

Abstract
Conjugation of small molecule drugs to specific sites on the antibody molecule has been increasingly used for the generation of relatively homogenous preparations of antibody-drug conjugates (ADCs) with physicochemical properties similar or identical to those of the naked antibody. Previously a method for conjugation of small molecules to glycoproteins through existing glycans by using an engineered glycotransferase and a chemically reactive sugar as a handle was developed. Here, for the first time, we report the use of this method with some modifications to generate an ADC from a monoclonal antibody, m860, which we identified from a human naïve phage display Fab library by panning against the extracellular domain of human HER2. M860 bound to cell surface-associated HER2 with affinity comparable to that of Trastuzumab (Herceptin®), but to a different epitope. The m860ADC was generated by enzymatically adding a reactive keto-galactose to m860 using an engineered glycotransferase and conjugating the reactive m860 to aminooxy auristatin F. It exhibited potent and specific cell-killing activity against HER2 positive cancer cells, including trastuzumab-resistant breast cancer cells. This unique ADC may have utility as a potential therapeutic for HER2 positive cancers alone or in combination with other drugs. Our results also validate the keto-galactose/engineered glycotransferase method for generation of functional ADCs, which could potentially also be used for preparation of ADCs targeting other disease markers.
Keywords: antibody-drug conjugates, ADC, site-specific ADC, glycoengineering, anti-HER2
Introduction
The two commonly-used conjugation approaches for generation of ADCs (through cysteines forming interchain disulfide bonds and through surface-exposed lysines) result in heterogeneous preparations and possible adverse effects on antigen binding and other physicochemical properties. In the case of attachment through cysteines, the conjugation could result in a distribution of 0 to 8 drugs per antibody because most human IgG molecules have four interchain disulfide bonds. Partial reduction of disulfide bonds and variations in solvent-exposure of cysteines normally lead to heterogeneous mixtures of ADCs with many different combinations of cysteine attachment sites.1 Conjugation through lysines results in much more sites of conjugation and possible combinations, e.g., in the huN901-DM1 40 modification sites were identified which is about 47% of the 86 lysine residues in the antibody.2 Therefore, both conjugation methods allow variable drug load and conjugations sites, which lead to heterogeneous distribution of ADCs and may result into unpredictable pharmacokinetic properties and lot-to-lot variations.
Various site-specific antibody conjugation approaches that may minimize such potential problems have been recently reviewed.3,4 These include site-specific conjugations through engineered cysteines,5 unnatural amino acids,6 and selenocysteines,7 and enzymatically, e.g., by using transglutaminase.8 For example, the use of monoclonal antibodies (mAbs) with engineered reactive cysteine residues results in homogeneous ADCs with defined drug-to-antibody ratio (DAR) of 2 or more, better stability and larger therapeutic window.9 However, even for this engineered cysteine method, selection of appropriate site for optimal maleimide-thiol conjugate stability in vivo could be a challenge.10
An approach for site-specific conjugation of fluoroprobes to engineered Fc N-glycans of recombinant antibodies using a mutant glycosyltransferase11,12 was previously described.13 Recently this enzymatic method of bio-conjugation via glycan chains of the prostate specific membrane antigen-targeting antibody J591 with the positron-emitting 89Zr was used for detection of tumors in nude mice in PET imaging experiments.14 In the study reported here, we explored the feasibility of using this approach for site-specific antibody-drug conjugation for targeted cancer therapy by using a newly identified mAb to HER2. HER2 is a validated target for antibody-based cancer therapy as demonstrated by the clinical success of trastuzumab (Herceptin®),15 and ado-trastuzumab emtansine (Kadcyla®, T-DM1), an antibody-drug conjugate of trastuzumab and maytansinoid DM1, for therapy of HER2-overexpressing breast cancer and other cancers.16 We identified a new HER2-specific human antibody from a naïve antibody phage display library and used it as a model for the validation of site-specific antibody-drug conjugation based on the antibody Fc N-glycans. The ADC derived from this antibody by glycoengineering technology involving a mutant glycosyltransferase and a reactive galactose exhibited specific binding and killing of HER2-overexpressing cancer cells, and has potential as a cancer therapeutic. The enzymatic and chemical reactions leading to the glycan specific ADC is illustrated in Figure 1.
Figure 1.

Schematic diagram of the site-specific glycoconjugation for generating IgG1 m860-nAF ADC is illustrated. The native IgG1 m860 with desialylated N-glycan attached to Asn 297 of the CH2 domain (a close-up molecular surface view of a CH2 domain with carbohydrates in sticks) was treated with β-galactosidase to obtain a homogeneous G0F glycoform population. The fully degalactosylated IgG1 contains four GlcNAc residues per IgG1 molecule at the terminal positions available for the transfer of C2-keto-Gal from its UDP-derivative by Y289L-b4Gal-T1. The chemically reactive C2-keto-gal sugar handle is used to conjugate the drug molecule AF via a non-cleavable linker as shown.
Results
Identification, expression and characterization of IgG1 m860
Five different Fabs with unique sequences were identified after three rounds of panning against a recombinant HER2 protein ectodomain. All Fabs showed specific binding activity to recombinant HER2 ectodomain with various affinities (Fig. 2A). One of the antibodies, m860, with the highest binding activity, was converted into full-length human IgG1. The IgG1 m860 was produced from a Chinese hamster ovary (CHO)-based stable clone and was used for the ADC generation. Competition ELISA showed that m860 Fab does not compete with Trastuzumab for binding to the HER2 ectodomain (Fig. 2B).
Figure 2.
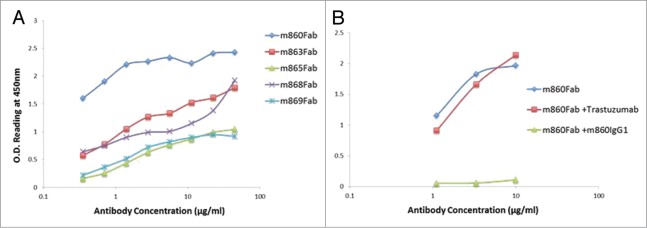
Binding assay of HER2-specific Fabs isolated from the library. (A) ELISA screening of the five Fabs (m860, m863, m865, m868 and m869) isolated from the naïve library on coated HER2 ectodomain. (B) Competition ELISA showed m860 Fab could not be competed by Trastuzumab at 20μg/ml, while m860 IgG1 at the same concentration potently outcompeted the binding of m860 Fab to HER2.
