

ABSTRACT
Relatively little is known about the phages that infect agriculturally important nitrogen-fixing rhizobial bacteria. Here we report the genome and cryo-electron microscopy structure of the Sinorhizobium meliloti-infecting T4 superfamily phage ΦM9. This phage and its close relative Rhizobium phage vB_RleM_P10VF define a new group of T4 superfamily phages. These phages are distinctly different from the recently characterized cyanophage-like S. meliloti phages of the ΦM12 group. Structurally, ΦM9 has a T=16 capsid formed from repeating units of an extended gp23-like subunit that assemble through interactions between one subunit and the adjacent E-loop insertion domain. Though genetically very distant from the cyanophages, the ΦM9 capsid closely resembles that of the T4 superfamily cyanophage Syn9. ΦM9 also has the same T=16 capsid architecture as the very distant phage SPO1 and the herpesviruses. Despite their overall lack of similarity at the genomic and structural levels, ΦM9 and S. meliloti phage ΦM12 have a small number of open reading frames in common that appear to encode structural proteins involved in interaction with the host and which may have been acquired by horizontal transfer. These proteins are predicted to encode tail baseplate proteins, tail fibers, tail fiber assembly proteins, and glycanases that cleave host exopolysaccharide.
IMPORTANCE Despite recent advances in the phylogenetic and structural characterization of bacteriophages, only a small number of phages of plant-symbiotic nitrogen-fixing soil bacteria have been studied at the molecular level. The effects of phage predation upon beneficial bacteria that promote plant growth remain poorly characterized. First steps in understanding these soil bacterium-phage dynamics are genetic, molecular, and structural characterizations of these groups of phages. The T4 superfamily phages are among the most complex phages; they have large genomes packaged within an icosahedral head and a long, contractile tail through which the DNA is delivered to host cells. This phylogenetic and structural study of S. meliloti-infecting T4 superfamily phage ΦM9 provides new insight into the diversity of this family. The comparison of structure-related genes in both ΦM9 and S. meliloti-infecting T4 superfamily phage ΦM12, which comes from a completely different lineage of these phages, allows the identification of host infection-related factors.
INTRODUCTION
Rhizobia are among the most important bacteria found in soils because of the specific nitrogen-fixing partnerships they form with host legume plants (1). Despite the fundamental importance of these bacteria to agriculture, phages of nitrogen-fixing rhizobia and other soil bacteria are not yet well characterized compared with phages of enteric bacteria or phages of cyanobacteria. The relatively small number of complete genomes of phages that infect rhizobia (see Table S1 in the supplemental material) suggests that this field of study has only scratched the surface. Many of the most virulent known phages of rhizobia have been isolated from commercial plant growth-promoting inocula (2), which suggests that our limited knowledge of these phages is detrimental to agricultural productivity. One of these virulent phages from alfalfa host plant inoculum is the Sinorhizobium meliloti myovirus ΦM9 (2). Here we present the 149,218-bp genome of myovirus ΦM9 and its T=16 capsid structure at 8 Å resolution.
Myoviruses are double-stranded DNA (dsDNA) phages with long, contractile tails, and T4 is the prototypical myovirus (3). In recent years, the incredible diversity of the T4 superfamily of phages has become apparent. In addition to T4 and its close relatives that infect enteric bacteria (4), several other groups of T4 superfamily phages that have a core set of genes in common (4) have been identified. These include T4-like phages of cyanobacteria (5); the “Viuna-like” phages, of which Salmonella phage ViI is the prototype (6); the Campylobacter-infecting T4-like phages (7); the cyanophage-like T4 superfamily phages of alphaproteobacteria (8–10); and the Far-T4 phage clades that have been identified through metagenomics but whose hosts remain unknown (11).
Despite the diversity of the T4 superfamily, the members of this group largely retain a vertically transmitted common core set of genes such as the T4 gp23-like major capsid protein and the T4 gp20-like portal protein (4, 5, 12), with accessory genome components acquired by lateral gene transfer from the host bacterial genome (13) or from other phages (14). The assembly of phage capsids with different icosahedral architectures from the largely conserved HK97-like capsid protein subunits depends upon subtle differences in protein sequence and subunit assembly (15).
Our analysis of DNA sequences from ΦM9 and observation of its morphology by cryogenic transmission electron microscopy (EM) indicate that it is a myovirus related to phage T4. Further, our genome analysis of ΦM9 demonstrates that it is a member of a completely new group of T4 superfamily phages. On the basis of our structural analysis, we have found that the ΦM9 capsid has a T=16 architecture, the same as that of unrelated cyanophage Syn9 (16). By comparing the core genes of ΦM9 with those of S. meliloti phage ΦM12 (10), we have shown that, despite having a host in common, these phages are not closely related. However, a small number of noncore genes are common to ΦM9 and ΦM12 and are located in a cluster of structural genes. On the basis of the conservation of these predicted structural genes across these diverse phage lineages, we propose four groups of “rhizophage conserved proteins.”
MATERIALS AND METHODS
Bacterial strains, phage isolates, and growth conditions.
S. meliloti 1021 (17) was grown at 30°C in LBMC medium (18) or tryptone yeast medium (0.5% tryptone, 0.3% yeast extract, 10 mM CaCl2) supplemented with 500 μg/ml streptomycin. Optimal production of ΦM9 virions was obtained by inoculating 10 μl of crude phage preparation into 25 ml of S. meliloti 1021 at an optical density at 600 nm (OD600) of 0.1 to 0.2. The infected culture was incubated at 30°C overnight or until lysis was apparent, at which point it was centrifuged at 3,800 × g for 30 min to remove cellular debris. The supernatant was extracted twice with chloroform (2). The phage lysate was stored over 1/5 volume of chloroform at 4°C until further purification. Phage titers were monitored by plaque assay (2).
Phage purification for genomic DNA sequencing.
Chloroform-extracted phage ΦM9 was concentrated and washed in an Amicon concentrator (Millipore, Billerica, MA) with a 50-kDa molecular mass cutoff. Concentrated phages were suspended in 10 ml of buffer EX from the Large Construct kit (Qiagen, Valencia, CA) and treated twice with DNase by using 1 U (80 μg) of ATP-dependent exonuclease (Qiagen) to remove S. meliloti genomic DNA. Prior to capsid lysis, the ATP was removed to inactivate the exonuclease by washing in an Amicon concentrator with a 50-kDa molecular mass cutoff. Phages were lysed at 65°C for 1 h in phage buffer with 0.5 M EDTA, 0.5% SDS, and 25 mg/ml proteinase K. Phage DNA was isolated by standard methods (19). After resuspension, phage DNA was treated with 1 mg of RNase A (Qiagen).
Illumina sequencing of the ΦM9 genome and genome assembly.
Two separate ΦM9 DNA samples, each from a plaque-purified phage sample, were sheared to an ∼800-bp average size with a Diagenode Bioruptor. Indexed libraries were constructed with an NEBNext Ultra DNA Library Prep kit for Illumina (NEB, Ipswich, MA) in accordance with the manufacturer's instructions. For each library, 1 μg of DNA was end repaired and ligated to NEBNext adapters. Fragments of ∼800 bp were isolated by electrophoresis on Bio-Rad Low Range Ultra Agarose and amplified for 8 to 10 cycles with NEB High-Fidelity 2× master mix and NEBNext multiplex oligonucleotides. Library size distribution was measured on an Agilent Bioanalyzer high-sensitivity chip, and quantity was determined with the KAPA Biosystems Library Quantification kit. Paired-end 300-base sequence reads were generated on an Illumina MiSeq with a 600-cycle MiSeq v3 Reagent kit. Genome assembly from MiSeq reads was performed with Lasergene SeqMan Pro v. 11.2.1.25 (DNAStar, Madison, WI).
ORF prediction and analysis.
Open reading frames (ORFs) were predicted with GeneMark.hmm for prokaryotes (version 2) (20), MyRast (21), and the NCBI ORF Finder (22). The genome was searched for tRNA sequences with tRNAScan-SE (23).
Construction of genomic alignments, amino acid sequence alignments, and phylogenetic trees.
The alignment of the ΦM9 (GenBank accession no. KP881232) and P10VF (GenBank accession no. NC_025429) genomes shown in Fig. 1 was performed with the Mauve (24) plugin in Geneious (25).
FIG 1.
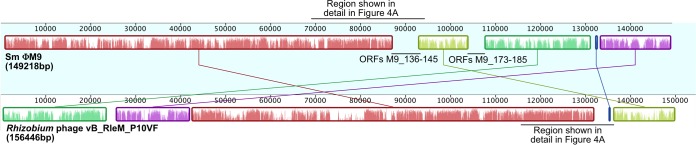
Whole-genome alignment generated in Mauve (24) showing synteny between ΦM9 and Rhizobium phage P10VF. Each block of synteny between the two phages is rendered in a different color. Two regions of ΦM9 that lack synteny with P10VF are labeled. The first of these regions, containing ORFs M9_136 to M9_145, is shown in greater detail in Fig. 4A.
For the phylogenetic trees, a MUSCLE multiple amino acid sequence alignment was performed on a 179-amino-acid internal fragment of the ΦM9 gp20 portal protein sequence and the 104 other sequences shown in Table S2 in the supplemental material with Geneious (25, 26). The maximum number of iterations selected was eight, with the anchor optimization option. The trees from iterations 1 and 2 were not retained. The distance measure for iteration 1 was kmer6_6, and that for subsequent iterations was pctid_kimura. The clustering method used for all iterations was UPGMB (which is based on a combination of both the unweighted-pair group method using average linkages and neighbor joining). Unrooted PhyML trees were constructed from the MUSCLE alignments with the PhyML plugin within Geneious (27, 28). PhyML was performed with the LG amino acid substitution matrix (29) with the proportion of invariable sites fixed and four substitution rate categories. The fast nearest-neighbor interchange tree topology search (30) was used, and 100 bootstraps were performed.
