

Abstract
Cytotoxic therapeutic monoclonal antibodies (mAbs) often mediate target cell-killing by eliciting immune effector functions via Fc region interactions with cellular and humoral components of the immune system. Key functions include antibody-dependent cell-mediated cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), and complement-dependent cytotoxicity (CDC). However, there has been increased appreciation that along with cell-killing functions, the induction of antibody-dependent cytokine release (ADCR) can also influence disease microenvironments and therapeutic outcomes. Historically, most Fc engineering approaches have been aimed toward modulating ADCC, ADCP, or CDC. In the present study, we describe an Fc engineering approach that, while not resulting in impaired ADCC or ADCP, profoundly affects ADCR. As such, when peripheral blood mononuclear cells are used as effector cells against mAb-opsonized tumor cells, the described mAb variants elicit a similar profile and quantity of cytokines as IgG1. In contrast, although the variants elicit similar levels of tumor cell-killing as IgG1 with macrophage effector cells, the variants do not elicit macrophage-mediated ADCR against mAb-opsonized tumor cells. This study demonstrates that Fc engineering approaches can be employed to uncouple macrophage-mediated phagocytic and subsequent cell-killing functions from cytokine release.
Keywords: Fc gamma receptors, cytokine release, interferon gamma, interleukin 10, monocyte-derived macrophages, natural killer cells, antibody-dependent cellular phagocytosis
Abbreviations
- mAbs
monoclonal antibodies
- ADCC
antibody-dependent cell-mediated cytotoxicity
- ADCP
antibody-dependent cellular phagocytosis
- CDC
complement-dependent cytotoxicity
- ADCR
antibody-dependent cytokine release
- FcγR
Fc gamma receptor
- PBMC
peripheral blood mononuclear cell
- NK
natural killer
- IFN
interferon
- IL
interleukin
- TNF
tumor necrosis factor
- APCs
antigen-presenting cells
- DC
dendritic cell
Introduction
There are multiple mechanisms by which cytotoxic monoclonal antibodies (mAbs) function to destroy targeted cells, and a key goal for pre-clinical studies is to provide insights into which mechanisms influence efficacy. Commonly, in vitro studies focus on determining the antibody-dependent cell-mediated cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), or complement-dependent cytotoxicity (CDC) capacity of mAbs intended for therapeutic intervention. Innate immune effector cells mediate ADCC and ADCP, whereas CDC is mediated by humoral components of the immune system. However, several recent studies demonstrated both in pre-clinical animal models and in human clinical trials that anti-tumor mAbs can, in some cases, elicit adaptive immune responses,1-5 suggesting that mAbs could serve as a link between the innate and adaptive immune responses. The mechanism(s) by which anti-tumor mAbs elicit adaptive immune responses against targeted tumors have not been definitively characterized, although the ability of tumor-targeting mAbs to elicit cytokines via interactions with Fc gamma receptors (FcγRs) have been implicated in shaping the overall tumor microenvironment.5 Furthermore, it has become increasingly apparent that antibody engagement of select FcγRs can drive efficacy for both pro-apoptotic and immune-modulatory antibodies.6-11
Human FcγRs are typically divided into the activating FcγRs (e.g., FcγRI, FcγRIIa, and FcγRIIIa), which contain immunoreceptor tyrosine-based activation motifs (ITAM), and the inhibitory FcγRIIb, which contains an immunoreceptor tyrosine-based inhibitory motif (ITIM).12 Both FcγRIIa and FcγRIIIa are further subdivided into high affinity polymorphisms (i.e., H131 and V158, respectively) and low affinity polymorphisms (i.e., R131 and F158, respectively). Of the 3 human IgG subclasses most commonly adopted for mAb-based therapeutics - IgG1, IgG2, and IgG4 - each displays distinct binding patterns to the human activating FcγRs. FcγRI, the highest affinity receptor, binds to IgG1 and to a slightly lesser extent IgG4,13 but does not show appreciable affinity for IgG2.14 The FcγRIIa and FcγRIIIa receptors bind to IgGs with lower affinities, and typically need to engage Fc domains under avidity-based conditions to facilitate productive interactions. IgG2 and IgG4 engage FcγRIIa with slightly weaker binding than IgG1, whereas IgG2 and IgG4 display much weaker binding to FcγRIIIa compared to IgG1.14 The inherent differences in FcγR engagement between the human IgG subclasses have proven useful in determining which FcγRs influence cell-mediated cytotoxic activities.15-17
It has long been observed that triggering FcγRIIIa on natural killer (NK) cells promotes interferon (IFN)γ secretion.18 In terms of anti-tumor mAbs, it was shown that trastuzumab-opsonized tumor cells could drive IFNγ secretion in human NK cells, particularly in the presence of interleukin (IL)-12, and the elicitation of IFNγ was considered an important factor for anti-tumor efficacy.19 Investigators recently correlated mAb ADCC capacity with antibody-dependent cytokine release (ADCR) of IFNγ, monocyte chemoattractant protein-1, IL-6, and tumor necrosis factor (TNF).20 MAb-mediated induction of these pro-inflammatory cytokines was postulated to influence other immune cells in close proximity. IFNγ in particular is a pro-inflammatory cytokine that can affect immune function via enhancing macrophage tumor cell-killing by increasing reactive nitrogen intermediates, augmenting cross-presentation by professional antigen-presenting cells (APCs), increasing expression of co-stimulatory molecules, including major histocompatibility complex (MHC) I and MHC II, and support Th1-cell differentiation.21 It was recently demonstrated that mAb-mediated induction of IFNγ resulted in dendritic cell (DC) maturation and increased antigen presentation, which was hypothesized to result in increased cross-presentation potential to CD8 T cells, thereby linking the innate and adaptive immune responses.5
In contrast to inducing the production of pro-inflammatory cytokines, mAb/immune cell interactions can also result in the release of immunosuppressive and tumor-promoting cytokines. Monocytes and macrophages play a key role in orchestrating the immune response. When monocytes encounter danger signals, which include pro-inflammatory cytokines and bacterial products (e.g., pathogen-associated molecular patterns), they often differentiate into M1 macrophages.22 These cells are thought to aid tumor suppression and microbial clearance by promoting Th1-like immune responses. However, these potent drivers of pro-inflammatory immune responses are rigorously regulated during the progression of immune responses in order to limit tissue destruction.23 It was recognized over 15 years ago that antibodies could provide a means of inducing a macrophage immune checkpoint by influencing macrophage polarization. Mosser and colleagues demonstrated that antibody triggering of FcγRI on macrophages converted pro-inflammatory macrophages into regulatory macrophages, and a hallmark of this conversion was the release of the anti-inflammatory cytokine IL-10.24 The authors concluded that the secretion of IL-10 could alleviate the potential of bystander tissue damage within disease microenvironments caused by pro-inflammatory macrophages. It was recently demonstrated that co-culture of macrophages with cetuximab-opsonized tumor cells resulted in the release of the anti-inflammatory cytokine IL-10 as well as IL-8.25 Moreover, induction of macrophage IL-10 release by cetuximab, which targets epidermal growth factor receptor, was postulated to influence the lower rates of progression free survival of some patients undergoing combined treatment with cetuximab, the vascular endothelial growth factor-targeted agent bevacizumab and chemotherapy.25,26 Therefore, IgG1-mediated induction of regulatory macrophages and the subsequent elaboration of anti-inflammatory cytokines are now proposed by independent researchers to be problematic for the treatment of tumors with cell-targeting mAbs.27
We recently reported a novel Fc engineered platform technology that resulted in selective cell-killing functions.28 Most conventional Fc engineering approaches are aimed at either silencing immune effector functions29 or augmenting cell-killing functions such as ADCC,30-32 ADCP,33 or CDC34-36 without taking into consideration downstream effects such as ADCR or subsequent macrophage polarization. Here we report an Fc engineering approach that dissociates cell-killing functions from cytokine-release by utilizing mAb Fc variants that selectively engage specific FcγRs.