Mass spectrometry analysis of glycans in native and β-galactosidase-treated IgG1 m860
To evaluate the N-glycosylation profile of the IgG1 m860 produced from CHO cells, the N-glycans were released by PNGase F treatment as described in Materials and Methods. MALDI-TOF analysis of oligosaccharides released after the PNGase F treatment of the CHO cell-expressed IgG1 m860 showed two main peaks at m/z 1484 and 1648 corresponding to the G0F and G1F glycoforms, respectively (Fig. 3A), indicating that IgG1 m860 mainly carries G0F and G1F glycoforms corresponding to 0 and 1 galactose residue at the tip of the N-glycan core structure, respectively. No sialylated structures were observed in this case, unlike other mAbs produced from CHO cells. To selectively remodel the oligosaccharide of the IgG1 m860 at Asn 297 in the Fc domain and to obtain a homogeneous population of G0F glycoform-bearing antibody, IgG1 m860 was degalactosylated by β1,4-galactosidase from S. pneumoniae for 24 h. After galactosidase treatment, the degree to which the IgG1 m860 glycans were degalactosylated was confirmed by MALDI-TOF analysis of the N-glycans after PNGase F treatment. Incubation for 24 h of 5 mg/ml−1 of IgG1 m860 with 50 mU units of the galactosidase in 50 μl incubation mixture completely converted the N-glycans on IgG1 m860 into G0F glycoform (Fig. 3B).
Figure 3.
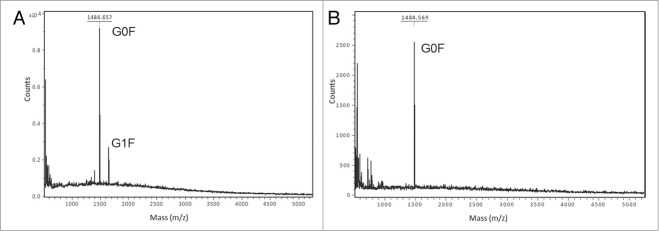
Glycoforms of IgG1 m860 produced from CHO cells. MS analysis of N-glycans after β-galactosidase and sialydase treatment of IgG1 m860. (A) MALDI-TOF MS analysis of N-glycans released by PNGase F treatment of native IgG1 m860. G0F glycoform with a peak at 1485.6 m/z, the G1F glycoform with a peak at 1647.6 m/z, and G2F glycoform was not detected in this preparation. IgG1 m860 was treated with β-galactosidase to obtain a homogeneous G0F glycoform population (B) MALDI-TOF analysis of N-glycans released by PNGase F treatment of β-galactosidase treated IgG1 m860. Only the G0F glycoform with a peak at 1485.6 m/z is seen after treatment.
Modification of the Fc N-glycan of IgG1 m860 through mutant β1,4Gal-T1-Y289L enzyme-mediated reaction using UDP-keto-Gal as a sugar donor
To attach the keto group onto the Fc-N-glycans, as previously described,11 a mutant β1,4Gal-T1-Y289L enzyme was used. Figure 4A shows the MALDI-TOF profile of the oligosaccharide after the transfer of the keto-galactose by the mutant enzyme to the free GlcNAc residues on the G0F glycoforms of IgG1 m860. The transfer of the UDP-keto-Gal by the mutant enzyme β1,4Gal-T1-Y289L to both arms of the G0F glycoform was observed as a main peak at m/z 1889 corresponding to modified G2F glycoform carrying the keto groups (Fig. 4A).
Figure 4.

(A) Reglycosylation of G0F glycoform using the mutant enzyme β1,4Gal-T1-Y289L and UDP-2-keto-Gal as sugar donor. A peak at 1889.6 m/z corresponds to the G2F glycoform, indicating the β-galactosidase treated IgG1 m860 having a G0F glycoform are fully galactosylated to the G2F glycoform after transfer of C2-keto-Gal moiety to the terminal GlcNAc residues. (B) Antibody-drug conjugation through oximation reaction. A peak at 3637 m/z corresponds to conjugated glycan with both arms attached with nAF.
Preparation of m860ADC and its characterization
To validate this site-specific antibody-drug conjugation, we chose auristatin F (AF), one of the most commonly used cytotoxic drugs in clinical ADC development. ADCs developed using AF with a non-cleavable linker at the C-terminus have shown potent cell killing activity and improved pharmacological profile in vivo.6 The non-cleavable ethylene glycol linker derivatized with an alkoxy-amine was synthesized and attached to the AF as previously described.6 The keto-containing m860 IgG was coupled to the alkoxy-amine linker-derivatized AF (3 mM) in 100 mM sodium acetate buffer, pH 4.5, at 37 °C for 60 h, followed by purification with a size exclusion column. Analysis by ESI-MS revealed that the heavy chain of the ADC has a mass 1747 Da larger than the keto group carrying IgG1 m860 heavy chain, which corresponds to the mass of two drug molecules with linker (nAF) per heavy chain (Fig. 5); this result is also consistent with the MALDI-TOF profile of the released oligosaccharide from the m860ADC (Fig. 4B).
Figure 5.

Mass spectrometry analysis of the light chain and heavy chain of IgG1 m860 before (A) and after the keto-modification (B) and after conjugation with nAF (C). The molecular weight of the light chains is not affected after the conjugation. The change in the molecular weight of the heavy chain after modification and subsequent conjugation indicated the efficient conjugation of two small molecule drugs per heavy chain.
Purified m860ADC was tested for binding to cell surface-expressed HER2 and compared with naked antibody IgG1 m860 and also Trastuzumab. The binding activity of of IgG1 m860 and Trastuzumab to SKBR3 is very similar (Fig. 6A). Also the binding activity of m860ADC to HER2 on SKBR3 cells was very well preserved (Fig. 6A). SDS-PAGE analysis of the purified m860ADC and naked IgG1 m860 under reducing and on-reducing conditions showed that the antibody is stable after the modification and conjugation treatment at 37° for 6 d (Fig. 6B). Furthermore, to evaluate whether the conjugation of the AF to glycans on the IgG1 m860 may sterically interfere with its binding activity to FcγRIIIa and FcγRI, Biacore analysis was performed to measure the binding affinities of m860ADC and IgG1 m860 to FcγRIIIa and FcγRI. As shown in Table 1, their affinity constants of binding to FcγRIIIa and FcγRI are similar before and after the N-glycan-specific conjugation, indicating that antibody FcγR effector functions are well preserved.
Figure 6.
