

Abstract
The response of the peripheral nervous system (PNS) to injury may go together with alterations in epigenetics, a conjecture that has not been subjected to a comprehensive, genome-wide test. Using reduced representation bisulfite sequencing, we report widespread remodeling of DNA methylation in the rat dorsal root ganglion (DRG) occurring within 24 h of peripheral nerve ligation, a neuropathy model of allodynia. Significant (P < 10−4) cytosine hyper- and hypo-methylation was found at thousands of CpG sites. Remodeling occurred outside of CpG islands. Changes affected genes with known roles in the PNS, yet methylome remodeling also involved genes that were not linked to neuroplasticity by prior evidence. Consistent with emerging models relying on genome-wide methylation and RNA-seq analysis of promoter regions and gene bodies, variation of methylation was not tightly linked with variation of gene expression. Furthermore, approximately 44% of the dynamically changed CpGs were located outside of genes. We compared their positions with the intergenic, tissue-specific differentially methylated CpGs (tDMCs) of an independent experimental set consisting of liver, spleen, L4 control DRG, and muscle. Dynamic changes affected those intergenic CpGs that were different between tissues (P < 10−15) and almost never the invariant portion of the methylome (those CpGs that were identical across all tissues). Our findings—obtained in mixed tissue—show that peripheral nerve injury leads to methylome remodeling in the DRG. Future studies may address which of the cell types found in the DRG, such as specific groups of neurons or non-neuronal cells are affected by which aspect of the observed methylome remodeling.
Keywords: dorsal root ganglion, methylation, peripheral nervous system, rat, RRBS, spinal nerve ligation, pain
Abbreviations
- dDMCs
dynamically differentially methylated CpGs
- DRG
dorsal root ganglion
- HCP
high-CpG promoter
- LCP
low-CpG promoter
- RRBS
reduced representation bisulfite sequencing
- SNL
spinal nerve ligation
- tDMCs
tissue-specific differentially methylated CpGs
- tIMCs
tissue-invariant methylated CpGs
Introduction
Injury to the peripheral nervous system (PNS) is a clinical cause (and laboratory model) of neuropathic pain and, unless regeneration occurs, neurological debility. PNS injury elicits a dynamic genome response in affected cells reflected in the alteration of hundreds of RNA transcripts in the dorsal root ganglion (DRG).1-3 Whether PNS injury leads to epigenetic remodeling on a grand scale has not been determined but candidate gene studies suggest the possibility.4-7
Denk and McMahon8 suggested in a recent review article that "direct evidence that epigenetic mechanisms could be involved in the development and/or maintenance of chronic pain conditions is only just beginning to surface, and [that] the field is in its infancy;" [that] "the currently available data suggest that epigenetic mechanisms may be important contributors to chronic pain states;" and that "descriptive studies, for instance examination of genome-wide … methylation in various models of chronic pain, will be useful." Here we provide such a study reporting, to the best of our knowledge, the largest corpus of data available to date to describe epigenomic events in a model of chronic pain.
Methylation of cytosines at CpG dinucleotide sites (mCpG) is the prototypic epigenetic modifier of neural genomes capable of encoding the effect of environmental factors on lifelong persisting behavioral traits.9 DNA methylation is seen in all regions of the genome and its different patterns demarcate the tissue specific “methylome." While organ differences—because they are stark and persistent—highlight the lifelong stability of the methylome, recent studies in the CNS have demonstrated dynamic modification of CpG sites in response to neural activity 10 akin to the remodeling of other chromatin marks, which can occur rapidly.
We tested the plasticity of the PNS methylome by performing an L5 spinal nerve ligation (SNL), a common model of PNS injury and neuropathic pain 11 by determining the methylation level at 1.4 × 106 CpG sites in the L5 DRG with reduced representation bisulfite sequencing (RRBS), a current technology providing digital quantification of CpG methylation levels.12 We found widespread remodeling of DNA methylation in the PNS. Methylation changes were most marked in promoters with an average density of CpG rather than at CpG islands (CGI); were highly significant across gene bodies; and extended to gene deserts (intergenic regions) demarcating regions within the genome with suggested roles in neurobiology.
Results
Nerve injury induces genome-wide methylation changes
Of 1.4 × 106 CpG sites analyzed by RRBS (Fig. 1A), 917,097 were located within genes or promoters covering 15,808 of the 22,920 rat genes (known protein encoding). Within 24 h, methylomes differed characteristically between SNL- and sham control groups separating animals with the 2 conditions into separate clusters (Fig. 1B). Statistical comparison of methylation levels at individual CpG between SNL and control animals identified highly significant (P < 10−4) alterations at 14,965 sites, which we termed dynamically differentially methylated CpGs (dDMCs).
Figure 1 (See previous page).