Results
IgG1-opsonized tumor cells drive peripheral blood mononuclear cell-based ADCC and ADCR, whereas IgG2 and IgG4 had limited activity
We previously showed that an anti-CD142 (tissue factor) human IgG1 mAb was capable of eliciting ADCC and ADCP in vitro and suppressing tumor growth in vivo against the triple-negative breast cancer cell line MDA-MB-231.37-39 To assess the ability of different IgG subclasses to elicit peripheral blood mononuclear cell (PBMC)-mediated ADCC and ADCR against MDA-MB-231 cells, we engineered variants of the IgG1 mAb to contain human IgG2 and IgG4 constant regions, respectively.40 As shown in Figure 1A, only the IgG1 subclass was capable of eliciting robust ADCC against MDA-MB-231 target cells, whereas the IgG2 and IgG4 subclasses each elicited low to undetectable ADCC in a 2 h assay. 48 h ADCR assays were next set-up both in the presence and absence of exogenous IL-12 since IL-12 has been previously shown to influence PBMC-based ADCR.19 Consistent with the ADCC results, in the absence of exogenous IL-12, IFNγ secretion by PBMCs in response to mAb-opsonized tumor cells was highest with the IgG1 subclass since IgG1 elicited a mAb concentration-dependent increase in IFNγ, whereas the IgG2 and IgG4 subclasses each elicited low to undetectable increases in IFNγ (Fig. 1B). In the presence of IL-12, the total amount of mAb concentration-dependent increase in IFNγ induced by IgG1 was higher than that observed in the absence of IL-12 (e.g., ∼8000 pg/ml vs. 1200 pg/ml, respectively, for IgG1 at 5 μg/ml).
Figure 1.
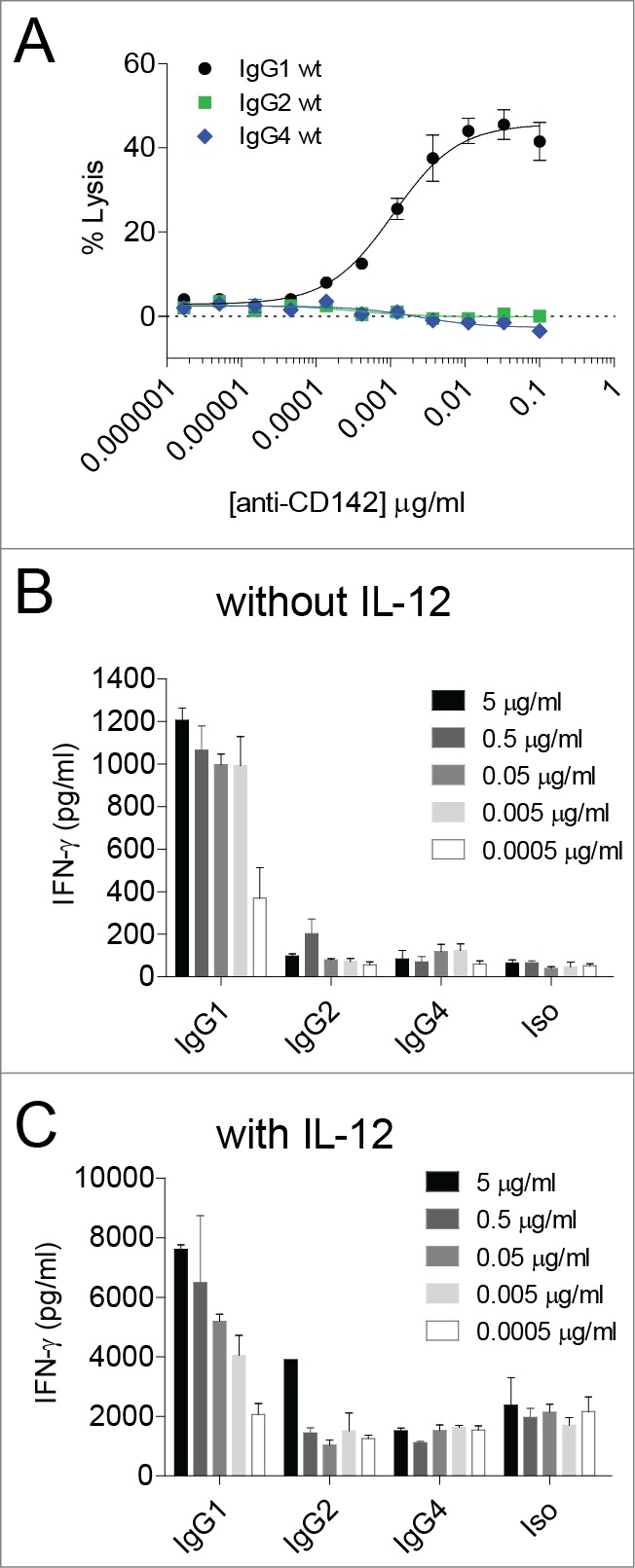
IgG1 antibodies mediate cytotoxicity and IFNγ cytokine secretion by PBMCs. (A) ADCC was performed using human PBMC as effector cells and MDA-MB-231 cells as targets with the indicated concentrations of anti-CD142 antibodies. Data are representative of 2 independent experiments (duplicate measurements per experiment). (B–C) ADCR assays were performed using PBMC effector cells and MDA-MB-231 target cells in the absence (B) or the presence (C) of IL-12. After 48-h supernatants were collected and IFNγ secretion was measured using ELISA. Data are representative of 2 independent experiments (duplicate measurements per experiment).
In contrast, tumor cells opsonized with the IgG2 or IgG4 subclasses resulted in low to undetectable increases in IFNγ over the background signal observed with the isotype control (Fig. 1B–C). In the presence of IL-12, the background levels of IFNγ secreted were augmented such that there was a roughly 10-fold increase in the baseline levels of IFNγ detected relative to ADCR assays performed in the absence of IL-12 (Fig. 1B), indicating that IL-12 treatment alone resulted in increased IFNγ secretion independent of the presence of tumor-targeting mAbs. These results demonstrated that IgG1 could elicit ADCC and IFNγ in a PBMC-based ADCR assay, whereas IgG2 and IgG4 had limited ability to drive ADCC and IFNγ secretion.
Macrophage-based ADCP and ADCR are distinct processes and IgG subclass-dependent
Macrophages are considered key regulators of the tumor microenvironment,27 and both pre-clinical animal studies41-44 and human clinical data45 have demonstrated that macrophages can facilitate mAb-mediated suppression of tumor cells in vivo. Additionally, unlike PBMC-mediated ADCC, multiple IgG subclasses can facilitate macrophage-mediated ADCP.16,28,46 We have previously reported a macrophage-mediated tumor cell-killing assay that assesses the total loss of tumor cells over a 24-h period as opposed to the more commonly employed 4-h assessments of macrophage internalization of mAb-opsonized tumor cells.28 For the purposes of this study, we performed 24-h macrophage-mediated cell-killing assays and also collected supernatants to assess the levels of IL-10 at the 24-h time point. Macrophages were differentiated from purified monocytes by incubating the monocytes with 25 ng/ml macrophage colony-stimulating factor (M-CSF) for 7 days, including the addition of 50 ng/ml IFNγ for the final 24 h. The relative expression levels of FcγRI, FcγRIIa, FcγRIIb, and FcγRIIIa are shown in Figure 2A. In contrast to ADCC, where only IgG1 was effective, all 3 human subclasses tested (i.e., IgG1, IgG2, and IgG4) were capable of killing mAb-opsonized tumor cells (Fig. 2B). However, there was a discrepancy between cell-killing and IL-10 production such that IgG1 and IgG4 elicited IL-10, whereas IgG2 induced substantially lower levels of IL-10 (Fig. 2C). These results demonstrated that, dependent on the IgG subclass, macrophages were capable of killing tumor cells while having limited secretion of IL-10.
Figure 2.
