

Abstract
Full-length IgG antibodies cannot cross cell membranes of living cells; this limits their use for direct targeting of cytosolic proteins. Here, we describe a general strategy for the generation of intact, full-length IgG antibodies, herein called cytotransmabs, which internalize into living cells and localize in the cytosol. We first generated a humanized light chain variable domain (VL) that could penetrate into the cytosol of living cells and was engineered for association with various subtypes of human heavy chain variable domains (VHs). When light chains with humanized VL were co-expressed with 3 heavy chains (HCs), including 2 HCs of the clinically approved adalimumab (Humira®) and bevacizumab (Avastin®), all 3 purified IgG antibodies were internalized into the cytoplasm of living cells. Cytotransmabs primarily internalized into living cells by the clathrin-mediated endocytic pathway through interactions with heparin sulfate proteoglycan that was expressed on the cell surface. The cytotransmabs escaped into the cytosol from early endosomes without being further transported into other cellular compartments, like the lysosomes, endoplasmic reticulum, Golgi apparatus, and nucleus. Furthermore, we generated a cytotransmab that co-localized with the targeted cytosolic protein when it was incubated with living cells, demonstrating that the cytotransmab can directly target cytosolic proteins. Internalized cytotransmabs did not show any noticeable cytotoxicity and remained in the cytosol for more than 6 h before being degraded by proteosomes. These results suggest that cytotransmabs, which efficiently enter living cells and reach the cytosolic space, will find widespread uses as research, diagnostic, and therapeutic agents.
Keywords: cell-penetrating antibody, cytosolic protein targeting, cellular internalization, endosomal release, IgG antibody, intracellular trafficking, next-generation antibody
Abbreviations
- IgG
immunoglobulin G
- VL
light chain variable domain
- VH
heavy chain variable domain
- LC
light chain
- HC
heavy chain
- CDR
complementarity-determining region
Introduction
Full-length immunoglobulin G (IgG) antibodies that specifically bind to a target molecule with high affinity have been extensively developed as research tools, as well as for diagnostic and therapeutic purposes. However, their targets are primarily the proteins expressed on the cell-surface and some secreted proteins because antibodies normally cannot cross intact cellular or subcellular membranes in living cells due to their large size and hydrophilicity.1,2 In this context, stepwise cellular fixation and permeabilization are necessary to make research antibodies to recognize intracellular targeted proteins. Furthermore, disease-associated protein–protein interactions occur mainly in the cytosol of cells, and the inhibition of these interactions can be more efficiently achieved by antibodies than by small chemical agents because of the large or flat interaction surfaces of antibodies.3 Therefore, there is an increasing demand for efficient methods of antibody delivery into the cytosol of living cells for use in a diverse array of applications.2
Many attempts have been made to directly deliver antibodies into intracellular compartments; some of the methods used include microinjection, electroporation, and protein transfection (profection).2,4,5 Although these methods are successful for delivering antibodies into the cytoplasm of cultured living cells, many issues, including cytotoxicity, loss of antibody stability, and difficulty of systemic administration, remain unresolved.1,2 Other approaches include using targeted receptor-mediated endocytosis and genetic or chemical conjugation with protein transduction domains that directly penetrate into living cells.1,6 However, large molecules, including antibodies that enter epithelial cells via receptor-mediated endocytosis, are usually retained in endosomes and are then recycled out of the cells or are degraded in lysosomes without being released into the cytosol.1,2,7 Conjugation with protein transduction domains, which are represented by cell-penetrating peptides (CPPs) such as the HIV-1 TAT peptide, has been extensively attempted in order to facilitate the intracellular delivery of antibodies formatted as single chain variable fragments (scFvs), antigen-binding fragments (Fabs), and full-length IgGs.8-10 However, most of the CPP-conjugated antibodies inherited the intrinsic intracellular trafficking of the parent CPPs, which were either entrapped inside endocytic vesicles, translocated into the nucleus, or eventually degraded in lysosomes without efficient endosomal release into the cytosol.1,10,11
Some anti-DNA polyclonal antibodies or monoclonal antibodies (mAbs) predominantly found in humans and mice with autoimmune diseases have been shown to possess the ability to penetrate into living cells in their IgG format.12,13 Most cell-penetrating anti-DNA antibodies in the IgG or scFv formats eventually localized in cell nucleus.12,13 Taking advantage of its ability to penetrate cells and localize inside the nucleus, the murine anti-DNA 3E10 scFv has been exploited for use as one arm of the bispecific antibody in the scFv-scFv format to target MDM2 protein in the nucleus.14 We previously reported the murine anti-DNA m3D8 scFv, which, unlike other anti-DNA antibodies, internalized into living cells and was localized in the cytosolic regions without further trafficking into other subcellular compartments, including the nucleus.15 The ability of m3D8 scFv to penetrate cells and then localize into the cytosol (referred to herein as ‘cytosol-penetrating ability’) was derived from the m3D8 VL.16,17 Thus, m3D8 VL is an ideal moiety for the generation of cytosol-penetrating antibodies that can be used for diverse purposes.
In this study, we aimed to generate a cytosol-penetrating antibody, which we called ‘cytotransmab’, in the form of a full-length IgG by incorporating a cytosol-penetrating VL into light chains (LCs) and then co-expressing the LCs with the heavy chains (HCs). For this, we first generated humanized VLs that retained the cytosol-penetrating ability of m3D8 VL and contained the consensus residues at the interface with the heavy chain variable domain (VH) for proper association with the VH partner. When the LCs containing the humanized VL were co-expressed with the HCs carrying various subtypes of VHs, the purified IgG mAbs internalized into living cells through a physiological endocytic pathway and then reached the cytosol regions. Furthermore, we show that our strategy facilitates the generation of a cytotransmab that can directly target cytosolic proteins when it is incubated with living cells under normal culture conditions.
Results
Generation of a cytosol-penetrating antibody containing humanized VL single domain
We previously generated a humanized VL, named hT0, by grafting the complementarity-determining regions (CDRs) of the m3D8 VL (first found in mice) into the corresponding regions of a human framework region of Vκ3 and Jκ1 gene segments.18 However, in live HeLa cells (human cervix carcinoma), hT0 failed to bind to cell surfaces, leading to complete loss of the cell-penetrating ability of m3D8 VL16,17 (Fig. 1A). The cell-penetrating ability of m3D8 VL was conferred by VL-CDR1 with a unique cationic patch that is composed of Arg27f, Arg29, and Lys30.16 Superimposition of the model of the homologous hT0 structure using the WAM algorithm19 and the reported crystal structure of m3D8 VL20 revealed that the regions from Phe27c to Lys30 of hT0-CDR1 have deviated from their positions on m3D8 VL; an approximately 5.3 Å shift was revealed between the 2 Cα atoms of Arg27f at the tip of the loop, while the other CDRs were largely well superimposed on each other (Fig. 1B). Some residues in the β-sheet framework regions that closely underlie CDRs, called Vernier-zone residues, play a critical role in adjustment of the loop structures of CDRs, thus affecting CDR loop conformation.21 Of the 14 amino acid residues that were determined to be Vernier-zone residues,21,22 only 2, the Ile2 and Leu4 residues of hT0 (which were positioned right below CDR1), were different from Leu2 and Met4 residues of m3D8 (Fig. S1A). Back mutations of these 2 residues with the corresponding murine residues (I2L and L4M) in hT0 predicted a similar CDR1 conformation to that of m3D8-CDR1 (Fig. 1B). Thus, we generated an hT0 variant, named hT2, with I2L and L4M substitutions (Fig. S1A). Similar to m3D8 VL, the soluble, purified hT2 VL internalized into cytoplasmic regions of HeLa cells without further trafficking into the nucleus (Fig. 1A), indicating that the CDR1 conformation, rather than the linear amino acid sequence, is a determining factor for cell-penetrating activity.
Figure 1.
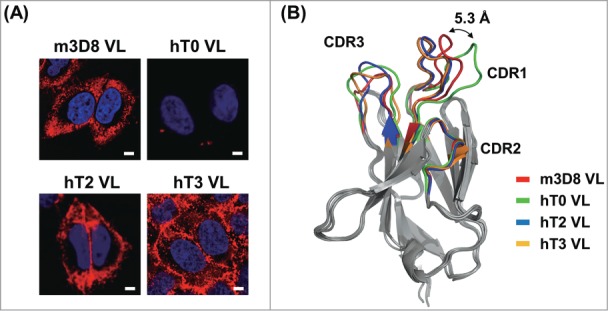
Generation of humanized, cytosol-penetrating VL single domain antibodies by closely maintaining the CDR1 conformation of m3D8 VL. (A) Cellular internalization and localization of mouse m3D8 VL and humanized VLs (hT0, hT2, and hT3) in HeLa cells that were treated with VLs (10 μM) for 6 h at 37°C. The cells were then analyzed for internalized VLs by confocal fluorescence microscopy. Images show the merging of VLs (red) and Hochest33342-stained nuclei (blue) at the centered single confocal section. (B) Superposition of the homology modeled hT0, hT2, and hT3 structures with the crystal structure of m3D8 VL (PDB 3BD3)20 to compare their CDR conformations with those of m3D8 VL. Double-headed arrow indicates the distance between 2 Cα atoms of Arg-27f in the tip of CDR1 of m3D8 VL and hT0 VL. The image was generated using PyMol software (DeLano Scientific LLC). The primary sequences of all VLs are shown in Fig. S1A.