All other protein alignment and percent identity comparisons were performed with whole protein sequences (see Tables S3, S4, and S6 in the supplemental material) and the MUSCLE parameters described above.
Structural modeling of conserved ORFs.
PHYRE was used for comparison of ORFs based on structural prediction of primary amino acid sequence (31). For the comparison of T4 gp33 with ΦM9 ORFs, each ORF of unknown function in the ΦM9 genome was threaded onto the gp33 crystal structure (3TBI chain A) (K. A. Twist, E. A. Campbell, S. A. Darst, E. P. Geiduschek, A. Hochschild, and P. Deighan, unpublished data, 2011) by using PHYRE (31), and no highly probable matches were found (data not shown).
Phage purification for cryo-EM.
Phage was prepared by inoculating 1 ml of crude phage preparation into 700 ml of S. meliloti 1021 in a stirred flask at an OD600 of 0.1 to 0.2. Lysate was cleared by centrifugation at 8,000 × g. Phage was collected first by filtration on a 0.45-μm mixed cellulose ester filter (GE Healthcare, Pittsburgh, PA), followed by a polyethersulfone filter with a 10-kDa cutoff (Pall, Port Washington, NY). The retentate from each filter was chloroform extracted. Phages were further purified via a two-step centrifugation procedure. Samples (suspended in 10 mM phosphate buffer, pH 7, 5 mM MgSO4) were layered onto a continuous density gradient of 10 to 50% OptiPrep density gradient medium (Sigma-Aldrich) in gradient buffer (20 mM Tris-HCl, pH 7, 100 mM KCl, 5 mM MgSO4) and centrifuged at 200,000 × g for 2 h. The peak fraction, as determined by phage titers (2), at approximately 40% OptiPrep, was then diluted in gradient buffer to approximately 20% OptiPrep, layered onto a discontinuous gradient of 35 and 50% OptiPrep, and centrifuged at 200,000 × g for 3 h. Purified phage was collected at the interface between the 35 and 50% OptiPrep layers. Gradient medium was removed by buffer exchanging the sample on a GE PD MiniTrap G25 column into OptiPrep-free gradient buffer, and the presence of phage was confirmed by EM of samples negatively stained with 1% uranyl formate.
Cryo-EM sample preparation and data collection.
Phages concentrated to approximately 3 × 1010 PFU/ml were applied to glow-discharged EM grids (Quantifoil 2/2). Grids were quickly blotted with filter paper and plunged into liquid ethane with a Vitrobot (FEI, Hillsboro, OR) at a chamber temperature of 4°C and 100% relative humidity.
Data collection was performed with a Titan Krios transmission electron microscope (FEI) equipped with a DE20 direct electron detector (Direct Electron, San Diego, CA). Phages were imaged at a nominal magnification of ×22,500, a defocus range of −3.5 to −1.0 μm, and an electron dose of 60 e−/Å2 via the Leginon automation package (32–34). Each field was imaged as 20 individual frames (each 3 e−/Å2), which were processed with DE image-processing software (Direct Electron, San Diego, CA) to correct for drift and compensate for radiation damage (35, 36).
Image processing and three-dimensional (3D) reconstruction.
Early processing tasks were performed within the Appion package (37). A hand-picked initial data set of 903 particles was aligned, classified, and averaged with EMAN and XMIPP with the “starticos” EMAN module to calculate an initial model (37, 38). The resulting model was then used as a template to automatically pick 6,741 packed viral capsids through the FindEM module (39). Defocus estimation for contrast transfer function correction was performed with the Ace2 and CTFFind functions in Appion (40, 41).
The capsid single-particle data set was initially aligned, classified, and reconstructed with the Relion package (42), followed by Frealign (43). After 46 rounds of refinement in Frealign, the icosahedral reconstruction has a resolution of 7.53 Å at a Fourier shell correlation (FSC) of 0.143 and a resolution of 8.97 Å at an FSC of 0.5 (see Fig. S1 in the supplemental material). The final map was sharpened by applying a B factor of −610.88, as calculated by EM-BFACTOR (44). Densities corresponding to individual gp23 subunits were segmented with the Segger extension of UCSF Chimera (45, 46). Local resolution estimation was prepared with the two half-maps from the final stage of Frealign refinement via ResMap (47).
Modeling of gp23 structure.
Six initial models of atomic coordinates were created with the S. meliloti phage ΦM9 gp23 protein sequence and the I-TASSER and RaptorX structure prediction packages (48, 49). These six models were then compared to the nonicosahedrally averaged capsid map, and the best-matching domains were selected from among these six models and combined. This model was then further refined against the cryo-EM map manually in Coot (50). The final model was produced by molecular dynamics flexible fitting with VMD and NAMD (51–53).
Nucleotide sequence accession numbers.
The S. meliloti phage ΦM9 genome has been deposited in GenBank (http://www.ncbi.nlm.nih.gov/nuccore) under accession no. KP881232, and the cryo-EM reconstruction has been deposited in the online Electron Microscopy Data Bank (http://www.ebi.ac.uk/pdbe/emdb/) under accession no. EMD-3036.
RESULTS
A phylogenetic tree based on the gp20 portal protein suggests that ΦM9 and P10VF define a new group of T4 superfamily phages.
ΦM9 has a 149,218-bp dsDNA genome predicted to encode 271 ORFs. The genome is very similar to the sequenced but unpublished genome of Rhizobium leguminosarum phage vB_RleM_P10VF (P10VF here; GenBank accession no. NC_025429) (54). Figure 1 shows the blocks of genomic synteny common to ΦM9 and P10VF and the gaps between those blocks. To further elucidate the position of ΦM9 in a broad phylogenetic context, we have examined diverse members of the T4 superfamily, including close relatives of T4 (4), cyanophages (5), the “Viuna-like” phages (6), and Campylobacter phages (7). The PhyML consensus tree of gp20 portal proteins supports the assessment that ΦM9 and P10VF are very closely related (Fig. 2; see Table S2 in the supplemental material for sequences). Furthermore, the gp20 protein of ΦM9 is more similar to that of Stenotrophomonas phage Smp14 and those of Campylobacter phage types Cp220/Cpt10 and Cp81/CPX than to those of the cyanophages or the T-even phages. However, the overall lack of genomic synteny between ΦM9 and Campylobacter phage Cp220 (data not shown) suggests that ΦM9 and P10VF define a new group of T4 superfamily phages distinct from the Campylobacter phages.
FIG 2.

An unrooted gp20 (portal vertex protein) tree generated by PhyML from a MUSCLE alignment of a 179-amino-acid internal sequence from 105 sequences (see Table S2 in the supplemental material). The bootstrap percentage for each branch is shown, and the bar indicates branch distance.
ΦM9 T4 core proteins are poorly conserved compared with those of other T4 superfamily phages.
ΦM9 lacks some of the large blocks of synteny (Fig. 3) that S. meliloti phage ΦM12 has in common with other T4 superfamily phages (10). However, the order of the T4 core genes is strikingly conserved between ΦM9 and P10VF in most regions (Fig. 3). Despite this overall conservation, the gene contents of these two phages are quite different in the regions outside the blocks of synteny. Unusual for T4 superfamily phages but not unique is the absence of any predicted tRNA genes in the ΦM9 genome. Additionally, there are no primary amino acid sequence homologs in the ΦM9 genome corresponding to gp33, the essential late transcription accessory factor of phage T4, nor were any candidates identified by structural modeling. The ΦM9 genome also lacks a homolog for another member of the T4 core set of genes, the translational repressor encoded by the regA gene (55).
FIG 3.

Global ORF cluster synteny of ΦM9 with selected T4 superfamily phages, showing T4 core protein ORFs. Also shown are Rhizobium phage P10VF, S. meliloti phage ΦM12, Caulobacter phage Cr30, and phage T4. The genomes have been positioned so that the sequence begins with the reverse complement of the gp41 ORF, which encodes a DNA primase/helicase.
Almost all of the ΦM9 core proteins are quite similar to their homologs in P10VF (Table 1; see Table S3 in the supplemental material for accession numbers and references), but overall, the percent identity of the ΦM9 and P10VF core proteins to those of other T4 superfamily phages, including Campylobacter phage Cp220, is low (Table 1) compared with the percent identity of ΦM12 core proteins to those of the cyanophages (10). This suggests that ΦM9 and P10VF are more distant from any previously sequenced T4 superfamily phages than ΦM12 is from the cyanophages. Notably, ΦM9 has only three of the ORFs defined as the “cyanophage core” gene set by Sullivan et al. (5) (Table 2, top). It has seven of the ORFs defined as the “noncyanophage core” gene set (Table 2, middle) and an additional five less commonly conserved T4 ORFs (Table 2, bottom). Another unusual feature that ΦM9 has in common with only P10VF is a gp5 baseplate hub protein that is split into two separate proteins (Table 1). The ΦM9 gp5.2 N terminus has the N-terminal gp27-binding OB domain of T4 gp5 (56). The ΦM9 gp5.1 C-terminal region has the β-helix-containing domain of T4 gp5 (56).
TABLE 1.
