

Abstract
Domain swapping occurs when identical proteins exchange segments in reciprocal fashion. Natural swapping mechanisms remain poorly understood and engineered swapping has the potential for creating self-assembling biomaterials that encode for emergent functions. We demonstrate that induced swapping can be used to regulate function of a target protein. Swapping is triggered by inserting a ‘lever’ protein (ubiquitin) into one of four loops of the ribose binding protein (RBP) target. The lever splits the target, forcing RBP to refold in trans to generate swapped oligomers. Identical RBP-ubiquitin fusions form homo-swapped complexes with the ubiquitin domain acting as the hinge. Surprisingly, some pairs of non-identical fusions swap more efficiently with each other than they do with themselves. NMR experiments reveal that the hinge of these hetero-swapped complexes maps to a region of RBP distant from both ubiquitins. This design is expected to be applicable to other proteins to convert them into functional switches.
Graphical abstract
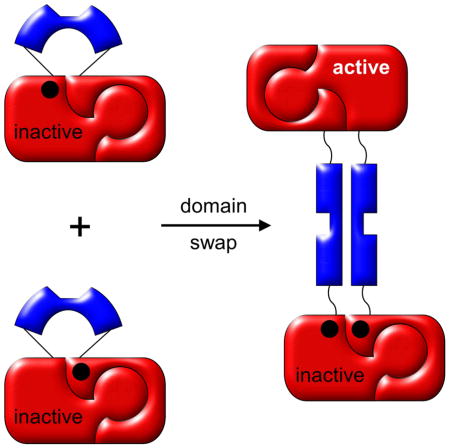
Domain swapping is one mechanism by which proteins evolved the ability to oligomerize. Natural swapping generates dimers that can have physiological advantages over their constitutive monomers (Cafaro et al., 1995; Di Donato et al., 1995; Gotte et al., 2012; Josephson et al., 2001; Liu and Eisenberg, 2002; Tsitsanou et al., 2013), as well as polymers that can contribute to pathogenic states (Bennett et al., 2006; Rousseau et al., 2012; van der Wel, 2012). Engineered swapping has the potential to create self-assembling materials that retain and integrate the activities of the parent proteins or encode for emergent functions. The new functionality that we explore in this study is the capability of using induced domain swapping as an on/off switch for protein activity.
Why do certain proteins naturally swap and how might others be induced to do so? The answers to these questions can be framed by Eisenberg’s original definition of a domain swap (Bennett et al., 1994; Bennett et al., 1995). For a protein to be classically swapped it must exist in equilibrium with its monomeric form, and the two structures should be identical except at the hinge region (typically a surface loop or turn) where the polypeptide segments cross over to generate the dimer or oligomer. By these criteria any protein is capable of swapping but relatively few do (Gronenborn, 2009; Huang et al., 2012). It stands to reason that the swapped protein must somehow be more stable than the non-swapped protein, despite the fact that they are stabilized by nearly identical interactions.
To explain swapping, researchers have focused on the properties of the hinge region in swapped vs. non-swapped states and on the concept of conformational strain in the latter. At minimum the hinge must be compatible, both energetically and sterically, with both structures. A compelling strategy for introducing a swap is to combine the above ideas by modifying the putative hinge region so that it’s strained in the monomeric conformation. Examples include shortening a surface turn (Green et al., 1995; Murray et al., 1998; Pica et al., 2013; Rousseau et al., 2001), placing residues in a turn that are forced to adopt unusual dihedral angles (Kuhlman et al., 2001) or are otherwise unfavorable for turn formation (Orlikowska et al., 2011), and replacing a surface loop with an α-helix that forms a coiled-coil (Reis et al., 2014). These alterations drove the turn or loop to adopt a more extended conformation that was better accommodated when it became the hinge region in a swapped structure.
We previously introduced the mutually exclusive folding mechanism—which proved to be an extreme version of the conformational strain model—by fusing a ‘lever’ protein into an internal position of a target protein. The fusion protein undergoes a tug-of-war in which the target compresses and unfolds the lever or the lever stretches and rips apart the target, depending on which domain is more stable (Cutler and Loh, 2007; Cutler et al., 2009; Ha et al., 2006; Radley et al., 2003). When the ubiquitin (Ub) lever was inserted into one of six surface loops of the barnase (Bn) target, strain was relieved in the fusion protein by the Ub domain unfolding the Bn domain, followed by intermolecular refolding of Bn domains to generate a domain-swapped, linear polymer as shown schematically in Fig. 1B (Ha et al., 2012). In this arrangement, the lever comprises the hinge and as such is effectively placed outside of the target structure. This mechanism offers a unique advantage over other approaches in that conformational stress has been shown to be proportional to the stability of the lever protein (Cutler and Loh, 2007). Swapping can therefore be controlled in theory by modulating lever stability using well-established principles (ligand binding, mutation, temperature/pH change, etc.).
Figure 1. Schematics of lever-target design and disulfide crosslinking and RBP activity tests for domain swapping.

(A) A lever protein (Ub, red) with a long (≥25 Å) N-to-C distance is inserted into a target protein (RBP, blue) at a point in a surface loop of the target (position 34 of RBP is shown as an example; red circle). Residues 1–34 of RBP are colored cyan. (B) The lever domain forces the target domain apart at the insertion site, separating it into two pieces that cannot refold within the same molecule. Each end of the bisected target can then refold via domain swap to generate a closed dimer (shown), longer closed oligomers, or a long linear polymer. (C) For the disulfide crosslinking assay, RU-Cys2 proteins are denatured and reduced, then allowed to refold and oxidize. If they dimerize via domain swapping, with the hinge region between the two Cys groups, then the disulfide bond will form intermolecularly, thereby covalently linking the two monomers. Similarly, if the protein swaps to generate longer polymers then the disulfide bonds will covalently link all subunits together allowing polymer size to be determined by SDS-PAGE. If the protein instead forms complexes by conventional interactions (i.e. surface binding without strand exchange) then the disulfide bonds will form intramolecularly and run as monomers on SDS-PAGE. Two identical RU-Cys2 variants are shown but the method works equally well for two different RU-Cys2 species. (D) For the ribose binding activity test, inactive NBM and CBM variants of the same RU (shown) or different RU variants are mixed, denatured, and allowed to refold. Domain swapping results in one inactive complex that contains both NBM+CBM mutations and one complex that’s free of mutations and fully active.