Most commercialized therapeutic mAbs have a Vκ1 subgroup of the VL gene family rather than Vκ3 subgroup for pairing with various VH subgroups.23 Accordingly, we generated an hT3 VL, which had the same consensus framework sequence of the human Vκ1 and Jκ1 genes and the same CDR sequences as hT2 VL (Fig. 1A and S1A). Soluble, purified hT3 VL also internalized and dominantly accumulated in the cytoplasmic regions of HeLa cells (Fig. 1A). Investigation of cellular internalization mechanisms revealed that the humanized hT2 and hT3 VLs retained the caveolae-mediated endocytosis pathway of m3D8 VL rather than by clathrin-mediated endocytosis or macropinocytosis (Fig. S1B and S1C).
Design of cytotransmabs by incorporation of humanized, cytosol-penetrating VL
We sought to develop a general strategy for the generation of cytosol-penetrating IgG-format cytotransmabs by replacing the VL in conventional IgG1 format with the humanized cytosol-penetrating VL (Fig. 2A). As an HC partner, we chose IgG1-isotype HC with humanized h3D8 VH, generated by grafting the 3 CDRs of m3D8 VH into the corresponding regions of human framework with VH3–23 (DP-47), JH6, and DH3 gene segments.18 To test the general applicability of this strategy, we further chose 2 HCs from clinically approved mAbs: adalimumab (Humira®) against tumor necrosis factor (TNF)24 and bevacizumab (Avastin®) against vascular endothelial growth factor A (VEGF).25 Adalimumab (IgG1 with VH3-Vκ1) and bevacizumab (IgG1 with VH7-Vκ1) were selected because they could not internalize into cells since they bind to the secreted cytokines, but not to any cell surface-expressed receptors. Furthermore, they have the most predominant VH-VL subtype pairing among the clinically approved mAbs with the same Vκ1 subtype as hT3.23
Figure 2.

Generation of cytosol-penetrating intact IgG-format cytotransmabs, which internalize into living cells and then localize in the cytosol without cytotoxicity. (A) Schematic diagram showing the general strategy for generation of cytotransmabs by replacing endogenous VL of conventional IgG with the humanized cytosol-penetrating hT4 VL. (B and C) Biochemical characterizations of the purified parent mAbs and cytotransmabs by reducing and non-reducing SDS-PAGE (B) and SEC (C) analyses. In (B), the purified antibodies (5 μg) were separated on 12% SDS polyacrylamide gels and visualized by Coomassie brilliant blue staining (R-250). In (C), the purified antibodies were injected at 200 μg/mL in 70 μL of sample volume and monitored at 280 nm. Arrows indicate the elution positions of molecular weight standards. (D) Cellular internalization and localization of cytotransmabs (TMab4, HuT4, and AvaT4) and control mAbs (adalimumab and bevacizumab) in HeLa cells that were treated with the antibodies (1 μM) for 6 h at 37°C and then analyzed by confocal immunofluorescence microscopy. (E) Concentration-dependent internalization of TMab4 in HeLa cells that were treated with the indicated concentrations of TMab4 for 6 h at 37°C prior to confocal immunofluorescence microscopy analysis. (F) Time-dependent internalization of TMab4 in HeLa cells that were treated with TMab4 (1 μM) at 37°C for the indicated periods. In (D–F), after removing cell-surface bound antibodies with low pH glycine buffer (pH 2.5), internalized antibodies were visualized with FITC-conjugated anti-human IgG Fc antibody (green). Images show the merging of antibodies (green) and Hochest33342-stained nuclei (blue) at the centered single confocal section. Image magnification, 630×; scale bar, 10 μm. (G and H) Cytotoxicity of internalized cytotransmabs in HeLa cells that were treated with the antibodies (1 μM) at 37°C for 24 h or 48 h. In (G), the morphological features of cells were observed by phase-contrast microscopy after 48 h. Image magnification, 200×; scale bar, 10 μm. In (H), the cell viability was determined by MTT assay after 24 h or 48 h. The data represent mean ± SD compared with the PBS-treated controls.
To evaluate whether hT3 (Vκ1) can be properly associated with the chosen VHs for IgG-format expression, we first analyzed the VH-interacting residues of hT3 VL (Fig. S2). Previous structural analyses of 23 crystallized VH-VL interfaces identified 10 positions of VL that were primarily involved in VH interactions, with the highest contact frequencies within a 4.1 Å distance from the corresponding VH residue atoms.26 hT3 VL had amino acid residues observed most frequently at all of the VH-interacting positions, except for 2 residues of K89 and S91 in VL-CDR3 that commonly interact with 103–3 and 95 residues in VH-CDR3, respectively (Fig. S2A).27,28 Improper association between VH and VL could impair the assembly between HCs and LCs in the IgG-format antibodies, leading to a decrease in the expression, stability, or antigen binding.27 Indeed, co-expression of LC carrying hT3 VL with the chosen HCs to generate IgG-format antibodies resulted in decreased IgG production yield compared with the parent mAbs (Table 1). Hence, we designed an hT3 variant, named hT4, with 2 substitutions of K89Q and S91Y to have the most frequently observed amino acid residues at the 2 positions such that hT4 can be broadly associated with various human VH gene family members (Fig. S2B).
Table 1.
Purification yields of modeled mAbs and cytotransmabs that were obtained by transient expression in HEK293F cells
| IgG clones | VH of HCa | VL of LCa | IgG purification yield (mg/1 L of transfected cells)b,c |
|---|---|---|---|
| TMab3 | h3D8 | hT3 | 8.2 ± 0.5 |
| TMab4 | h3D8 | hT4 | 10.8 ± 1.0 |
| Adalimumab | Adalimumab | Adalimumab | 11.6 ± 0.3 |
| HuT3 | Adalimumab | hT3 | 3.5 ± 0.8 |
| HuT4 | Adalimumab | hT4 | 10.9 ± 0.8 |
| Bevacizumab | Bevacizumab | Bevacizumab | 8.8 ± 0.4 |
| AvaT3 | Bevacizumab | hT3 | 4.5 ± 0.8 |
| AvaT4 | Bevacizumab | hT4 | 8.0 ± 1.1 |
| C20 | C20 | C20 | 22.5 ± 2.5 |
| KT4 | C20 | hT4 | 21.3 ± 0.9 |
aThe constant domains of HC and LC were the IgG1-isotype and Cκ, respectively.
bThe 2 plasmids that encode the HC and LC of each IgG antibody were co-transfected with the equivalent molar ratio into HEK293F cells in 1 L of culture media following standard protocol.50
cAfter 7 d of culture, antibodies were purified from the cell culture supernatant using a protein-A affinity column. The values represent mean ± SD of at least 3 independent experiments.
Generation of cytosol-penetrating cytotransmabs
Both plasmids that encode HCs, which were composed of the chosen VH-IgG1 HC constant domains (CH1-hinge-CH2-CH3) and LCs of the hT4 VL-Cκ constant domain, were transiently co-transfected into HEK293F cells to express the IgG1-format cytotransmab, generating TMab4 (h3D8 HC-hT4 LC), HuT4 (Humira HC-hT4 LC), and AvaT4 (Avastin HC-hT4 LC) (Table 1). All of the cytotransmabs were well expressed in correctly assembled form and showed production yields (∼8–10 mg/L of culture) comparable to those of the parent mAbs (Fig. 2B and Table 1). The purified cytotransmabs existed in the intact monomeric form without soluble oligomers as assessed by size-exclusion chromatography (SEC) analyses (Fig. 2C). Because of the replacement of the original VLs with hT4 VL, HuT4 and AvaT4 lost their associated antigen binding activity (Fig. S3A). None of the cytotransmabs showed any nuclease activity (Fig. S3B).
When the 3 cytotransmabs were incubated at 37°C for 6 h with live HeLa cells under normal culture conditions in a medium with 10% fetal bovine serum (FBS), they internalized into the cytoplasmic regions of cells with little accumulation in the nucleus (Fig. 2D). Internalized cytotransmabs were seen as diffuse fluorescence throughout the cytosol and intense punctate fluorescence within the cytoplasm, indicating that some of them reached the cytosol and others remained entrapped within endocytic vesicles. As expected, the parent adalimumab and bevacizumab mAbs failed to internalize into the cells (Fig. 2D). The cytotransmabs also efficiently penetrated into other human cell lines, including PANC-1 (pancreatic carcinoma), HT-29 (colorectal carcinoma), and MCF-7 (breast carcinoma) cells, showing similar diffusive and punctate distributions in the cytoplasmic regions compared to those of HeLa cells (Fig. S4A). The internalized cytotransmabs were detected in all of the cells as revealed by confocal fluorescence microscopy (Fig. S4B), indicating that the internalization efficiency almost reached 100%. The internalizing efficiency of cytotransmabs was further confirmed by flow cytometric analysis (Fig. S4C).