Gene methylation response to nerve injury. (A) Genome-wide quantification of CpG methylation by RRBS. Genomic DNA from the L5 dorsal root ganglion (DRG) of Brown Norway rats was isolated 24 h after spinal nerve ligation (SNL) or a sham procedure (negative control). Genomic DNA (gDNA) was isolated and subjected to a restriction digest with MspI. DNA fragments were ligated to adapters, bisulfite treated converting unmethylated cytosines to uracils, and sequenced. Resulting paired-end reads—1.1 billion in total from 8 independent libraries analyzed in the present study—were aligned to the rat genome. Cytosine methylation levels were called only for CpG sites covered in a given library by ≥10 independent sequence reads. (B) Nerve injury eliciting methylome alterations: Evidence at the whole-data set level. DNA methylation was markedly altered after SNL. Hierarchical clustering—using all methylation levels measured at 917,097 CpG sites within genes—clearly separated control animals (left) from SNL animals (right). (C) Nucleotide-resolution analysis of the methylation profile of HCN2. Top panel: Changes in methylation were noted in clusters of juxtaposed CpGs. Shown as an example is HCN2, an ion channel modulating inflammatory and neuropathic pain 34. The x-axis indicates the position of captured CpG sites within a gene. RRBS captured 216 CpGs located in gene body of HCN2, while no CpGs were captured in the promoter region for this gene. The negative log-p value of the significance level computed by a likelihood ratio test using a generalized linear model (GLM) is shown on the y-axis. Higher values indicate stronger significance. Differences with a P > 10−3 were considered non-significant (CpG positions shown below the dotted line). Differences at individual CpG sites were highly significant ranging from P < 10−3 to P < 10−14. The bars in the colored band above the scatterplot indicate for each CpG whether the mean methylation level was higher (red) or lower (blue) in the SNL group compared with controls. The direction of change is shown regardless of significance at the level of a specific CpG. Regions that are significantly changed (P < 10−3) according to a sign test of the direction of juxtaposed CpGs are represented by brown stars. Bottom panel: Random permutation of group assignment and CpG positions confirmed that both statistical testing procedures were robust as indicated by low false-positive rates of 0.004 for single CpG testing of HCN2 (1/216 CpG above significance threshold) and of 0 (no false-positive) for regions. Additional examples are shown in Figure S1. (D) Gene types and regions undergoing hyper- and hypo-methylation. The fraction of CpGs with significantly altered methylation was calculated across different gene regions for the entire dataset. Low CpG content promoter (LCP) genes and high CpG content promoter (HCP) genes differed. LCP genes were altered in the promoter, exon, and intron regions. HCP genes harbored a comparable fraction of altered methylation sites only in exons and introns, while HCP gene promoters were unaffected. Hypermethylation (red) accounted for a greater fraction of changes than hypomethylation (blue) in all regions. (E) Axon guidance pathway genes differentially methylated after SNL. The most significantly enriched molecular mechanism in an unbiased global analysis of genes undergoing differential methylation after SNL was the axon guidance pathway (P < 10−11). Depicted are 35 differentially methylated genes with dense connectivity. Variable methylation predominantly occurred in the gene body. Only FES and CDK5 showed methylation alterations in their promoter (black stars). A total of 98 out of 468 axon guidance pathway genes were differentially methylated (complete list provided as Table S1).
Genic dDMCs altered by spinal nerve ligation
The majority of dDMCs (8,311) were found in promoters, exons, or introns, where they typically formed clusters of juxtaposed CpG locations. A total of 2,479 genes harbored dDMCs in their promoter or gene body. Affected genes highlighted neurobiologically relevant molecular mechanisms known to be involved in the response of the PNS to injury or development of neuropathic pain (Fig. 1C and Fig. S1) but also many others with unknown direct implication (full gene list provided as Table S2). Analysis of all genic dDMCs showed that 82% were affected by hypermethylation and 18% by hypomethylation. Promoters are the regions in the genome that are best-studied in regards to methylation, because of their established role in gene regulation. Promoters can be separated into low CpG promoters (LCP) and high CpG promoters (HCP) based on the frequency of the CpG motif. We compared the number of dDMCs at LCP and HCP genes. dDMCs were frequent in LCP and rare in HCP (Fig. 1D), echoing the conclusion from our previous study (on the baseline characteristics of the uninjured PNS) that methylation has a stronger role at LCP.13 Exons and introns of both gene classes contained similar fractions of dDMCs (Fig. 1D). An integrated analysis of all dDMCs was performed through pathway analysis. Among all molecular functions, axon guidance was the top enriched pathway with 98 genes (21% of pathway members) found to be differentially methylated, a highly significant result (P < 10−11) depicted in Figure 1E. For 97 of these genes, CpG methylation was available in the gene body. Of these, 93 harbored methylation changes within the gene body. Of 75 genes, for which promoter CpG information was available, 15 genes harbored changes in the promoter (9 of these genes harboring changes in the promoter and the gene body and 6 genes harboring changes only in the promoter). These results suggest that CpGs located in the gene body were more susceptible to methylation changes than those located in the promoter region.
Relationship between methylome and transcriptome changes
To explore a possible relationship of dDMCs with the dynamic regulation of gene expression, RNA-seq was performed in DRG harvested 24 h after SNL or a control procedure (same as for RRBS above). We profiled 22,908 genes out of which 10,315 genes were found to be expressed in the DRG (see methods). The distribution is provided in Figure S2. As expected, a considerable fraction of genes were induced or suppressed in response to SNL, 2,938 (28.5%) in the present study, including genes such as AFT3, A2m, NGF, SOCS3, SOX11, and STAT3, which are known to show expression changes following nerve injury.14-18
As has been proposed in the past,19 we found genes in which promoter methylation was associated with gene downregulation (29 genes). However, a similar number of genes (36) were upregulated when the promoter was hypermethylated. Conclusions that are based on individual genes are therefore not sufficient when attempting to find a systematic association between methylome and transcriptome changes.
In order to address whether a genome-wide trend relating methylome and transcriptome alterations could be found, we performed an integrated analysis of dDMCs and gene expression changes. Based on prior observations by us and others suggesting that (1) HCP genes and LCP genes were regulated differently 13,20,21 and (2) the effects of promoter and gene body methylation diverged,22,23 we analyzed these scenarios separately. We identified genes that were up- or down-regulated by nerve damage and harbored dDMCs in their promoter or gene body. Figure 2 illustrates the relationship between gene expression and methylation changes in promoters and gene bodies of HCP (A and B) and LCP genes (C and D), respectively. The promoter regions of HCP genes were poorly enriched for dDMCs (see Fig. 1D). The dDMCs that were associated with an up- or down-regulation of the corresponding gene are shown in Figure 2A and represent 33% of dDMCs in this genomic location. A similar number of genes were found to be up- and downregulated when a dDMC was hypermethylated (21 and 16 genes, respectively). We found 152 dysregulated HCP genes that harbored dDMCs in their gene body. The majority of these dDMCs gained methylation after SNL (Fig. 2B). This went along with changes of gene expression in both possible directions. Similar results were obtained for dDMCs that lost methylation.