Here we test the generality of the lever-target design by fusing the Ub lever into one of four surface loops of the ribose binding protein (RBP) target (Fig. 1A). RBP was chosen because it’s relatively large (277 AA), offers numerous surface loops for lever insertion, and possesses a readily-assayable biological function (ribose binding) that we can attempt to switch on and off via domain swapping. To effect functional switching we knocked out ribose binding activity by introducing ribose binding mutations at positions either N-terminal or C-terminal to the Ub insertion sites. Either mutant alone cannot bind ribose; only by swapping with each other can function be restored.
We find that: (i) all fusion proteins homo-swap, suggesting that swapping is a general response to lever-induced strain; (ii) some combinations of fusion proteins swap with each other (hetero-swap) more efficiently than they swap with themselves (homo-swap), indicating that hetero-swapping generates different protein-protein binding interfaces that interact with increased propensity; (iii) NMR structural analysis of a hetero-swapped dimer reveals the existence of a preferred hinge region distant from either Ub domain that may represent the optimal crossover point for swapping of RBP; and (iv) induced domain swapping can be used to regulate function of RBP.
Results
Design strategy for fusion proteins
The lever-target design entails inserting the lever into the target at a site that’s compatible with folding of the latter in both its swapped and non-swapped states. Surface loops are typically selected for insertion sites, as placing the lever in a secondary structural element or at a buried position would likely so destabilize the target as to prevent it from folding. In order to introduce strain, the N-to-C distance of the lever should be at least twice as long as the Cα-Cα distance between terminal residues of the surface loop in the target. RBP has ten surface loops or turns that point away from the central ribose binding cleft, toward the “outside” of the molecule (Fig. 1A). We targeted four of these for Ub insertion: two on the N-terminal domain (centered at positions 34 and 60) and two on the C-terminal domain (centered at positions 125 and 210). The resulting RBP-Ub fusion proteins are designated RU34, RU60, RU125, and RU210 (see Table 1 for nomenclature of all constructs created for this study). The location of the insertion point at position 34 is shown in Fig. 1A and the resulting domain swap is illustrated in Fig. 1B.
Table 1.
Proteins created for this study.
| Variants | Additional mutations | Comments |
|---|---|---|
| RU34, RU60, RU125, RU210 | None | Ubiquitin inserted into RBP at indicated positions in RBP |
| RU34-Cys2, RU60-Cys2, RU125-Cys2, RU210-Cys2 | K28C+S245C | Forms inter-SS or intra-SS bonds when RBP domain is swapped or non-swapped, respectively |
| RU125-CyPet, RU125-YPet | CyPet or YPet fused to N- terminus of RU125 | FRET donor and acceptor groups |
| RU60-ΔN | Residues 1–59 deleted from RU60 | Can’t homo-swap; can only form hetero-swapped dimer |
| RU125-ΔC | Residues 125–277 deleted from RU125 | Can’t homo-swap; can only form hetero-swapped dimer |
| RU34-NBM, RU60-NBM, RU125-NBM, RU210-NBM | F17A+F18A | N-terminal ribose binding mutants |
| RU34-CBM, RU60-CBM, RU125-CBM, RU210-CBM | F217A+D218S | C-terminal ribose binding mutants |
Structural characterization by circular dichroism
To determine the effect of Ub insertion on the structure of RBP we performed circular dichroism (CD) wavelength scans of the RU-Cys2 variants (see below for description of Cys mutations). The CD signal arises mainly from the α-helices of the large RBP domain, consequently, CD data do not reveal the conformation of the smaller, mostly β-sheet Ub domain. CD spectra of all four RU-Cys2 constructs are similar to each other and to that of free RBP-Cys2, suggesting that the RBP domains are folded (Fig. S1). The CD signal is slightly more negative for free RBP-Cys2, possibly reflecting a small loss of helical structure in the RU-Cys2 proteins.
Disulfide crosslinking reveals homo- and hetero-swapping
We previously developed the native disulfide test to differentiate between swapped and non-swapped target domains and to determine the sizes of the swapped polymers (Ha et al., 2012). The strategy is to identify a pair of residues (K28 and S245) that, when mutated to Cys, readily form a disulfide bond in the native structure of wild-type (WT) RBP. K28C+S245C double mutants are designated by the -Cys2 suffix (Table 1). Because the native interactions present in a domain-swapped protein are essentially identical to those present in the non-swapped monomer (Bennett et al., 1994), the presence of an intermolecular disulfide bond (inter-SS) means that C28 has cross linked to C245 across a domain-swapped interface, locking the complex together with a covalent bond (Fig. 1C). By contrast, if RU-Cys2 proteins associate by conventional means (i.e. surface binding without exchange of polypeptide strands) then disulfide bonds will form intramolecularly (intra-SS) within each monomer. Denaturing but non-reducing polyacrylamide gel electrophoresis (SDS-PAGE) is used to distinguish inter-SS bonded oligomers from intra-SS bonded monomers. An inter-SS bond indicates: (i) that a swap has occurred, (ii) that the hinge is somewhere between C28 and C245 in sequence, and (iii) the approximate size of the swapped complex.
RBP-Cys2 and RU-Cys2 variants (20 μM) were denatured in GdnHCl, reduced with DTT, and refolded under oxidizing conditions. As expected, the majority (~95 %) of the RBP-Cys2 positive control runs as the intra-SS monomer on SDS-PAGE with a minor fraction appearing as an inter-SS dimer (Fig. 2A). Cys2 variants of RU34, RU60, RU125, and RU210 exhibit more extensive laddering with RU125-Cys2 displaying the most. These results indicate that all RU variants swap with themselves (homo-swap) and that RU125 has the greatest propensity for doing so. Inter-SS bonding along with our previous X-ray structure of the analogous barnase-Ub fusion protein (Ha et al., 2012) suggests that the structure of each RU complex consists of homo-swapped RBP domains with the hinge regions comprising the Ub domains at their various points of insertion, as shown for RU34 in Fig. 1B and Fig. 2B. Once an RU initially swaps to form a homodimer, it can either close on itself (Fig. 1B) or additional RU’s can homo-swap at either end to generate a linear polymer (Fig. 2B).
Figure 2. Pairwise mixing of RU-Cys2 variants reveals homo- and hetero-swapping.