MUSCLE multiple alignment percent identity scores of full-length amino acid sequences of ΦM9 T4-like phage universal core proteins and nearly universal core proteins with other T4 superfamily phagesa
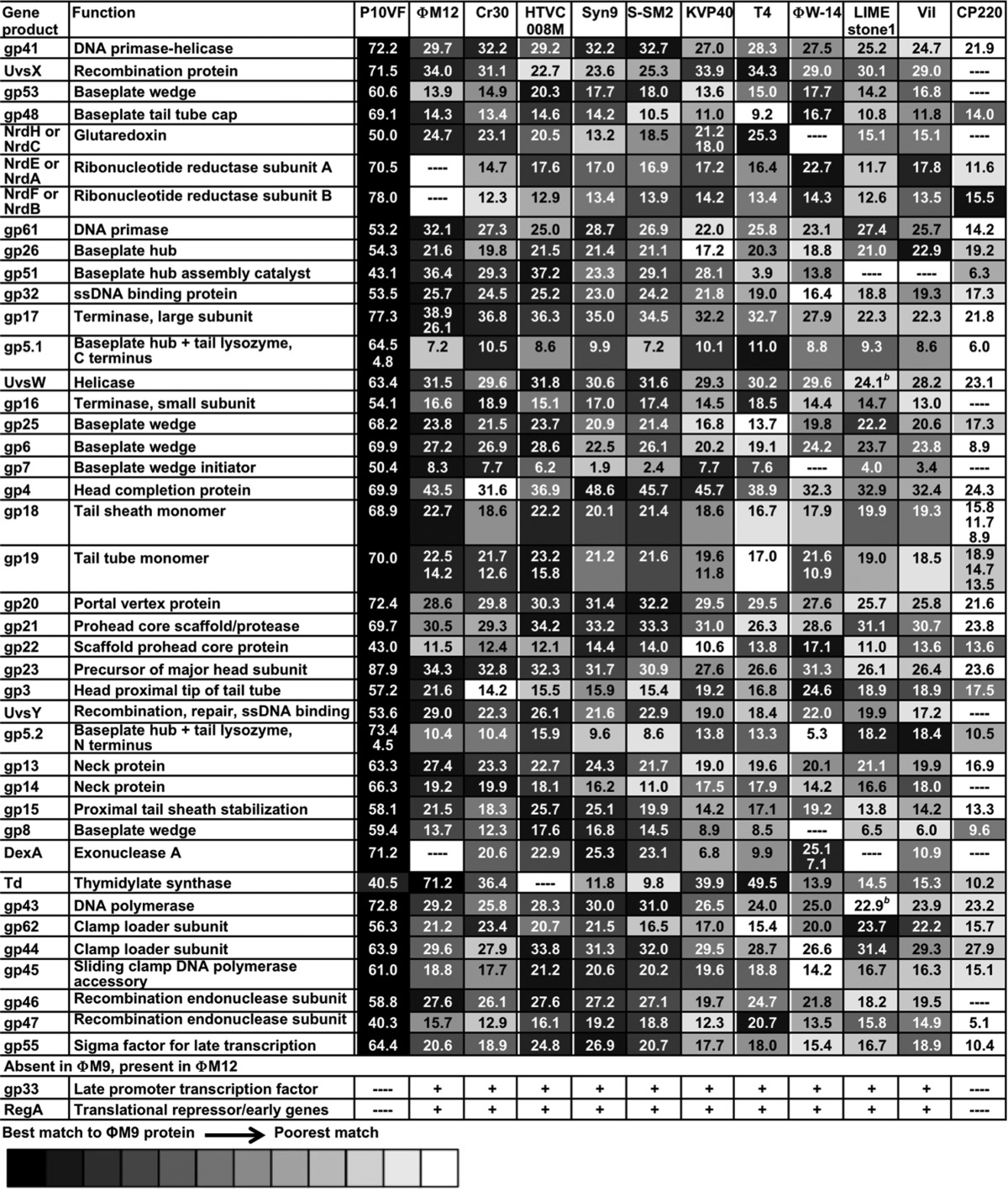
Only core proteins found in S. meliloti phage ΦM9 or ΦM12 were included in this analysis. See Table S3 in the supplemental material for sequences and accession numbers.
b Sequences for which two ORFs separated by an intron have been unified.
TABLE 2.
MUSCLE multiple alignment percent identity scores of full-length amino acid sequences of ΦM9 “T4-like cyanophage core” proteins and “T4-like non-cyanophage core” proteins with other T4 superfamily phagesa
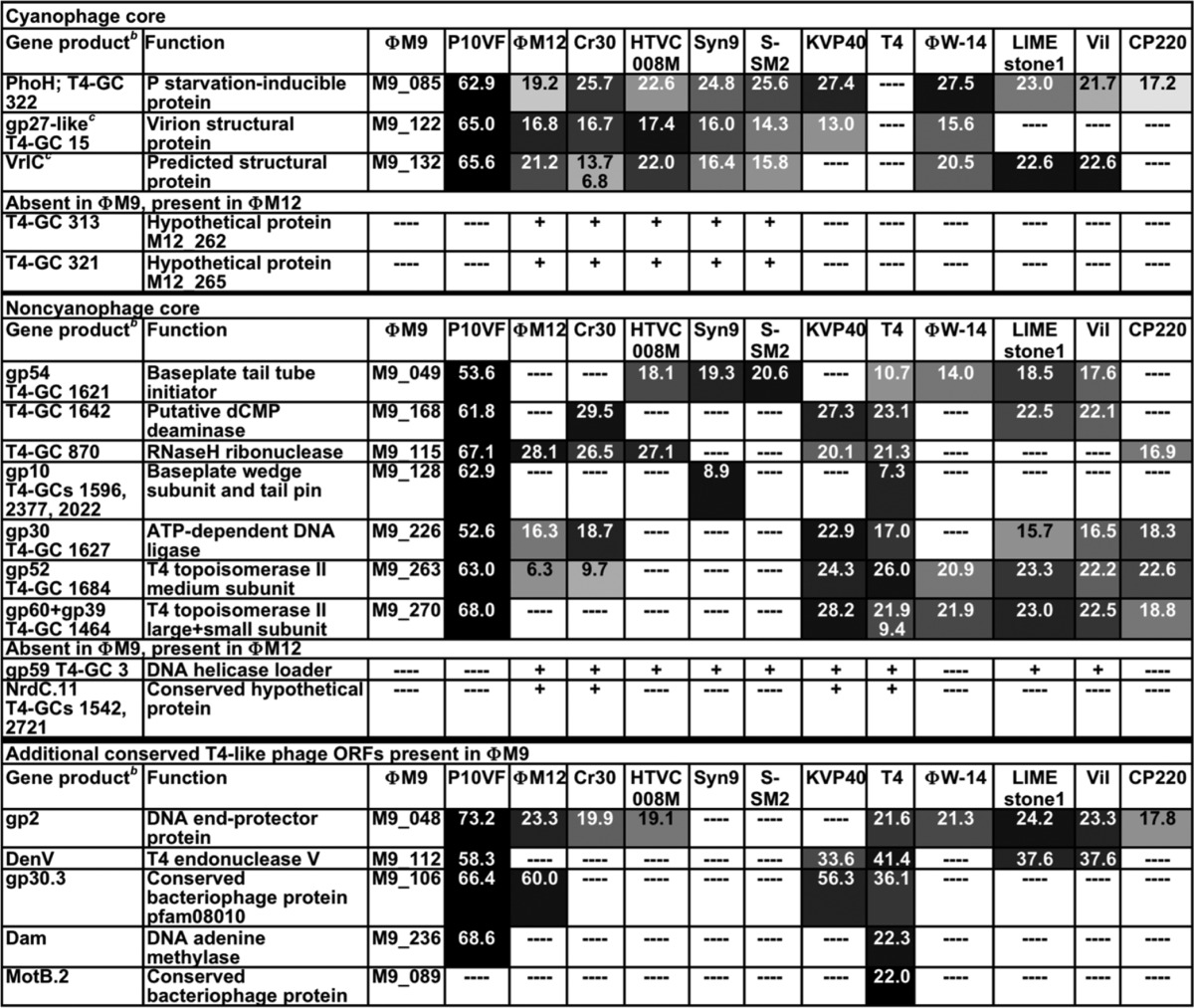
Only core proteins found in S. meliloti phage ΦM9 or ΦM12 were included in this analysis. See Table S3 in the supplemental material for sequences and accession numbers. See Table 1 for the color code legend.
b T4-GC, T4 gene cluster.
c Proteins for which the ΦM12 homolog was detected in the proteome (10).
Comparisons and structural modeling of conserved ΦM9 and ΦM12 ORFs.
There are only 14 ORFs (Table 3) conserved between ΦM9 and S. meliloti phage ΦM12 that are neither part of the T4 core gene set nor classed as genes that encode homing endonucleases (see Tables S4 and S5 in the supplemental material for accession numbers). Six of these 14 ORFs are located in a cluster of phage baseplate and neck structural protein ORFs (Fig. 4A). One of the ORFs in this cluster, ΦM9_121, is predicted to encode a glycanase/laminarinase that is 60.6% identical to its ΦM12 homolog (10) and 21.3% identical to the exopolysaccharide-cleaving ExsH glycanase produced by the host bacterium S. meliloti 1021 (57). Other conserved ORFs in this region (Fig. 4A) are predicted by primary sequence comparisons or by structural modeling to encode structural proteins. These can be organized into distinct groups (Fig. 4B to E; see Discussion).
TABLE 3.
ΦM9 predicted proteins with homologs in phage ΦM12a
| ΦM9 ORF | Predicted function or domain | % Identity |
Similar to ORFs in other phages of rhizobia | |||
|---|---|---|---|---|---|---|
| Other ΦM9 ORF | P10VF | ΦM12c | Cr30 | |||
| M9_035 | ATPase | 47.0 (P10VF_146) | 18.4 (M12_456) | |||
| M9_038 | Hypothetical protein | 43.5 (M12_363) | ||||
| M9_121 | Putative glycanase/laminarinase | 60.6 b (M12_182) | ||||
| M9_136 | Predicted tail fiber protein | 19.9 (M9_134) | 30.2 (P10VF_049), 17.9 (P10VF_051) | 18.8 b (M12_124) | + | |
| M9_137 | Predicted tail fiber assembly protein | 36.6 (M9_138) | 17.3 (M12_122) | + | ||
| M9_138 | Predicted tail fiber assembly protein | 36.6 (M9_137) | 17.9 (M12_122) | + | ||
| M9_141 | Hypothetical protein | 45.2 (M12_431) | ||||
| M9_149 | Predicted glycoprotein | 36.9 (P10VF_039) | 22.7 (M12_410) | + | ||
| M9_176 | 5′ nucleotidase, deoxy (pyrimidine), cytosolic type C protein (NT5C) | 38.2 (P10VF_004) | 38.8 (M12_197) | 21.7 | + | |
| M9_179 | Hypothetical protein | 30.9 (P10VF_252) | 31.2 (M12_173) | |||
| M9_210 | Hypothetical protein | 58.6 (P10VF_233) | 27.1 (M12_384) | |||
| M9_213 | Hypothetical protein, DUF3820 superfamily | 8.1 (M9_216) | 49.2 (P10VF_231) | 46.0 (M12_381) | + | |
| M9_216 | Hypothetical protein | 8.1 (M9_213) | 18.8 (P10VF_231) | 10.4 (M12_381) | + | |
| M9_264 | Hypothetical protein | 26.8 (M12_235) | + | |||
Not including T4 core proteins or homing endonucleases. See Table S4 in the supplemental material for sequence accession numbers.
