA, Young KD. 2012. Escherichia coli low-molecular-weight penicillin-binding proteins help orient septal FtsZ, and their absence leads to asymmetric cell division and branching. Mol Microbiol 84:203–224. doi: 10.1111/j.1365-2958.2012.08023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.de Pedro MA, Young KD, Holtje JV, Schwarz H. 2003. Branching of Escherichia coli cells arises from multiple sites of inert peptidoglycan. J Bacteriol 185:1147–1152. doi: 10.1128/JB.185.4.1147-1152.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ursell TS, Nguyen J, Monds RD, Colavin A, Billings G, Ouzounov N, Gitai Z, Shaevitz JW, Huang KC. 2014. Rod-like bacterial shape is maintained by feedback between cell curvature and cytoskeletal localization. Proc Natl Acad Sci U S A 111:E1025–E1034. doi: 10.1073/pnas.1317174111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bramkamp M. 2015. Following the equator: division site selection in Streptococcus pneumoniae. Trends Microbiol 23:121–122. doi: 10.1016/j.tim.2015.02.001. [DOI] [PubMed] [Google Scholar]
- 86.Fleurie A, Lesterlin C, Manuse S, Zhao C, Cluzel C, Lavergne JP, Franz-Wachtel M, Macek B, Combet C, Kuru E, VanNieuwenhze MS, Brun YV, Sherratt D, Grangeasse C. 2014. MapZ marks the division sites and positions FtsZ rings in Streptococcus pneumoniae. Nature 516:259–262. doi: 10.1038/nature13966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ramamurthi KS, Losick R. 2009. Negative membrane curvature as a cue for subcellular localization of a bacterial protein. Proc Natl Acad Sci U S A 106:13541–13545. doi: 10.1073/pnas.0906851106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Eswaramoorthy P, Erb ML, Gregory JA, Silverman J, Pogliano K, Pogliano J, Ramamurthi KS. 2011. Cellular architecture mediates DivIVA ultrastructure and regulates Min activity in Bacillus subtilis. mBio 2(6):e00257-11. doi: 10.1128/mBio.00257-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Johnston C, Bootsma HJ, Aldridge C, Manuse S, Gisch N, Schwudke D, Hermans PW, Grangeasse C, Polard P, Vollmer W, Claverys JP. 2015. Co-inactivation of GlnR and CodY regulators impacts pneumococcal cell wall physiology. PLoS One 10:e0123702. doi: 10.1371/journal.pone.0123702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Potluri L, Karczmarek A, Verheul J, Piette A, Wilkin JM, Werth N, Banzhaf M, Vollmer W, Young KD, Nguyen-Distèche M, den Blaauwen T. 2010. Septal and lateral wall localization of PBP5, the major D,D-carboxypeptidase of Escherichia coli, requires substrate recognition and membrane attachment. Mol Microbiol 77:300–323. doi: 10.1111/j.1365-2958.2010.07205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.