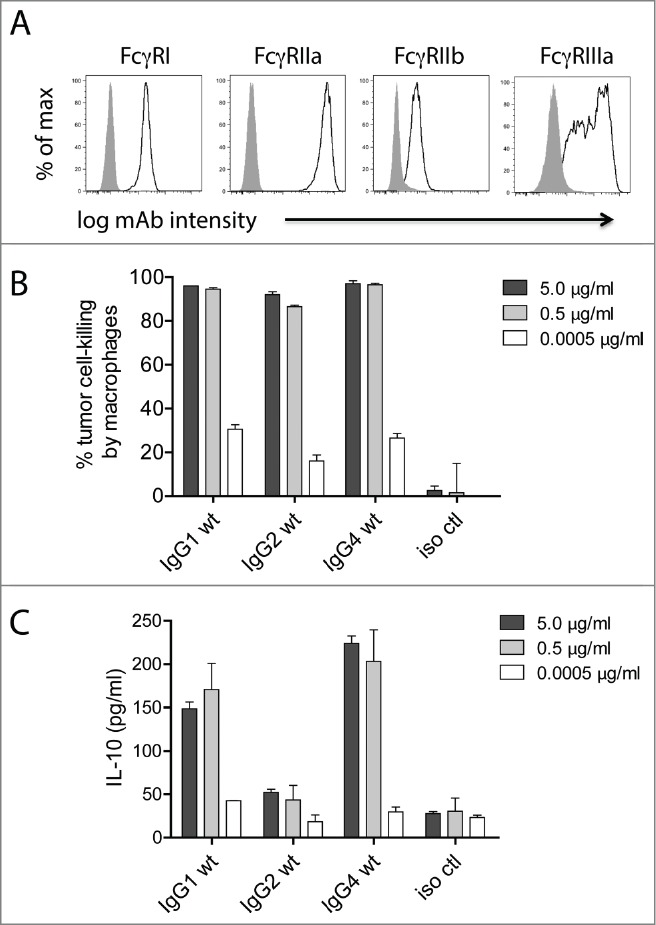
Macrophage tumor-cell-killing is uncoupled from ADCR. (A) Flow cytometry analysis for the cell surface expression of macrophage FcγRI, FcγRIIa, FcγRIIb, and FcγRIIIa. Isotype controls are shown as solid gray and FcγRs are shown as open lines. 24 h macrophage tumor cell-killing (B) and ADCR from the same experiment (C) are shown. Supernatants were collected after 24 h, and the IL-10 levels were measured using ELISA. Data are representative of 4 independent experiments (duplicate measurements per experiment).
Characterization of Fc engineered variants that engage FcγRIIa and FcγRIIIa but have low to undetectable binding to FcγRI
We previously reported a protease-resistant IgG1 platform with selectable cell-killing functions.28 This was accomplished first by engineering the lower hinge of IgG1 to contain the corresponding amino acids of IgG2 (i.e., E233P-L234V-L235A with G236-deleted, designated 2h), which conferred protease-resistance but rendered the IgG silent in terms of eliciting ADCC and CDC. To restore ADCC/ADCP or CDC, we selectively incorporated mutations into the CH2 region. The variants were cloned onto the anti-CD142 variable region, and we previously demonstrated that the variants displayed identical binding to CD142 as the parental mAb.28 For this study, we assessed the binding of 2 of these variants (i.e., 2h-DAA containing S239D/K326A/E333A and 2h-AEA containing K326A/I332E/E333A) to 3 activating FcγRs: FcγRI, FcγRIIa, and FcγRIIIa. As shown in Figure 3A, IgG4 had slightly reduced binding to FcγRI compared to IgG1. In contrast, IgG2 and the variants 2h-DAA and 2h-AEA had undetectable binding to FcγRI. All of the variants tested bound to FcγRIIa (R131) within 5-fold of human IgG1 (Fig. 3B). The variants 2h-DAA and 2h-AEA bound to FcγRIIIa (V158) within 2-fold of human IgG1, whereas IgG2 and IgG4 had greater than 30-fold decreased binding to FcγRIIIa compared to IgG1 (Fig. 3C). These results revealed 2 Fc variants with low to undetectable binding to FcγRI that maintained binding to FcγRIIa and FcγRIIIa comparable to IgG1.
Figure 3.
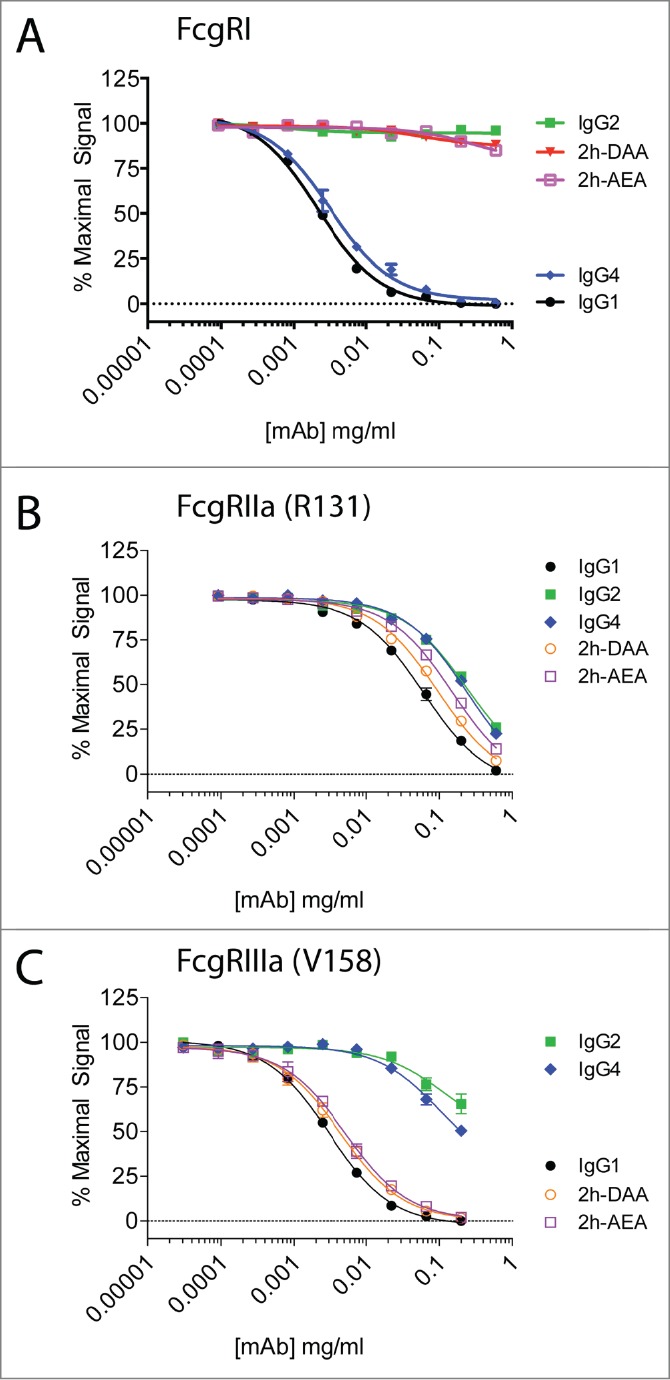
Two engineered variants demonstrate binding to FcγRIIa and FcγRIIIa, but lack binding to FcγRI. The binding of IgG1 (solid black circles), IgG2 (solid green squares), IgG4 (solid blue diamonds), 2h-DAA (solid down red triangle), and 2h-AEA (open purple squares) to FcγRI (A), FcγRIIa (R131) (B), and FcγRIIIa (V158) (C) were performed by competition AlphaScreen. Data are representative of 2 independent experiments (duplicate measurements per experiment).
Engineered variants that lack FcγRI binding elicit similar cytokines as IgG1 in a PBMC-based ADCR assay
To determine which other cytokines in addition to IFNγ were secreted by PBMCs in response to antibody-opsonized tumor cells, PBMCs from multiple donors were incubated with anti-CD142 antibody variants bound to MDA-MB-231 target cells. After 48 h, supernatants were collected and analyzed by Luminex Multi-Analyte Profile in the presence or absence of IL-12. Multiple cytokines were induced by IgG1, 2h-DAA, and 2h-AEA opsonized tumor cells including IFNγ, IL-1α, IL-1β, as well as matrix metalloproteinase 3 (MMP-3) (Fig. 4A). Additional cytokines were detected including IL-6, IL-8, GM-CSF, MIP-1α, MIP-1β, RANTES and TNF among others (data not shown). Unlike the macrophage-mediated ADCR described in Figure 2, IL-10 was not secreted in substantial amounts by PBMCs. The IgG2 and IgG4 subclasses elicited limited to undetectable cytokine secretion. The fold-change in cytokine secretion was higher in the absence of IL-12, presumably because the background levels were lower. In addition, the total amount of cytokines secreted was elevated in the presence of IL-12 (Fig. 4B).