(A) Non-reducing SDS-PAGE indicates that all RU-Cys2 variants swap with themselves (lanes labeled homo-swap), but some pairs swap more extensively with each other than with themselves (lanes labeled hetero-swap). The RU34+RU210 and RU60+RU210 lanes contain larger swapped polymers than the RU34, RU60, or RU210 alone lanes. By contrast, the RU34+RU60 lane shows a banding pattern similar to that of the sum of the RU34 and RU60 alone lanes, suggesting that RU34 and RU60 prefer to swap with themselves rather than with each other. (B) and (C) diagram the amino acid connectivities and hinge regions of the proposed homo- and hetero-swapped structures in panel A. The amino acid sequence of the RBP domain is shown as a thick colored arrow and the amino acid connectivity of the swapped complex is traced by a dashed white arrow. The hinge region is a dashed magenta arrow. In the hetero-swapped complex formed by RU34 and RU210 (panel C), the potential hinge region (magenta box) extends from positions 34 to 210. Once the RU34/RU210 hetero-swapped dimer forms, additional RU34 and RU210 proteins can add to the N-terminal and C-terminal ends (respectively; panel C) via the homo-swapping interactions depicted in panel B. (D) Addition of subunits to the heterodimer can be prevented by truncating the amino acids to either side of the Ub domain in the RU hetero-pair.
The classic domain swap generates a highly specific protein-protein binding interface. Indeed, one of its known purposes in nature is to establish a mechanism by which only identical proteins interact (Patel et al., 2003; Shapiro and Weis, 2009). With this model in mind we hypothesized that if two or more non-identical lever-target insertion variants were to be denatured and mixed, they would spontaneously self-segregate upon refolding by reforming the original homo-swapped oligomers. Surprisingly, several hetero-pairs produce more extensive laddering on SDS-PAGE than either of the individual proteins (Fig. 2A). This is most clearly seen for RU34+RU210 and RU60+RU210. Neither RU34 nor RU60 swap particularly efficiently by themselves, but when either is mixed with RU210 we observe the most widespread laddering of all combinations. By contrast, mixing RU34 and RU60 results in a banding pattern similar to the sum of the individual proteins.
The above findings demonstrate that RU34 and RU60 form heterocomplexes with RU210 but not with each other. One possible mechanism entails a hetero-swap as illustrated in Fig. 2C for RU34+RU210. Like a homo-swap, a hetero-swap produces a dimer consisting of a swapped RBP domain at the center with ends capable of further homo-swapping. The key difference with a hetero-pair is that a portion of the RBP sequence is duplicated (e.g. amino acids 34–210 for RU34 and RU210). This redundancy allows the hinge region of a hetero-swapped complex to be at either of the Ub insertion sites or anywhere in between. Thus, the proteins are free to choose the hinge that is the most energetically or sterically favorable (the ‘preferred hinge’), rather than be limited to the Ub domains as hinges in a homo-swap. By this reasoning, there does not seem to be a preferred hinge between residues 34 and 60, since RU34 and RU60 only homo-swap and do not hetero-swap (Fig. 2A). The observation that RU34+RU210 and RU60+RU210 swap more efficiently than RU34 or RU60 alone hints that a preferred hinge exists between residues 60 and 210.
We note that all lanes exhibit more bands than would be anticipated for a simple monomer-dimer-trimer etc. equilibrium. The likely reason is that each oligomeric species can be linear or closed which doubles the number of expected bands. In addition, RBP is an extremely stable protein that resists complete SDS denaturation, and residual structure within the oligomers may slightly shift their mobility.
Domain swapping monitored by FRET
To further test for hetero-swapping we developed a FRET-based assay in which CyPet or YPet fluorescent proteins (Nguyen and Daugherty, 2005) were fused to the N-terminus of RU125 (Table 1). Folded RU125-CyPet and RU125-YPet were mixed (20 μM each, in native buffer) in the presence of different concentrations of unlabeled RU34, RU60, RU125, or RU210 and the solutions were allowed to equilibrate under native conditions (0.5 M GdnHCl, 37 °C, 2 days) so as not to unfold CyPet or YPet. If the proteins only homo-swap the RU125-CyPet will only bind to RU125-YPet and FRET efficiency will remain constant at all concentrations of unlabeled competitor, except for the positive control (unlabeled RU125). Instead, we observe that FRET efficiency decreases with increasing concentration of all RU variants (Fig. 3). RU125 exhibits a slight positive preference for swapping with RU34 and RU60 (compared to itself), and a considerable negative preference for swapping with RU210. According to the model in Fig. 2C these findings suggest that a preferred hinge exists between residues 60 and 125.
Figure 3. FRET competition assay for domain swapping.
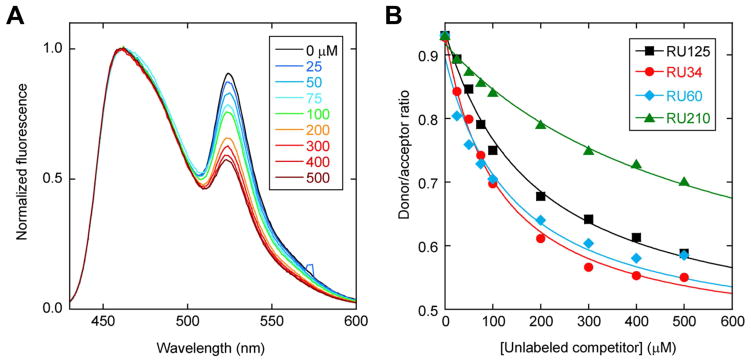
(A) Homo-swapping control in which RU125-CyPet and RU125-YPet are challenged with unlabeled RU125 competitor (concentrations in inset). Fluorescence spectra are normalized to CyPet emission for clarity. (B) The same data in panel A are plotted as CyPet/YPet emission ratio, with hetero-swapping results obtained with unlabeled RU34, RU60, and RU210 competitors included. Lines are best fit to the one-site binding equation.
NMR structural analysis of a hetero-swapped complex
To structurally validate the domain swap model and to identify the location of the hinge, we created two truncated RU proteins that reproduce the swapping interaction of their full-length counterparts but can only form dimers. The main challenge for structural determination is that RU proteins are large (360 AA) and self-assemble into oligomers of mixed composition and length, making crystallization difficult and NMR analysis out of the question. Our solution was to delete the residues N-terminal to the Ub domain in one construct and delete the residues C-terminal to the Ub domain in a second construct (Fig. 2D). These truncations preclude the proteins from homodimerizing as well as prevent additional subunits from adding to the heterodimer as they do in Fig. 2C. We deleted residues 1–59 from RU60 (RU60-ΔN) and residues 125–277 from RU125 (RU125-ΔC) (Table 1) so as to create the smallest possible dimer for NMR analysis (56,509 Da).