Proteins for which the ΦM12 homolog was detected in the proteome (10).
FIG 4.

(A) Detailed synteny in the neck/baseplate/tail region of the genomes of ΦM9, Rhizobium phage P10VF, and ΦM12. ORFs filled in black do not have homologs in either of the other two genomes. ORFs filled in white in ΦM9 and P10VF encode hypothetical proteins conserved at the same position in P10VF. The genomes are oriented in the same direction with respect to baseplate protein gp27. All three genomes have baseplate proteins gp5 and gp8 and neck proteins gp13, gp14, and gp15 shaded in gray. (Asterisks next to ΦM12 ORF names mean that these proteins were detected in the ΦM12 proteome [10].) There is nearly complete synteny between ΦM9 and P10VF, except for the absence from P10VF of the region from the middle of ΦM9_136 to the beginning of the glf gene (encodes UDP-galactopyranose mutase) and the absence from P10VF of the glycanase encoded by ΦM9_121. Four ΦM9 ORFs in the region shown lack homologs in P10VF but have homologs in ΦM12, i.e., ΦM9_121 (glycanase), ΦM9_141, and ΦM9_137, and ΦM9_138, which are partial duplicates of one another. (B) Rhizophage-conserved ORF group 1. Shown are the predicted VrlC proteins from the three phage genomes with regions of homology in red and gaps in white. VrlC is one of the more abundant proteins in the ΦM12 proteome (10). (C) Rhizophage-conserved ORF group 2, which is structurally similar to collagen (31) and consists of one ΦM9 ORF and two P10VF ORFs. Regions of homology are solid green. The two nearly adjacent P10VF ORFs may have arisen from a gene duplication (see panel A). (D) Rhizophage group 3, predicted tail fiber assembly proteins, is composed of two very similar adjacent ORFs in ΦM9, a homolog in ΦM12, and ORFs from five other phages of rhizobia (not pictured) (see the text and Table S4 in the supplemental material). (E) Rhizophage predicted tail fiber proteins, group 4. ΦM12_124 is the third most abundant protein in the ΦM12 proteome and is predicted to encode a T4 gp12-like/short-tail fiber/phage tail collar protein. ΦM9_136 shows 13% identity with ΦM12_124 in its central domain and has a C-terminal domain that is 37% identical to Sinorhizobium phage PBC5 protein 14 (GenBank accession no. NC_003324.1) and an N-terminal domain that is 56% identical to phage P10VF_049. ORFs ΦM9_136 and P10VF_049 are ≥36% identical in their first 130 amino acids to ΦM9_134 and P10VF_051, which are structurally similar to B. subtilis short-tailed phage neck appendage proteins (75, 76). These ORFs may have arisen from gene duplication events in the ΦM9 and P10VF genomes or in a common progenitor strain.
Tail.
ΦM9 has a 125-nm-long contractile tail with visible striations due to the helical repeat of the gp18/gp19 subunits (Fig. 5). The neck that attaches the tail to the capsid is simple, with no large protrusions or densities. The baseplate is also simple and bell shaped with protruding, sparse irregular tail fibers that are visible in both cryogenic and negatively stained images (Fig. 5).
FIG 5.
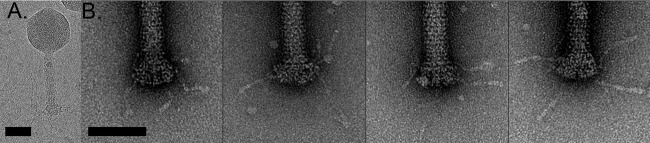
ΦM9 tail and baseplate. (A) Cryogenic image of a full-tailed ΦM9 capsid. (B) Individual negatively stained tail base plates show irregular and sparse protruding tail fibers.
Structure of ΦM9 capsid.
ΦM9 is an icosahedral, T4-like bacteriophage with a DNA-filled capsid and a long, contractile tail (Fig. 5A). Six thousand seven hundred forty-one particles were excised from 2,184 motion- and dose-corrected images and aligned to show a smooth, thin-shelled virion with small, unobtrusive turrets at the 5-fold vertices and a radius of about 56 nm (Fig. 6). At 7.5 to 9.0 Å resolution, judged by 3D mapping of the local resolution (47) (Fig. 7A) and the 0.5/0.143 FSC criteria (see Fig. S1 in the supplemental material), the shape of the 16-mer asymmetric unit is readily identifiable (Fig. 7B and C), where 15 of the monomers make up 2.5 hexamers (Fig. 8). The 16th monomer is distinct, one subunit of the pentamer (Fig. 8). No gp24-like molecule is encoded in the genome, so this distinct subunit is likely made of gp23 but in a 5-fold, not 6-fold, symmetric environment; however, extra density at the tip of each pentamer suggests that an additional helical protein factor caps the turrets (Fig. 8, left inset). Likewise, extra density in the center of the hexamer that does not appear to be 6-fold symmetric may be due to an additional protein factor(s) (Fig. 8, right inset).
FIG 6.
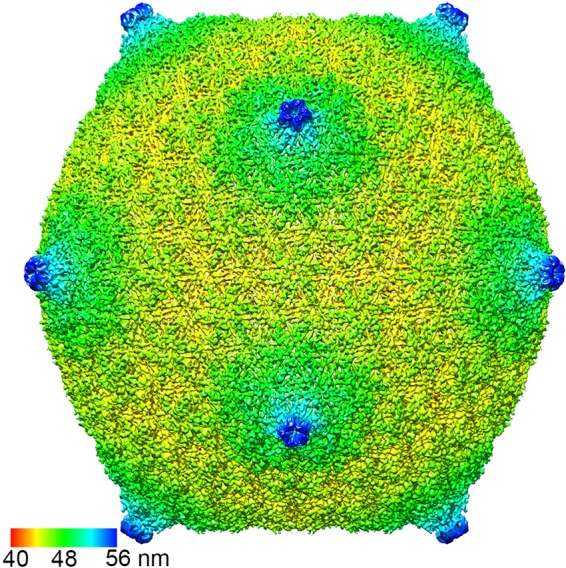
Overall ΦM9 topology. The capsid is colored radially.
FIG 7.
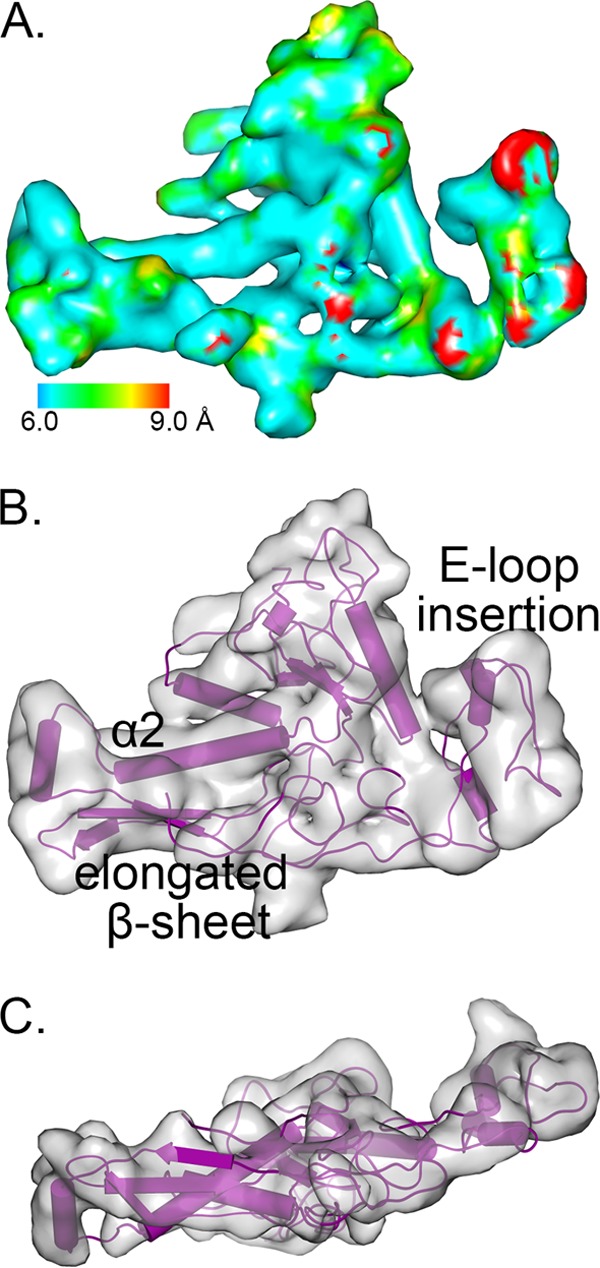
The ΦM9 capsid monomer. (A) One subunit from the asymmetric unit, colored by local resolution calculated by ResMap (47). (B) The modeled structure fits the density well, showing a variable E-loop insertion that accommodates the T=16 symmetry. (C) The side view of the coat protein demonstrates the elongated structure.
FIG 8.