Figure 2.
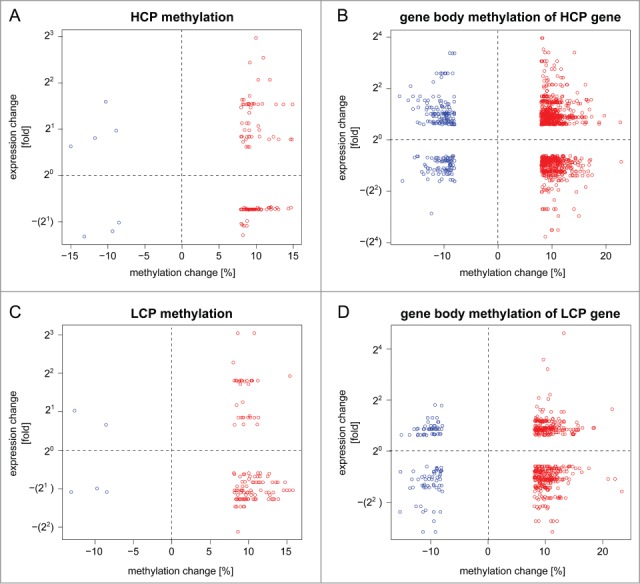
Transcriptome up- and down-regulation can co-exist with CpG hyper- or hypo-methylation in the promoter and gene body. An integrated transcriptome-methylome analysis supported a negative finding, notably the absence of any tight correlation between transcriptome- and methylome alterations following SNL. Shown is the direction of CpG methylation change and gene expression change following nerve injury. Genes were divided into 2 groups: high CpG promoter (HCP; panel A and B) and low CpG promoter (LCP) genes (panel C and D). Promoter- (left, panel A and C) and gene body methylation (right, panel B and D) were analyzed separately. The methylation values of dDMCs (x-axis) were plotted against the expression value of their corresponding gene (y-axis). Each circle represents a differentially methylated CpG (dDMC), thereby allowing multiple pairings of individual dDMCs with the same gene. The analysis was performed on significantly changed CpG sites located in genes that underwent a significant change in expression. The results shown demonstrate that all possible scenarios exist: hyper- and hypo-methylation of CpG associated with increase or decrease in RNA levels from the same gene. Red: hypermethylated dDMC, blue: hypomethylated dDMC.
As expected, a lower absolute number of dDMCs associated with gene up- or downregulation was found in the promoter and gene body of LCP genes (Figs. 2C and D). LCP genes contain few CpGs, which are therefore less likely to be captured by RRBS, as compared to CpGs located in HCP genes. Yet, CpGs captured in LCP were more frequently differentially methylated than those located in HCP (Fig. 1D). Differential methylation of LCP was associated with gene expression changes of 30% of the affected genes. Hypermethylation was predominant but no directional trend for transcriptome alteration was observed (Fig. 2C). When next considering dDMCs located in the gene body of LCP genes, both methylation gain and loss were observed in association with gene up- and down-regulation (Fig. 2D).
Plasticity extends to gene deserts
Methylome alterations in the nervous system have, to date, been interpreted in the context of genes. Yet the present study—thanks to its unbiased genome-wide ascertainment of methylation levels—found 44% of dDMCs in gene deserts, dDMCs in non-annotated regions of the genome (Fig. 3A). Analyzing intergenic regions, a CpG was equally likely to be a dDMC regardless of its distance to the most nearby gene, supporting the classification of this dDMC subset as "desert" (Fig. 3A). We next investigated whether the intergenic alterations were randomly distributed across the gene deserts or followed a specific pattern. Therefore, dDMCs were aggregated into regions. This step was implemented by sliding an in silico window of 100 bp over the gene deserts and extended as soon as a dDMC was reached. This procedure revealed that dDMCs occurred in clusters and that inside a cluster, neighboring dDMCs varied in the same direction. This result is shown in Figure 3B and suggests a well-organized structure of dDMC events despite the lack of a gene structure.
Figure 3 (See previous page).

Methylome remodeling of gene deserts. (A) Desert dDMCs. Nearly half of the CpGs undergoing statistically significant hyper- or hypo-methylation in response to nerve injury, dDMCs, were discovered in intergenic regions, 44% of dDMCs in the study (left panel). The distribution was even across the intergenic span, as seen from the combined analysis of the location of 5,042 hypermethylated and 1,612 hypomethylated sites (right panel). The likelihood of a CpG to be differentially methylated (after SNL) was independent of its distance from the closest gene, thereby authenticating the classification of all these sites as "desert dDMCs." (B) Uniformity of desert dDMC clusters. Neighboring dDMCs in gene deserts underwent unidirectional methylation change forming clusters. All clusters consisting of ≥5 dDMC members were examined. The largest clusters (unselected) are shown. Non-randomness was significant with P < 0.05 to P < 2 × 10−6 for sizes ranging from 7 to 21. Red bar: hypermethylated dDMC. Blue: hypomethylated dDMC. A complete depiction of clusters is provided in Figure S3. (C) DRG-specific partitioning of the gene desert methylome. Cytosine methylation sites were dichotomized into tissue-differentially methylated CpGs (tDMCs) and tissue-invariant methylated CpGs (tIMCs), demarcating CpGs that are DRG specific, tDMCs, and others that were equally methylated in all organs, tIMCs. Rat control DRG (new replicates), spleen, muscle, and liver were compared. Sample regions with tDMCs and tIMCs are shown (left). The absolute methylation level is represented on the y-axis. A principal components analysis (PCA) confirmed organ-specific methylation of gene deserts (right). (D) Desert remodeling after SNL (dDMCs) occurs at CpG with a DRG-specific methylation level (tDMCs). CpG undergoing methylation changes in response to SNL, dDMCs, were a subset of tDMCs. The enrichment of dDMCs in the tDMC vs. tIMC subsets of CpG sites was highly significant (P < 10−15). Gene desert methylation can be divided according to a dual model: The majority of CpG methyl-marks remain stable organism-wide regardless of tissue type or pathology; a substantial minority of CpG methyl-marks, tDMCs, are DRG-specific and encompass all sites capable of responding to changes in the environmental such as at the CpG sites identified in the present study as dDMCs through their response to nerve injury.