We labeled RU60-ΔN and RU125-ΔC uniformly with 15N and recorded heteronuclear single-quantum coherence (HSQC) spectra of each protein in the absence of the other. 15N-RU60-ΔN alone exhibits relatively well-dispersed peaks with some degeneracy toward the center of the spectrum, consistent with a structure composed of mostly folded but some unfolded regions (Fig. S2A). By contrast, 15N-RU125-ΔC displays a small set of intense, resolved peaks with a large cluster of broad, overlapped peaks near the center of the spectrum (Fig. S2B). This same set of intense, resolved peaks is observed in the spectrum of 15N-RU60-ΔN, and moreover, both sets align with the majority of peaks in the WT Ub spectrum (Fig. S3). Thus, the Ub domain is folded and native-like in both RU constructs, while the truncated RBP domain appears to be mostly unstructured in RU125-ΔC.
To test for binding we mixed each labeled protein with its unlabeled partner. Nearly all of the non-Ub resonances of 15N-RU60-ΔN shift on addition of unlabeled RU125-ΔC (Fig. S2A), and nearly all of the non-Ub peaks of 15N-RU125-ΔC shift on addition of unlabeled RU60-ΔN (Fig. S2B). The Ub peaks in both cases remain stationary (Fig. S3). Most of the expected RBP peaks (~125 and ~217 in the 15N-RU125-ΔC and 15N-RU60-ΔN labeled samples, respectively) are now observed in the complex of RU60-ΔN+RU125-ΔC. These results indicate that: (i) the two proteins bind; (ii) binding involves significant folding of RU125-ΔC; and (iii) the complex is likely a dimer, as any higher-order oligomer would exceed 80,000 Da and would result in very broad peaks.
To assess the structure of the dimer we overlaid the HSQC spectra of WT 15N-RBP (red) and WT 15N-Ub (black) on the spectra of 15N-RU60-ΔN (blue) and 15N-RU125-ΔC (green) (Fig. 4). It is immediately apparent that the two labeled RU constructs alone align poorly with WT RBP (Fig. 4A), but the majority of peaks shift into alignment in the complex (Fig. 4B). Nearly all of the resolved red peaks are covered by either blue or green peaks in Fig. 4B, but not by both. This finding suggests that the RBP fragments of RU60-ΔN and RU125-ΔC combine to form a structure similar to that of WT RBP, and that there is no region in the reconstituted RBP domain in which the same residue from RU60-ΔN and RU125-ΔC adopts the WT RBP conformation. This mutual exclusivity argues that there is only a single major hinge in the dimer. For example, if half the dimers formed by crossing over at position 60 and the other half by crossing over at position 125, then residues 60–125 from RU60-ΔN would take on the WT RBP structure in one population and the same residues from RU125-ΔC would adopt the WT RBP conformation in the other population.
Figure 4. The hinge region of the hetero-swapped dimer maps to residues 94–101.

Panels A and B reveal that RU60-ΔN and RU125-ΔC bind to regenerate native-like RBP structure. (A) HSQC cross peaks of 15N-RU60-ΔN alone (blue) and 15N-RU125-ΔC alone (green) show little superposition with resonances of WT RBP (red). (B) When 15N-RU60-ΔN is mixed with unlabeled RU125-ΔC (blue), and when 15N-RU125-ΔC is mixed with unlabeled RU60-ΔN (green), most of the blue and green peaks shift into alignment with the WT RBP peaks (red). WT Ub resonances are colored black; these black peaks overlay well with blue and green peaks in both panel A and panel B, indicating that Ub is folded and native-like in all samples. (C) The process of mapping the hinge region is demonstrated using a zoomed region of panel B. Resonance assignments of WT RBP are indicated with residue numbers colored blue if the WT RBP peak superimposes with a peak from 15N-RU60-ΔN (complexed with unlabeled RU125-ΔC), green if the WT RBP peak superimposes with a peak from 15N-RU125-ΔC (complexed with unlabeled RU60-ΔN), and gray if no superposition is observed with either protein. Ub assignments are in black.
The finding that nearly all peaks in the dimer superimpose with either WT RBP or WT Ub enables the hinge region to be deduced by chemical shift change using cross assignments. We assigned the 15N, 13C, and 1H backbone resonances of WT RBP (30,944 Da). Figure 4C demonstrates the strategy for mapping the hinge region using a zoomed portion of Fig. 4B for clarity. The WT RBP peaks (red) are denoted by their residue number, colored according to whether they superimpose with resonances from 15N-RU60-ΔN (blue), 15N-RU125-ΔC (green), or neither (gray). The hinge (being the region of the domain-swapped structure that’s most different from WT RBP) is expected to map to a segment of gray residues, but not to any gray segment. It must be flanked by a trail of green residues at its N-terminus and a trail of blue residues at its C-terminus. This pattern signifies that the chain comprising the RBP domain has crossed over from RU125-ΔC to RU60-ΔN (Fig. 2D). A gray section embedded in a stretch of green residues or a gray section surrounded by blue residues cannot be the hinge. In the area shown in Fig. 4C, all WT RBP residues are green from 6–69 and blue from 128–215, revealing the hinge to be between residues 69 and 128.
The chemical shift mapping data, summarized in Fig. 5, rule out the hinge region being at either Ub insertion site. The RBP domain instead opts to swap by crossing over somewhere between residues 94–101 (magenta), well away from the Ub domains. These residues comprise an extended surface loop that connects two β-strands in the β-sheet of the N-terminal lobe of RBP (Fig. 5). Interestingly, the longest stretch of gray residues, which implies the region of greatest difference between WT RBP and the RBP domain of the swapped dimer, maps to amino acids 71–82. These residues form an α-helix that abuts the putative hinge and chemical shifts in this helix may be perturbed upon swapping by virtue of this contact.
Figure 5. Location of the hinge region on RBP.