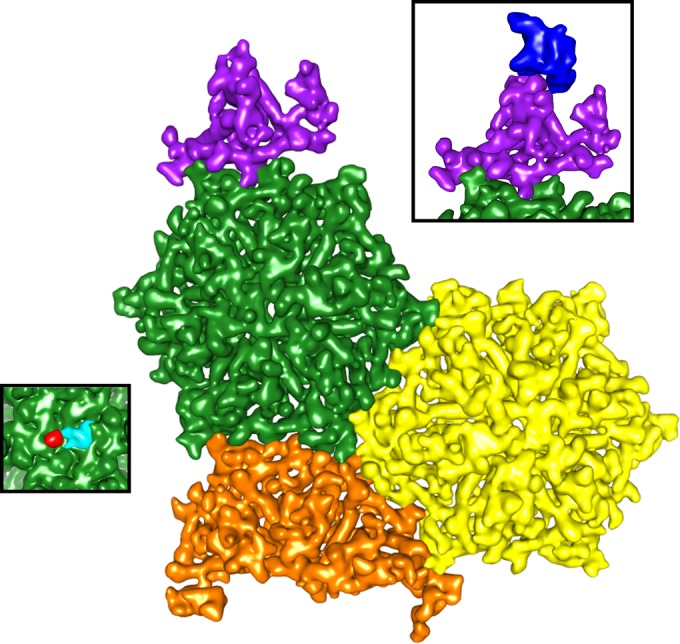
ΦM9's T=16 asymmetric unit. All 16 unique HK97-like subunits fit together to make the tightly packed capsid. The structural elements of the T=16 capsid include a monomer from the pentameric cap (purple), two full hexamers (green and yellow), and half a hexamer (orange) (colors are as in reference 16). ΦM9's reduced E-loop insertion domain reaches over to an adjacent monomer to make the hexameric repeat. Tight interactions at the 2- and 3-fold symmetry axes complete the asymmetric unit. (Left inset) Additional density appearing α-helical in nature forms the turret at the pentamer. (Right inset) Additional density at the center of the hexamer appears to break the 6-fold symmetry.
Modeling of the T=16 coat assembly.
ΦM9 ORF ΦM9_100 was easily identified as the T4 gp23 homolog on the basis of the predicted amino acid sequence, with 26.6% identity and varying most significantly at amino acids 125 to 180, which comprise the E-loop insertion domain. Structurally, the E-loop density varies the most from gp24 of phage T4 (Protein Data Bank code 1YUE) because it is smaller and positioned in an elongated way to accommodate the T=16 symmetry (Fig. 7).
DISCUSSION
Genome and phylogeny.
Our sequence data indicate that ΦM9 is part of a new group (along with phages ΦM10, ΦM14 [K. M. Jones, unpublished results], and P10VF [54]) of S. meliloti T4 superfamily phages. The ΦM9 group is distinct from the ΦM12 group (10), which also includes S. meliloti phages ΦM7, ΦM19 (2) (GenBank accession no. KR052480 and KR052481, respectively) (Jones, unpublished), and ΦN3 (GenBank accession no. KR052482) (58). Phages of the ΦM9 and ΦM12 groups interact with the S. meliloti host cell surface by different mechanisms. Phages of the ΦM9 group are dependent upon the presence of an outer membrane lipopolysaccharide containing a complete core oligosaccharide (59, 60). In contrast, phages of the ΦM12 group are dependent upon the outer membrane protein encoded by ropA1 for infection of S. meliloti (60).
There are significant gaps in the data available for phylogenetic comparison of T4 superfamily phages of alphaproteobacteria. Currently, the only T4 superfamily phages that infect alphaproteobacteria to have been completely sequenced are ΦM9, P10VF, ΦM12/ΦM7/ΦM19, ΦN3, “Candidatus Pelagibacter” phage HTVC008M (8), and Caulobacter phage Cr30 (9). (Sphingomonas phage PAU was previously considered part of this group, but its host bacterium, Sphingomonas paucimobilis strain HER 1398, has been reclassified as a member of the Bacteroidetes phylum [61]). Despite the dearth of genomic information, it is clear that there are at least two distinct groups of these phages: the more cyanophage-like ΦM12/ΦM7/ΦM19, ΦN3, HTVC008M, and Cr30 and the more Campylobacter phage-like ΦM9 and P10VF. However, ΦM9 and P10VF are distant enough from the Campylobacter phages that they define a new group of T4 superfamily phages. The precise position of ΦM9 and P10VF relative to the Campylobacter phages or the Viuna-like phages cannot be determined on the basis of the phage sequences that are currently available.
In contrast to ΦM9, ΦM12 (10) is more closely related to the T4 superfamily cyanophages than to other T4 superfamily phages. The position of S. meliloti phage ΦM12 in the tree in Fig. 2 is different from its position in the gp20 tree we previously presented (10). This is due to the inclusion of the gp20 sequence from Caulobacter phage Cr30 in this study and the omission of the sequences of most uncultured phages. The tree presented in Fig. 2 suggests a close relationship between ΦM12 and Cr30 (discussed more thoroughly by Ely et al. [9]). A PhyML tree constructed without Caulobacter phage Cr30 but otherwise with the same parameters as the tree in Fig. 2 produced a tree in which the positions of both ΦM12 and “Candidatus Pelagibacter” phage HTVC008M were altered with respect to the cyanophages (see Fig. S2 in the supplemental material). This suggests that the inclusion of Caulobacter phage Cr30 supplies important information that clarifies the relatedness of the phages on this branch of the tree.
One of the most surprising features of the ΦM9 and P10VF genomes is the lack of a homolog for T4 gp33, which encodes the late transcription accessory factor that acts in concert with gp55 to fully activate transcription instead of a bacterial RNA polymerase sigma factor (62, 63). In phage T4, the sigma70 domain 4-like activity of gp33 is essential for complete activation of late transcription and for production of phage particles (64, 65). Comparisons of predicted structures of all ΦM9 ORFs with the gp33 crystal structure failed to identify any proteins with clear structural similarity to gp33 (data not shown). ΦM9 gp55 may function with an accessory factor with a structure very different from that of gp33. Another possibility is that ΦM9 gp55 participates in basal transcription, as it also does in phage T4 (65), but that it does not partner with another protein to function as a complete sigma factor analog. Unusual for phages, both the ΦM9 and P10VF genomes have an ORF (ΦM9_087 and P10VF_093) predicted to encode a canonical RpoE stress response sigma factor (COG1595) (66–68). The alignments shown in Fig. S3 in the supplemental material demonstrate that ΦM9_087 and P10VF_093 are much more similar to RpoE sigma factors of alphaproteobacteria than to sigma factors found in other phages (see Table S6 in the supplemental material for GenBank accession numbers and references). These phage RpoE proteins encoded by ΦM9 and P10VF are candidates for late transcription sigma factors that might function in place of gp55/gp33.
The ΦM9 genome also lacks a homolog for another member of the T4 core set of genes, the translational repressor encoded by the regA gene (55). Although RegA is not essential for the production of T4 phage particles (12), the absence of regA in ΦM9 and P10VF is unusual for T4 superfamily phages. In contrast, ΦM9 does have members of the T4 core gene set that are missing from S. meliloti phage ΦM12: the dexA gene, encoding exonuclease A, and the nrd genes, encoding the two subunits of a class I oxygen-dependent ribonucleotide reductase (Table 1).
Conserved structural gene clusters allow the prediction of “rhizophage-conserved ORF groups.”
Beyond the T4-like phage core genes, there are very few ORFs that are common to ΦM9 and ΦM12, despite their common host. In spite of these differences, there is strong conservation of a small number of genes predicted to encode phage structural proteins or proteins involved in infection-related functions. Given that our phylogenetic analysis suggests that ΦM9 and ΦM12 derive from very different lineages within the T4 superfamily, the most likely scenario is that these ORFs have been acquired by horizontal gene transfer events. These ORFs are also conserved in ΦM9-like S. meliloti phages ΦM10 and ΦM14 (Jones, unpublished), in ΦM12-like phages ΦM7 (GenBank accession no. KR052480) and ΦM19 (GenBank accession no. KR052481), and with one exception (Table 3), in ΦN3 (GenBank accession no. KR052482). Although this is a limited number of phage genomes, the conservation of these ORFs across the sequenced members of two very diverse S. meliloti-infecting T4 superfamily lineages suggests that these ORFs may be critical for the successful infection of S. meliloti. For example, the strong conservation of the glycanase that is encoded in both ΦM9 and ΦM12 with the host exopolysaccharide-cleaving glycanase ExsH suggests the fundamental importance of this enzymatic function in phage-host interactions. ExsH cleaves the S. meliloti exopolysaccharide succinoglycan that is required to establish a symbiosis with its host plant (69). ExsH-like glycanases are found in many bacteria but very few phages. The ΦM12 homolog of this ORF is abundant in the ΦM12 proteome (10). If these glycanases are, like ExsH, able to cleave the S. meliloti exopolysaccharide succinoglycan, they might help these phages gain access to the S. meliloti cell surface.
Six of the 14 noncore ORFs that are common to ΦM9 and ΦM12 are located in the cluster of phage baseplate and neck structural protein ORFs shown in Fig. 4A.
On the basis of the clustering of these ORFs, comparisons of the ORFs with other phages that infect rhizobia, and our previous analysis of the ΦM12 proteome (10), we have now proposed a set of four groups of rhizophage-conserved, predicted structural proteins. We hypothesize that proteins in these groups encode baseplate proteins, tail fibers, and tail fiber assembly proteins.
Group 1 contains the VrlC ORF (Fig. 4B), which, in ΦM12, encodes one of the most abundant proteins in the proteome (10), suggesting that it is a structural protein. VrlC is found in the T4-like cyanophages (5), the Viuna-like phages (6) of enteric bacteria, and in many phages of Gram-positive bacteria (70) but not in T4 or its closest relatives (12). The function of VrlC has not yet been established, but the Listeria phage A511 VrlC homolog has been immunolocalized to that phage's baseplate (70). The correlation between the presence of vrlC in a phage genome and that phage having a large, double-ringed baseplate structure has led to the hypothesis that the VrlC protein is responsible for forming these structures (70).