Cellular internalization of TMab4 was readily detected above 100 nM and increased in proportion to concentration increase (Fig. 2E), indicating that the internalizing activity of cytotransmab is improved by ∼100-fold compared with that of hT3 VL (Fig. 1A). Time-course analyses revealed that TMab4 internalization was significantly detected as early as 30 min, gradually increased with incubation time with plateaued levels between 2 h and 12 h, and subsequently decreased at 24 h with almost disappearance after 48 h (Fig. 2F). Noticeably, internalization of TMab4, HuT4, and AvaT4 cytotransmabs did not cause any noticeable cytotoxicity when tested in HeLa and PANC-1 cells for either 24 h or 48 h of incubation (Fig. 2G and H; Fig. S5).
Endocytic mechanisms underlying cytotransmab internalization
To elucidate the internalization mechanism(s) of cytotransmabs, we first tested their temperature dependence. Cellular internalization of TMab4 and HuT4 into HeLa cells at 37°C was totally blocked at 4°C (Fig. 3A), which indicates that the internalization occurs through an energy-dependent, endocytic pathway.29 We previously reported that cellular uptake of m3D8 VL was initiated by non-specific electrostatic interactions with negatively-charged glycosaminoglycan heparan sulfate (HS), which was linked as a side chain to cell surface core proteins to form HS proteoglycan (HSPG).16,17 The presence of exogenous heparin, which is a close structural homolog of HS, significantly inhibited the cellular internalization of TMab4 and HuT4 (Fig. 3B). Additionally, TMab4 and HuT4 efficiently internalized into the cytoplasmic compartments of wild type CHO-K1 cells, but completely failed to internalize into HS-deficient pgsD-677 CHO-K1 mutant cells (Fig. 3C). In accordance with the above results with adherent cells, TMab4 entered non-adherent K562 (human erythroleukemia) cells with HSPG expression, but did not enter non-adherent Ramos (human Burkitt's lymphoma) cells, which are deficient in cell-surface HSPGs30 (Fig. 3D). Overall, these results provide biochemical and genetic evidence that cell surface HSPGs are the primary cellular receptors for cell-surface binding prior to the internalization of cytotransmabs.
Figure 3.

Cytotransmabs internalize into living cells primarily by energy-dependent, clathrin-mediated endocytosis through interactions with cell-surface HSPG. (A) Effect of temperature on the internalization of cytotransmabs into HeLa cells that were incubated with TMab4 or HuT4 (1 μM) for 6 h at 4°C or 37°C. (B) Effect of an external heparin competitor on cytotransmab internalization into HeLa cells that were treated with heparin (300 IU/mL) for 30 min at 37°C before incubating TMab4 or HuT4 (1 μM) for an additional 6 h. (C) Internalization of cytotransmabs into CHO-K1 wild type and pgsD-677 mutant (HSPG-deficient) cells that were incubated with TMab4 or HuT4 (1 μM) for 6 h at 37°C. (D) Internalization of cytotransmabs into non-adherent K562 (HSPG-expressed) and Ramos (HSPG-deficient) cells that were treated with TMab4 (1 μM) for 6 h at 37°C. In (A–D), internalized cytotransmabs were observed by confocal microscopy after staining with FITC-conjugated anti-human IgG Fc antibody (green) and nuclei with Hoechst33342 (blue). (E) Co-localization of internalized cytotransmabs with endocytosis markers. HeLa cells were co-incubated with cytotransmabs (1 μM) and endocytosis markers including 10 μg/mL of Alexa Fluor 488-transferrin (TF, green), FITC-cholera toxin B (Ctx-B, green), and Oregon green-dextran (Dextran, green) for 30 min at 37°C. Subsequently, internalized cytotransmabs were stained with TRITC-conjugated anti-Human IgG Fc antibody (red). The regions in the white box were magnified for better imaging of co-localization. In (A–E), centered single confocal sections are shown. Image magnification, 630×; scale bar, 5 μm. (F) Effect of endocytosis inhibitors on TMab4 internalization. HeLa cells were pre-treated for 30 min with the indicated endocytosis inhibitors, and then incubated with TMab4 for additional 2 h, followed by immunofluorescence staining, as shown Fig. S6. The internalization levels of TMab4 in the presence of inhibitors are represented as mean percentage (%) ± SEM by comparing the fluorescence intensity from inside of cells (n > 100 cells per group) with that of the untreated ′control′. ***P < 0.001 compared with the control.
To determine the specific endocytosis pathway that functions to internalize cytotransmabs, internalized TMab4 and HuT4 were co-stained with specific endocytic markers of the 3 main endocytosis pathways, including transferrin (TF) for clathrin-mediated endocytosis, cholera toxin B (Ctx-B) for caveolae-mediated endocytosis, and dextran for macropinocytosis.7,15,31 Cytotransmab-containing punctate vesicles mainly co-localized with TF and partly with Ctx-B, but did not localize at all with dextran (Fig. 3E). Accordingly, the cytotransmabs seemed to primarily internalize via clathrin-mediated endocytosis and partly via caveolae-mediated endocytosis. This result was further confirmed by studies with pharmacological inhibitors of this endocytosis pathway (Fig. 3F; Fig. S6). Chlorpromazine (CPZ) and dansylcadaverine (inhibitors of clathrin-mediated endocytosis) remarkably inhibited cellular uptake of TMab4. In contrast, methyl-β-cyclodextrin (MβCD) and nystatin (inhibitors of caveolae-mediated endocytosis), as well as 5-(N-ethyl-N-isopropyl) amiloride (EIPA) and wortmannin (inhibitors of macropinocytosis), negligibly affected TMab4 internalization. Actin dynamics are required for clathrin-mediated endocytosis, and dynamin is essential for the fission of endocytic vesicles from the plasma membrane in both clathrin- and caveolae-mediated endocytosis.7 As expected, cytochalasin D (Cyt-D) (inhibitor of actin-dependent endocytosis) and dynasore (dynamin inhibitor) also modestly impaired the internalization.
Intracellular trafficking and stability of internalized cytotransmab
To determine intracellular trafficking routes and the stability of cytotransmabs, a pulse-chase experiment was performed. After HeLa cells were incubated with TMab4 (3 μM) for 30 min, the cells were completely washed with low pH glycine buffer to remove non-internalized, surface-bound cytotransmabs. Thereafter, time-course intracellular routes of pulsed TMab4 were tracked by co-staining with endosome/sub-organelle markers, including EEA-1 for early endosomes, LysoTracker for lysosomes, caveolin-1 for caveosomes, calnexin for endoplasmic reticulum (ER), and 58K Golgi for Golgi apparatus. Alexa Fluor 488-labeled TF, which was destined to a recycling pathway or to lysosomal degradation after clathrin-mediated endocytosis, was also traced with TMab4 to compare intracellular stability.
After 0 h of tracing (i.e., after 30 min of internalization), TMab4 was detected in a dispersive pattern throughout the cytosol, as well as in small punctate, cytoplasmic vesicular structures that were dominantly co-localized with co-treated TF and partially with caveolin-1 and EEA-1, but not with the other sub-organelle markers of LysoTracker, calnexin, and 58K Golgi (Fig. 4A). At 2 h, TMab4 was substantially detected with similar patterns to those at 0 h. However, co-treated TF totally disappeared at 2 h, which is indicative of rapid recycling back out of cells by trafficking into recycling endosomes or lysosomal degradation, which is consistent with the findings of a previous study.32 This indicates that the endocytic vesicles containing TMab4 are more sustainable in the cytoplasmic spaces than those containing TF, even though both TMab4 and TF are initially similarly internalized by clathrin-mediated endocytosis. At 6 h, TMab4 signals were significantly reduced and the remaining punctate vesicles were still partially co-localized with EEA-1 without transporting to the lysosomes, ER, and Golgi apparatus. At 18 h, TMab4 completely disappeared. These results indicate that TMab4 mainly internalizes via clathrin-mediated endocytosis, localizes in early endosomes, and is then directly released into the cytosol from the early endosomes without further trafficking into the lysosomes, ER, Golgi apparatus, and nucleus, thereby persisting in the cytosol for more than 6 h prior to degradation.
Figure 4.