The structure of WT RBP is shown at top and the amino acid sequence at bottom. Color coding is the same as in Fig. 4; namely, green and blue indicate backbone resonances of WT RBP that superimpose with those of 15N-RU125-ΔC and 15N-RU60-ΔN, respectively, gray denotes resonances of WT RBP that do not superimpose with any peak from either 15N-RU125-ΔC or 15N-RU60-ΔN, and black indicates lack of assignment or Pro. The potential hinge (magenta) is located between residues 94–101, in the longest surface loop on RBP.
Induced domain swapping as a mechanism for functional switching
As final proof of the domain swap mechanism, and to test whether it can be used to regulate the function of RBP, we knocked out ribose binding activity of each RU construct in such a way that function can only be restored by a domain swap. We generated ribose binding knockout variants by creating one set of RU proteins with binding mutations (F17A+F18A) N-terminal to the Ub insertion sites, and a second set with binding mutations (F217A+D218S) C-terminal to the Ub insertion sites (Boas and Harbury, 2008; Ha et al., 2013; Vercillo et al., 2007). Variants containing the N-terminal or C-terminal binding mutations are designated with the -NBM and -CBM suffixes (Table 2). Similar to the native disulfide experiment, the only way that the functional RBP amino acid sequence can be restored is if two RBP chains cross over with the hinge region somewhere between residues 18 and 217 (Fig. 1D). The product of this swap contains one RBP with the WT sequence and one RBP with the NBM/CBM double-knockout sequence (Fig. 1D).
Table 2. Activation of ribose binding activity by domain swapping.
FA and FA,max values are shown with FA,max in parentheses. Homo-pairs appear on the diagonal, positive binding orientation hetero-pairs appear above the diagonal, and negative binding orientation hetero-pairs appear below the diagonal. Errors are standard deviations of three measurements.
| RU34-CBM | RU60-CBM | RU125-CBM | RU210-CBM | |
|---|---|---|---|---|
| RU34-NBM | 0.073 ± 0.0080 (0.11 ± 0.0080) | 0.18 ± 0.0081 (0.16 ± 0.0082) | 0.30 ± 0.0087 (0.37 ± 0.0090) | 0.23 ± 0.0071 (0.41 ± 0.0074) |
| RU60-NBM | 0.011 ± 0.0094 (0.028 ± 0.0094) | 0.070 ± 0.0087 (0.075 ± 0.0086) | 0.21 ± 0.011 (0.27 ± 0.011) | 0.17 ± 0.010 (0.32 ± 0.011) |
| RU125-NBM | 0.0055 ± 0.0092 (0.010 ± 0.010) | 0.0099 ± 0.0094 (−0.0037 ± 0.0093) | 0.18 ± 0.0092 (0.17 ± 0.0092) | 0.095 ± 0.0091 (0.32 ± 0.0097) |
| RU210-NBM | 0.0050 ± 0.0085 (0.010 ± 0.0091) | 0.0092 ± 0.0091 (0.010 ± 0.0081) | 0.0087 ± 0.0093 (0.025 ± 0.0091) | 0.049 ± 0.0078 (0.070 ± 0.0082) |
All possible homo- and hetero-pairs of RU-NBM and RU-CBM were mixed, denatured, and refolded. The fraction of molecules competent to bind ribose was then determined by a kinetic unfolding assay monitored by CD. Upon addition of 7 M GdnHCl, ribose-free RBP denatures within the ~30 s dead time of CD experiments whereas ribose-bound RBP unfolds with a half-time of hours (Fig. S4). The fraction of active RBP (FA) can then be calculated using the equation , where θI is the initial ellipticity and θN and θU are the ellipticities of the native and unfolded states, respectively. As a positive control we added ribose to a second set of samples prior to denaturation so that the binding energy of ribose helps drive domain swapping. The fraction of active RBP obtained under positive control conditions (FA,max) can be considered the maximum value for each RU combination under the assay conditions. We note that the theoretical maximum of FA is 0.5 even if 100 % of the molecules swap, since only up to one-half of the RBP domains will contain neither NBM nor CBM mutations.
Ribose binding results are summarized in Table 2 with the homo-pairs appearing on the diagonal (light gray) and the hetero-pairs above the diagonal (white). Ribose binding is restored in all homo-pairs with RU125-NBM+RU125-CBM showing the greatest activity (FA = 0.18). The remaining three bind ribose to significant but lesser extents. For the hetero-pairs, all six exhibit substantial activation upon mixing, with FA values ranging from 0.095 to 0.30. FA generally correlates well with the extent of inter-SS bonded species in Fig. 2A. One exception is RU34-NBM+RU60-CBM which is as active as RU60-NBM+RU210-CBM but does not oligomerize as extensively in the native disulfide assay.
As a negative control we reversed the orientation of the binding mutations such that hetero-swapping produces the NBM/CBM double knockout mutant instead of the WT RBP sequence. Results of the ribose binding assay for the reversed binding mutants are shown below the diagonal (dark gray) in Table 2. In agreement with the structural model, FA and FA,max values of all six hetero-pairs are close to zero, indicating that few if any RBP domains in any of the RU polymers are capable of binding ribose in the negative binding orientation.
Discussion
Structural model of domain swapping
The combination of ribose binding, NMR, and disulfide cross linking experiments provide conclusive evidence for our domain swap model. The only way that RU-NBM and RU-CBM variants can regain biological activity is by generating a native, WT binding site from the two proteins. NMR experiments demonstrate that RU60-ΔN and RU125-ΔC form a heterodimer in which the native RBP structure is largely reconstituted. This structure forms the core of the domain swapped oligomer proposed in Fig. 2C. The sole reasonable scenario that takes into account these results, plus the oligomerization and native disulfide bonding observed in Fig. 2A, is a domain swap.
According to our model, the lever protein introduces conformational strain into the monomeric target which is relieved when the target unfolds and refolds as a swapped dimer or oligomer. That this phenomenon occurs when Ub is inserted into four sites in RBP (and in six sites in barnase (Ha et al., 2012), suggests that swapping is a common response when a lever is inserted into many, if not most, surface loops of a target protein.