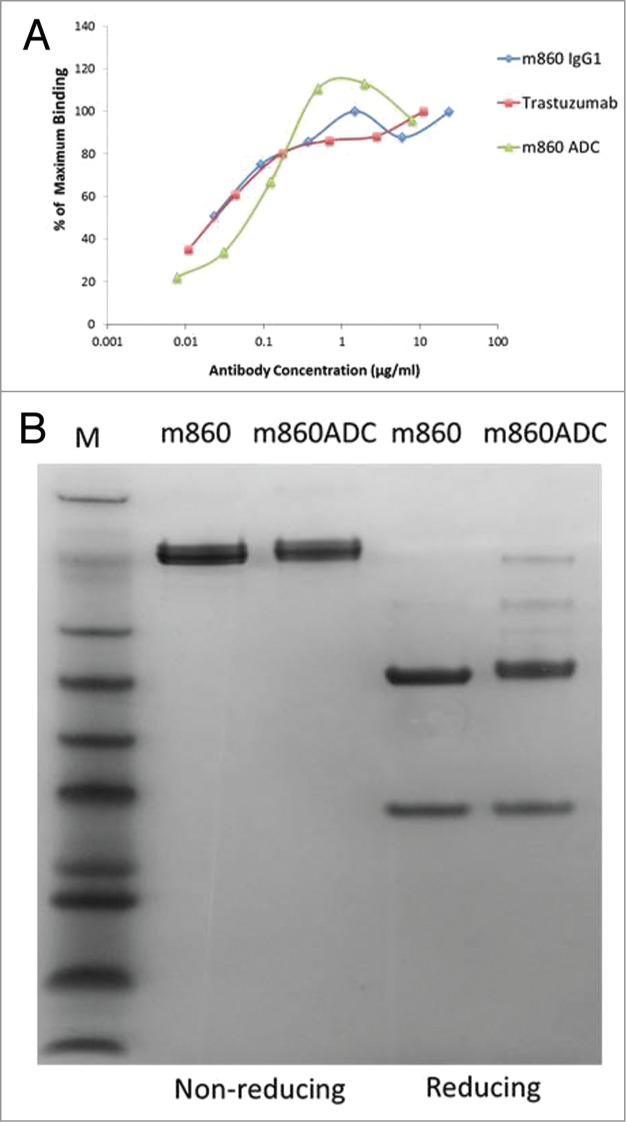
Characterization of m860ADC. (A) Flow cytometry analysis of the binding of IgG1 m860, Trastuzumab and m860ADC to SK-BR-3 cells. As expected, the binding activity of IgG1 m860 to HER2 on SK-BR-3 was not affected by the drug (nAF) conjugation to N-glycans attached to Fc. (B) SDS-PAGE gel analysis of m860ADC and naked IgG1 m860 under reducing and non-reducing conditions.
Table 1.
Site-specific antibody-drug conjugation on N-glycan of Fc does not affect the antibody binding activity to FcγRI and FcγRIIIa
| ka[1/Ms] |
kd[1/s] |
KD[M] |
||||
|---|---|---|---|---|---|---|
| Antibody | FcγRI | FcγRIIIa | FcγRI | FcγRIIIa | FcγRI | FcγRIIIa |
| m860 IgG1 | 4.5E+4 | 1.1E+4 | 0.001007 | 0.01608 | 2.2E-8 | 1.5E-6 |
| m860ADC | 4.4E+4 | 0.9E+4 | 0.001078 | 0.01752 | 2.5E-8 | 1.9E-6 |
| Control IgG1 | 7.6E+4 | 2.2E+4 | 0.001372 | 0.01484 | 1.8E-8 | 6.7E-7 |
m860ADC specifically kills HER2-overexpressing cancer cells
Purified m860ADC was tested with the HER2-overexpressing breast cancer cell line SK-BR-3 cells for cytotoxicity, and the cell line MCF-7 cells with minimal HER2 expression level for specificity. m860ADC potently killed SK-BR-3 cells (Fig. 7A), but only moderately killed MCF-7 cells at relatively high concentrations (Fig. 7B). Potent cell-killing activity of m860ADC was also observed with JIMT-1 cells (Fig. 7C), a cell line isolated from a patient with Trastuzumab-resistant breast cancer. The free drug with linker killed both SK-BR-3 cells and MCF-7 cells with similar potency. These data indicate that the active drug was efficiently released through degradation of the ADC inside the cells. To compare the cell killing potency of m860ADC and T-DM1, the same cell killing assay was also performed with SK-BR-3 cells using serially diluted m860ADC and T-DM1. The results showed that both m860ADC and T-DM1 can kill the target cells with similar potency (Fig. 7D).
Figure 7.

In vitro cytotoxicity assays. (A) m860ADC is more cytotoxic (IC50 0.032nM) than the free drug (nAF) (IC50 0.096nM) to HER2-overexpresing SK-BR-3 cells, while the WT IgG1 m860 does not show any cytotoxicity. (B) m860ADC shows cytotoxicity only at relatively high concentration to MCF-7 cells, while the free drug with linker alone shows significant cytotoxicity to MCF-7 cells (IC50 0.26nM). (C) Cytotoxicity assay on JIMT-1 cells. The JIMT-1 cell line was isolated from a Trastuzumab resistant patient, this cell line shows moderate HER2 expression compared with SK-BR-3. m860ADC can still potently kill JIMT-1 cells (IC50 0.087nM), while the IgG1 m860 alone has little effect on the cells. (D) Cell killing activities of m860ADC and T-DM1 were compared side by side on SK-BR-3 cells, the IgG1 m860 and an irrelevant ADC were used as control. The IC50s for m860ADC and T-DM1 are respectively 0.038nM and 0.095nM under these test conditions. (E and F) Serum stability of m860ADC was evaluated through cell killing assay using m860ADC samples collected after incubation in human serum for up to 30 d on HER2-positive SK-BR-3 cells and samples collected at day 0, 5 and 9 were assayed on HER2-negative Kelly cells. (G) Serum stability of T-DM1 was also evaluated and compared side by side with m860ADC.
Serum stability comparison of m860ADC with T-DM1
Recently, plasma/serum stability of an ADC was shown to have an impact on its efficacy.10 Therefore, the serum stability of m860ADC and T-DM1 was evaluated by incubating with human serum at 37 °C for up to 30 d, ADC samples collected after 0, 5, 9, 20 and 30 d were tested on HER2-positive SK-BR-3 cells and m860ADC samples collected at day0, 5 and 9 d were also tested on HER2-negative Kelly cells. The results indicate that there was no activity loss or specificity change after 30 d incubation in human serum (Figs. 7E and 7F). In contrast, Kadcyla exhibited significant loss of activity even after 5 Days of incubation (Fig. 7G). One should note that we did not purify the ADC from drug released from the ADC (see also, e.g.,6). The rationale is that the free drug is bound to serum protein and cannot exert the same level of toxicity. In addition, we showed that when incubated with HER2-negative cells there is no any toxicity (Fig. 7F). Therefore, if there was free drug released from the ADC and not associated with serum proteins it would have had toxicity. Also this assay can clearly distinguish between relatively unstable ADCs, e.g., T-DM1 and more stable ones, e.g., m860ADC (Figs. 7E,G).