Intracellular trafficking and cytosolic release of internalized TMab4. (A) Pulse-chase intracellular trafficking of internalized TMab4 monitored by co-localization with TF, early endosome marker EEA-1, caveolin-1, late endosome/lysosome marker LysoTracker, ER marker calnexin, or Golgi marker 58K Golgi protein, as visualized by confocal immunofluorescence microscopy. Insets, enlarged images of the regions in the boxes. (B and C) Intracellular stability of internalized TMab4 with or without the proteasome inhibitor MG132 (30 μM) was analyzed by confocal microscopy (B) and western blotting (C). HeLa cells were pulsed with 3 μM of TMab4 for 30 min and incubated for the indicated times in the presence or absence of MG132 (30 μM) prior to analysis. In (B), TMab4 was visualized with FITC-conjugated anti-human IgG Fc antibody (green). In (C), equal amounts of cell lysates were loaded, and retained TMab4 was detected by protein gel blotting, using p53 as a positive control (revealing proteasome-involved degradation) and β-actin as a loading control. In (A–B): magnification, 630×; scale bar, 5 μm. (D) Cytotransmab-mediated cytosolic release of calcein. HeLa cells were untreated (′control′) or treated with 5 μM of TMab4, adalimumab, or HuT4 for 4 h and then incubated with 50 μM of calcein for additional 2 h at 37°C, followed by confocal analysis. Image magnification, 630×; scale bar, 20 μm. In (A, B, and D), images show the merging of markers/antibodies (indicated colors) and Hochest33342-stained nuclei (blue) at the centered single confocal section.
To elucidate the degradation mechanism of internalized TMab4, we examined the effects of MG132, an inhibitor to proteasomal degradation of ubiquitin-conjugated proteins,33 on the time-course intracellular stability of TMab4 after treatment for 30 min with HeLa cells. Confocal fluorescence microscopic analyses showed that the presence of MG132 made TMab4 substantially persist in the cytosol after 6 h and even slightly persist after 18 h (Fig. 4B), which was also confirmed by western blotting (Fig. 4C). Since the ubiquitin-proteasome proteolytic machinery operates in the cytosol of cells,34 the above results strongly support the cytosolic localization of TMab4 and its degradation by the cytosolic proteasomes.
Cytosolic release activity of cytotransmabs was further assessed by monitoring cytosolic release of calcein that was co-treated with cytotransmabs. Calcein, a membrane-impermeable fluorophore, was employed as a cargo tracer for cytosolic release from endosomes after endocytic uptakes by co-treated internalizing agents, because it cannot penetrate into cytosol from endosomes for itself.35 Treatment of calcein alone with live HeLa cells did not display any significant fluorescence in the cytosol while showing very dim fluorescence in the punctate endosomal pattern (Fig. 4D). However, the co-treatments of calcein with either TMab4 or HuT4, but not adalimumab, induced diffused calcein fluorescence throughout the cytoplasm of cells in addition to the punctate endosomal fluorescence (Fig. 4D), indicating that the cytotransmabs induced cytosolic release of calcein from endosomes.
We further monitored the cellular uptake and cytosolic localization of TMab4 using live-cell time-lapse fluorescence imaging for 2 h by incubating Alexa Fluor 488-labeled TMab4 with HeLa cells. Internalization of TMab4 occurred very rapidly, as indicated by the appearance of small punctate, cytoplasmic vesicles (Fig. S7A and Movie S1), which seem to be the early endosomes (Fig. 4A). The number of cytoplasmic vesicles increased over time in the cytosolic regions without further trafficking into the nucleus (Fig. S7A and Movie S1), which is consistent with the above pulse-chase trafficking results (Fig. 4A). Simultaneously, diffuse fluorescence signals steadily accumulated throughout the cytosolic regions (Fig. S7B), which is indicative of the direct release of TMab4 into the cytosol from early endosomes.
Cytotransmab penetrates and directly binds to a targeted cytosolic protein
To test whether cytotransmabs can directly target cytosolic proteins from outside of living cells, we generated a cytotransmab that specifically binds to human lysyl-tRNA synthetase (KRS), a cytosolic protein as a component of the multi-tRNA synthetase complex.36 We first generated a fully human C20 mAb, which specifically bound to KRS, primarily via the VH domain, with an apparent dissociation equilibrium constant (KD) of ∼0.58 nM (Fig. S8). Then, the VL of C20 was replaced with hT4 VL, which generated a KRS-targeting cytotransmab that was named KT4 (Fig. 5A).
Figure 5.
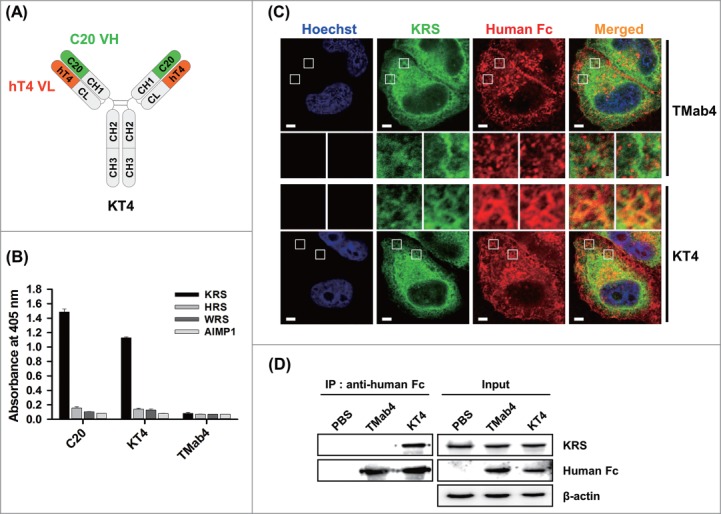
Anti-KRS KT4 internalizes into living cells and binds to the cytosolic KRS. (A) Diagram of IgG-format KT4 carrying KRS-binding C20 VH domain and cytosol-penetrating hT4 VL domain. (B) Direct ELISA to determine the binding specificity of C20 and KT4 IgG antibodies (100 nM) to plate-coated antigens (1 μg/mL), KRS, histidyl-tRNA synthetase (HRS), tryptophanyl-tRNA synthetase (WRS), or ARS-interacting multifunctional protein 1 (AIMP1). TMab4 was employed as a negative control. (C) Co-localization of internalized KT4 with KRS in GFP-fused KRS expressing HeLa cells, treated KT4 or TMab4 (20 μM) for 2 h at 37°C, and then determined by confocal microscopy. KT4 and TMab4 were visualized with TRITC-conjugated anti-Human IgG Fc antibody. Image magnification, 630×; scale bar, 5 μm. Images show the KRS (green), antibodies (red), and Hochest33342-stained nuclei (blue) at the centered single confocal section. Middle insets, enlarged images of the boxed regions. (D) Western blot analysis of immunoprecipitation (IP) of KRS by internalized KT4. The cell lysates prepared from HeLa cells that were treated with PBS (control), KT4 (6 μM), or TMab4 (6 μM) for 12 h at 37°C were precipitated using protein A agarose and analyzed by western blotting with β-actin as a loading control.
KT4 was well expressed, showing a similar purification yield to that of C20 (Table 1). KT4 specifically bound to KRS with an apparent KD of ∼36 nM, which is about 62-fold lower than that of C20 (Fig. S8), but did not cross-react with other aminoacyl-tRNA synthetases (ARSs) (Fig. 5B). In order to determine the direct cytosolic KRS targeting ability of KT4 after cellular internalization, KT4 was incubated with HeLa cells expressing green fluorescent protein (GFP)-fused KRS. KT4, but not TMab4, successfully co-localized with KRS in the cytosolic regions (Fig. 5C). However, a substantial portion of KT4 that was detected in punctate endocytic vesicles did not co-localize with KRS, indicating that KT4, which is sequestered into early endosomes, needs to be released into the cytosol in order to recognize the cytosolic target protein. The poor cytosolic release from early endosomes were also observed for cell-penetrating cationic proteins, which showed about 1 to 5% cytosolic access efficiency from their endocytosed vesicles.37 The specific interaction between KT4 and KRS in the cytosol was further confirmed by immunoprecipitation experiments for the lysates of HeLa cells that were pre-treated with KT4 for internalization prior to cell lysis. KRS co-precipitated with internalized KT4, but not internalized TMab4 (Fig. 5D).
Discussion
Innovative technology that enables full-length IgG antibodies to directly target cytosolic proteins is in high demand for a diverse array of applications, and these antibodies can be used as analytical and functional research agents in living cells, as well as imaging and therapeutic agents that target intracellular proteins.2 Here, we demonstrated a proof-of-concept for cytosol-penetrating IgG-format cytotransmabs that were generated by incorporation of a humanized, cytosol-penetrating VL into the VL position of conventional IgG mAbs. Simple pairing of LCs that contained the cytosol-penetrating VL with HCs transforms conventional mAbs into cytotransmabs, as evidenced by pairing with HCs of adalimumab and bevacizumab. Current approaches for intracellular delivery of antibodies, such as CPP-conjugated and anti-DNA scFv-fused antibodies, are almost restricted to nuclear targets because of the intrinsic nuclear targeting property of the carrier.1,2,8,10,14 On the other hand, cytotransmabs penetrated into the cytosol of living cells, enabling its direct targeting of cytosolic proteins.