Position of non-genic dDMCs can be predicted by an organism-wide methylome model
To further test whether dDMCs were randomly or systematically spread across gene deserts, we sought to determine whether functional methylome plasticity in desert regions occurring in response to PNS damage—dDMCs—can be predicted from a genome-biology guided, organism-wide analysis of the methylome. We bioinformatically overlaid non-genic dDMCs with a tiling map dichotomizing the genome into CpGs that were similarly methylated throughout the organism vs. CpGs that were methylated at a DRG specific level. To create the map, additional RRBS was performed on liver, muscle, spleen, and an independent set of DRG. Organism-wide comparisons showed that a substantial number of CpGs (40%) were tissue differentially methylated CpGs (tDMCs), while the remaining (60%) were tissue invariant methylated CpGs (tIMCs), identical in every organ. The DRG methylome could be readily distinguished from other tissues (Fig. 3C). We then tested the hypothesis that the methylome response to SNL, dDMCs, would occur only in one of the 2 types of regions defined by tissue comparison, i.e., either in the tDMC or the tIMC region. A very marked level of agreement was found between dDMCs and tDMCs (Fig. 3D): CpGs remodeled after SNL, i.e., dDMCs, were co-identified through the organism-wide methylome comparison as DRG specific sites, tDMCs. The vast majority (>97%) of gene desert dDMCs were also tDMCs. The odds ratio (OR) for the enrichment was 58, which was highly significant (P < 10−15). Using the new classification of intergenic CpGs, we defined 2 categories of non-genic regions: "Dead deserts" where methylation levels are irrevocably set regardless of the tissue type or pathology (demarcated by tIMCs) and "alive deserts" consisting of the intergenic regions that were methylated in a DRG-specific pattern (tDMCs) and encompass all sites capable of responding to environmental changes such as nerve injury (dDMCs).
Alive and dead deserts differ in their binding site motif
The new distinction raised the possibility that the 2 types of gene deserts might differ in regard to the genomic sequence patterns. We tested the possibility by performing an analysis for known DNA binding site sequence motifs. Alive deserts were found to be markedly different from dead deserts. Interestingly, while the analysis took an unbiased approach considering the entire universe of eukaryotic DNA binding proteins, the top enriched DNA motifs matched transcription factors with important roles in PNS development, regeneration, and sensory dysfunction. Hypomethylated alive deserts were most markedly enriched for ETS binding (P < 10−38), which was noteworthy because ETS binding controls the transcription of axotomy-responsive genes in the DRG.24 Binding sites of the RUNX gene family of transcription factors was the other top enriched motif (P < 10−31) with Runx1 sites driving the result. Runx1 determines the nociceptive sensory neuron phenotype and is required for thermal and neuropathic pain.25 Other Runx proteins have been reported to control the axonal projection of proprioceptive DRG neurons.26 Hypermethylated alive deserts were enriched for SOX gene family (P < 10−51) and neurofibromin 1 (NF1) binding sites (P < 10−22). SOX genes are involved in neuronal development 27 and axon regeneration 28 and NF1 has long been recognized for its critical role in the PNS.
Discussion
The present study examined DNA methylation in the rat PNS in a rodent model of neuropathic pain. Using RRBS we provide results at a genome-wide scale with nucleotide resolution examining methylome alterations in both genic and non-genic regions. While the methylome remodeling affected genes with known roles in the PNS and thus matches predictions by some authors over the past years,8 the plasticity further extended to genes with undefined direct implication and to intergenic regions.
To date, the only previous publication reporting genome-wide dynamic methylome alterations in the field of neurobiology is, to the best of our knowledge, the report by Guo et al.10 Guo et al. studied CpGs in genic regions using mixed tissue from the mouse hippocampus after electrostimulation. Our study found, similarly to the observation by Guo et al., that methylome alteration in the adult nervous system can occur rapidly. Interestingly, we also found, like Guo et al., that hypermethylation was more common than hypomethylation. In contrast, Tajerian et al., who assessed methylation globally in the prefrontal mouse cortex after pain induction using the Luminometric Methylation Assay, found a global methylation decrease 6 months after nerve injury.29 Furthermore, 2 single-gene methylation studies are available in the context of pain. Tajerian et al. analyzed the methylation status of 6 CpGs located in the promoter region of the SPARC gene in mice.5 The authors found that lumbar disc degeneration was associated with an increase of methylation at these sites, resulting in an increased expression of SPARC. In the present study, 9 CpGs were captured in the SPARC gene; yet, all were located in the gene body. None of them got significantly modified after spinal nerve ligation. Moreover, no expression changes were observed 24 h after SNL for this gene. Viet et al. suggested that EDNRB promoter methylation leads to cancer-induced pain by gene downregulation.30 While we also found that EDNRB expression was reduced after spinal nerve ligation, no methylation data was available for this gene in the present report.