Three-dimensional (3D) structure modeling of m860ADC
To identify putative structural features and the possible effector and complement functions of m860ADC, we generated 3D structural models of full-length IgG1 m860 and nAF using the procedures described in the “Material and Methods” section. Four of the nAF molecules were attached to the four modeled C2-keto-gal sugar moieties of m860 without any significant steric clashes. Thus, the m860ADC model has a maximum drug to antibody ratio of 4 (DAR = 4) as depicted in Figure 8. IgG1 residues comprising the binding sites of FcRn,17,18 FcγIIIR,17,19 and C1q17,20,21 (in blue, magenta and pink, respectively) were mapped in the 3D m860ADC model as shown in Figure 8.
Figure 8.
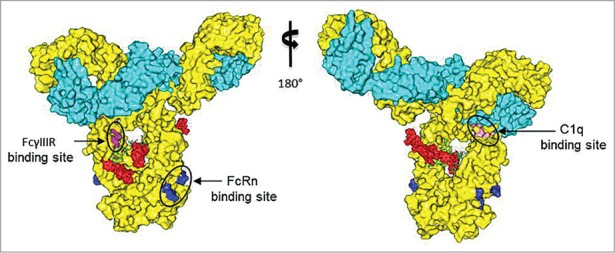
A three-dimensional molecular model of the IgG1 m860-nAF ADC. Heavy and light chains of IgG1 m860 are shown in yellow and cyan, respectively, and four nAF molecules are shown as red spheres. Mutational data along with the structural analyses from previous studies revealed amino acids critical for binding to FcRn (blue), C1q (pink) and FcγRIII (magenta) which are mapped in the m860ADC model.
Discussion
Site-specific modifications of the Fc oligosaccharide moieties on the antibodies have been studied for decades.22-25 However, the chemistry-based oxidation of oligosaccharides is hard to control, and also may potentially modify the amino acid side chains on the antibody peptide backbone and cause formation of unpredictable metabolite products in vivo. The enzyme-based approach for the modification of the Fc N-glycans could be advantageous over traditional approaches,13 as the mild reaction conditions will most likely leave the other part of the antibody intact and may not have any negative effect on the native conformation, thereby, retaining the stability and binding activity of antibody as shown in a recent study.14,26
As shown from the MS analysis of the intact light chain and heavy chain of the m860ADC without any deglycosylation treatment, the homogeneity of the heavy chain was dramatically improved, which also indicates that most of the end product ADC had 4 drug molecules per antibody molecule, i.e., the DAR was 4. A very small portion of the antibody heavy chain bearing mannose only glycan formats may exist in the antibody produced from CHO cells, which will not be modified during the enzyme-based modification step, and will also be spared during the final conjugation step. Stochastic combination of heavy chains bearing mannose only glycans and heavy chains bearing other glycans such as G0F, G0NF in full-length IgG could generate a small percentage of ADCs with 0, 1, 2, and 3 drugs. However, the percentage of full-length antibodies with mannose-only glycans, i.e., antibodies without drug molecules, should be less than 1%.
It has been proposed that the mechanism of the off-target toxicities for ADC biotherapeutics is at least in part driven by the carbohydrates, specifically agalactosylated glycans, such as G0F, their interactions with mannose receptor and resulting glycan-derived cellular uptake of ADCs.27 However, the ADCs prepared using our approach have all the G0F re-glycosylated with modified galactose and conjugated with small molecule drugs, thus inactivating the binding of ADCs to the mannose receptor, which could potentially lower the glycan-driven off-target toxicity of ADCs.
We used a 3D molecular model of the m860ADC, as shown in a surface view (Fig. 8) to predict possible structural features and map the binding sites for effector molecules. The ADC model suggested that four molecules of the nAF could be occupied at the cleft between the CH2 domains without any steric clashes. This indicated that the maximum DAR of 4 may be accommodated for m860ADC. The total nonpolar surface area for one nAF molecule was calculated to be 1266 Å2. Interestingly, according to the 3D model of the m860ADC described in this particular approach of glycoconjugation, the part of the AF including the hydrophobic phenylalanine moiety might be hidden inside the cleft between the two CH2 domains, and kept in close proximity to hydrophilic regions of carbohydrates, which could decrease the aggregation tendency of the m860ADC. Structural and mutational analyses of human IgG1 variants for mapping the binding sites of effector and complement molecules had been extensively performed in the past. We mapped in the ADC model those amino acids identified as critical from the previous studies (Fig. 8) including the IgG1 residues S239, D265, D270, E293, Y296 and N297 which when altered can abrogate or reduce the ability of IgG1 to bind to FcγRIIIa.17,19 The surface view of the ADC model showed no overlapping or interference between the FcγRIIIa binding site and nAF. This further reinforced the binding data of the ADC to FcγRIIIa and FcγRI, indicating that the nAF conjugation may not disrupt this effector function of IgG1 m860.
The binding of the C1q subunit to the Fc region is the first step of complement activation. Although crystal structures of the individual molecules are known, the complex crystal structure of those molecules is not yet available.28 It has been shown that the binding sites on the Fc region for the C1q and the FcγR molecules are overlapping. Also, the C1q binding enhancing residues on the Fc region have been identified to be in the CH2 domain near the upper hinge region.21 The four residues D270, P329, P331 and K322 of IgG1 that had been already reported17,20,21 to constitute the C1q binding site are displayed in pink (Fig. 8). We found that the Gal moiety of the Asn297 N-glycan is far away from the C1q binding site, and did not overlap with the nAF. Therefore, it could be possible that the presence of the drug conjugate at the Gal moiety of the Asn297 N-glycan may not affect the binding of IgG1 m860 to C1q. We also tried to predict the possible FcRn binding properties of m860ADC. The interaction between FcRn and the Fc portion of IgG is critical for the serum half-life of therapeutic mAbs.18 Mapping of the FcRn binding site, including the amino acids I253,S254,H435 and Y436 that are important in mediating the binding to human FcRn at pH 6.0,17,29,30 on the ADC model as shown in blue (Fig. 8) suggested that the drug conjugation may not disrupt those amino acids at the FcRn binding site. Therefore, 3D modeling and mapping of C1q and FcRn binding sites as shown in the surface view of m860ADC (Fig. 8) suggested that the drug conjugation may not disrupt the secondary effector and complement function. However, further experimental studies are needed to confirm the predictions. Taken together, these results suggest that Fc N-glycan-specific antibody-drug conjugation using engineered enzymes combined with reactive sugars can be a useful approach for site-specific ADC preparation. The new mAb-based ADC could also have potential as a cancer therapeutic.