Figure 4.

(See previous page). Multiple cytokines are triggered by engagement of FcγRs on PBMCs in response to antibody-opsonized tumor cells. ADCR assays with PBMC effector cells were performed in the absence (A) or presence (B) of IL-12. After 48 h, supernatants were collected and analyzed by a quantitative Luminex Multi-Analyte Profile assay. Dot plots on top represent the concentration of cytokines detected from a single donor, and the bar graphs below represent the mean +/− SEM fold-change in cytokine levels normalized to the isotype control from 4 independent PBMC donors. (C) Shown are flow cytometric histograms of IFNγ secretion elicited by IgG1 (black) compared to negative control (gray) of NK Cells (CD56pos, CD3neg), NKT cells (CD56posCD3pos), CD16pos monocytes (CD14posCD16pos), CD16neg monocytes (CD14posCD16neg), T cells (CD3pos). (D) NK and NKT cells were the primary producers of IFNγ in ADCR assays using PBMCs. ADCR assays were performed with PBMCs, MDA-MB-231-GFP target cells and 5 µg/ml of anti-CD142 antibody variants in the absence or presence of IL-12. Bar graphs summarize the percent of each population secreting IFNγ elicited by each of the antibody variants. Data are representative from 3 donors in 2 independent experiments.
Human PBMC populations consist of T cells, B cells, NK cells, NKT cells, and monocytes. The relative levels of FcγRs on NK cells and monocytes in PBMCs are shown in Figure S1. To assess which populations of cells within PBMCs were secreting cytokines in response to antibody-opsonized tumor cells, intracellular cytokine staining for IFNγ was utilized. PBMCs were co-incubated with MDA-MB-231 target cells opsonized with anti-CD142 variants. Flow cytometry was used to identify the cell populations within PBMCs and to determine the percentage of each cell population secreting IFNγ (Fig. 4C-D). In the absence of IL-12, only NK cells and NKT cells expressed IFNγ in the presence of IgG1, 2h-DAA, and 2h-AEA. Consistent with the cytokine secretion results, minimal intracellular IFNγ was observed with IgG4, and treatment with IgG2 resulted in slightly lower frequencies of intracellular IFNγ positive cells relative to isotype control treated cells. In the presence of IL-12, the background levels of IFNγ positive cells increased, but tumor cells opsonized with IgG1, 2h-DAA, and 2h-AEA demonstrated an increased frequency of IFNγ positive NK cells. These results indicated that although PBMCs contain effector cells expressing FcγRI and FcγRIIa (e.g., monocytes) and FcγRIIb (e.g., monocytes and B cells), the ability of mAbs to drive cytokine production in a 48 h ADCR assay was largely dependent upon interactions with FcγRIIIa because only IgG1, but not IgG2 or IgG4, mAbs possesses the capability to mediate these effects.
Macrophage-mediated ADCR is dependent on interactions with FcγRI
We next determined if the engineered variants were capable of eliciting ADCP without inducing ADCR. As shown in Figure 5A, IgG1, IgG4, 2h-DAA, and 2h-AEA all elicited similar levels of tumor cell-killing after 24 h, indicating that Fc interactions with FcγRI were dispensable for eliciting ADCP, since 2h-DAA and 2h-AEA displayed weak to undetectable binding to FcγRI (Fig. 3A). In contrast to the PBMC-mediated ADCR, IgG1 macrophage-mediated ADCR elicited minimal levels of IFNγ, IL-1α, and IL-1β. However, the IgG1 and IgG4 opsonized tumor-cells resulted in ∼15-fold increased IL-8, ∼10-fold increased IL-10, ∼12-fold increased MIP-1α, ∼30-fold increased MIP-1β, ∼10-fold increased RANTES, ∼ 25-fold increased TNF, and ∼7-fold increased MMP-3, whereas the variants 2h-DAA and 2h-AEA elicited low to undetectable levels of these cytokines (Fig. 5B), despite the ability to kill variant-opsonized tumor cells. Because only IgG1 and IgG4 elicited the most robust ADCR and, among the mAbs tested, these were the only 2 mAbs that showed appreciable binding to FcγRI (Fig. 3A), the results implicate Fc interactions with FcγRI as a key requirement for macrophage-mediated ADCR under the conditions tested. Collectively, these results demonstrate that an Fc engineering approach can be employed that uncouples macrophage-mediated tumor cell-killing from the induction of cytokines, while still maintaining the ability to elicit cytokines by PBMCs.
Figure 5.

Macrophage-mediated ADCR is dependent on interactions with FcγRI. (A) Macrophage 24-h tumor cell-killing is depicted for 4 independent donors (duplicate measurements per experiment). (B) At the end of the 24-h incubation in the tumor cell-killing assay, supernatants were collected and analyzed for cytokine levels. Dot plots on top represent the concentration of cytokines detected from a single donor (duplicate measurements per experiment), and the bar graphs below represent the mean +/− SEM fold-change in cytokine levels normalized to the isotype control from 4 independent PBMC donors.
Discussion
Isotype selection and the employment of Fc engineered variants for therapeutic mAbs is often driven by the intent to match the appropriate immune effector functions with any given disease microenvironment.47,48 For example, IgG1 or Fc-enhanced backbones are typically chosen when the intent is to kill tumor cells via immune effector functions, whereas engineered Fc “silent” platforms are employed when immune effector functions are not warranted or could prove detrimental. We show here that isotype selection also influences the secretion of cytokines. IgG1 - the most common subclass used for mAb-based therapies - is capable of eliciting pro-inflammatory cytokines by PBMCs (e.g., IFNγ, IL-1α, IL-1β). Furthermore, intracellular flow cytometry demonstrated that IgG1 PBMC-based IFNγ ADCR was limited to NK and NKT cells. Since both NK and NKT49 cells predominantly express FcγRIIIa (a subset of NK cells can also express FcγRIIc),50 IFN-γ secretion via PBMC-based ADCR was regulated in large part by FcγRIIIa and potentially FcγRIIc. In contrast, neither IgG2 nor IgG4 elicited robust cytokine secretion from PBMC effector cells. The intracellular cytokine studies were limited to detection of IFNγ. Future studies will explore if other populations within the PBMCs were secreting other cytokines detected in the supernatants.
When macrophages were used as effector cells in the ADCP/ADCR assays, tumor opsonization with IgG1 resulted in both tumor cell-killing and elicitation of a mix of pro-inflammatory (e.g., TNF, RANTES) and anti-inflammatory cytokines (e.g., IL-10). IgG4 opsonized tumor cells in the macrophage-mediated ADCP/ADCR assays were similar to IgG1 in that IgG4 drove both tumor cell-killing and cytokine secretion. In contrast, IgG2 was capable of driving macrophage-mediated ADCP without eliciting cytokine secretion. Since IgG2 has weak to undetectable binding to FcγRI,14 these results corroborated previous findings that mAb-induced secretion of IL-10 was largely dependent on interactions with FcγRI.24 The 2 engineered variants, 2h-DAA and 2h-AEA, had reduced to undetectable binding to FcγRI but maintained binding to both FcγRIIa and FcγRIIIa, and, as such, displayed unique properties in that they could facilitate macrophage-mediated cell-killing without eliciting cytokines from macrophages (Fig. 5A-B) while still maintaining the ability drive PBMC-based ADCR (Fig. 4A-B). To our knowledge, these are the only 2 IgG variants reported in the literature that display these properties.