Like other neural tissues, the DRG consists of several cell populations, presumably characterized by different methylation profiles. Analysis of such mixed tissues has been routinely performed previously, but results need to be interpreted with appropriate caution. Spinal nerve damage leads to the activation of satellite cells in the DRG,16,31 thus potentially contributing to the observed methylome alterations. As expected, the glial fibrillary acidic protein (GFAP), an astroglial marker that is also expressed by satellite cells,32 was upregulated, emphasizing that glial activation took place within 24 h after nerve injury. In the present study, methylome remodeling affected genes that were previously shown to be neuron-specific, such as OZD2 and PCSK2.33 Similarly, HCN2, whose CpG methylation changes are depicted in Figure 1C is strongly expressed in primary sensory neurons.34 These findings—along with the highly significant enrichment of axonal guidance genes—suggest that methylome remodeling occurred in neurons as well as, possibly, in glial cells.
Using RRBS, we captured ∼3% of all genomic CpGs in the rat. Comprehensiveness of analysis, resolution of exact genome position, and experimental resource requirements are recognized competing features in the design of unbiased epigenomics studies. RRBS is an efficient approach to methylome profiling that has yielded critical progress in the field because it provides nucleotide resolution data. While RRBS is most efficient for CpG rich regions, its “reduced representation” has been found repeatedly to provide highly informative genome-wide extrapolation, e.g., as shown in the comparison of RRBS by Ziller et al. (2013) with whole genome bisulfite sequencing (WGBS). Furthermore, over 70% of the reads captured by WGBS either do not contain any CpG or may suffer from a low coverage depth.35 RRBS, therefore, enables highly reproducible, quantitative comparisons of methylation levels at single-base resolution at reasonable sequencing cost, making it the technology of choice for CpG methylation analysis. The bisulfite sequencing methodology underlying RRBS could not distinguish methylcytosine (mC), the principal modification of CpG sites from hydroxymethyl cytosine (hmC), a rare variant found in up to 2% of modified CpGs in certain tissues. The first step of gaining a cytosine modification in vivo is addition of the methyl mark, C→mC. Hydroxymethylation can only occur as a subsequent step, mC→hmC. Therefore, any new CpG modification that was detected in the present study by RRBS (following SNL) consisted of an obligatory modification C→mC. While some of the newly formed mC might have subsequently been further changed to hmC, the conclusion that gain of mC occurred after SNL is not affected. The opposite case, CpG demethylation, also needs to be considered. In this case, RRBS determined the loss of a CpG modification, which could have occurred at a site that was a mC (most likely) or a hmC (possibly at few sites) at baseline before SNL. Therefore, while RRBS could not resolve whether the DRG methylome consists only of mC marks or includes some hmC modifications, the presented data can be interpreted without ambiguity in regard to the dynamic methylome changes observed after SNL.
The old paradigm relating methylome and transcriptome suggested a negative correlation between methylation and gene expression.19 Recently, a positive correlation, especially when methylation affected the gene body, was also described.22 An increasing number of reports have further proposed that the methylome-transcriptome relationship cannot be represented by a simple model when considering large, high quality datasets.10,21,23,36,37 In these studies, positive and negative correlations between methylation alterations and transcriptome changes were found only for a minority of CpGs. It was also suggested that the relationship depends on the CpG-content of the promoter region. Ng et al. (2013) observed that DNA methylation in CpG-rich promoters was anticorrelated with transcription, while increased or decreased methylation in CpG-poor promoters could result either in enhanced or diminished expression.21 In the present study, we examined promoter regions and gene bodies of HCP and LCP genes separately. The results of our integrated analysis of dDMCs and RNA alterations suggested that the old model (promoter methylation up-transcription down) fell short at least when attempting to explain the data set in its entirety. Following nerve injury, a gain of methylation in the gene body was not associated with an upregulation of the corresponding gene. Overall, our observations are in line with some of the above-mentioned reports and suggest the absence of a systematic relationship between variation in DNA methylation and variation in gene expression. Pitfalls in the field awaiting resolution in order to better characterize the methylome-transcriptome relationship include several recognized issues, such as the limitations from using tissues rather than single cells, the role of other epigenetic modifications like histone methylation and acetylation and the role of other DNA modifications, such as hydroxymethylation, that remain difficult to assess with precision at nucleotide resolution. Furthermore, the literature might be affected by a recurrent narrative bias to report an association of methylation gain with gene repression regardless of the biological context and might underreport results that do not fit with any of the most straightforward models. The present study does not resolve this issue for peripheral nerve injury or the DRG but may provide additional data that can be used in the future for comparative analyses or to devise further experiments.
The present study also pointed to a role of non-genic regions, which was consistent with published reports showing that CpG methylation changes can affect regulatory elements throughout the genome including sites that are far distant from annotated promoter regions.35,38 To gain some understanding of the changes occurring across gene deserts in the DRG after SNL, we took a detour into organism-wide genome-biology: performing extensive methylation sequencing of various organs with the goal of better understanding what is happening in the PNS. Our classification of non-genic regions into alive and dead deserts is based on the new type of methylome data we obtained. Prediction of alive desert CpGs was supported statistically highly significantly—excluding the risk of a chance observation—and further supported as functionally important by the highly significant binding site enrichment patterns observed. These findings suggest that methylation of gene deserts may co-determine the function of genes by modulating the configuration of regulatory regions. Recently, Stadler et al. identified low methylated, CpG-poor regions that were enriched for DNA binding factors sites in the mouse. By means of 2 transcription factors, REST and CTCF, the authors showed that binding within these active regulatory regions results in the demethylation of the site and suggested that this process could in turn facilitate the binding of additional binding factors that are sensitive to DNA methylation.38 Similarly, Ng et al. showed that the loss of binding of certain transcription factors was strongly associated with increased DNA methylation.21 Whether DNA methylation changes precede the binding of transcription factors or are its consequence need to be understood.39 Our data are consistent with these reports by others. In the present study, the HOMER algorithm40 was used to detect the enriched consensus motifs for transcription factor binding sites in the SNL vs. control animals. The motif discovery includes base position variability, thereby not permitting the identification of the exact genomic position of these sequences. To confirm that RUNX, SOX, ETS and NF1 are truly bound to regions of dynamically altered DNA methylation, genome-wide chromatin immunoprecipitation sequencing (ChIP-Seq) would be required, which was beyond the scope of the present study.