Group 2 rhizophage-conserved ORFs are structurally similar to type I collagen (structure template 1YGV chain A) (31). This group consists of ΦM9_135, P10VF_050, and P10VF_048 (Fig. 4A and C). Collagen-like motifs in tail fiber proteins have been described in several phages (71–73), supporting our hypothesis that the group 2 ORFs encode tail fibers in ΦM9 and P10VF.
Group 3 ORFs have similarity to a family of phage tail fiber assembly proteins (pfam02413) (14). This rhizophage group consists of two adjacent, very similar ΦM9 ORFs, ΦM9_137 and ΦM9_138; an ORF from ΦM12, ΦM12_122 (Fig. 4A and D; Table 3); and ORFs from several other rhizophages (listed in Table S4 in the supplemental material but not pictured in Fig. 4). There is no group 3 ORF in phage P10VF.
The interrelationships among the group 4 ORFs are complex, and these ORFs may be examples of phage tail fiber ORFs that have developed a mosaic composition (74) because of domain swapping. ΦM9_136 appears to be a very complex mosaic of sequences from other rhizophages. It has a central domain that is 13% identical to ORF ΦM12_124 (Fig. 4A and E), which is predicted to encode a T4 gp12-like/short-tail fiber/phage tail collar protein, the third most abundant protein in the ΦM12 proteome (10). Other domains of ΦM9_136 appear to have come from other rhizophages. It has a C-terminal domain that is 37% identical to Sinorhizobium phage PBC5 protein 14 (GenBank accession no. NC_003324.1), and its N-terminal domain is 56% identical to phage P10VF ORF P10VF_049 (Fig. 4E).
Adding further complexity to group 4 is the fact that ΦM9_136 and P10VF_049, as well as ORFs ΦM9_134 and P10VF_051, are 36% identical in their first 130 amino acids (Fig. 4E). ΦM9_134 and P10VF_051 are structurally similar to the “neck appendage” protein found in Bacillus subtilis short-tailed phage GA-1 (structure template 3GUD chain B) (75, 76), which acts as a receptor-binding tail fiber protein (77). The genomic context for these ORFs in the ΦM9 and P10VF genomes (Fig. 4A) shows that they may have been formed by gene duplication events in these phages or in a common progenitor strain.
In the ΦM9, P10VF, and ΦM12 genomes, the group 4 ORFs are close to the ORF encoding VrlC. In Listeria phage A511, the host cell receptor-binding tail fiber protein gp108 is also located nearly adjacent to VrlC and immunolocalization assays suggest that A511 gp108 is directly bound to VrlC in the A511 phage particle (70). On the basis of genomic context, primary sequence, and structural comparisons, we hypothesize that ΦM9_136 and ΦM9_134, ΦM12_124, and P10VF_049 and P10VF_051 encode tail fibers in their respective genomes. If this is the case, it could mean that both ΦM9 and P10VF have multiple tail fibers. Simultaneously displaying multiple receptor-binding tail fiber proteins and thereby increasing the phage host range has been observed in the Escherichia coli myovirus phi92 (78).
Another ORF in this ΦM9 cluster of phage baseplate and neck structural protein ORFs that is conserved in ΦM12 is ΦM9_141, which has an alpha-helical coiled-coil structure similar to that of the T4 fibritin neck-whisker protein (structure template 1OX3 chain A) (79), although it is much smaller than T4 fibiritin. This ORF is 45% identical to the ΦM12 ORF ΦM12_431 but has no clear homolog in P10VF. Part of the role of the neck whiskers is to interact with the long tail fibers of T4-like phages and control their assembly and retraction (79), but not all myoviruses have collar whiskers (78). It has not yet been possible to determine from our structural analysis if ΦM9 or ΦM12 has neck whiskers.
Overall ΦM9 morphology and T=16 capsid.
ΦM9 is a T=16 contractile bacteriophage with a long tail that shows the striations of the helically twisting subunits (Fig. 5). The neck, which joins a 5-fold of the icosahedral capsid and the tail, has a simple architecture, whereas the smooth plate at the base of the tail (Fig. 5A) is quite different from the other known T=16 phage of cyanobacteria, Syn9, whose baseplate is strikingly bushy (16). In raw images, however, the fibers appear to be sparsely distributed on the surface of the plate (Fig. 5) and are also structurally quite different from those of S. meliloti phage ΦM12 (80) and the well-studied T4 enteric bacteriophage (81).
ΦM9 is a T=16 phage like Syn9, formed by a single T4 gp23-like capsid protein (Fig. 6 and 7). In contrast, the well-studied T=16 herpesvirus capsid is made from multiple peptides, including an asymmetric trimeric assembly that spans its 3-fold symmetry axis (82). The gp23 homologs from ΦM9 and Syn9 show 31.7% sequence identity, and their capsids are virtually superimposable at similar resolutions. ΦM9's 149,218-bp-long genome is somewhat shorter than Syn9's 177,300-bp-long genome. In this way, ΦM9 is less efficient than Syn9 in preparing a capsid that maximizes the subunit number for the enclosed volume, unlike its cousin ΦM12, whose T=19 capsid is particularly tightly packed and efficient at maximizing its internal volume (80). Nonetheless, the shared T number and similar genome size suggests that ΦM9 could have also evolved from the same T=13 capsid progenitor as Syn9, T4, and T5 (16).
Subunit interactions in T=16 capsids.
The common fold for T4-like phage coat proteins is defined by the first instance of its structural determination, gp5 in the HK97 virus (gp23/24 in T4) (83, 84). Its structural core is L shaped, where the stem of the L is made from the peripheral (P) domain and the arm is composed of the axial (A) domain (Fig. 8C) (84). Individual isotypes are made unique by insertion (I) domains and extended (E) loops, which determine the surface texture, symmetry, and size of any capsid (15). ΦM9 has a fairly smooth surface (Fig. 6), not unlike that of ΦM12 (80) but quite distinct from that of T4 (85), which has an extended E-loop insertion domain, in contrast to the minimal domain in ΦM12 and the slightly bigger but still minimal E-loop insertion in ΦM9.
The capsid protein from the monomers excised from ΦM9's hexamers is extended relative to that from its pentamers, which echoes T4's gp24 protein (86). The stretched fold stems from a core structural feature that define how the T=16 capsid assembles; the central β-sheet from the P domain is longer than it is in T4 gp24. Specifically, in ΦM9, the central β strand is made of amino acids 402 to 420, whereas in T4 it is made from amino acids 370 to 384 (Fig. 7B). Consequently, α2 protrudes from the central core and α3 runs straight through the center of the molecule to give it the extended conformation (Fig. 7C). At 7.5 to 9.0 Å resolution, α-helical density is clearly visible, so these secondary structural elements are reliably mapped; indeed, these regions of the map represent the highest resolution (Fig. 7A). In T4, the corresponding central α-helix bends at Met206, compressing the structure and allowing it to form the pentamer (86). The capsid resulting from ΦM9's extended capsid protein is thin, without significant surface features, but expansive (Fig. 6B).
The pentamer density exhibits the same conformation and 2-fold symmetry interactions with its neighboring hexamer as the hexamers do with one another (Fig. 8). There is additional non-gp23 density at the tip of the penton, consisting of five small helical bundles each of which protrudes from a different asymmetric unit (Fig. 7B, left inset). There is also extra density at the center of the hexamers that breaks the pseudo-6-fold symmetry within the asymmetric unit, which is unenforced in the capsid map (Fig. 5C and 7B, right inset). This extra density appears 2-fold symmetric, perhaps formed by a dimer of an accessory protein(s) (Fig. 7B, right inset).
Conclusions.
ΦM9, along with Rhizobium phage P10VF, defines a new group of T4 superfamily phages. It is not closely related to any previously characterized T4-like phage groups. The closest relationship appears to be with the T4-like Campylobacter phages. Despite the genetic novelty of ΦM9, its gp23-like capsid subunits assemble to form a T=16 capsid like that previously observed in the phages Syn9 and SPO1 and the herpesviruses. Assembly into this common capsid form occurs despite the large phylogenetic distance between these groups of viruses. Both the genome and structure of ΦM9 show surprising new ways in which the basic building blocks of T4 superfamily phages can be assembled.
Supplementary Material
ACKNOWLEDGMENTS
We are very grateful to Robert Haselkorn and E. Peter Geiduschek for helpful discussions and suggestions.
This work was funded by Agriculture and Food Research Initiative award 2014-67013-21579 from the USDA National Institute of Food and Agriculture to K.M.J. and NSF award MCB-1149763 to M.E.S.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01353-15.