Previous reports have often concentrated on detecting a fairly limited set of cytokines secreted from macrophages.24,25 Our results corroborate that mAb opsonized tumors can elicit IL-10 in an FcγRI-dependent manner,24 but we also detected cytokines typically considered to be pro-inflammatory (e.g., IL-8, MIP-1α, MIP-1β, TNF, and RANTES). It has yet to be determined how this cytokine mixture will influence disease microenvironments, particularly the maturation state or polarization of immune effector cells. It has been previously demonstrated that among a mixture of cytokines, one individual cytokine can have a strong influence on cell fate decisions. Ferris and colleagues showed that in tri-cultures of NK cells, mAb-opsonized tumor cells, and dendritic cells, the solitary immuno-depletion of IFNγ had a profound effect on the maturation state of the DCs in the culture.5 It is intriguing to speculate which cytokines elicited from macrophages will have the strongest effect on the tumor microenvironment. Our engineered variants have the unique ability to elicit cell-killing from both PBMCs and macrophages, but only induce the pro-inflammatory milieu secreted by PBMCs without inducing any detected anti-inflammatory cytokines. Future studies will investigate if these variants can tip the balance of the tumor microenvironment away from pro-tumor to a more pro-inflammatory, Th1-like response. The ability to test this at present is limited by in vivo mouse models containing murine FcγRs that also lack the adaptive immune system. However, the advent of fully humanized FcγR mice on a non-immune compromised mouse background may provide an acceptable model system.51
An additional consideration for macrophage-mediated ADCR concerns the context in which the antibody Fc is presented to the macrophage. It was previously demonstrated that IL-10 was not elicited from macrophages when an IgG1 mAb was coated directly onto plates.25 Rather, the IL-10 secretion was dependent on the Fc being presented in the context of mAb-opsonized tumor cells. Furthermore, whereas monomeric IgG2 and anti-F(ab’)2 aggregated IgG2 immune complexes do not bind to FcγRI,14 higher-order IgG2 immune complexes have demonstrated binding to FcγRI.52 Although IgG2 opsonized on the surface of tumor cells did not elicit cytokines from macrophages in our studies, it will be of interest to test if IgG2 in other contexts influences macrophage cytokine secretion (e.g., high-order immune complexes or opsonized onto different sized tumor cells or bacteria). Additionally, as it is becoming increasingly understood that cellular responses, particularly with regards to immune effector cells, are a result of multiple signaling pathways acting in concert,53 it is intriguing to speculate that the mAb-opsonized tumor cells are stimulating multiple pathways in macrophages to elicit cytokine secretion. Ferris and colleagues demonstrated that NKG2D/MICA interactions influenced IFNγ-dependent DC maturation in the presence of NK cells and mAb-opsonized tumor cells.5 Future studies will be needed to define which interactions, in addition to Fc:FcγR engagement, influence cytokine secretion in macrophage-based ADCR.
Our group and others previously demonstrated that MMP-3 is capable of rapidly cleaving human IgG1 antibodies.28,39,54,55 We show here that both PBMC-mediated and macrophage-mediated ADCR resulted in the secretion of MMP-3. One potential consequence of this MMP-3 secretion is the cleavage and inactivation of mAbs within the tumor microenvironment. We had previously detected an accumulation of cleaved IgGs at the invasive front of human head and neck squamous cell carcinoma tumors,28 a location where antibodies are enriched within the tumor microenvironment due to proximity to the perivascular space and high interstitial tumor pressure.56 This raises the possibility that ADCR-mediated MMP-3 secretion contributes to the detection of cleaved IgGs at the tumor-stromal interface. We and others have shown that a single cleavage in one heavy chain of an IgG1 mAb abrogates tumor cell-killing function in vitro and in vivo.39,57 As the 2 variants described herein are also resistant to MMP-3-mediated cleavage,28 the use of these variants in the tumor setting could also eliminate cleavage by a highly-relevant protease induced through ADCR.
In summary, we engineered variants with a unique FcγR-binding profile that resulted in an uncoupling of macrophage-mediated cell-killing from ADCR without compromising PBMC-mediated pro-inflammatory immune responses. Future studies will address the effect of competing IgGs on the differential FcγR-binding, expand the studies to include other tumor-targeting mAbs and tumor cell lines, as well as test the variants in appropriate genetically engineered mouse models.
Materials and Methods
Antibodies
The IgG1 anti-CD142 mAb (CDR grafted, humanized) contains the same V-region as the murine anti-human CD142 mAb TF8-5G9, which came from the Scripps Research Institute and has been previously described.28,58 The heavy and light chains of the anti-CD142 mAb were engineered onto the IgG2 and IgG4 subclasses and the 2h-DAA and 2h-AEA variant constant regions by molecular cloning. All antibodies used in the ADCR assays were assessed for the presence of endotoxin, because endotoxin-contaminated preparations of recombinant proteins can profoundly influence cytokine secretion.59 All mAbs used in the ADCR assays had less than 2 EU/mg.
ADCC assays
A final number of 1.0 × 104 BATDA-labeled (PerkinElmer, cat.no. C136-100) MDA-MB-231 target cells and 0.5 × 106 human PBMC effector cells (1:50 target:effector ratio) were added to the wells of a 96-well plate and incubated with increasing concentrations of the anti-CD142 mAbs, centrifuged for 2 minutes at 200 g and incubated at 37°C. The medium for the assay was DMEM containing glutamax (Life Technologies, cat.no. 10569), 1X non-essential amino acids (Life Technologies, cat.no. 11140), and 10% FBS (Life Technologies, cat.no. 10437). After 2 h, plates were centrifuged for 5 minutes at 200 g, and 50 μl of supernatant were mixed with 200 μl of DELPHIA Europium-based reagent (PerkinElmer, cat.no. C135-100). Relative Fluorescence Units were measured using an Envision 2101 Multilabel Reader (Perkin Elmer). Percent lysis was calculated using the following equation: (Sample Release – Spontaneous Release)/(Maximal Release (from Triton X-100 lysis) – Spontaneous Release) × 100%. Data were log transformed and fitted to a sigmoidal dose-response curve using GraphPad Prism v5.
PBMC-based ADCR
ADCR assays with PBMC effector cells and MDA-MB-231 target cells (from American Type Culture Collection) were performed using an effector: target ratio of 10:1 (200,000 PBMC: 20,000 target cells) in a 96-well plate in the presence or absence of 10 ng/ml IL-12 (R&D Systems, cat.no. 219-IL-005) with the indicated antibody concentrations. After 48 h, the plates were centrifuged and the supernatant was collected. IFNγ cytokine concentrations were determined using Quantikine ELISA kits (R&D Systems, cat.no. SIF50). Luminex quantitative multi-analyte profile was performed at Myriad Rules-Based Medicine (cat.no. Human InflammationMAP®). Data was graphed using GraphPad Prism and fold-changes were normalized relative to the isotype control.
24h macrophage-based ADCP and ADCR
Human PBMCs were isolated from leukopaks (Biologics Specialty Corp) using Ficoll gradient centrifugation. CD14pos monocytes were purified from PBMCs by negative depletion using a CD14 isolation kit that did not deplete CD16pos monocytes (Stem Cell Technologies, cat.no. 19058). Monocytes were plated at 0.1 × 106 cells/cm2 in X-VIVO-10 medium (Lonza, cat.no. 04-380Q) containing 10% FBS. Macrophages were differentiated by the addition of 25 ng/ml of M-CSF (R&D Systems, cat.no. 216-MC-025/CF) for 7 days. Fifty ng/ml of IFNγ (R&D Systems, cat.no. 285-IF-100/CF) was added for the final 24 h of differentiation. The relative expression of FcγRs was monitored by flow cytometry. Briefly, 0.15 × 106 cells in bovine serum albumin (BSA)-containing FACS buffer (BD Biosciences, cat.no. 554657) were incubated with anti-FcγRI FITC clone 10.1 (BD Biosciences, cat.no 55527), anti-FcγRIIa clone IV.3 (StemCell Technologies, cat.no. 01470 conjugated to Alexa Fluor 647 per the manufacturer's instructions (Invitrogen, cat.no. A20186)), anti-FcγRIIb clone 2B6 (mouse IgG2a constructed by gene synthesis conjugated to Pacific Blue per the manufacturer's instructions (Invitrogen, cat.no. P30013)), and anti-FcγRIII PE-Cy7 clone 3G8 (BD Biosciences, cat.no. 557744). Cells were then washed in BSA-containing FACS buffer and were acquired on an LSRFortessa (BD Biosciences). Target cells for the assay were GFP-expressing MDA-MB-231 cells.42 -24 h ADCP assays were performed as previously described.28 At the end of the incubation, supernatants were collected and IL-10 levels were assayed using a plate-based ELISA (R&D Systems, cat.no. S1000B), whereas the larger cytokine panels were performed by Rules-Based Medicine (cat.no. Human InflammationMAP→). Data was graphed using GraphPad Prism v5 and fold-changes were normalized relative to the isotype control.