Although RU34, RU60, RU125, and RU210 all homo-swap, the sizes of the resulting oligomers vary depending on the site of Ub insertion (Fig. 2A). We speculate that the extent to which the target protein swaps is determined at least in part by steric crowding at the hinge region. When two identical target-lever fusion proteins swap, the levers (being the hinges) necessarily become close to each other and to the target domains. One particular combination of lever and insertion site (e.g. RU125) may tolerate this arrangement. For example, the positioning of the Ub hinge domains in our X-ray structure of a barnase-Ub domain fusion protein, while close, is sterically compatible with the juxtaposition of successive swapped barnase domains to generate a polymer. Depending on its size and shape, another lever at the same position, or the same lever at a different position (e.g. RU34, RU60, and RU210), may result in steric clash that limits the extent of swapping. It is difficult to predict the extent of interdomain crowding based on simple inspection of lever and target structures, but it should be possible to do so using molecular simulations of simplified protein models, as Chong and coworkers did for barnase-Ub fusion proteins (Cutler et al., 2009; Mills and Chong, 2011).
The most surprising discovery from this study is that some combinations of proteins prefer to hetero-swap while others prefer to homo-swap. Given the recognized importance of the hinge region to swapping, this observation implies that a hetero-swapped complex has found a preferred hinge that is more favorable than those employed by the corresponding homo-swapped complexes. RU34+RU210 and RU60+RU210 display the largest increase in hetero-swapping compared to homo-swapping by SDS-PAGE (Fig. 2A), and RU34-NBM+RU125-CBM exhibits the greatest ribose binding activity of all pairs tested (Table 2). These observations suggest that at least one preferred hinge resides between residues 60–125. Using RU60-ΔN and RU125-ΔC as a model of the hetero-swapped dimer the NMR data map the hinge region to an extended loop (residues 94–101) that connects two β-strands (Fig. 5). The ArchDB database (Bonet et al., 2014) classifies this loop to be the second longest in the nearly identical E. coli RBP, and likely the longest loop in the present T. tengcongensis RBP. It may be that the longest loops make the most favorable hinges because they afford maximal flexibility and accommodation of steric clashes.
The finding of a preferred hinge that is distant from the Ub insertion sites changes the role of the lever in our mechanism and has implications toward domain swapping in natural proteins. We originally conceived the lever’s first role as tearing the target protein in two pieces at the point of insertion and physically preventing them from refolding in the same molecule. The lever’s second role is to serve as the hinge region for subsequent swapping. This must be true for homo-swapped complexes because if the hinge were elsewhere the lever would remain internal to the 3D structure of the target domain, and conformational strain would not be relieved. Our results argue that the second role needs to be expanded. If levers are inserted into non-identical positions, then the chains can cross over at either lever domain or anywhere in between. Thus, the role of the lever is to destabilize the monomeric target as originally envisioned, but then to simply allow the target to swap at the best position available given the placement of the levers. We have identified one such preferred hinge between residues 94–101, in one of the longest loops in RBP. Based on all available data it seems reasonable to speculate that this may represent the best possible hinge for domain swapping of RBP, and the site that RBP would choose if it were to naturally swap. This hypothesis can be tested by determining the structure of the RU34-ΔN+RU210-ΔC complex and asking whether the same hinge is chosen, although the increased size of the dimer may preclude the use of NMR. Structural investigation of the dimer of WT RBP-Cys2 observed on SDS-PAGE may also reveal whether it’s swapped, and if so, the location of the hinge.
Induced domain swapping as an on/off switch
The main attraction of our mechanism is that levers and targets can potentially be combined in a modular fashion, integrating the properties of both proteins and generating emergent properties. One such property may be the ability to turn on and off the function of the target using a stimulus to which the lever responds, such as ligand binding or change in pH/temperature. We demonstrate here that RBP function can be regulated by domain swapping of non-functional monomers. We previously established that swapping is in turn controlled by the stability of the lever domain: when the lever is unfolded, or when it’s stable but joined to the target by long, structureless linker peptides, conformational strain is decoupled and the target does not swap (Cutler and Loh, 2007; Cutler et al., 2009). It is therefore possible in theory to trigger domain swapping, and hence biological activity of the target, by stabilization of a suitable lever through binding of a small molecule or protein ligand. While this mechanism is new it is not without analogy. It is reminiscent of that of receptor tyrosine kinases, in which ligand binding causes the inactive receptor monomers to dimerize and activate each other. We are currently testing the feasibility of this design with other lever/target combinations.
Significance
Prevalent in nature yet poorly understood, domain swapping provides protein engineers with a heretofore underutilized tool to manipulate protein structure, function, and self-assembly. We demonstrate here that domain swapping can be rationally introduced into a target protein, and that by doing so the biological activity of that protein can be switched on and off. The principles that underlie our mechanism draw from relatively well-understood properties of protein structure and stability. It therefore seems possible to convert many types of proteins into molecular switches using induced domain swapping.
Experimental Procedures
Gene construction and protein purification
Human Ub DNA sequences were inserted into the specified position of the Thermoanaerobacter tengcongensis RBP gene according to the procedure of Geiser at al. (Geiser et al., 2001). All genes were fully sequenced. Proteins were expressed in E. coli BL21(DE3) with IPTG induction occurring at 20 °C for 12–15 h. For purification, cell pellets were resuspended in 10 mM Tris (pH 7.5), 0.3 M NaCl, and lysed using a small amount of lysozyme followed by sonication. The soluble fraction of the lysate was loaded onto a nickel-NTA column (Bio-Rad) and washed extensively with 10 mM Tris (pH 7.5), 6 M GdnHCl to denature the proteins and remove bound ribose. Proteins were eluted using 6 M GdnHCl (pH 3.8), dialyzed against double-distilled H2O, and lyophilized. -NBM, -CBM, -CyPet, and -YPet variants were purified as above except GdnHCl was omitted from the column washing and elution steps. All proteins were judged to be >95 % pure by SDS-PAGE with coomassie staining.
Disulfide experiments
RBP is extremely stable and slow to unfold (Ha et al., 2013). For this reason we denatured the proteins by adding GdnHCl and/or heating to 90 °C prior to disulfide and ribose binding experiments. RU-Cys2 variants (20 μM) were denatured in 6 M GdnHCl, reduced with 10 mM DTT, and refolded under oxidizing conditions by dialyzing against 20 mM sodium citrate (pH 5.0), 0.15 M NaCl for 3 h, then against 20 mM Tris (pH 8.0), 0.15 M NaCl for an additional 16–20 h at room temperature. Proteins were loaded onto 5–15 % gradient SDS-PAGE gels (Bio-Rad) and gels were stained with coomassie brilliant blue.