Material and Methods
Reagents
Recombinant β1,4 galactosidase from Streptococcus pneumoniae was from Calbiochem (San Diego, CA). Peptide N-glycoside F (PNGase F) was from New England Biolabs (Ipswich, MA). Microcon Ultracel YM-50 centrifugation devices came from Millipore Corporation, (Bedford, MA). Protein A-Sepharose 4B Conjugate was from Invitrogen (Eugene, OR). Trastuzumab and Kadcyla (T-DM1) were purchased from NIH pharmacy (Bethesda, MD). UDP-C2-keto-Gal was synthesized by Allichem LLC (Baltimore, MD). Auristatin F with noncleavable linker (nAF) attached at C-terminus,6 which was synthesized by Concortis Biosystems.
Isolation of HER2-specific antibody
A naıve human Fab phage display library (a total of about 1010 members), constructed from peripheral blood B cells of 10 healthy donors,31 was used for selection of Fabs against purified recombinant HER2 ectodomain mainly following the process as previously described; the isolated Fabs were expressed, purified and tested for binding to the HER2 ectodomain through ELISA and the best binder was converted to full-length IgG1 as previously described.32 A CHO cell-based stable cell line was generated as previously described and was used for the production of full-length IgG1.32
ELISA and competition ELISA
HER2 ectodomain Fc fusion protein was diluted in PBS at 2μg/ml and coated on 96-well plate at 4 °C overnight. Serially diluted purified Fabs (m860, 863, m865, m868 and m869) were added to the coated wells after blocking and washing. Bound Fabs with Flag tag were detected by mouse anti-Flag HRP conjugate(Sigma-Aldrich, St. Louis, MO). The TMB substrate (Sigma-Aldrich, St. Louis, MO) was added, and A450 was measured. For competition ELISA, m860 IgG1 and Trastuzumab were pre-mixed with the diluted m860 Fab with final concentration at 20 μg/ml before adding to the target antigen coated wells, bound m860 Fab was detected and signal was recorded as described above.
β1,4 galactosyltransferase expression and folding from bacterial inclusion body
Expression and purification of the enzyme β1,4Gal-T1-Y289L used in this study have been previously described.11
Degalactosylation of IgG1 m860
IgG1 m860 was washed with 50 mM sodium phosphate pH 6.0, using a Microcon Ultracel YM-50 centrifugation device. The samples at 8 mg/ml were incubated with 100 mU of recombinant Streptococcus pneumoniae β1,4 galactosidase for 24 h at 37 °C. Removal of terminal galactose residues was confirmed by analysis of the N-glycans released after PNGase F treatment by MALDI TOF spectrometry. Approximately 3 μg of mAbs were incubated in the presence of PNGase F (2500 Units), 16 h at 37 °C in PBS. Digested sample was analyzed by mass spectrometry. Degalactosylated IgG1 m860 was then purified by protein A affinity chromatography.
Transfer of C-2 keto Galactose from its UDP-derivative to free GlcNAc residues on IgG1 m860 using the mutant β1,4Gal-T1-Y289L and conjugate with nAF
Two mg of degalactosylated IgG1 m860 was incubated with 2 mM UDP-C2 keto-Gal and 0.5 mg of the mutant β1,4Gal-T1-Y289L in 2 ml 50 mM TRIS-HCl (pH 8.0) containing 10 mM MnCl2 at 37 °C for 48 h. The ketone-labeled antibody was subsequently dialyzed into 50 mM NaOAc (pH 4.5) through filtration and mixed with nAF with mole ratio at 1 to 5. The conjugation reaction was performed at 37 °C for 72 h. A portion of the sample was treated with PNGase F for the MALDI-TOF analysis of the glycan chain.
Matrix-assisted Laser Desorption/Ionization Mass Spectrometry
Typically, 1–2 μl of sample were mixed with an equal volume of 2,5-dihydrobenzoic acid (DHB) matrix solution (Agilent, part #G2039A). All spectra were acquired on a Bruker Ultraflex III TOF/TOF mass spectrometer (Bruker Daltonics, Billerica, MA). Spectra were acquired in positive ion reflector mode, with no ion extraction delay. Ion source voltage 1 was 25 kV, ion source voltage 2 was 21.85 kV and lens voltage was 8.55 kV. Reflectron and reflectron 2 were 26.30 and 13.75 kV, respectively. Deflection was set at 600 Da. Monoisotopic masses were determined using FlexAnalysis 3.0 (Bruker Daltonics) with the SNAP peak picking algorithm. External calibration in reflector mode was performed using peptide standards of the Bruker peptide calibration kit (Bruker Daltonics, Billerica, MA).
Cell surface HER2 binding activity comparison of IgG1 m860, m860ADC and Trastuzumab by flow cytometry analysis
To determine if the modifications in IgG1 m860 after the drug conjugation influenced its ability to bind to the cellular HER2 receptor, FACS analysis was performed to compare IgG1 m860 binding activity to HER2 on SK-BR-3 cells before and after the drug conjugation. HER2 receptor expressing human breast adenocarcinoma cells (SK-BR-3) was purchased from the American Type Culture Collection (ATCC, Manassas, VA). The cells were cultured in DMEM medium supplemented with 10% (v/v) heat inactivated fetal bovine serum (FBS) and penicillin/streptomycin as antibiotics at 37 °C in a humidified atmosphere containing 5% CO2. SK-BR-3 cells were harvested using a PBS-based, enzyme-free cell dissociation buffer, and suspended to a concentration of 107 cells per ml in PBS containing 0.5% BSA (PBSA). IgG1 m860, m860ADC and Trastuzumab were serially diluted in PBSA and mixed with cells. The mixtures were incubated for 1 h on ice, centrifuged and washed twice with cold PBSA. PE-conjugated goat anti-human IgG was mixed with all the samples and incubated on ice for 30 min. Control samples were incubated without any primary antibody, with only the PE-conjugated goat anti-human IgG. All samples were washed twice with cold PBSA and then analyzed using a FACS Calibur flow cytometer (Becton Dickinson, San Jose, CA).