We generated the humanized, cytosol-penetrating hT4 VL based on the consensus framework sequence of Vκ1 family germline segments with consensus residues at the interface with VH for proper association with VH. In this study, hT4 VL was well associated with VHs that were derived from either VH3 or VH7 family germline segments showing that the assembled cytotransmabs were well expressed in correctly assembled form without any compromise in the production yield compared to the parent mAbs. It is not certain whether hT4 VL is well associated with the other VH family germline segments. However, based on the successful generation of cytosol-penetrating VLs with Vκ3 (hT3) or Vκ1 (hT4) subtypes, we believe that cytosol-penetrating VLs can be rationally designed according to other VH subtypes for cytotransmab generation.
Cytotransmabs did not show noticeable cytotoxicity, which is indicative of their internalization via a physiological endocytosis pathway without plasma membrane disruption. The inability of cytotransmabs to internalize into HSPG-deficient cells demonstrated that their cellular uptake initiated via HSPGs as cell surface receptors, like the parent VLs,16,17 though the precise HSPG molecule remains to be identified. HSPGs commonly serve as internalizing receptors for many macromolecules, including CPPs, anti-DNA antibodies, polycation–nucleic acid complexes, viruses, and growth factors.29,38 Notably, endocytosis that is initiated by HSPG interactions does not follow one particular pathway, but instead varies depending on the cellular context and type of extracellular binding ligand.39 Previous studies have shown that, after binding of HSPG at the cell surface, some proteins or CPPs are internalized by clathrin-mediated endocytosis,7,40 whereas other CPPs, such as HIV1-TAT, are internalized via caveolae-mediated endocytosis.41 Intriguingly, cytotransmabs with the embedded cytosol-penetrating VL are primarily internalized by clathrin-mediated endocytosis, while the parent VLs retained the caveolae-mediated endocytic mechanism. Thus, HSPG acts as cell surface receptor, but does not determine the subsequent endocytic pathway. Size of clathrin-coated vesicles and caveosomes are ∼200 nm and 50 nm–60 nm in diameter, respectively, both of which are large enough to contain an approximately 10 nm IgG antibody and smaller VL, indicating that the antibody size is not a determining factor.7 The Fcγ receptors that interact with the Fc region of IgG are exclusively expressed in immune cells, but are not normally expressed in cancer cells,42 excluding their involvement in the cellular uptake of cytotransmabs in cancer cells. Although neonatal Fc receptor is ubiquitously expressed in epithelial cells, endothelial cells and many other cell types, it only interacts with the Fc portion of IgG in acidic endosomal compartments,43 excluding their involvement as cell-surface receptors for the internalization of cytotransmabs. We speculate that bivalent engagement of HSPG by cytotransmab induces different conformations of HSPG from those induced by VL monovalent binding to preferentially recruit proteins that are involved in clathrin-mediated endocytosis compared with those that are involved in caveolae-mediated endocytosis. The bivalency of cytotransmab might also mediate more efficient cellular uptake and early endosome escape by being readily detected, even at ∼100-fold lower concentrations compared with the monovalent VLs, as was seen for multivalent CPPs.11
Although cargo proteins that are internalized by clathrin-mediated endocytosis often route to late endosomes and subsequently undergo lysosomal degradation (or routes to recycling endosomes),7,11 this is not always the case, as exemplified by cytosolic release of some CPPs or toxins after cellular uptake by clathrin-mediated endocytosis.9,37,44,45 Several lines of evidence, such as intracellular distribution and trafficking studies by confocal microscopy, direct access of KT4 to cytosolic KRS proteins, and calcein release assay, indicate that endocytosed cytotransmab is directly released to cytosol from early endosomes, passing over lysosome, ER, Golgi, and nucleus trafficking. However, the underlying molecular mechanism by which cytotransmab escapes from early endosomes into the cytosol is not clear. The cytotransmab carrying hT4 VL with multiple basic residues on the CDR1 might destabilize early endosomes or interfere with further maturation to late endosomes/lysosomes, and this is supported by content releases from vesicles via interactions of oligoarginines with anionic vesicles.46
Taking advantage of the cytosol-penetrating ability of cytotransmabs, we generated anti-KRS KT4 that directly accesses the cytosolic antigen KRS after incubation with living cells under normal cell culture conditions. KT4 is an example of a dual-specific cytotransmab that directly targets cytosolic proteins of living cells, which are generated by the combination of the KRS-binding VH domain and cytosol-penetrating VL domain into the IgG framework. Recently developed antibody engineering techniques can generate VH to specifically recognize targeted antigens with high affinity without the help or interference of VL, as evidenced by VH single domain antibodies47,48 and dual-specific antibodies.49 Thus, we envision that cytotransmab technology will have practical applicability for direct targeting of cytosolic proteins.
The intact IgG-format cytotransmab designed in this study can be produced in large-scale using conventional mAb production techniques and holds many desirable features of the conventional IgG mAbs, including the pharmacokinetic behavior and immune cells-recruiting ability. However, many issues should be first addressed for practical in vivo applications of cytotransmabs, including more efficient cellular uptake at the achievable physiological concentration after their systemic administration and more rapid and sufficient cytosolic access from the endocytosed vesicles. Although internalized cytotransmab did not show any noticeable cytotoxicity, lack of tissue specificity of cytotransmab, which is due to internalization through HSPG that is ubiquitously expressed in epithelial cells, is another issue to be addressed for the systemic applications of cytotransmabs. Nonetheless, the ability of cytotransmab to directly access cytosolic proteins after physiological endocytosis makes it an ideal candidate for construction of next-generation therapeutic antibodies that directly target cytosolic proteins that are associated with many human diseases, including tumors. Furthermore, degradation of cytosolic cytotransmabs by the ubiquitin-mediated proteasome pathways makes them suitable carriers for the cytosolic delivery of therapeutic agents such as chemical drugs and toxins. In conclusion, cytotransmab technology has the potential to generate full-length IgG-format antibodies for detection of cytosolic proteins as research and diagnostic agents, as well as inhibition of cytosolic protein-protein interactions as therapeutic agents.
Materials and Methods
Cell lines
The cell lines, human cervix carcinoma HeLa, pancreatic carcinoma PANC-1, colorectal carcinoma HT-29, breast carcinoma MCF-7, erythroleukemia K562, Burkitt's lymphoma Ramos, Chinese hamster ovary CHO-K1, and proteoglycan-deficient CHO-K1 mutant pgsD-677, were purchased from American Type Culture Collection (ATCC). HeLa and PANC-1 cells were maintained in Dulbecco's Modified Eagle Medium (DMEM, Welgene, LM 001–11), and HT-29, MCF-7, K562, Ramos, CHO-K1, and pgsD-677 cells were maintained in Roswell Park Memorial Institute medium (RPMI, Welgene, LM 011–01). The pgsD-677 cell line lacks both N-acetylglucosaminyltransferase and glucuronyltransferase activities that are required for synthesis of HS, causing selective depletion of HS without affecting chondroitin sulfate (CS) production.17 All cells were cultured in growth media that was supplemented with 10% (v/v) heat-inactivated FBS (Welgene, S 001–07), 100 U/mL penicillin, and 100 μg/mL streptomycin (Welgene, LS 202–02). All of the cell lines were maintained at 37°C in a humidified 5% CO2 incubator and routinely screened for Mycoplasma contamination (CellSafe).
Modeling of humanized VL single domain antibodies
Modeling of the 3-dimensional structure of humanized VLs from the primary amino acid sequence was performed using the web antibody modeling (WAM) algorithm (http://antibody.bath.ac.uk/).19 WAM offers an improved algorithm for homology CDR modeling of VH and VL by aligning the submitted sequence with the most similar framework regions and CDRs of the same canonical class, respectively, from the Brookhaven Protein Data Bank of known antibody structures.
Construction, expression, and purification of humanized VL single domain antibodies
The hT2 VL was generated by introducing 2 point mutations (I2L, L4M) into hT0 VL by overlapping PCR. The hT3 VL was constructed by grafting CDRs of hT2 VL into the human 4D5 VL framework with Vκ1–39 and Jκ1 (PDB 1fvc), which conserves the Vernier zone and N-terminal D1 to M4 residues in hT2 VL. The hT4 VL was constructed by introducing 2 point mutations (K89Q, S91Y) into hT3 VL using overlapping PCR. The amino acid sequences of all VLs are shown in the supplementary data (Figa. S1A and S2A). The genes that encode the hT VL variants were cloned into the NheI/BamHI site of the pIg20 vector, which had a protein A tag at the C-terminus.18 m3D8 and hT VL variants containing the protein-A tag were purified from the supernatant of bacterial cultures using IgG-Sepharose affinity chromatography, as previously described.18
Construction of intact IgG cytotransmabs
The VH and VL genes for anti-TNF adalimumab24 (PDB 3wd5) (DrugBank Accession No. DB00051) and anti-VEGF bevacizumab 25 (PDB 1bj1) (DrugBank Accession No. DB00112) were prepared by DNA synthesis (Bioneer, Inc.) using the sequence from the published crystal structure. The VH and VL genes of TMab4 were amplified from the bacterial plasmids or prepared by DNA synthesis. The VH and VL genes were subcloned in-frame without additional amino acids into the Not1/ApaI sites of the pcDNA 3.4-HC vector carrying human IgG1 constant domain sequence (CH1-hinge-CH2-CH3, residues 118–447 in EU numbering) and NotI/BsiWI sites of the pcDNA 3.4-LC vector with the Cκ constant domain sequence (residues 108–214 in EU numbering), respectively.50
Generation of anti-KRS KT4 cytotransmab
Anti-KRS C12 scFv antibody was isolated from a synthetic human scFv phage library.51 Using yeast surface display technology,52 the C12 Fab was further engineered by randomization of VH-CDR3 to isolate a high affinity anti-KRS scFv, the binding of which was primarily mediated via the VH domain. The resulting C20 Fab with the engineered VH was formatted into IgG1 by subcloning the VH and VL genes into the plasmids of pcDNA 3.4-HC and pcDNA 3.4-LC, respectively, generating C20 IgG mAb. KT4 cytotransmab was generated by co-expression of HC of C20 mAb and LCs that were composed of hT4 VL and Cκ.