The present study leveraged a current technology, RRBS, providing precise quantification of methylation levels not only for individual genes but specific CpG sites. The approach demonstrated widespread methylome remodeling in the DRG affecting 2 portions of the genome: 1) well annotated genic regions, such as those of the axon guidance pathway genes and; 2) alive deserts, regions that were previously unstudied in the context of PNS biology, yet appeared to undergo equally marked epigenomic remodeling. The majority of genes and most of gene deserts were invariant among all experimental conditions. Thereby, the present study demonstrated methylome stability as well as genome-wide, targeted methylome remodeling in the PNS.
Materials and Methods
Animal experiments, tissue procurement of tissues, and animal group sizes
Male Brown-Norway rats were used for all experiments. This strain was chosen because it is the reference strain of the publicly available rattus norvegicus reference genome. None of the animals had a previous history such as prior drug administration, surgery, behavioral testing or other. All procedures involving live animals were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC).
Rats were purchased from Charles River Laboratories and housed 2 per cage prior to the start of the experiment. Animals had a body weight of 250–300 g. All experiments and tissue harvesting were performed during the dark cycle of a 24 h period. L5 spinal nerve ligation (SNL) was performed as described by Chung et al.41 and previously performed in the author's laboratory.42 In brief, L5 SNL was performed under deep anesthesia achieved isoflurane inhalation. L5 SNL consisted of ligation of the left spinal nerve immediately distally to the L5 dorsal root ganglion (DRG) followed by cutting the nerve distally to the ligature. Control animals in the present study underwent a sham procedure consisting of isoflurane anesthesia and a skin incision followed by surgical wound closure without L5 SNL.
L5 DRG for analysis of dDMCs (by RRBS) were harvested 24 h after the L5 SNL or sham procedure. L4 DRG, liver, skeletal muscle, and spleen for organism-wide identification of tDMCs and tIMCs (by RRBS) were harvested from animals sacrificed without a prior procedure. All DRG or other tissues were flash-frozen and stored at −80°C.
Group sizes were n = 4 for the SNL group and n = 4 for the control group in the experiment determining DNA methylation levels. The group size was chosen based on our previous study characterizing the DRG methylome in control animals,13 which had demonstrated that RRBS of the DRG could be performed highly reproducibly by rigorously standardizing procedures for tissue procurement and library construction. The experimental design thereby matched the minimum group size to the resource intensity of RRBS and the multiple strengths of the technology: reproducibility, genome-wide reach, single CpG resolution, and methylation level quantification from digital data (read counts), which can be analyzed by tests with high statistical power.
Characterization of organ specific methylomes was performed for each tissue in duplicate. This design was based on successful organ comparisons in recent reports by others 43 and further justified by the marked organ differences detectable by a principal component analysis (PCA; see below).
Reduced representation bisulfite sequencing (RRBS)
RRBS was performed by an investigator who was blinded toward the experimental groups. Tissues were thawed on ice. Genomic DNA (gDNA) was extracted from whole DRG and aliquots of liver, skeletal muscle, and spleen using the QIAmp DNA Micro Kit (Qiagen). RRBS sequencing libraries were prepared as described previously.13 In brief, 250 ng of genomic DNA from each sample were digested with 200 U of MspI, a methylation-insensitive restriction enzyme. DNA fragments were purified. Sticky ends resulting from the restriction digest were converted to blunt ends by end-repair using T4 DNA polymerase and Klenow enzyme (NEB). An 'A' nucleotide was added to the 3'-end of the blunted fragments and distinct adaptor sequences were ligated at both ends of the DNA fragments. Fragments between 30–300 bp were selected and gel-extracted. Libraries were then bisulfite treated using the EZ DNA Methylation Kit (Zymo Research) following the manufacturer's instructions. Subsequently, 15 cycles of PCR amplification were performed followed by purification of the amplification products with Ampure XP magnetic beads. The resulting libraries were quality controlled and quantified on an Agilent 2,100 micofluidic analyzer. Sequencing was performed on an Illumina HiSeq 2,000 platform genome analyzer for 50 cycles in paired-end mode (2 × 50 bp).
Determining CpG methylation levels at nucleotide resolution by bioinformatics processing of RRBS reads
Sequence reads were mapped in the 3-nucleotide space (A, G, T) to MspI fragments (30–300 bp length) predicted from the forward and reverse strand of the rat reference genome (rn4) using Bowtie244 allowing for a maximum of 2 mismatches and retaining only uniquely aligning reads. Methylation levels were then determined as previously described13 for all cytosines occurring within a CpG dinucleotide motif in the rat genome by computing the fraction of cytosines that was chemically protected from bisulfite conversion to uracil. A minimum coverage of ≥10 reads was required for each library to declare a methylation level; CpG sites covered by fewer reads in any of the replicates were excluded from subsequent analyses.
The bisulfite conversion rate, an important quality control parameter of RRBS experiments, was determined by computing the conversion rate of cytosines to uracil occurring outside of CpG motifs, where DNA methylation is expected to be absent and therefore bisulfite conversion of cytosines to uracils complete. Sequencing libraries were only accepted for downstream analysis if the bisulfite conversion rate was found to be >99 % thereby assuring that the fraction of unmethylated CpGs could be underestimated in the present study only by <1 %.