High-resolution mass spectrometry
Modified or unmodified IgG1 m860 were mixed with buffer (7.5M guanidine-HCl, 0.1 M TRIS-HCl and 1mM EDTA) in the presence of 20 mM DTT and incubated at 70 °C for 15 min. Mass spectrometry data were acquired on an Agilent 6520 Accurate-Mass Q-TOF LC/MS System, (Agilent Technologies, Inc., Santa Clara, CA) equipped with a dual electro-spray source, operated in the positive-ion mode. Separation was performed on Zorbax 300SB-C3 Poroshell column (2.1 mm × 75 mm; particle size 5 μm). The analytes were eluted at a flow rate of 1 ml/min with a 1 to 90% organic gradient over 5 min and holding organic for 1 min. Both mobile phases, water and acetonitrile, contained 0.1% formic acid. The instrument was used in a full-scan TOF mode. MS source parameters were set with a capillary voltage of 4 kV, the fragmentor voltage of 220 V and skimmer 65 V. The gas temperature was 350 °C, drying gas flow 12 l/min and nebulizer pressure 55 psig. Data were acquired at high resolution (3,200 m/z), 4 GHz. TOF-MS mass spectra were recorded across the range 100–3,200 m/z. To maintain mass accuracy during the run time, an internal mass calibration sample was infused continuously during the LC/MS runs. Data acquisition was performed using Mass Hunter Workstation (version B.02.00). Mass Hunter Qualitative Analysis software (version B.03.01) with Bioconfirm Workflow was used for data analysis and deconvolution of mass spectra.
Size exclusion chromatography
m860ADC was purified and analyzed using a Superdex200 10/300 GL column (GE Healthcare, Piscataway, NJ) calibrated with protein molecular mass standards of 14-kDa RNase A, 25-kDa chymotrypsin, 44-kDa ovalbumin, 67-kDa albumin, 158-kDa aldolase, 232-kDa catalase, 440-kDa ferritin, and 669-kDa thyroglobulin. Purified m860ADC in phosphate-buffered saline (PBS) was loaded onto the pre-equilibrated column and eluted with PBS at 0.5 ml/min.
Surface plasmon resonance
Binding kinetics of IgG1 m860 and m860ADC with human FcγRIIIa and FcγRI were measured using surface plasmon resonance analysis on a Biacore X100 apparatus (GE Healthcare) and using a single-cycle approach according to the manufacturer's instructions. Briefly, human recombinant FcγRIIIa and FcγRI were diluted in sodium acetate (pH 5.0) and immobilized directly onto a CM5 sensor chip via the standard amine coupling method. The reference cell was injected with N-hydroxysuccinimide–1-ethyl-3-(3Dimethyaminopropy) carbodiimide and ethanolamine without injection of gp140. Purified IgG1 m860 and m860ADC were diluted with running buffer HBS-EP (100 mM HEPES [pH 7.4], 1.5 M NaCl, 30 mM EDTA, 0.5% surfactant 20). All analytes were tested at 500, 100, 20, 4, and 0.8 nM concentrations. Kinetic constants were calculated from the sensorgrams fitted with the monovalent binding model of the BiacoreX100 Evaluation software 2.0.
Cell killing assay
SK-BR-3 and MCF-7 cell lines were obtained from ATCC. Cells were cultured and assayed in DMEM (Life technology) with 10% FBS (Gibco), 100 IU/ml penicillin, and 100 μg/ml streptomycin (Gibco) for SK-BR-3, JIMT-1,Kelly and MCF-7. For the cell killing assays, the cancer cell line of interest was plated in 96-well plates at 1,000 cells in 75 μL per well in cell culture media and incubated overnight at 37 °C and 5% CO2. All testing reagents were sterilized through filtration (0.22 μm; Millipore) and 4 × solutions were prepared in cell culture medium as serial dilutions. Twenty-five μL from the 4 × stocks were added to the 75 μL of cells in duplicate. Wells with PBS only and 5 μM nAF were used as 100% viability and 0% viability controls, respectively. The wells on the outermost edges of the plate were filled with 250 μL of cell culture media. After 3 d of incubation, 20 μL of CellTiter 96® AQueous One Solution (Promega) was added and incubated for 4 h before reading at 490 nm. Relative readings were normalized to percent viability between PBS and free drug controls.
Serum stability assay
m860ADC and T-DM1 stock solutions at 1mg/ml were diluted and incubated in human serum (Sigma) at 150ug/ml at 37 °C, samples were collected at day 0, 5, 9, 20 and 30 and stored at −80 °C before the cytotoxicity assay on SK-BR-3 cells and HER2 negative Kelly cells to evaluate the cell killing potency and specificity of the ADCs in the collected samples after the serum incubation.
3D molecular modeling of ADC
To build a 3D molecular model of m860ADC, we performed molecular modeling procedures involving homology modeling of the full-length IgG1 m860, 3D atomic modeling of drug molecule with linker and manual molecular building of ADC. We used the PIGS,33 a homology modeling server for constructing the 3D structure of the variable fragment (Fv) regions of m860. Modeled CDRs L1-L3, H1 and H2 were from the same canonical structures, and grafting of the CDR-H3 made with similar length and composition of the target as available from the database of known structures. We applied different criteria for light/heavy chain template selection such as “Same Antibody” and “Best H and L chains.” The conformations of side chains were maintained as long as those residues were conserved between the target and the template, while side chains from other residues were modeled using SCWRL 3.0 as implemented. The final model Fv m860 was selected based on the best stereochemical parameters, including the minimal steric clashes at the VH/VL interface. We then built the full-length IgG1 m860 based on the known crystal structure of the human IgG1 b12 (PDB accession code: 1HZH).34 The Fv region of the model generated from PIGS was replaced with both arms of the IgG1 b12 Fv regions by 3D-aligning of structures. Then, the κ light chain portion of CH1 domain in the b12 was replaced with that of the λ light chain of a known antibody structure (PDB accession code: 2DD8),35 and the Fc portion of b12 was retained to create a complete model with the correct constant regions. To further improve the 3D model of IgG1 m860, we performed molecular refinement and energy minimization procedures using Chiron36 and 3D-Refine.37 The atomic model of nAF was built using the “Ligand Builder” module of COOT,38 which produced the 3D coordinates with optimized geometry. Discovery Studio version 2.5 (Accelrys Inc., San Diego, CA) was used to compute the molecular properties of the linker-drug molecule. The nAF was manually connected to all four of the C2-keto-gal sugar moiety from the two CH2 domains. The orientations of four molecules of nAF attached to IgG1 m860 were selected such that they did not have any clashes among themselves and with antibody. PyMOL39 was used for further visual examination of the ADC and molecular rendering.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank members of our group and Sergey Tarasov from the Structural Biology Laboratory, Center for Cancer Research, National Cancer Institute, National Institutes of Health for helpful discussions.