Competition AlphaScreen assays for FcγR-binding
AlphaScreen (PerkinElmer) was used to assess competitive binding of IgG1, IgG2, IgG4, 2h-DAA, and 2h-AEA as was previously described.39 Human FcγRI (cat.no. 1257-FC-050), FcγRIIa R131 (cat.no. 1330-CD-050/CF), and FcγRIIIa V158 (cat.no. 4325-FC-050) were purchased from R&D Systems. Briefly, competition binding studies were carried out in half-well volume 96-well opaque plates (Corning, cat. no. 3693) in PBS, 0.05% BSA, 0.01% Tween-20 at pH 7.4. Competition studies were performed against a biotinylated IgG1 at a set concentration of 0.2 μg/mL in the final assay concentration and varying levels of competing mAbs. FcγR concentrations were 0.2 μg/mL in final concentration of the assays. A 1/50 diluted nickel chelate (Ni)-acceptor beads (PerkinElmer, custom order) and streptavidin (SA)-donor beads (PerkinElmer, cat.no. 6760002) were added in the final set-up step in the assay prior to incubation for 30-60 minutes. For each assay, Relative Fluorescence Units were measured using an Envision 2101 Multilaber Reader (PerkinElmer). Data were normalized for maximal signal and were log-transformed and fitted to a sigmoidal dose-response curve using GraphPad Prism v5.
Flow cytometry for intracellular cytokines
For intracellular cytokine staining, PBMC effector cells, 5 µg/ml of anti-CD142 antibody variants and MDA-MB-231-GFP target cells were incubated for 16 h prior to a 4 h addition of GolgiPlug (BD Biosciences, cat.no. 555029). Cells were then stained using a fixable Near IR live/dead marker (Invitrogen, cat.no. L10119) and fixed using CytoFix/CytoPerm solution (BD Biosciences, cat.no. 554714). Cells were washed in Perm/Wash (BD Biosciences, cat.no. 554714) and stained using the following antibodies: anti-CD11b-PE (BD PharMingen, cat.no. 555388), anti-CD16-PECy7 (BD PharMingen, cat. no. 557744), anti-CD3-AlexaFluor700 (BD PharMingen cat. no. 561027), anti-IFNγ Horizon V450 (BD PharMingen, cat.no. 560371), anti-CD56-AlexaFluor647 (Biolegend, cat.no. 304612) and anti-CD14 QDot605 (Invitrogen, cat.no. Q10013). Cells were acquired on a BD Fortessa and data was analyzed using Flowjo (TreeStar).
Disclosure of Potential Conflicts of Interest
MK, ARG, WRS, REJ, and RJB are all employees of Janssen Research and Development, LLC.
Acknowledgments
We thank Zhiqiang An and Ningyan Zhang for thoughtful discussions and Michael Naso, Jill Carton, and Katharine Heeringa in the Molecular Protein and Biosciences group at Janssen Research and Development, LLC for cloning, expression, purification, and quality control of mAb variants.
Supplementary Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Park S, Jiang Z, Mortenson ED, Deng L, Radkevich-Brown O, Yang X, Sattar H, Wang Y, Brown NK, Greene M, et al.. The therapeutic effect of anti-HER2/neu antibody depends on both innate and adaptive immunity. Cancer Cell 2010; 18:160-70; PMID:20708157; http://dx.doi.org/ 10.1016/j.ccr.2010.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taylor C, Hershman D, Shah N, Suciu-Foca N, Petrylak DP, Taub R, Vahdat L, Cheng B, Pegram M, Knutson KL, et al.. Augmented HER-2 specific immunity during treatment with trastuzumab and chemotherapy. Clin Cancer Res 2007; 13:5133-43; PMID:17785568; http://dx.doi.org/ 10.1158/1078-0432.CCR-07-0507 [DOI] [PubMed] [Google Scholar]
- 3.Hilchey SP, Hyrien O, Mosmann TR, Livingstone AM, Friedberg JW, Young F, Fisher RI, Kelleher RJ Jr., Bankert RB, Bernstein SH. Rituximab immunotherapy results in the induction of a lymphoma idiotype-specific T-cell response in patients with follicular lymphoma: support for a “vaccinal effect” of rituximab. Blood 2009; 113:3809-12; PMID:19196657; http://dx.doi.org/ 10.1182/blood-2008-10-185280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abes R, Gelize E, Fridman WH, Teillaud JL. Long-lasting antitumor protection by anti-CD20 antibody through cellular immune response. Blood 2010; 116:926-34; PMID:20439625; http://dx.doi.org/ 10.1182/blood-2009-10-248609 [DOI] [PubMed] [Google Scholar]
- 5.Srivastava RM, Lee SC, Andrade Filho PA, Lord CA, Jie HB, Davidson HC, Lopez-Albaitero A, Gibson SP, Gooding WE, Ferrone S, et al.. Cetuximab-activated natural killer and dendritic cells collaborate to trigger tumor antigen-specific T-cell immunity in head and neck cancer patients. Clin Cancer Res 2013; 19:1858-72; PMID:23444227; http://dx.doi.org/ 10.1158/1078-0432.CCR-12-2426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim JM, Ashkenazi A. Fcgamma receptors enable anticancer action of proapoptotic and immune-modulatory antibodies. J Exp Med 2013; 210:1647-51; PMID:23980122; http://dx.doi.org/ 10.1084/jem.20131625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bulliard Y, Jolicoeur R, Windman M, Rue SM, Ettenberg S, Knee DA, Wilson NS, Dranoff G, Brogdon JL. Activating Fc gamma receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J Exp Med 2013; 210:1685-93; PMID:23897982; http://dx.doi.org/ 10.1084/jem.20130573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, Korman AJ. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res 2013; 1:32-42; PMID:24777248; http://dx.doi.org/ 10.1158/2326-6066.CIR-13-0013 [DOI] [PubMed] [Google Scholar]
- 9.Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, Roddie C, Henry JY, Yagita H, Wolchok JD, et al.. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med 2013; 210:1695-710; PMID:23897981; http://dx.doi.org/ 10.1084/jem.20130579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.White AL, Chan HT, Roghanian A, French RR, Mockridge CI, Tutt AL, Dixon SV, Ajona D, Verbeek JS, Al-Shamkhani A, et al.. Interaction with FcgammaRIIB is critical for the agonistic activity of anti-CD40 monoclonal antibody. J Immunol 2011; 187:1754-63; PMID:21742972; http://dx.doi.org/ 10.4049/jimmunol.1101135 [DOI] [PubMed] [Google Scholar]
- 11.Furness AJ, Vargas FA, Peggs KS, Quezada SA. Impact of tumour microenvironment and Fc receptors on the activity of immunomodulatory antibodies. Trends Immunol 2014; 35:290-8; PMID:24953012; http://dx.doi.org/ 10.1016/j.it.2014.05.002 [DOI] [PubMed] [Google Scholar]
- 12.Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol 2008; 8:34-47; PMID:18064051; http://dx.doi.org/ 10.1038/nri2206 [DOI] [PubMed] [Google Scholar]
- 13.Woof JM, Partridge LJ, Jefferis R, Burton DR. Localisation of the monocyte-binding region on human immunoglobulin G. Mol Immunol 1986; 23:319-30; PMID:3487030; http://dx.doi.org/ 10.1016/0161-5890(86)90059-3 [DOI] [PubMed] [Google Scholar]
- 14.Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, Daeron M. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood 2009; 113:3716-25; PMID:19018092; http://dx.doi.org/ 10.1182/blood-2008-09-179754 [DOI] [PubMed] [Google Scholar]