Hierarchical clustering
CpG sites were included if they were covered by ≥10 RRBS reads in each sample. Hierarchical clustering was executed in R using the Ward method with the Manhattan distance executing the command:
hclust(dist(t(methylation_data_matrix), method = 'manhattan'), method = 'ward');
Statistical testing of individual CpG sites: dDMCs, tDMCs, and tIMCs
To test for differences in methylation at individual CpG sites, either between conditions (SNL vs. control) or between multiple tissues (DRG, liver, muscle, spleen), each site was treated as a separate logistic regression. A generalized linear model (GLM) using a logistic link function was used. GLM uses a likelihood ratio test (LRT), which makes use of the raw counts directly and so correctly calibrates the P-value between sites with high or low depth of coverage.
Implementation in R was as follows:
# Data for one CpG
meth <- c(73, 39, 102, 199, 135, 41, 197, 151);
nmeth <- c(2, 3, 3, 5, 3, 1, 12, 9);
grp <- factor(c(1, 1, 1, 1, 0, 0, 0, 0));
# model
fit <- glm(cbind(meth, nmeth) ∼ grp, family = 'binomial');
aa <- anova(fit, 'chisq');
In our special case of a single predictor the LRT of the glm function can be computed more directly. This was done using the Java code below, resulting in a 10x speedup over the R code.
A subset of cases was evaluated with both to validate the computation.
// glm
publicstatic class GLM {
// compute deviance
publicstatic double deviance(List<List<Integer>> samp_mread_lst,
List<List<Integer>> samp_nread_lst) {
// sample level, grp and total level methylation estimation
List<Double> grp_meth_lst = new ArrayList<Double>();
int all_mread = 0, all_nread = 0;
for (int i = 0; i < samp_mread_lst.size(); i++) {
int grp_mread_tot = 0, grp_nread_tot = 0;
for (int j = 0; j < samp_mread_lst.get(i).size(); j++) {
// sample
int samp_mread = samp_mread_lst.get(i).get(j);
int samp_nread = samp_nread_lst.get(i).get(j);
// group
grp_mread_tot += samp_mread;
grp_nread_tot += samp_nread;
// all
all_mread += samp_mread;
all_nread += samp_nread;
}
grp_meth_lst.add(grp_mread_tot
/ (grp_mread_tot + grp_nread_tot * 1.0));
}
double all_meth = all_mread / (all_mread + all_nread * 1.0);
// model deviance
double dev_model = 0, dev_null = 0;
for (int i = 0; i < grp_meth_lst.size(); i++) {
double grp_meth = grp_meth_lst.get(i);
for (int j = 0; j < samp_mread_lst.get(i).size(); j++) {
int samp_mread = samp_mread_lst.get(i).get(j);
int samp_nread = samp_nread_lst.get(i).get(j);
int samp_tot = samp_mread + samp_nread;
// expected meth counts
double grp_est_samp_mread = grp_meth * samp_tot;
double all_est_samp_mread = all_meth * samp_tot;
// deviance
if (samp_mread != 0) {
dev_model += samp_mread
* Math.log(samp_mread / grp_est_samp_mread);
dev_null += samp_mread
* Math.log(samp_mread / all_est_samp_mread);
}
if (samp_nread != 0) {
dev_model += samp_nread
* Math.log(samp_nread
/ (samp_tot - grp_est_samp_mread));
dev_null += samp_nread
* Math.log(samp_nread
/ (samp_tot - all_est_samp_mread));
}
}
}
// done
return 2 * (dev_null - dev_model);
}
}
dDMCs were determined by requiring a minimum absolute difference of 0.08 and a significance level by the above test of P < 10−4. The difference cut-off was deliberately chosen less stringent than previously selected by others, as we analyzed the same tissue type at an early time-point and thus expected the variation to be less pronounced than between distinct tissues.
Methylation changes across regions of juxtaposed CpG sites
A complementary assessment of the methylation was based on a clustering metric, testing whether changes occurred in contiguous blocks. For the set of genes highlighted in Figure 1C and Figure S1, we found all runs of 10 sequential CpG sites in the gene with the same sign, where “+” = mean methylation of the SNL group was larger than the control and “-” = the converse. This was then compared to the same result after randomly permuting the order of the CpG sites within the gene. In the overall data set, the likelihood of “+" and "-" were identical; thus, a run of 10 "+" or 10 "-" each had a frequency of approximately 0.001. The permutation comparison provides an appropriate reference for the overall count of runs. This run test lacks statistical power compared to the likelihood ratio test-based GLM above, but gives a second confirmation of systematic changes.
Annotation of CpG sites relative to genes
The gene annotation Ensembl68 of the rat reference genome assembly version rn4 was used. Genes were divided into the high CpG promoter density (HCP) and low CpG promoter density (LCP) groups as described previously,13 i.e., genes with a promoter-CpG-content of ≥3.2% were defined as HCP genes; genes with a promoter-CpG-content of <3.2% were defined as low-density CpG promoter (LCP) genes. This definition was adopted from the classification of human promoters by Saxonov et al.20 with minor modifications for the rat genome as previously described.13 Similarly to the human genome, the promoter CpG density in the rat genome follows a bimodal distribution, which allowed dichotomization of promoters into 2 classes, HCP and LCP, with a cut-off at the inter-peak minimum (as shown in Figure 1B of our previous publication).13 CpG sites were annotated as "promoter" if they were located with within 1,000 bp 5' or 3' of the transcription start site (TSS). Non-promoter CpGs within protein encoding genes were then assigned to the bins "exon" or "intron" according to the gene annotation. Remaining CpGs were assigned to the bin "gene desert."