FRET experiments
RU125-CyPet and RU125-YPet were prepared separately in 20 mM sodium phosphate (pH 7.0), 0.15 M NaCl and mixed to a final concentration of 20 μM each in the same buffer. Unlabeled RU proteins (prepared in the same buffer) were added at the indicated concentrations, GdnHCl was added to a final concentration of 0.5 M, and samples were incubated at 37 °C for 48 h. Samples were diluted 1:50 and fluorescence spectra were recorded with excitation at the CyPet wavelength (433 nm) on a Horiba Fluoromax-4 instrument. FRET data are expressed as the ratio of CyPet emission (460 nm) to YPet emission (525 nm).
NMR experiments
Proteins were expressed using standard minimal media protocols with 13C-glucose and/or 15NH4Cl (both 99 atom %) as the sole sources of carbon and nitrogen. Protein samples were prepared in 20 mM sodium phosphate (pH 7.0), 0.15 M NaCl, 5 % 2H2O. Protein concentrations were as follows: 1.24 mM WT 15N/13C-RBP, 1.5 mM 15N-Ub, 0.25 mM 15N-RU60-ΔN and 15N-RU125-ΔC, and 0.30 mM unlabeled RU60-ΔN and RU125-ΔC. All RBP and RU samples contained 5 mM ribose. NMR spectra were acquired at 60 °C on a Bruker Avance III HD 800 MHz spectrometer equipped with a 5 mm TCI triple resonance cryogenic probe. 15N, 13C, and 1H backbone assignments of WT RBP were determined using HSQC, HNCACB (Wittekind and Mueller, 1993), and CBCACONH (Grzesiek and Bax, 1992) experiments. Spectra were processed with NMRPipe (Delaglio et al., 1995) and analyzed using CCPNMR (Vranken et al., 2005).
Ribose binding activity assays
RU-NBM and RU-CBM (20 μM each) were mixed in 10 mM sodium phosphate (pH 7.0), heated at 90 °C for 5 m with or without 5 mM ribose, and cooled to room temperature over the course of ~1 h. The total concentration of the RU34-NBM + RU210-CBM mixture was approximately half that of the others due to precipitation during heating. Samples were transferred to a 22 °C bath and CD data were collected at 22 °C on an Aviv model 420 instrument. 1/50 volume ribose (5 mM final concentration) or water was added to samples that were heated without or with ribose (respectively), 5 m prior to initiating unfolding by 6.1-fold dilution into 8.3 M GdnHCl, 25 mM sodium phosphate (pH 7.0). CD ellipticity was recorded at 226 nm.
Supplementary Material
Highlights.
We have developed a technology for causing monomeric proteins to domain swap.
Engineered swapping is a novel mechanism for regulating protein function.
Non-identical fusion proteins can swap more efficiently than identical proteins.
Non-identical proteins swap by crossing over at a unique, ‘hot-spot’ hinge.
Acknowledgments
We thank A. Blanden for discussions and D. Kiemle for assistance with NMR experiments. This work was supported by NIH grants R01 GM069755 and R01 GM115762 to S.N.L. and NIH shared instrumentation grant 1S10OD012254 for purchase of the 800 MHz NMR spectrometer.
Footnotes
Author contributions
J.-H.H., J.M.K., and S.N.L designed the experiments. J.-H.H. constructed all genes and carried out ribose binding assays. J.M.K. performed NMR experiments and analyzed NMR data along with C.A.C. N.W.-K. conducted FRET experiments. S.N.L carried out disulfide crosslinking assays and wrote the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bennett MJ, Choe S, Eisenberg D. Domain swapping: entangling alliances between proteins. Proc Natl Acad Sci U S A. 1994;91:3127–3131. doi: 10.1073/pnas.91.8.3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MJ, Sawaya MR, Eisenberg D. Deposition diseases and 3D domain swapping. Structure. 2006;14:811–824. doi: 10.1016/j.str.2006.03.011. [DOI] [PubMed] [Google Scholar]
- Bennett MJ, Schlunegger MP, Eisenberg D. 3D domain swapping: a mechanism for oligomer assembly. Protein Sci. 1995;4:2455–2468. doi: 10.1002/pro.5560041202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boas FE, Harbury PB. Design of protein-ligand binding based on the molecular-mechanics energy model. J Mol Biol. 2008;380:415–424. doi: 10.1016/j.jmb.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonet J, Planas-Iglesias J, Garcia-Garcia J, Marin-Lopez MA, Fernandez-Fuentes N, Oliva B. ArchDB 2014: structural classification of loops in proteins. Nucleic Acids Res. 2014;42:D315–319. doi: 10.1093/nar/gkt1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cafaro V, De Lorenzo C, Piccoli R, Bracale A, Mastronicola MR, Di Donato A, D’Alessio G. The antitumor action of seminal ribonuclease and its quaternary conformations. FEBS Lett. 1995;359:31–34. doi: 10.1016/0014-5793(94)01450-f. [DOI] [PubMed] [Google Scholar]
- Cutler T, Loh SN. Thermodynamic analysis of an antagonistic folding-unfolding equilibrium between two protein domains. J Mol Biol. 2007;371:308–316. doi: 10.1016/j.jmb.2007.05.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutler T, Mills BM, Lubin DJ, Chong LT, Loh SN. Effect of interdomain linker length on an antagonistic folding-unfolding equilibrium between two protein domains. J Mol Biol. 2009;386:854–868. doi: 10.1016/j.jmb.2008.10.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Di Donato A, Cafaro V, Romeo I, D’Alessio G. Hints on the evolutionary design of a dimeric RNase with special bioactions. Protein Sci. 1995;4:1470–1477. doi: 10.1002/pro.5560040804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiser M, Cebe R, Drewello D, Schmitz R. Integration of PCR fragments at any specific site within cloning vectors without the use of restriction enzymes and DNA ligase. Biotechniques. 2001;31:88–92. doi: 10.2144/01311st05. [DOI] [PubMed] [Google Scholar]
- Gotte G, Mahmoud Helmy A, Ercole C, Spadaccini R, Laurents DV, Donadelli M, Picone D. Double domain swapping in bovine seminal RNase: formation of distinct N- and C-swapped tetramers and multimers with increasing biological activities. PLoS One. 2012;7:e46804. doi: 10.1371/journal.pone.0046804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green SM, Gittis AG, Meeker AK, Lattman EE. One-step evolution of a dimer from a monomeric protein. Nat Struct Biol. 1995;2:746–751. doi: 10.1038/nsb0995-746. [DOI] [PubMed] [Google Scholar]