Funding
This project was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, and by federal funds from the National Cancer Institute, NIH, under contract N01-CO-12400, HHSN261200800001E as well as by the SU2C-St.Baldrick's Foundation.
References
- 1. Hamblett KJ, Senter PD, Chace DF, Sun MM, Lenox J, Cerveny CG, Kissler KM, Bernhardt SX, Kopcha AK, Zabinski RF, et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin Cancer Res 2004; 10:7063-70; PMID:15501986; http://dx.doi.org/ 10.1158/1078-0432.CCR-04-0789 [DOI] [PubMed] [Google Scholar]
- 2. Wang L, Amphlett G, Blättler WA, Lambert JM, Zhang W. Structural characterization of the maytansinoid-monoclonal antibody immunoconjugate, huN901-DM1, by mass spectrometry. Protein Sci 2005; 14:2436-46; PMID:16081651; http://dx.doi.org/ 10.1110/ps.051478705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Behrens CR, Liu B. Methods for site-specific drug conjugation to antibodies. MAbs 2014; 6:46-53; PMID: 24135651; http://dx.doi.org/ 10.4161/mabs.26632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Panowksi S, Bhakta S, Raab H, Polakis P, Junutula JR. Site-specific antibody drug conjugates for cancer therapy. MAbs 2014; 6:34-45; PMID:24423619; http://dx.doi.org/ 10.4161/mabs.27022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Junutula JR, Raab H, Clark S, Bhakta S, Leipold DD, Weir S, Chen Y, Simpson M, Tsai SP, Dennis MS, et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat Biotechnol 2008; 26:925-32; PMID:18641636; http://dx.doi.org/ 10.1038/nbt.1480 [DOI] [PubMed] [Google Scholar]
- 6. Axup JY, Bajjuri KM, Ritland M, Hutchins BM, Kim CH, Kazane SA, Halder R, Forsyth JS, Santidrian AF, Stafin K, et al. Synthesis of site-specific antibody-drug conjugates using unnatural amino acids. Proc Natl Acad Sci U S A 2012; 109:16101-6; PMID:22988081; http://dx.doi.org/ 10.1073/pnas.1211023109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hofer T, Skeffington LR, Chapman CM, Rader C. Molecularly defined antibody conjugation through a selenocysteine interface. Biochemistry 2009; 48:12047-57; PMID:19894757; http://dx.doi.org/ 10.1021/bi901744t [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jeger S, Zimmermann K, Blanc A, Grünberg J, Honer M, Hunziker P, Struthers H, Schibli R. Site-specific and stoichiometric modification of antibodies by bacterial transglutaminase. Angew Chem Int Ed Engl 2010; 49:9995-7; PMID:21110357; http://dx.doi.org/ 10.1002/anie.201004243 [DOI] [PubMed] [Google Scholar]
- 9. Junutula JR, Flagella KM, Graham RA, Parsons KL, Ha E, Raab H, Bhakta S, Nguyen T, Dugger DL, Li G, et al. Engineered thio-trastuzumab-DM1 conjugate with an improved therapeutic index to target human epidermal growth factor receptor 2-positive breast cancer. Clin Cancer Res 2010; 16:4769-78; PMID:20805300; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-0987 [DOI] [PubMed] [Google Scholar]
- 10. Shen BQ, Xu K, Liu L, Raab H, Bhakta S, Kenrick M, Parsons-Reponte KL, Tien J, Yu SF, Mai E, et al. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat Biotechnol 2012; 30:184-9; PMID:22267010; http://dx.doi.org/ 10.1038/nbt.2108 [DOI] [PubMed] [Google Scholar]
- 11. Ramakrishnan B, Qasba PK. Structure-based design of beta 1,4-galactosyltransferase I (beta 4Gal-T1) with equally efficient N-acetylgalactosaminyltransferase activity: point mutation broadens beta 4Gal-T1 donor specificity. J Biol Chem 2002; 277:20833-9; PMID:11916963; http://dx.doi.org/ 10.1074/jbc.M111183200 [DOI] [PubMed] [Google Scholar]
- 12. Khidekel N, Arndt S, Lamarre-Vincent N, Lippert A, Poulin-Kerstien KG, Ramakrishnan B, Qasba PK, Hsieh-Wilson LC. A chemoenzymatic approach toward the rapid and sensitive detection of O-GlcNAc posttranslational modifications. J Am Chem Soc 2003; 125:16162-3; PMID:14692737; http://dx.doi.org/ 10.1021/ja038545r [DOI] [PubMed] [Google Scholar]
- 13. Boeggeman E, Ramakrishnan B, Pasek M, Manzoni M, Puri A, Loomis KH, Waybright TJ, Qasba PK. Site specific conjugation of fluoroprobes to the remodeled Fc N-glycans of monoclonal antibodies using mutant glycosyltransferases: application for cell surface antigen detection. Bioconjug Chem 2009; 20:1228-36; PMID:19425533; http://dx.doi.org/ 10.1021/bc900103p [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zeglis BM, Davis CB, Aggeler R, Kang HC, Chen A, Agnew BJ, Lewis JS. Enzyme-mediated methodology for the site-specific radiolabeling of antibodies based on catalyst-free click chemistry. Bioconjug Chem 2013; 24:1057-67; PMID:23688208; http://dx.doi.org/ 10.1021/bc400122c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dinh P, de Azambuja E, Piccart-Gebhart MJ. Trastuzumab for early breast cancer: current status and future directions. Clin Adv Hematol Oncol 2007; 5:707-17; PMID:17982412 [PubMed] [Google Scholar]
- 16. Burris HA, 3rd, Tibbitts J, Holden SN, Sliwkowski MX, Lewis Phillips GD. Trastuzumab emtansine (T-DM1): a novel agent for targeting HER2+ breast cancer. Clin Breast Cancer 2011; 11:275-82; PMID:21729661; http://dx.doi.org/ 10.1016/j.clbc.2011.03.018 [DOI] [PubMed] [Google Scholar]
- 17. Shields RL, Namenuk AK, Hong K, Meng YG, Rae J, Briggs J, Xie D, Lai J, Stadlen A, Li B, et al. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J Biol Chem 2001; 276:6591-604; PMID:11096108; http://dx.doi.org/ 10.1074/jbc.M009483200 [DOI] [PubMed] [Google Scholar]
- 18. Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol 2007; 7:715-25; PMID:17703228; http://dx.doi.org/ 10.1038/nri2155 [DOI] [PubMed] [Google Scholar]