REFERENCES
- 1.Fowler D, Coyle M, Skiba U, Sutton MA, Cape JN, Reis S, Sheppard LJ, Jenkins A, Grizzetti B, Galloway JN, Vitousek P, Leach A, Bouwman AF, Butterbach-Bahl K, Dentener F, Stevenson D, Amann M, Voss M. 2013. The global nitrogen cycle in the twenty-first century. Philos Trans R Soc Lond B Biol Sci. 368:20130164. doi: 10.1098/rstb.2013.0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Finan TM, Hartweig E, LeMieux K, Bergman K, Walker GC, Signer ER. 1984. General transduction in Rhizobium meliloti. J Bacteriol 159:120–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hatfull GF, Hendrix RW. 2011. Bacteriophages and their genomes. Curr Opin Virol 1:298–303. doi: 10.1016/j.coviro.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Comeau AM, Bertrand C, Letarov A, Tétart F, Krisch HM. 2007. Modular architecture of the T4 phage superfamily: a conserved core genome and a plastic periphery. Virology 362:384–396. doi: 10.1016/j.virol.2006.12.031. [DOI] [PubMed] [Google Scholar]
- 5.Sullivan MB, Huang KH, Ignacio-Espinoza JC, Berlin AM, Kelly L, Weigele PR, DeFrancesco AS, Kern SE, Thompson LR, Young S, Yandava C, Fu R, Krastins B, Chase M, Sarracino D, Osburne MS, Henn MR, Chisholm SW. 2010. Genomic analysis of oceanic cyanobacterial myoviruses compared with T4-like myoviruses from diverse hosts and environments. Environ Microbiol 12:3035–3056. doi: 10.1111/j.1462-2920.2010.02280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Adriaenssens EM, Ackermann HW, Anany H, Blasdel B, Connerton IF, Goulding D, Griffiths MW, Hooton SP, Kutter EM, Kropinski AM, Lee JH, Maes M, Pickard D, Ryu S, Sepehrizadeh Z, Shahrbabak SS, Toribio AL, Lavigne R. 2012. A suggested new bacteriophage genus: “Viunalikevirus”. Arch Virol 157:2035–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Javed MA, Ackermann HW, Azeredo J, Carvalho CM, Connerton I, Evoy S, Hammerl JA, Hertwig S, Lavigne R, Singh A, Szymanski CM, Timms A, Kropinski AM. 2014. A suggested classification for two groups of Campylobacter myoviruses. Arch Virol 159:181–190. doi: 10.1007/s00705-013-1788-2. [DOI] [PubMed] [Google Scholar]
- 8.Zhao Y, Temperton B, Thrash JC, Schwalbach MS, Vergin KL, Landry ZC, Ellisman M, Deerinck T, Sullivan MB, Giovannoni SJ. 2013. Abundant SAR11 viruses in the ocean. Nature 494:357–360. doi: 10.1038/nature11921. [DOI] [PubMed] [Google Scholar]
- 9.Ely B, Gibbs W, Diez S, Ash K. 2015. The Caulobacter crescentus transducing phage Cr30 is a unique member of the T4-like family of myophages. Curr Microbiol 70:854–858. doi: 10.1007/s00284-015-0799-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brewer TE, Stroupe ME, Jones KM. 2014. The genome, proteome and phylogenetic analysis of Sinorhizobium meliloti phage ΦM12, the founder of a new group of T4-superfamily phages. Virology 450–451:84–97. [DOI] [PubMed] [Google Scholar]
- 11.Roux S, Enault F, Ravet V, Pereira O, Sullivan MB. 2015. Genomic characteristics and environmental distributions of the uncultivated Far-T4 phages. Front Microbiol 6:199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller ES, Kutter E, Mosig G, Arisaka F, Kunisawa T, Ruger W. 2003. Bacteriophage T4 genome. Microbiol Mol Biol Rev 67:86–156. doi: 10.1128/MMBR.67.1.86-156.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ignacio-Espinoza JC, Sullivan MB. 2012. Phylogenomics of T4 cyanophages: lateral gene transfer in the ‘core’ and origins of host genes. Environ Microbiol 14:2113–2126. doi: 10.1111/j.1462-2920.2012.02704.x. [DOI] [PubMed] [Google Scholar]
- 14.Haggård-Ljungquist E, Halling C, Calendar R. 1992. DNA sequences of the tail fiber genes of bacteriophage P2: evidence for horizontal transfer of tail fiber genes among unrelated bacteriophages. J Bacteriol 174:1462–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suhanovsky MM, Teschke CM. 2015. Nature's favorite building block: deciphering folding and capsid assembly of proteins with the HK97-fold. Virology 479–480:487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weigele PR, Pope WH, Pedulla ML, Houtz JM, Smith AL, Conway JF, King J, Hatfull GF, Lawrence JG, Hendrix RW. 2007. Genomic and structural analysis of Syn9, a cyanophage infecting marine Prochlorococcus and Synechococcus. Environ Microbiol 9:1675–1695. doi: 10.1111/j.1462-2920.2007.01285.x. [DOI] [PubMed] [Google Scholar]
- 17.Meade HM, Long SR, Ruvkun GB, Brown SE, Ausubel FM. 1982. Physical and genetic characterization of symbiotic and auxotrophic mutants of Rhizobium meliloti induced by transposon Tn5 mutagenesis. J Bacteriol 149:114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glazebrook J, Walker GC. 1991. Genetic techniques in Rhizobium meliloti. Methods Enzymol 204:398–418. doi: 10.1016/0076-6879(91)04021-F. [DOI] [PubMed] [Google Scholar]
- 19.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 20.Lukashin AV, Borodovsky M. 1998. GeneMark.hmm: new solutions for gene finding. Nucleic Acids Res 26:1107–1115. doi: 10.1093/nar/26.4.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O. 2008. The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sayers EW, Barrett T, Benson DA, Bolton E, Bryant SH, Canese K, Chetvernin V, Church DM, DiCuccio M, Federhen S, Feolo M, Fingerman IM, Geer LY, Helmberg W, Kapustin Y, Landsman D, Lipman DJ, Lu Z, Madden TL, Madej T, Maglott DR, Marchler-Bauer A, Miller V, Mizrachi I, Ostell J, Panchenko A, Phan L, Pruitt KD, Schuler GD, Sequeira E, Sherry ST, Shumway M, Sirotkin K, Slotta D, Souvorov A, Starchenko G, Tatusova TA, Wagner L, Wang Y, Wilbur WJ, Yaschenko E, Ye J. 2011. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 39:D38–51. doi: 10.1093/nar/gkq1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lowe TM, Eddy SR. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25:955–964. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Darling AC, Mau B, Blattner FR, Perna NT. 2004. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res 14:1394–1403. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Drummond AJ, Ashton B, Buxton S, Cheung M, Cooper A, Duran C, Field M, Heled J, Kearse M, Markowitz S, Moir R, Stones-Hayes S. 2012. Geneious 5.6.6. Biomatters Ltd., Auckland, New Zealand. [Google Scholar]
- 26.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guindon S, Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 28.Lefort V, Heled J, Guindon S. 2012. Geneious 5.6.6, PhyML plugin. Biomatters Ltd., Auckland, New Zealand. [Google Scholar]
- 29.Le SQ, Gascuel O. 2008. An improved general amino acid replacement matrix. Mol Biol Evol 25:1307–1320. doi: 10.1093/molbev/msn067. [DOI] [PubMed] [Google Scholar]
- 30.Desper R, Gascuel O. 2002. Fast and accurate phylogeny reconstruction algorithms based on the minimum-evolution principle. J Comput Biol 9:687–705. doi: 10.1089/106652702761034136. [DOI] [PubMed] [Google Scholar]
- 31.Kelley LA, Sternberg MJ. 2009. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc 4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 32.Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, Carragher B. 2005. Automated molecular microscopy: the new Leginon system. J Struct Biol 151:41–60. doi: 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 33.Carragher B, Kisseberth N, Kriegman D, Milligan RA, Potter CS, Pulokas J, Reilein A. 2000. Leginon: an automated system for acquisition of images from vitreous ice specimens. J Struct Biol 132:33–45. doi: 10.1006/jsbi.2000.4314. [DOI] [PubMed] [Google Scholar]
- 34.Shrum DC, Woodruff BW, Stagg SM. 2012. Creating an infrastructure for high-throughput high resolution cryogenic electron microscopy. J Struct Biol 180:254–258. doi: 10.1016/j.jsb.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li X, Mooney P, Zheng S, Booth CR, Braunfeld MB, Gubbens S, Agard DA, Cheng Y. 2013. Electron counting and beam-induced motion correction enable near-atomic resolution single-particle cryo-EM. Nat Methods 10:584–590. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Z, Hryc CF, Bammes B, Afonine PV, Jakana J, Chen DH, Liu X, Baker ML, Kao C, Ludtke SJ, Schmid MF, Adams PD, Chiu W. 2014. An atomic model of brome mosaic virus using direct electron detection and real-space optimization. Nat Commun 5:4808. doi: 10.1038/ncomms5808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lander GC, Stagg SM, Voss NR, Cheng A, Fellmann D, Pulokas J, Yoshioka C, Irving C, Mulder A, Lau PW, Lyumkis D, Potter CS, Carragher B. 2009. Appion: an integrated, database-driven pipeline to facilitate EM image processing. J Struct Biol 166:95–102. doi: 10.1016/j.jsb.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ludtke SJ, Baldwin PR, Chiu W. 1999. EMAN: semiautomated software for high resolution single-particle reconstructions. J Struct Biol 128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- 39.Roseman AM. 2004. FindEM—a fast, efficient program for automatic selection of particles from electron micrographs. J Struct Biol 145:91–99. doi: 10.1016/j.jsb.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 40.Mallick SP, Carragher B, Potter CS, Kriegman DJ. 2005. ACE: automated CTF estimation. Ultramicroscopy 104:8–29. doi: 10.1016/j.ultramic.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 41.Mindell JA, Grigorieff N. 2003. Accurate determination of local defocus and specimen tilt in electron microscopy. J Struct Biol 142:334–347. doi: 10.1016/S1047-8477(03)00069-8. [DOI] [PubMed] [Google Scholar]