Expression and purification of IgG antibodies
The plasmids that encode HC and LC were transiently co-transfected in pairs at an equivalent molar ratio into 200 mL – 1 L of HEK293F cell cultures in Freestyle 293F media (Invitrogen) following a previously described standard protocol.50 Culture supernatants were harvested after 7 d at 37°C in a humidified 8% CO2 incubator by centrifugation and filtration (pore size 0.22 μm, cellulose acetate, Corning, CLS430521). Antibodies were purified from the culture supernatants using a protein-A agarose chromatography column (GE Healthcare) and dialyzed to achieve a final buffer composition of PBS with pH 7.4. Prior to cell treatments, antibodies were sterilized by filtration using a cellulose acetate membrane filter (pore size 0.22 μm, Corning). Antibody concentrations were determined using a Bicinchoninic Acid (BCA) Kit (Pierce, 23225).
Cell viability assay
Cytotoxicity of cytotransmab on tumor cells was determined as previously described.50 Briefly, cells were seeded at a density of 1 × 104 cells per well in 96-well plates and incubated at 37°C with indicated antibodies (1 μM) for 24 h or 48 h. Then, cell viability was analyzed using a colorimetric MTT assay (Sigma-Aldrich, TOX1); the absorbance was then measured at 570 nm and read by a microplate reader (Molecular Devices).
Confocal immunofluorescence microscopy
Confocal microscopy was performed for detection of internalized VLs and cytotransmabs in cultured cells.15,17,50 Briefly, cells (5 × 104) that were grown on 12-mm diameter coverslips in 24-well culture plates were treated with indicated antibodies, as specified in the Figure Legends. For non-adherent cell lines, cells (2 × 105) were grown on 12-mm diameter coverslips that were pre-coated with poly-L-lysine (Sigma-Aldrich, P8920). After 2× washes with PBS, the cells were washed 2× for 30 s at 25°C with low-pH glycine buffer (200 mM glycine, 150 mM NaCl, pH 2.5), followed by 2 additional washes with PBS to remove non-internalized and nonspecifically surface-bound antibodies.17 After fixation with 4% paraformaldehyde (PFA) in PBS for 10 min at 25°C, permeabilization with PERM-buffer (0.1% saponin, 0.1% sodium azide, 1% BSA in PBS) for 10 min at 25°C, and then blocking with 2% BSA in PBS for 1 h at 25°C, the internalized cytotransmabs were detected with FITC- or TRITC-conjugated goat anti-human IgG antibodies for 2 h at 25°C. Protein-A-tagged VLs were detected with rabbit IgG and subsequently with TRITC-conjugated goat anti-rabbit antibody.16 The nucleus was stained with Hoechst 33342 for 5 min in PBS. After mounting the coverslips onto glass slides with Perma Fluor aqueous mounting medium (Thermo Scientific, TA-030-FM), center-focused single z-section images were obtained on a Zeiss LSM710 system with ZEN software (Carl Zeiss). In case of using 63× objective lens in confocal microscope, zoom factor 3 was applied for better resolution.
Heparin competition assay
Cells were pretreated with 300 IU/mL heparin (Sigma-Aldrich, H3149) for 30 min at 37°C prior to the addition of cytotransmabs.17
Experiments with endocytosis markers and inhibitors
For co-localization with endocytosis markers, cells were co-treated with cytotransmabs (1 μM) and either Alexa Fluor 488-transferrin (TF, 10 μg/mL), FITC-cholera toxin B (Ctx-B, 10 μg/mL), or Oregon green-dextran (10 μg/mL) prior to cell fixation and permeabilization.15 In experimentation with inhibitors for endocytic pathways, cells were pretreated for 30 min at 37°C with CPZ (10 μg/mL), dansylcadaverine (200 μM), MβCD (5 mM), nystatin (50 μg/mL), EIPA (10 μM), wortmannin (200 nM), Cyt-D (1 μg/mL), or dynasore (40 μM) and then incubated with TMab4 (1 μM) for an additional 2 h.15 Internalized TMab4 detected TRITC-conjugated anti-Human IgG Fc antibody. The internalization levels of TMab4 in the presence of inhibitors were represented as percentage (%) by comparing the fluorescence intensity from the inside of cells (n > 100 cells per group) with that of control cells that were not treated with the endocytosis inhibitors.
Analysis of calcein release from endosomes
For tracing endosomal release of cytotransmabs using calcein (Sigma-Aldrich, C0875), cells were incubated for 4 h with cytotransmabs (5 μM) in serum-free media at 37°C using adalimumab as a non-internalizing negative control. Subsequently, 50 μM of calcein was added for 2 h at 37°C.35 Cells were then washed 3 times with PBS and fixed. Calcein fluorescence was analyzed by confocal microscopy.
KT4 and KRS co-localization
HeLa cells were transfected with plasmids encoding GFP-tagged KRS (generously provided by Dae Gyu Kim, Seoul National University, Seoul, Korea) using Lipofectamine™ 2000 reagent (Invitrogen). After incubation for 24 h, the transfected cells were incubated with TMab4 or KT4 (20 μM) for 2 h and then analyzed by confocal microscopy.
Pulse-chase experiment to monitor intracellular trafficking of cytotransmab
Cells grown on coverslips in 24-well culture plates were incubated with 3 μM of cytotransmab for 30 min, quickly washed 3 times with PBS, and then incubated at 37°C for the indicated periods of time. After washing with PBS, stripping with low pH glycine buffer, fixation, permeabilization, and blocking of the cells, internalized cytotransmabs were stained with FITC- or TRITC-conjugated anti-human antibody, and subcellular organelles were stained with anti-EEA1, anti-caveolin-1, anti-calnexin, or anti-58K Golgi protein, followed by the appropriate FITC- or TRITC-conjugated secondary antibodies.17 Lysosomes were visualized with LysoTracker® Red DND-99 that was diluted in medium (1 μM) and directly added to the cell culture for 30 min at 37°C. To visualize clathrin-mediated endosomes, Alexa Fluor 488-transferrin (10 μg/mL) was added to cell culture for 30 min before fixing cells. For the inhibition of proteasomal degradation, a pulse-chase experiment was performed in the presence or absence of MG132 (30 μM).
Immunoprecipitation and protein gel blotting
Cells that were grown on 100 mm2 plate were incubated with TMab4 and KT4 for 12 h and then cell lysates were extracted using 20 mM Tris-HCl (pH 7.4) buffer containing 150 mM NaCl, 0.5% Triton X-100, 0.1% SDS, and protease inhibitors (Halt Protease Inhibitor Cocktail, Thermo Scientific, 78440). Subsequently, the cells were incubated with protein A agarose for 2 h at 4°C with agitation.50 After washing 3 times with cold lysis buffer, the precipitates were dissolved in the SDS sample buffer. Western blotting was then performed following the standard procedure, as previously described.50 Proteins were visualized using a PowerOpti-ECL western blotting detection reagent (Animal Genetics) and an ImageQuant LAS 4000 mini (GE Healthcare). Equal amount of lysates were analyzed by protein gel blotting with β-actin as a loading control.
Statistical analysis
Data are represented as mean ± SD of at least 3 independent experiments that were performed in triplicate, unless otherwise specified. Comparison between the experimental and control data was analyzed for statistical significance by a 2-tailed unpaired Student's t-test using Excel (Microsoft). A p value of less than 0.05 was considered statistically significant.
Details regarding the reagents and antibodies, SEC, ELISA, surface plasmon resonance (SPR), DNA hydrolyzing assay, flow cytometry, and live cell imaging are provided in the Supplementary Materials and Methods.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank Dr. Dae Gyu Kim (Medicinal Bioconvergence Research Center, Gyeonggi, Korea) and Prof. Hyunbo Shim (Ewha Womans University, Korea) for generously providing the plasmid expressing GFP-fused KRS and anti-KRS C12 scFv, respectively.
Supplemental Material
Supplemental data for this article can be accessed on thepublisher's website.
Funding
This work was supported by the Pioneer Research Center Program (2014M3C1A3051470), the Global Frontier Project (2013M3A6A4043874), and the Priority Research Center Program (2012–0006687) through the National Research Foundation of Korea, by the Ministry of Science, ICT & Future Planning.