- 19. Ferrara C, Grau S, Jäger C, Sondermann P, Brünker P, Waldhauer I, Hennig M, Ruf A, Rufer AC, Stihle M, et al. Unique carbohydrate-carbohydrate interactions are required for high affinity binding between FcgammaRIII and antibodies lacking core fucose. Proc Natl Acad Sci U S A 2011; 108:12669-74; PMID:21768335; http://dx.doi.org/ 10.1073/pnas.1108455108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Idusogie EE, Presta LG, Gazzano-Santoro H, Totpal K, Wong PY, Ultsch M, Meng YG, Mulkerrin MG. Mapping of the C1q binding site on rituxan, a chimeric antibody with a human IgG1 Fc. J Immunol 2000; 164:4178-84; PMID:10754313; http://dx.doi.org/ 10.4049/jimmunol.164.8.4178 [DOI] [PubMed] [Google Scholar]
- 21. Moore GL, Chen H, Karki S, Lazar GA. Engineered Fc variant antibodies with enhanced ability to recruit complement and mediate effector functions. MAbs 2010; 2:181-9; PMID:20150767; http://dx.doi.org/ 10.4161/mabs.2.2.11158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zara JJ, Wood RD, Boon P, Kim CH, Pomato N, Bredehorst R, Vogel CW. A carbohydrate-directed heterobifunctional cross-linking reagent for the synthesis of immunoconjugates. Anal Biochem 1991; 194:156-62; PMID:1867379; http://dx.doi.org/ 10.1016/0003-2697(91)90163-N [DOI] [PubMed] [Google Scholar]
- 23. Vogel CW. Preparation of immunoconjugates using antibody oligosaccharide moieties. Methods Mol Biol 2004; 283:87-108; PMID:15197304 [DOI] [PubMed] [Google Scholar]
- 24. Rakestraw SL, Tompkins RG, Yarmush ML. Preparation and characterization of immunoconjugates for antibody-targeted photolysis. Bioconjug Chem 1990; 1:212-21; PMID:1710935; http://dx.doi.org/ 10.1021/bc00003a006 [DOI] [PubMed] [Google Scholar]
- 25. Rodwell JD, Alvarez VL, Lee C, Lopes AD, Goers JW, King HD, Powsner HJ, McKearn TJ. Site-specific covalent modification of monoclonal antibodies: in vitro and in vivo evaluations. Proc Natl Acad Sci U S A 1986; 83:2632-6; PMID:3458222; http://dx.doi.org/ 10.1073/pnas.83.8.2632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Acchione M, Kwon H, Jochheim CM, Atkins WM. Impact of linker and conjugation chemistry on antigen binding, Fc receptor binding and thermal stability of model antibody-drug conjugates. MAbs 2012; 4:362-72; PMID:22531451; http://dx.doi.org/ 10.4161/mabs.19449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gorovits B, Krinos-Fiorotti C. Proposed mechanism of off-target toxicity for antibody-drug conjugates driven by mannose receptor uptake. Cancer Immunol Immunother 2013; 62:217-23; PMID:23223907; http://dx.doi.org/ 10.1007/s00262-012-1369-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schneider S, Zacharias M. Atomic resolution model of the antibody Fc interaction with the complement C1q component. Mol Immunol 2012; 51:66-72; PMID:22425350; http://dx.doi.org/ 10.1016/j.molimm.2012.02.111 [DOI] [PubMed] [Google Scholar]
- 29. Firan M, Bawdon R, Radu C, Ober RJ, Eaken D, Antohe F, Ghetie V, Ward ES. The MHC class I-related receptor, FcRn, plays an essential role in the maternofetal transfer of gamma-globulin in humans. Int Immunol 2001; 13:993-1002; PMID:11470769; http://dx.doi.org/ 10.1093/intimm/13.8.993 [DOI] [PubMed] [Google Scholar]
- 30. Dall’Acqua WF, Woods RM, Ward ES, Palaszynski SR, Patel NK, Brewah YA, Wu H, Kiener PA, Langermann S. Increasing the affinity of a human IgG1 for the neonatal Fc receptor: biological consequences. J Immunol 2002; 169:5171-80; PMID:12391234; http://dx.doi.org/ 10.4049/jimmunol.169.9.5171 [DOI] [PubMed] [Google Scholar]
- 31. Zhu Z, Dimitrov DS. Construction of a large naïve human phage-displayed Fab library through one-step cloning. [xv.]. Methods Mol Biol 2009; 525:129-42, xv; PMID:19252833; http://dx.doi.org/ 10.1007/978-1-59745-554-1_6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhu Z, Dimitrov AS, Bossart KN, Crameri G, Bishop KA, Choudhry V, Mungall BA, Feng YR, Choudhary A, Zhang MY, et al. Potent neutralization of Hendra and Nipah viruses by human monoclonal antibodies. J Virol 2006; 80:891-9; PMID:16378991; http://dx.doi.org/ 10.1128/JVI.80.2.891-899.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Marcatili P, Rosi A, Tramontano A. PIGS: automatic prediction of antibody structures. Bioinformatics 2008; 24:1953-4; PMID:18641403; http://dx.doi.org/ 10.1093/bioinformatics/btn341 [DOI] [PubMed] [Google Scholar]
- 34. Saphire EO, Parren PW, Pantophlet R, Zwick MB, Morris GM, Rudd PM, Dwek RA, Stanfield RL, Burton DR, Wilson IA. Crystal structure of a neutralizing human IGG against HIV-1: a template for vaccine design. Science 2001; 293:1155-9; PMID:11498595; http://dx.doi.org/ 10.1126/science.1061692 [DOI] [PubMed] [Google Scholar]
- 35. Prabakaran P, Gan J, Feng Y, Zhu Z, Choudhry V, Xiao X, Ji X, Dimitrov DS. Structure of severe acute respiratory syndrome coronavirus receptor-binding domain complexed with neutralizing antibody. J Biol Chem 2006; 281:15829-36; PMID:16597622; http://dx.doi.org/ 10.1074/jbc.M600697200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ramachandran S, Kota P, Ding F, Dokholyan NV. Automated minimization of steric clashes in protein structures. Proteins 2011; 79:261-70; PMID:21058396; http://dx.doi.org/ 10.1002/prot.22879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bhattacharya D, Cheng J. 3Drefine: consistent protein structure refinement by optimizing hydrogen bonding network and atomic-level energy minimization. Proteins 2013; 81:119-31; PMID:22927229; http://dx.doi.org/ 10.1002/prot.24167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 2004; 60:2126-32; PMID:15572765; http://dx.doi.org/ 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 39. The PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC. [Google Scholar]