- 42.Scheres SH. 2012. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grigorieff N. 2007. FREALIGN: high resolution refinement of single particle structures. J Struct Biol 157:117–125. doi: 10.1016/j.jsb.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 44.Fernández JJ, Luque D, Caston JR, Carrascosa JL. 2008. Sharpening high resolution information in single particle electron cryomicroscopy. J Struct Biol 164:170–175. doi: 10.1016/j.jsb.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 45.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 46.Pintilie GD, Zhang J, Goddard TD, Chiu W, Gossard DC. 2010. Quantitative analysis of cryo-EM density map segmentation by watershed and scale-space filtering, and fitting of structures by alignment to regions. J Struct Biol 170:427–438. doi: 10.1016/j.jsb.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kucukelbir A, Sigworth FJ, Tagare HD. 2014. Quantifying the local resolution of cryo-EM density maps. Nat Methods 11:63–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Källberg M, Wang HP, Wang S, Peng J, Wang ZY, Lu H, Xu JB. 2012. Template-based protein structure modeling using the RaptorX web server. Nat Protoc 7:1511–1522. doi: 10.1038/nprot.2012.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang JY, Yan RX, Roy A, Xu D, Poisson J, Zhang Y. 2015. The I-TASSER suite: protein structure and function prediction. Nat Methods 12:7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr Sect D Biol Crystallogr 60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 51.Humphrey W, Dalke A, Schulten K. 1996. VMD: visual molecular dynamics. J Mol Graph Model 14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 52.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. 2005. Scalable molecular dynamics with NAMD. J Comput Chem 26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trabuco LG, Villa E, Schreiner E, Harrison CB, Schulten K. 2009. Molecular dynamics flexible fitting: a practical guide to combine cryo-electron microscopy and X-ray crystallography. Methods 49:174–180. doi: 10.1016/j.ymeth.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Restrepo-Cordoba M, Halmillawewa AP, Perry B, Hynes MF, Yost CK. 2014. Isolation and characterization of Rhizobium leguminosarum phages from western Canadian soils and complete genome sequences of rhizobiophages vB_RleS_L338C and vB_RleM_P10VF. Department of Biological Sciences, University of Calgary, Calgary, Alberta, Canada. [Google Scholar]
- 55.Karam J, Gold L, Singer BS, Dawson M. 1981. Translational regulation: identification of the site on bacteriophage T4 rIIB mRNA recognized by the regA gene function. Proc Natl Acad Sci U S A 78:4669–4673. doi: 10.1073/pnas.78.8.4669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rossmann MG, Mesyanzhinov VV, Arisaka F, Leiman PG. 2004. The bacteriophage T4 DNA injection machine. Curr Opin Struct Biol 14:171–180. doi: 10.1016/j.sbi.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 57.York GM, Walker GC. 1997. The Rhizobium meliloti exoK gene and prsD/prsE/exsH genes are components of independent degradative pathways which contribute to production of low-molecular-weight succinoglycan. Mol Microbiol 25:117–134. doi: 10.1046/j.1365-2958.1997.4481804.x. [DOI] [PubMed] [Google Scholar]
- 58.Martin MO, Long SR. 1984. Generalized transduction in Rhizobium meliloti. J Bacteriol 159:125–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Campbell GR, Reuhs BL, Walker GC. 2002. Chronic intracellular infection of alfalfa nodules by Sinorhizobium meliloti requires correct lipopolysaccharide core. Proc Natl Acad Sci U S A 99:3938–3943. doi: 10.1073/pnas.062425699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Crook MB, Draper AL, Guillory RJ, Griffitts JS. 2013. The Sinorhizobium meliloti essential porin RopA1 is a target for numerous bacteriophages. J Bacteriol 195:3663–3671. doi: 10.1128/JB.00480-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.White RA III, Suttle CA. 2013. The draft genome sequence of Sphingomonas paucimobilis strain HER1398 (Proteobacteria), host to the giant PAU phage, indicates that it is a member of the genus Sphingobacterium (Bacteroidetes). Genome Announc 1(4):e00598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nechaev S, Kamali-Moghaddam M, Andre E, Leonetti JP, Geiduschek EP. 2004. The bacteriophage T4 late-transcription coactivator gp33 binds the flap domain of Escherichia coli RNA polymerase. Proc Natl Acad Sci U S A 101:17365–17370. doi: 10.1073/pnas.0408028101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nechaev S, Geiduschek EP. 2008. Dissection of the bacteriophage T4 late promoter complex. J Mol Biol 379:402–413. doi: 10.1016/j.jmb.2008.03.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Herendeen DR, Williams KP, Kassavetis GA, Geiduschek EP. 1990. An RNA polymerase-binding protein that is required for communication between an enhancer and a promoter. Science 248:573–578. doi: 10.1126/science.2185541. [DOI] [PubMed] [Google Scholar]
- 65.Geiduschek EP, Kassavetis GA. 2010. Transcription of the T4 late genes. Virol J 7:288. doi: 10.1186/1743-422X-7-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS, Kiryutin B, Galperin MY, Fedorova ND, Koonin EV. 2001. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res 29:22–28. doi: 10.1093/nar/29.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sauviac L, Philippe H, Phok K, Bruand C. 2007. An extracytoplasmic function sigma factor acts as a general stress response regulator in Sinorhizobium meliloti. J Bacteriol 189:4204–4216. doi: 10.1128/JB.00175-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jones KM, Kobayashi H, Davies BW, Taga ME, Walker GC. 2007. How rhizobial symbionts invade plants: the Sinorhizobium-Medicago model. Nat Rev Microbiol 5:619–633. doi: 10.1038/nrmicro1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Habann M, Leiman PG, Vandersteegen K, Van den Bossche A, Lavigne R, Shneider MM, Bielmann R, Eugster MR, Loessner MJ, Klumpp J. 2014. Listeria phage A511, a model for the contractile tail machineries of SPO1-related bacteriophages. Mol Microbiol 92:84–99. doi: 10.1111/mmi.12539. [DOI] [PubMed] [Google Scholar]
- 71.Desiere F, Lucchini S, Brussow H. 1998. Evolution of Streptococcus thermophilus bacteriophage genomes by modular exchanges followed by point mutations and small deletions and insertions. Virology 241:345–356. doi: 10.1006/viro.1997.8959. [DOI] [PubMed] [Google Scholar]
- 72.Duplessis M, Moineau S. 2001. Identification of a genetic determinant responsible for host specificity in Streptococcus thermophilus bacteriophages. Mol Microbiol 41:325–336. doi: 10.1046/j.1365-2958.2001.02521.x. [DOI] [PubMed] [Google Scholar]
- 73.Smith MC, Burns N, Sayers JR, Sorrell JA, Casjens SR, Hendrix RW. 1998. Bacteriophage collagen. Science 279:1834. [DOI] [PubMed] [Google Scholar]
- 74.Tétart F, Desplats C, Krisch HM. 1998. Genome plasticity in the distal tail fiber locus of the T-even bacteriophage: recombination between conserved motifs swaps adhesin specificity. J Mol Biol 282:543–556. doi: 10.1006/jmbi.1998.2047. [DOI] [PubMed] [Google Scholar]
- 75.Schulz EC, Dickmanns A, Urlaub H, Schmitt A, Muhlenhoff M, Stummeyer K, Schwarzer D, Gerardy-Schahn R, Ficner R. 2010. Crystal structure of an intramolecular chaperone mediating triple-beta-helix folding. Nat Struct Mol Biol 17:210–215. doi: 10.1038/nsmb.1746. [DOI] [PubMed] [Google Scholar]
- 76.Xiang Y, Leiman PG, Li L, Grimes S, Anderson DL, Rossmann MG. 2009. Crystallographic insights into the autocatalytic assembly mechanism of a bacteriophage tail spike. Mol Cell 34:375–386. doi: 10.1016/j.molcel.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Meijer WJ, Horcajadas JA, Salas M. 2001. Phi29 family of phages. Microbiol Mol Biol Rev 65:261–287. doi: 10.1128/MMBR.65.2.261-287.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schwarzer D, Buettner FF, Browning C, Nazarov S, Rabsch W, Bethe A, Oberbeck A, Bowman VD, Stummeyer K, Muhlenhoff M, Leiman PG, Gerardy-Schahn R. 2012. A multivalent adsorption apparatus explains the broad host range of phage phi92: a comprehensive genomic and structural analysis. J Virol 86:10384–10398. doi: 10.1128/JVI.00801-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Boudko SP, Strelkov SV, Engel J, Stetefeld J. 2004. Design and crystal structure of bacteriophage T4 mini-fibritin NCCF. J Mol Biol 339:927–935. doi: 10.1016/j.jmb.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 80.Stroupe ME, Brewer TE, Sousa DR, Jones KM. 2014. The structure of Sinorhizobium meliloti phage ΦM12, which has a novel T=19l triangulation number and is the founder of a new group of T4-superfamily phages. Virology 450–451:205–212. [DOI] [PubMed] [Google Scholar]
- 81.Leiman PG, Arisaka F, van Raaij MJ, Kostyuchenko VA, Aksyuk AA, Kanamaru S, Rossmann MG. 2010. Morphogenesis of the T4 tail and tail fibers. Virol J 7:355. doi: 10.1186/1743-422X-7-355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Newcomb WW, Trus BL, Booy FP, Steven AC, Wall JS, Brown JC. 1993. Structure of the herpes simplex virus capsid. Molecular composition of the pentons and the triplexes. J Mol Biol 232:499–511. [DOI] [PubMed] [Google Scholar]
- 83.Helgstrand C, Wikoff WR, Duda RL, Hendrix RW, Johnson JE, Liljas L. 2003. The refined structure of a protein catenane: the HK97 bacteriophage capsid at 3.44 A resolution. J Mol Biol 334:885–899. doi: 10.1016/j.jmb.2003.09.035. [DOI] [PubMed] [Google Scholar]
- 84.Wikoff WR, Liljas L, Duda RL, Tsuruta H, Hendrix RW, Johnson JE. 2000. Topologically linked protein rings in the bacteriophage HK97 capsid. Science 289:2129–2133. doi: 10.1126/science.289.5487.2129. [DOI] [PubMed] [Google Scholar]
- 85.Fokine A, Chipman PR, Leiman PG, Mesyanzhinov VV, Rao VB, Rossmann MG. 2004. Molecular architecture of the prolate head of bacteriophage T4. Proc Natl Acad Sci U S A 101:6003–6008. doi: 10.1073/pnas.0400444101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fokine A, Leiman PG, Shneider MM, Ahvazi B, Boeshans KM, Steven AC, Black LW, Mesyanzhinov VV, Rossmann MG. 2005. Structural and functional similarities between the capsid proteins of bacteriophages T4 and HK97 point to a common ancestry. Proc Natl Acad Sci U S A 102:7163–7168. doi: 10.1073/pnas.0502164102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.