References
- 1.Marschall AL, Frenzel A, Schirrmann T, Schungel M, Dubel S. Targeting antibodies to the cytoplasm. MAbs 2011; 3:3-16; PMID:21099369; http://dx.doi.org/ 10.4161/mabs.3.1.14110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marschall A, Zhang C, Frenzel A, Schirrmann T, Hust M, Perez F, Dübel S. Delivery of antibodies to the cytosol: Debunking the myths. MAbs 2014; 6:943-56; PMID:24848507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ivanov AA, Khuri FR, Fu H. Targeting protein-protein interactions as an anticancer strategy. Trends Pharmacol Sci 2013; 34:393-400; PMID:23725674; http://dx.doi.org/ 10.1016/j.tips.2013.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Freund G, Sibler AP, Desplancq D, Oulad-Abdelghani M, Vigneron M, Gannon J, Van Regenmortel MH, Weiss E. Targeting endogenous nuclear antigens by electrotransfer of monoclonal antibodies in living cells. MAbs 2013; 5:518-22; PMID:23765067; http://dx.doi.org/ 10.4161/mabs.25084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weill CO, Biri S, Adib A, Erbacher P. A practical approach for intracellular protein delivery. Cytotechnology 2008; 56:41-8; PMID:19002840; http://dx.doi.org/ 10.1007/s10616-007-9102-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ritchie M, Tchistiakova L, Scott N. Implications of receptor-mediated endocytosis and intracellular trafficking dynamics in the development of antibody drug conjugates. MAbs 2013; 5:13-21; PMID:23221464; http://dx.doi.org/ 10.4161/mabs.22854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doherty GJ, McMahon HT. Mechanisms of endocytosis. Annu Rev Biochem 2009; 78:857-902; PMID:19317650; http://dx.doi.org/ 10.1146/annurev.biochem.78.081307.110540 [DOI] [PubMed] [Google Scholar]
- 8.Avignolo C, Bagnasco L, Biasotti B, Melchiori A, Tomati V, Bauer I, Salis A, Chiossone L, Mingari MC, Orecchia P, et al. . Internalization via Antennapedia protein transduction domain of an scFv antibody toward c-Myc protein. FASEB J 2008; 22:1237-45; PMID:18048579; http://dx.doi.org/ 10.1096/fj.07-8865com [DOI] [PubMed] [Google Scholar]
- 9.Montrose K, Yang Y, Sun X, Wiles S, Krissansen GW. Xentry, a new class of cell-penetrating peptide uniquely equipped for delivery of drugs. Sci Rep 2013; 3:1661; PMID:23588666; http://dx.doi.org/ 10.1038/srep01661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu M, Chen P, Wang J, Scollard DA, Vallis KA, Reilly RM. 123I-labeled HIV-1 tat peptide radioimmunoconjugates are imported into the nucleus of human breast cancer cells and functionally interact in vitro and in vivo with the cyclin-dependent kinase inhibitor, p21(WAF-1Cip-1). Eur J Nucl Med Mol Imaging 2007; 34:368-77; PMID:17021818; http://dx.doi.org/ 10.1007/s00259-006-0189-0 [DOI] [PubMed] [Google Scholar]
- 11.Erazo-Oliveras A, Muthukrishnan N, Baker R, Wang TY, Pellois JP. Improving the endosomal escape of cell-penetrating peptides and their cargos: strategies and challenges. Pharm (Basel) 2012; 5:1177-209; PMID:24223492; http://dx.doi.org/ 10.3390/ph5111177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rivadeneyra-Espinoza L, Ruiz-Arguelles A. Cell-penetrating anti-native DNA antibodies trigger apoptosis through both the neglect and programmed pathways. J Autoimmun 2006; 26:52-6; PMID:16368224; http://dx.doi.org/ 10.1016/j.jaut.2005.10.008 [DOI] [PubMed] [Google Scholar]
- 13.Yanase K, Madaio MP. Nuclear localizing anti-DNA antibodies enter cells via caveoli and modulate expression of caveolin and p53. J Autoimmun 2005; 24:145-51; PMID:15829407; http://dx.doi.org/ 10.1016/j.jaut.2004.11.008 [DOI] [PubMed] [Google Scholar]
- 14.Weisbart RH, Gera JF, Chan G, Hansen JE, Li E, Cloninger C, Levine AJ, Nishimura RN. A cell-penetrating bispecific antibody for therapeutic regulation of intracellular targets. Mol Cancer Ther 2012; 11:2169-73; PMID:22863609; http://dx.doi.org/ 10.1158/1535-7163.MCT-12-0476-T [DOI] [PubMed] [Google Scholar]
- 15.Jang JY, Jeong JG, Jun HR, Lee SC, Kim JS, Kim YS, Kwon MH. A nucleic acid-hydrolyzing antibody penetrates into cells via caveolae-mediated endocytosis, localizes in the cytosol and exhibits cytotoxicity. Cell Mol Life Sci 2009; 66:1985-97; PMID:19373434; http://dx.doi.org/ 10.1007/s00018-009-9179-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee WR, Jang JY, Kim JS, Kwon MH, Kim YS. Gene silencing by cell-penetrating, sequence-selective and nucleic-acid hydrolyzing antibodies. Nucleic Acids Res 2010; 38:1596-609; PMID:20007602; http://dx.doi.org/ 10.1093/nar/gkp1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim A, Shin TH, Shin SM, Pham CD, Choi DK, Kwon MH, Kim YS. Cellular internalization mechanism and intracellular trafficking of filamentous M13 phages displaying a cell-penetrating transbody and TAT peptide. PLoS One 2012; 7:e51813; PMID:23251631; http://dx.doi.org/ 10.1371/journal.pone.0051813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim DS, Lee SH, Kim JS, Lee SC, Kwon MH, Kim YS. Generation of humanized anti-DNA hydrolyzing catalytic antibodies by complementarity determining region grafting. Biochem Biophys Res Commun 2009; 379:314-8; PMID:19103171; http://dx.doi.org/ 10.1016/j.bbrc.2008.12.051 [DOI] [PubMed] [Google Scholar]
- 19.Whitelegg NR, Rees AR. WAM: an improved algorithm for modelling antibodies on the WEB. Protein Eng 2000; 13:819-24; PMID:11239080; http://dx.doi.org/ 10.1093/protein/13.12.819 [DOI] [PubMed] [Google Scholar]
- 20.Park SY, Lee WR, Lee SC, Kwon MH, Kim YS, Kim JS. Crystal structure of single-domain VL of an anti-DNA binding antibody 3D8 scFv and its active site revealed by complex structures of a small molecule and metals. Proteins 2008; 71:2091-6; PMID:18338383; http://dx.doi.org/ 10.1002/prot.22011 [DOI] [PubMed] [Google Scholar]
- 21.Makabe K, Nakanishi T, Tsumoto K, Tanaka Y, Kondo H, Umetsu M, Sone Y, Asano R, Kumagai I. Thermodynamic consequences of mutations in vernier zone residues of a humanized anti-human epidermal growth factor receptor murine antibody, 528. J Biol Chem 2008; 283:1156-66; PMID:17947238; http://dx.doi.org/ 10.1074/jbc.M706190200 [DOI] [PubMed] [Google Scholar]
- 22.Lee SH, Park DW, Sung ES, Park HR, Kim JK, Kim YS. Humanization of an agonistic anti-death receptor 4 single chain variable fragment antibody and avidity-mediated enhancement of its cell death-inducing activity. Mol Immunol 2010; 47:816-24; PMID:19864027; http://dx.doi.org/ 10.1016/j.molimm.2009.09.041 [DOI] [PubMed] [Google Scholar]
- 23.Magdelaine-Beuzelin C, Kaas Q, Wehbi V, Ohresser M, Jefferis R, Lefranc MP, Watier H. Structure-function relationships of the variable domains of monoclonal antibodies approved for cancer treatment. Crit Rev Oncol Hematol 2007; 64:210-25; PMID:17624800; http://dx.doi.org/ 10.1016/j.critrevonc.2007.04.011 [DOI] [PubMed] [Google Scholar]
- 24.Hu S, Liang S, Guo H, Zhang D, Li H, Wang X, Yang W, Qian W, Hou S, Wang H, et al. . Comparison of the inhibition mechanisms of adalimumab and infliximab in treating tumor necrosis factor alpha-associated diseases from a molecular view. J Biol Chem 2013; 288:27059-67; PMID:23943614; http://dx.doi.org/ 10.1074/jbc.M113.491530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muller YA, Chen Y, Christinger HW, Li B, Cunningham BC, Lowman HB, de Vos AM. VEGF and the Fab fragment of a humanized neutralizing antibody: crystal structure of the complex at 2.4 A resolution and mutational analysis of the interface. Structure 1998; 6:1153-67; PMID:9753694; http://dx.doi.org/ 10.1016/S0969-2126(98)00116-6 [DOI] [PubMed] [Google Scholar]