- 15.Beenhouwer DO, Yoo EM, Lai CW, Rocha MA, Morrison SL. Human immunoglobulin G2 (IgG2) and IgG4, but not IgG1 or IgG3, protect mice against Cryptococcus neoformans infection. Infect Immun 2007; 75:1424-35; PMID:17220317; http://dx.doi.org/ 10.1128/IAI.01161-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schneider-Merck T, Lammerts van Bueren JJ, Berger S, Rossen K, van Berkel PH, Derer S, Beyer T, Lohse S, Bleeker WK, Peipp M, et al.. Human IgG2 antibodies against epidermal growth factor receptor effectively trigger antibody-dependent cellular cytotoxicity but, in contrast to IgG1, only by cells of myeloid lineage. J Immunol 2010; 184:512-20; PMID:19949082; http://dx.doi.org/ 10.4049/jimmunol.0900847 [DOI] [PubMed] [Google Scholar]
- 17.Steplewski Z, Sun LK, Shearman CW, Ghrayeb J, Daddona P, Koprowski H. Biological activity of human-mouse IgG1, IgG2, IgG3, and IgG4 chimeric monoclonal antibodies with antitumor specificity. Proc Natl Acad Sci U S A 1988; 85:4852-6; PMID:3387441; http://dx.doi.org/ 10.1073/pnas.85.13.4852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cassatella MA, Anegon I, Cuturi MC, Griskey P, Trinchieri G, Perussia B. Fc gamma R(CD16) interaction with ligand induces Ca2+ mobilization and phosphoinositide turnover in human natural killer cells. Role of Ca2+ in Fc gamma R(CD16)-induced transcription and expression of lymphokine genes. J Exp Med 1989; 169:549-67; PMID:2536067; http://dx.doi.org/ 10.1084/jem.169.2.549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parihar R, Dierksheide J, Hu Y, Carson WE. IL-12 enhances the natural killer cell cytokine response to Ab-coated tumor cells. J Clin Invest 2002; 110:983-92; PMID:12370276; http://dx.doi.org/ 10.1172/JCI0215950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kircheis R, Halanek N, Koller I, Jost W, Schuster M, Gorr G, Hajszan K, Nechansky A. Correlation of ADCC activity with cytokine release induced by the stably expressed, glyco-engineered humanized Lewis Y-specific monoclonal antibody MB314. MAbs 2012; 4:532-41; PMID:22665069; http://dx.doi.org/ 10.4161/mabs.20577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ikeda H, Old LJ, Schreiber RD. The roles of IFN gamma in protection against tumor development and cancer immunoediting. Cytokine Growth Factor Rev 2002; 13:95-109; PMID:11900986; http://dx.doi.org/ 10.1016/S1359-6101(01)00038-7 [DOI] [PubMed] [Google Scholar]
- 22.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol 2011; 11:723-37; PMID:21997792; http://dx.doi.org/ 10.1038/nri3073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol 2010; 11:889-96; PMID:20856220; http://dx.doi.org/ 10.1038/ni.1937 [DOI] [PubMed] [Google Scholar]
- 24.Sutterwala FS, Noel GJ, Salgame P, Mosser DM. Reversal of proinflammatory responses by ligating the macrophage Fcgamma receptor type I. J Exp Med 1998; 188:217-22; PMID:9653099; http://dx.doi.org/ 10.1084/jem.188.1.217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pander J, Heusinkveld M, van der Straaten T, Jordanova ES, Baak-Pablo R, Gelderblom H, Morreau H, van der Burg SH, Guchelaar HJ, van Hall T. Activation of tumor-promoting type 2 macrophages by EGFR-targeting antibody cetuximab. Clin Cancer Res 2011; 17:5668-73; PMID:21788356; http://dx.doi.org/ 10.1158/1078-0432.CCR-11-0239 [DOI] [PubMed] [Google Scholar]
- 26.Tol J, Koopman M, Cats A, Rodenburg CJ, Creemers GJ, Schrama JG, Erdkamp FL, Vos AH, van Groeningen CJ, Sinnige HA, et al.. Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. N Engl J Med 2009; 360:563-72; PMID:19196673; http://dx.doi.org/ 10.1056/NEJMoa0808268 [DOI] [PubMed] [Google Scholar]
- 27.De Palma M, Lewis CE. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell 2013; 23:277-86; PMID:23518347; http://dx.doi.org/ 10.1016/j.ccr.2013.02.013 [DOI] [PubMed] [Google Scholar]
- 28.Kinder M, Greenplate AR, Grugan KD, Soring KL, Heeringa KA, McCarthy SG, Bannish G, Perpetua M, Lynch F, Jordan RE, et al.. Engineered protease-resistant antibodies with selectable cell-killing functions. J Biol Chem 2013; 288:30843-54; PMID:23986451; http://dx.doi.org/ 10.1074/jbc.M113.486142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Labrijn AF, Aalberse RC, Schuurman J. When binding is enough: nonactivating antibody formats. Curr Opin Immunol 2008; 20:479-85; PMID:18577454; http://dx.doi.org/ 10.1016/j.coi.2008.05.010 [DOI] [PubMed] [Google Scholar]
- 30.Lazar GA, Dang W, Karki S, Vafa O, Peng JS, Hyun L, Chan C, Chung HS, Eivazi A, Yoder SC, et al.. Engineered antibody Fc variants with enhanced effector function. Proc Natl Acad Sci U S A 2006; 103:4005-10; PMID:16537476; http://dx.doi.org/ 10.1073/pnas.0508123103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shields RL, Namenuk AK, Hong K, Meng YG, Rae J, Briggs J, Xie D, Lai J, Stadlen A, Li B, et al.. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J Biol Chem 2001; 276:6591-604; PMID:11096108; http://dx.doi.org/ 10.1074/jbc.M009483200 [DOI] [PubMed] [Google Scholar]
- 32.Stavenhagen JB, Gorlatov S, Tuaillon N, Rankin CT, Li H, Burke S, Huang L, Vijh S, Johnson S, Bonvini E, et al.. Fc optimization of therapeutic antibodies enhances their ability to kill tumor cells in vitro and controls tumor expansion in vivo via low-affinity activating Fcgamma receptors. Cancer Res 2007; 67:8882-90; PMID:17875730; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-0696 [DOI] [PubMed] [Google Scholar]
- 33.Richards JO, Karki S, Lazar GA, Chen H, Dang W, Desjarlais JR. Optimization of antibody binding to FcgammaRIIa enhances macrophage phagocytosis of tumor cells. Mol Cancer Ther 2008; 7:2517-27; PMID:18723496; http://dx.doi.org/ 10.1158/1535-7163.MCT-08-0201 [DOI] [PubMed] [Google Scholar]
- 34.Idusogie EE, Wong PY, Presta LG, Gazzano-Santoro H, Totpal K, Ultsch M, Mulkerrin MG. Engineered antibodies with increased activity to recruit complement. J Immunol 2001; 166:2571-5; PMID:11160318; http://dx.doi.org/ 10.4049/jimmunol.166.4.2571 [DOI] [PubMed] [Google Scholar]
- 35.Moore GL, Chen H, Karki S, Lazar GA. Engineered Fc variant antibodies with enhanced ability to recruit complement and mediate effector functions. MAbs 2010; 2:181-9; PMID:20150767; http://dx.doi.org/ 10.4161/mabs.2.2.11158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Natsume A, In M, Takamura H, Nakagawa T, Shimizu Y, Kitajima K, Wakitani M, Ohta S, Satoh M, Shitara K, et al.. Engineered antibodies of IgG1/IgG3 mixed isotype with enhanced cytotoxic activities. Cancer Res 2008; 68:3863-72; PMID:18483271; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-6297 [DOI] [PubMed] [Google Scholar]