- Gronenborn AM. Protein acrobatics in pairs--dimerization via domain swapping. Curr Opin Struct Biol. 2009;19:39–49. doi: 10.1016/j.sbi.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzesiek S, Bax A. Correlating backbone amide and side chain resonances in larger proteins by multiple relayed triple resonance NMR. J Am Chem Soc. 1992;114:6291–6293. [Google Scholar]
- Ha J-H, Butler JS, Mitrea DM, Loh SN. Modular enzyme design: regulation by mutually exclusive protein folding. J Mol Biol. 2006;357:1058–1062. doi: 10.1016/j.jmb.2006.01.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha J-H, Karchin JM, Walker-Kopp N, Huang L-S, Berry EA, Loh SN. Engineering domain-swapped binding interfaces by mutually exclusive folding. J Mol Biol. 2012;416:495–502. doi: 10.1016/j.jmb.2011.12.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha JH, Shinsky SA, Loh SN. Stepwise conversion of a binding protein to a fluorescent switch: application to Thermoanaerobacter tengcongensis ribose binding protein. Biochemistry. 2013;52:600–612. doi: 10.1021/bi301105u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Cao H, Liu Z. Three-dimensional domain swapping in the protein structure space. Proteins. 2012;80:1610–1619. doi: 10.1002/prot.24055. [DOI] [PubMed] [Google Scholar]
- Josephson K, Logsdon NJ, Walter MR. Crystal structure of the IL-10/IL-10R1 complex reveals a shared receptor binding site. Immunity. 2001;15:35–46. doi: 10.1016/s1074-7613(01)00169-8. [DOI] [PubMed] [Google Scholar]
- Kuhlman B, O’Neill JW, Kim DE, Zhang KY, Baker D. Conversion of monomeric protein L to an obligate dimer by computational protein design. Proc Natl Acad Sci USA. 2001;98:10687–10691. doi: 10.1073/pnas.181354398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Eisenberg D. 3D domain swapping: as domains continue to swap. Protein Sci. 2002;11:1285–1299. doi: 10.1110/ps.0201402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills BM, Chong LT. Molecular simulations of mutually exclusive folding in a two-domain protein switch. Biophys J. 2011;100:756–764. doi: 10.1016/j.bpj.2010.12.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray AJ, Head JG, Barker JJ, Brady RL. Engineering an intertwined form of CD2 for stability and assembly. Nat Struct Biol. 1998;5:778–782. doi: 10.1038/1816. [DOI] [PubMed] [Google Scholar]
- Nguyen AW, Daugherty PS. Evolutionary optimization of fluorescent proteins for intracellular FRET. Nat Biotechnol. 2005;23:355–360. doi: 10.1038/nbt1066. [DOI] [PubMed] [Google Scholar]
- Orlikowska M, Jankowska E, Kolodziejczyk R, Jaskolski M, Szymanska A. Hinge-loop mutation can be used to control 3D domain swapping and amyloidogenesis of human cystatin C. J Struct Biol. 2011;173:406–413. doi: 10.1016/j.jsb.2010.11.009. [DOI] [PubMed] [Google Scholar]
- Patel SD, Chen CP, Bahna F, Honig B, Shapiro L. Cadherin-mediated cell-cell adhesion: sticking together as a family. Curr Opin Struct Biol. 2003;13:690–698. doi: 10.1016/j.sbi.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Pica A, Merlino A, Buell AK, Knowles TP, Pizzo E, D’Alessio G, Sica F, Mazzarella L. Three-dimensional domain swapping and supramolecular protein assembly: insights from the X-ray structure of a dimeric swapped variant of human pancreatic RNase. Acta Crystallogr D Biol Crystallogr. 2013;69:2116–2123. doi: 10.1107/S0907444913020507. [DOI] [PubMed] [Google Scholar]
- Radley TL, Markowska AI, Bettinger BT, Ha J-H, Loh SN. Allosteric switching by mutually exclusive folding of protein domains. J Mol Biol. 2003;332:529–536. doi: 10.1016/s0022-2836(03)00925-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis JM, Burns DC, Woolley GA. Optical Control of Protein-Protein Interactions via Blue Light-Induced Domain Swapping. Biochemistry. 2014;53:5008–5016. doi: 10.1021/bi500622x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau F, Schymkowitz J, Itzhaki LS. Implications of 3D domain swapping for protein folding, misfolding and function. Adv Exp Med Biol. 2012;747:137–152. doi: 10.1007/978-1-4614-3229-6_9. [DOI] [PubMed] [Google Scholar]
- Rousseau F, Schymkowitz JW, Wilkinson HR, Itzhaki LS. Three-dimensional domain swapping in p13suc1 occurs in the unfolded state and is controlled by conserved proline residues. Proc Natl Acad Sci U S A. 2001;98:5596–5601. doi: 10.1073/pnas.101542098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro L, Weis WI. Structure and biochemistry of cadherins and catenins. Cold Spring Harb Perspect Biol. 2009;1:a003053. doi: 10.1101/cshperspect.a003053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsitsanou KE, et al. Crystal and solution studies of the “Plus-C” odorant-binding protein 48 from Anopheles gambiae: control of binding specificity through three-dimensional domain swapping. J Biol Chem. 2013;288:33427–33438. doi: 10.1074/jbc.M113.505289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Wel PC. Domain swapping and amyloid fibril conformation. Prion. 2012;6:211–216. doi: 10.4161/pri.18987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vercillo NC, Herald KJ, Fox JM, Der BS, Dattelbaum JD. Analysis of ligand binding to a ribose biosensor using site-directed mutagenesis and fluorescence spectroscopy. Protein Sci. 2007;16:362–368. doi: 10.1110/ps.062595707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vranken WF, et al. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins. 2005;59:687–696. doi: 10.1002/prot.20449. [DOI] [PubMed] [Google Scholar]
- Wittekind M, Mueller L. HNCACB, a high-sensitivity 3D NMR experiment to correlate amide-proton and nitrogen resonances with the alpha- and beta-carbon resonances in proteins. J Magn Reson. 1993;B101:201–205. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.