- 26.Vargas-Madrazo E, Paz-Garcia E. An improved model of association for VH-VL immunoglobulin domains: asymmetries between VH and VL in the packing of some interface residues. J Mol Recognit 2003; 16:113-20; PMID:12833565; http://dx.doi.org/ 10.1002/jmr.613 [DOI] [PubMed] [Google Scholar]
- 27.Ewert S, Huber T, Honegger A, Pluckthun A. Biophysical properties of human antibody variable domains. J Mol Biol 2003; 325:531-53; PMID:12498801; http://dx.doi.org/ 10.1016/S0022-2836(02)01237-8 [DOI] [PubMed] [Google Scholar]
- 28.Pei XY, Holliger P, Murzin AG, Williams RL. The 2.0-A resolution crystal structure of a trimeric antibody fragment with noncognate VH-VL domain pairs shows a rearrangement of VH CDR3. Proc Natl Acad Sci U S A 1997; 94:9637-42; PMID:9275175; http://dx.doi.org/ 10.1073/pnas.94.18.9637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Richard JP, Melikov K, Brooks H, Prevot P, Lebleu B, Chernomordik LV. Cellular uptake of unconjugated TAT peptide involves clathrin-dependent endocytosis and heparan sulfate receptors. J Biol Chem 2005; 280:15300-6; PMID:15687490; http://dx.doi.org/ 10.1074/jbc.M401604200 [DOI] [PubMed] [Google Scholar]
- 30.Tuve S, Wang H, Jacobs JD, Yumul RC, Smith DF, Lieber A. Role of cellular heparan sulfate proteoglycans in infection of human adenovirus serotype 3 and 35. PLoS Pathog 2008; 4:e1000189; PMID:18974862; http://dx.doi.org/ 10.1371/journal.ppat.1000189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khalil IA, Kogure K, Akita H, Harashima H. Uptake pathways and subsequent intracellular trafficking in nonviral gene delivery. Pharmacol Rev 2006; 58:32-45; PMID:16507881; http://dx.doi.org/ 10.1124/pr.58.1.8 [DOI] [PubMed] [Google Scholar]
- 32.Magadan JG, Barbieri MA, Mesa R, Stahl PD, Mayorga LS. Rab22a regulates the sorting of transferrin to recycling endosomes. Mol Cell Biol 2006; 26:2595-614; PMID:16537905; http://dx.doi.org/ 10.1128/MCB.26.7.2595-2614.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feng SM, Muraoka-Cook RS, Hunter D, Sandahl MA, Caskey LS, Miyazawa K, Atfi A, Earp HS 3rd. The E3 ubiquitin ligase WWP1 selectively targets HER4 and its proteolytically derived signaling isoforms for degradation. Mol Cell Biol 2009; 29:892-906; PMID:19047365; http://dx.doi.org/ 10.1128/MCB.00595-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ciechanover A. The ubiquitin-proteasome proteolytic pathway. Cell 1994; 79:13-21; PMID:7923371; http://dx.doi.org/ 10.1016/0092-8674(94)90396-4 [DOI] [PubMed] [Google Scholar]
- 35.Salomone F, Cardarelli F, Di Luca M, Boccardi C, Nifosi R, Bardi G, Di Bari L, Serresi M, Beltram F. A novel chimeric cell-penetrating peptide with membrane-disruptive properties for efficient endosomal escape. J Control Release 2012; 163:293-303; PMID:23041543; http://dx.doi.org/ 10.1016/j.jconrel.2012.09.019 [DOI] [PubMed] [Google Scholar]
- 36.Kim DG, Lee JY, Kwon NH, Fang P, Zhang Q, Wang J, Young NL, Guo M, Cho HY, Mushtaq AU, et al. . Chemical inhibition of prometastatic lysyl-tRNA synthetase-laminin receptor interaction. Nat Chem Biol 2014; 10:29-34; PMID:24212136; http://dx.doi.org/ 10.1038/nchembio.1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Appelbaum JS, LaRochelle JR, Smith BA, Balkin DM, Holub JM, Schepartz A. Arginine topology controls escape of minimally cationic proteins from early endosomes to the cytoplasm. Chem Biol 2012; 19:819-30; PMID:22840770; http://dx.doi.org/ 10.1016/j.chembiol.2012.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O’Donnell V, Larocco M, Baxt B. Heparan sulfate-binding foot-and-mouth disease virus enters cells via caveola-mediated endocytosis. J Virol 2008; 82:9075-85; PMID:18614639; http://dx.doi.org/ 10.1128/JVI.00732-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Christianson HC, Belting M. Heparan sulfate proteoglycan as a cell-surface endocytosis receptor. Matrix Biol 2013; 35:51-5; PMID:24145152; http://dx.doi.org/ 10.1016/j.matbio.2013.10.004 [DOI] [PubMed] [Google Scholar]
- 40.Mercer J, Schelhaas M, Helenius A. Virus entry by endocytosis. Annu Rev Biochem 2010; 79:803-33; PMID:20196649; http://dx.doi.org/ 10.1146/annurev-biochem-060208-104626 [DOI] [PubMed] [Google Scholar]
- 41.Fittipaldi A, Ferrari A, Zoppe M, Arcangeli C, Pellegrini V, Beltram F, Giacca M. Cell membrane lipid rafts mediate caveolar endocytosis of HIV-1 Tat fusion proteins. J Biol Chem 2003; 278:34141-9; PMID:12773529; http://dx.doi.org/ 10.1074/jbc.M303045200 [DOI] [PubMed] [Google Scholar]
- 42.Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol 2008; 8:34-47; PMID:18064051; http://dx.doi.org/ 10.1038/nri2206 [DOI] [PubMed] [Google Scholar]
- 43.Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol 2007; 7:715-25; PMID:17703228; http://dx.doi.org/ 10.1038/nri2155 [DOI] [PubMed] [Google Scholar]
- 44.Vendeville A, Rayne F, Bonhoure A, Bettache N, Montcourrier P, Beaumelle B. HIV-1 Tat enters T cells using coated pits before translocating from acidified endosomes and eliciting biological responses. Mol Biol Cell 2004; 15:2347-60; PMID:15020715; http://dx.doi.org/ 10.1091/mbc.E03-12-0921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ratts R, Zeng H, Berg EA, Blue C, McComb ME, Costello CE, vanderSpek JC, Murphy JR. The cytosolic entry of diphtheria toxin catalytic domain requires a host cell cytosolic translocation factor complex. J Cell Biol 2003; 160:1139-50; PMID:12668662; http://dx.doi.org/ 10.1083/jcb.200210028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hitz T, Iten R, Gardiner J, Namoto K, Walde P, Seebach D. Interaction of alpha-and beta-oligoarginine-acids and amides with anionic lipid vesicles: a mechanistic and thermodynamic study. Biochemistry 2006; 45:5817-29; PMID:16669625; http://dx.doi.org/ 10.1021/bi060285d [DOI] [PubMed] [Google Scholar]
- 47.Reiter Y, Schuck P, Boyd LF, Plaksin D. An antibody single-domain phage display library of a native heavy chain variable region: isolation of functional single-domain VH molecules with a unique interface. J Mol Biol 1999; 290:685-98; PMID:10395823; http://dx.doi.org/ 10.1006/jmbi.1999.2923 [DOI] [PubMed] [Google Scholar]
- 48.Tanaka T, Williams RL, Rabbitts TH. Tumour prevention by a single antibody domain targeting the interaction of signal transduction proteins with RAS. EMBO J 2007; 26:3250-9; PMID:17568777; http://dx.doi.org/ 10.1038/sj.emboj.7601744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang H, Yun S, Batuwangala TD, Steward M, Holmes SD, Pan L, Tighiouart M, Shin HJ, Koenig L, Park W, et al. . A dual-targeting antibody against EGFR-VEGF for lung and head and neck cancer treatment. Int J Cancer 2012; 131:956-69; PMID:21918971; http://dx.doi.org/ 10.1002/ijc.26427 [DOI] [PubMed] [Google Scholar]
- 50.Shin TH, Sung ES, Kim YJ, Kim KS, Kim SH, Kim SK, Lee YD, Kim YS. Enhancement of the tumor penetration of monoclonal antibody by fusion of a neuropilin-targeting Peptide improves the antitumor efficacy. Mol Cancer Ther 2014; 13:651-61; PMID:24435448; http://dx.doi.org/ 10.1158/1535-7163.MCT-13-0748 [DOI] [PubMed] [Google Scholar]
- 51.Yang HY, Kang KJ, Chung JE, Shim H. Construction of a large synthetic human scFv library with six diversified CDRs and high functional diversity. Mol Cells 2009; 27:225-35; PMID:19277506; http://dx.doi.org/ 10.1007/s10059-009-0028-9 [DOI] [PubMed] [Google Scholar]
- 52.Baek DS, Kim YS. Construction of a large synthetic human fab antibody library on yeast cell surface by optimized yeast mating. J Microbiol Biotechnol 2014; 24:408-20; PMID:24394194; http://dx.doi.org/ 10.4014/jmb.1401.01002 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.