- 37.Brezski RJ, Luongo JL, Petrone D, Ryan MH, Zhong D, Tam SH, Schmidt AP, Kruszynski M, Whitaker BP, Knight DM, et al.. Human anti-IgG1 hinge autoantibodies reconstitute the effector functions of proteolytically inactivated IgGs. J Immunol 2008; 181:3183-92; PMID:18713989; http://dx.doi.org/ 10.4049/jimmunol.181.5.3183 [DOI] [PubMed] [Google Scholar]
- 38.Brezski RJ, Oberholtzer A, Strake B, Jordan RE. The in vitro resistance of IgG2 to proteolytic attack concurs with a comparative paucity of autoantibodies against peptide analogs of the IgG2 hinge. MAbs 2011; 3:558-67; PMID:22123056; http://dx.doi.org/ 10.4161/mabs.3.6.18119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brezski RJ, Vafa O, Petrone D, Tam SH, Powers G, Ryan MH, Luongo JL, Oberholtzer A, Knight DM, Jordan RE. Tumor-associated and microbial proteases compromise host IgG effector functions by a single cleavage proximal to the hinge. Proc Natl Acad Sci U S A 2009; 106:17864-9; PMID:19815504; http://dx.doi.org/ 10.1073/pnas.0904174106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brezski RJ, Kinder M, Grugan KD, Soring KL, Carton J, Greenplate AR, Petley T, Capaldi D, Brosnan K, Emmell E, et al.. A monoclonal antibody against hinge-cleaved IgG restores effector function to proteolytically-inactivated IgGs in vitro and in vivo. MAbs 2014; 6:1265-73; PMID:25517311; http://dx.doi.org/ 10.4161/mabs.29825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med 2000; 6:443-6; PMID:10742152; http://dx.doi.org/ 10.1038/74704 [DOI] [PubMed] [Google Scholar]
- 42.Grugan KD, McCabe FL, Kinder M, Greenplate AR, Harman BC, Ekert JE, van Rooijen N, Anderson GM, Nemeth JA, Strohl WR, et al.. Tumor-associated macrophages promote invasion while retaining Fc-dependent anti-tumor function. J Immunol 2012; 189:5457-66; PMID:23105143; http://dx.doi.org/ 10.4049/jimmunol.1201889 [DOI] [PubMed] [Google Scholar]
- 43.Oflazoglu E, Stone IJ, Brown L, Gordon KA, van Rooijen N, Jonas M, Law CL, Grewal IS, Gerber HP. Macrophages and Fc-receptor interactions contribute to the antitumour activities of the anti-CD40 antibody SGN-40. Br J Cancer 2009; 100:113-7; PMID:19066610; http://dx.doi.org/ 10.1038/sj.bjc.6604812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oflazoglu E, Stone IJ, Gordon KA, Grewal IS, van Rooijen N, Law CL, Gerber HP. Macrophages contribute to the antitumor activity of the anti-CD30 antibody SGN-30. Blood 2007; 110:4370-2; PMID:17909075; http://dx.doi.org/ 10.1182/blood-2007-06-097014 [DOI] [PubMed] [Google Scholar]
- 45.Taskinen M, Karjalainen-Lindsberg ML, Nyman H, Eerola LM, Leppa S. A high tumor-associated macrophage content predicts favorable outcome in follicular lymphoma patients treated with rituximab and cyclophosphamide-doxorubicin-vincristine-prednisone. Clin Cancer Res 2007; 13:5784-9; PMID:17908969; http://dx.doi.org/ 10.1158/1078-0432.CCR-07-0778 [DOI] [PubMed] [Google Scholar]
- 46.Nesspor TC, Raju TS, Chin CN, Vafa O, Brezski RJ. Avidity confers FcgammaR binding and immune effector function to aglycosylated immunoglobulin G1. J Mol Recognit 2012; 25:147-54; PMID:22407978; http://dx.doi.org/ 10.1002/jmr.2155 [DOI] [PubMed] [Google Scholar]
- 47.Jefferis R. Antibody therapeutics: isotype and glycoform selection. Expert Opin Biol Ther 2007; 7:1401-13; PMID:17727329; http://dx.doi.org/ 10.1517/14712598.7.9.1401 [DOI] [PubMed] [Google Scholar]
- 48.Strohl WR. Optimization of Fc-mediated effector functions of monoclonal antibodies. Curr Opin Biotechnol 2009; 20:685-91; PMID:19896358; http://dx.doi.org/ 10.1016/j.copbio.2009.10.011 [DOI] [PubMed] [Google Scholar]
- 49.Kim HY, Kim S, Chung DH. FcgammaRIII engagement provides activating signals to NKT cells in antibody-induced joint inflammation. J Clin Invest 2006; 116:2484-92; PMID:16917543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ernst LK, Metes D, Herberman RB, Morel PA. Allelic polymorphisms in the FcgammaRIIC gene can influence its function on normal human natural killer cells. J Mol Med (Berl) 2002; 80:248-57; PMID:11976734; http://dx.doi.org/ 10.1007/s00109-001-0294-2 [DOI] [PubMed] [Google Scholar]
- 51.Smith P, DiLillo DJ, Bournazos S, Li F, Ravetch JV. Mouse model recapitulating human Fcgamma receptor structural and functional diversity. Proc Natl Acad Sci U S A 2012; 109:6181-6; PMID:22474370; http://dx.doi.org/ 10.1073/pnas.1203954109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lux A, Yu X, Scanlan CN, Nimmerjahn F. Impact of immune complex size and glycosylation on IgG binding to human FcgammaRs. J Immunol 2013; 190:4315-23; PMID:23509345; http://dx.doi.org/ 10.4049/jimmunol.1200501 [DOI] [PubMed] [Google Scholar]
- 53.Kagan JC. “Complementing” toll signaling. Sci Signal 2010; 3:pe15; PMID:20442416 [DOI] [PubMed] [Google Scholar]
- 54.Gearing AJ, Thorpe SJ, Miller K, Mangan M, Varley PG, Dudgeon T, Ward G, Turner C, Thorpe R. Selective cleavage of human IgG by the matrix metalloproteinases, matrilysin and stromelysin. Immunol Lett 2002; 81:41-8; PMID:11841844; http://dx.doi.org/ 10.1016/S0165-2478(01)00333-9 [DOI] [PubMed] [Google Scholar]
- 55.Ryan MH, Petrone D, Nemeth JF, Barnathan E, Bjorck L, Jordan RE. Proteolysis of purified IgGs by human and bacterial enzymes in vitro and the detection of specific proteolytic fragments of endogenous IgG in rheumatoid synovial fluid. Mol Immunol 2008; 45:1837-46; PMID:18157932; http://dx.doi.org/ 10.1016/j.molimm.2007.10.043 [DOI] [PubMed] [Google Scholar]
- 56.Adams GP, Schier R, McCall AM, Simmons HH, Horak EM, Alpaugh RK, Marks JD, Weiner LM. High affinity restricts the localization and tumor penetration of single-chain fv antibody molecules. Cancer Res 2001; 61:4750-5; PMID:11406547 [PubMed] [Google Scholar]
- 57.Fan X, Brezski RJ, Fa M, Deng H, Oberholtzer A, Gonzalez A, Dubinsky WP, Strohl WR, Jordan RE, Zhang N, et al.. A single proteolytic cleavage within the lower hinge of trastuzumab reduces immune effector function and in vivo efficacy. Breast Cancer Res 2012; 14:R116; PMID:22873525; http://dx.doi.org/ 10.1186/bcr3240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ngo CV, Picha K, McCabe F, Millar H, Tawadros R, Tam SH, Nakada MT, Anderson GM. CNTO 859, a humanized anti-tissue factor monoclonal antibody, is a potent inhibitor of breast cancer metastasis and tumor growth in xenograft models. Int J Cancer 2007; 120:1261-7; PMID:17192924; http://dx.doi.org/ 10.1002/ijc.22426 [DOI] [PubMed] [Google Scholar]
- 59.Gao B, Tsan MF. Endotoxin contamination in recombinant human heat shock protein 70 (Hsp70) preparation is responsible for the induction of tumor necrosis factor alpha release by murine macrophages. J Biol Chem 2003; 278:174-9; PMID:12403778; http://dx.doi.org/ 10.1074/jbc.M208742200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.