Integrated analysis of dDMCs located in genes
Ingenuity Systems pathway analysis IPA was used for pathway analysis on differentially methylated genes defined by containing dDMCs in the promoter (2 kb centered around the transcription start site) or gene body. The Ingenuity IPA system was accessed through the website interface, the only currently available user interface http://www.ingenuity.com. The Ensembl IDs of genes were imported into IPA; a core analysis was conducted to identify the most enriched pathways; the list of differentially methylated genes occurring in the most significantly altered pathway, axon guidance, was downloaded and provided as Table S1; core pathway components were also represented graphically (Fig. 1E).
Gene expression analysis
L5 DRG for the primary transcriptome analysis were harvested 24 h after the L5 SNL or skin incision. Two biological replicates were available per condition. RNA isolation was performed using the TRIzol Reagent (Invitrogen) according to the manufacturer's protocol. For library construction the protocol for TruSeq RNA Sample Prep Kit v2 was followed. RNA-seq was performed on an Illumina HiSeq 2,000 platform and the 50 bp long reads were aligned to the rat genome (RGSC v3.4) by the Bowtie 2 algorithm. We discarded reads with more than 2 mismatches and kept only uniquely mapped reads (UMRs). ENSEMBL genome browser 67 was used to annotate the genes. A coverage ≥ 20 reads in each replicate of either the control or the SNL condition was required, resulting in 10,315 genes. To normalize for the total read length and the number of sequencing reads, RPKM (Reads Per Kilobase per Million mapped reads) was applied to each read;45 First, a pseudo-count of 1 was added, then RPKM was computed and the RPKM values were log2-transformed. Genes with a [log2 (fold change)] of ≥ 0.6 or ≤ −0.6 were defined as up- or downregulated (corresponding to a fold change of ≤ −1.5 or ≥ 1.5).
Distribution of dDMCs across gene deserts
Gene deserts (intergenic regions) differ in size. To analyze the distribution of dDMCs across gene deserts for the entire dataset, gene desert sizes were standardized to a length of 1. The span was then subdivided into 10 bins corresponding to an interval between percentile ranks as indicated in Figure 2A. The frequency of dDMCs in each bin relative to the number of assayed CpG sites in the bin was then computed.
Principal component analysis (PCA)
PCA was performed in R using the command princomp on the matrix of methylation levels for all CpG sites covered by ≥ 10 reads in all tissues indicated.
Motif enrichment analysis comparing alive and dead deserts
We performed a transcription factor motif enrichment analysis comparing alive deserts and dead deserts. Alive deserts were formed by joining juxtaposed hyper- or hypo-methylated dDMCs into respective regions using a sliding window of 100 bp size. Dead deserts were formed by applying the same procedure to intergenic tIMC. Comparisons between dead and alive desert regions were then made for 300 bp regions centered around each desert's interval midpoint. The window size and the demarcation of regions to be compared were chosen following the motif enrichment analysis methodology used by Ng et al. (2013).21 Enrichment analysis was then executed using the Hypergeometric Optimization of Motif EnRichment algorithm (HOMER) 46 version 4.2 (downloaded January 2013). Two comparisons were made: 1) hypermethylated deserts (foreground) against dead deserts (background); 2) hypomethylated deserts (foreground) against dead deserts (background).
The authors declare no competing financial interests.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
Support was provided by the National Institute of Neurological Disorders and Stroke (NINDS) (R21NS062271) and by the Schulze Family Foundation (to A.S.B.).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1. Costigan M, Belfer I, Griffin RS, Dai F, Barrett LB, Coppola G, Wu T, Kiselycznyk C, Poddar M, Lu Y, et al. . Multiple chronic pain states are associated with a common amino acid-changing allele in KCNS1. Brain 2010; 133:2519-27; PMID:20724292; http://dx.doi.org/ 10.1093/brain/awq195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hammer P, Banck MS, Amberg R, Wang C, Petznick G, Luo S, Khrebtukova I, Schroth GP, Beyerlein P, Beutler AS. mRNA-seq with agnostic splice site discovery for nervous system transcriptomics tested in chronic pain. Genome Res 2010; 20:847-60; PMID:20452967; http://dx.doi.org/ 10.1101/gr.101204.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Michaelevski I, Segal-Ruder Y, Rozenbaum M, Medzihradszky KF, Shalem O, Coppola G, Horn-Saban S, Ben-Yaakov K, Dagan SY, Rishal I, et al. . Signaling to transcription networks in the neuronal retrograde injury response. Sci Signal 2010; 3:ra53; PMID:20628157; http://dx.doi.org/ 10.1126/scisignal.2000952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhang Z, Cai Y-Q, Zou F, Bie B, Pan ZZ. Epigenetic suppression of GAD65 expression mediates persistent pain. Nat Med 2011; 17:1448-55; PMID:21983856; http://dx.doi.org/ 10.1038/nm.2442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tajerian M, Alvarado S, Millecamps M, Dashwood T, Anderson KM, Haglund L, Ouellet J, Szyf M, Stone LS. DNA methylation of SPARC and chronic low back pain. Mol Pain 2011; 7:65; PMID:21867537; http://dx.doi.org/ 10.1186/1744-8069-7-65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhu X-Y, Huang C-S, Li Q, Chang R-M, Song Z-B, Zou W-Y, Guo Q-L. p300 exerts an epigenetic role in chronic neuropathic pain through its acetyltransferase activity in rats following chronic constriction injury (CCI). Mol Pain 2012; 8:84; PMID:23176208; http://dx.doi.org/ 10.1186/1744-8069-8-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Uchida H, Matsushita Y, Ueda H. Epigenetic regulation of BDNF expression in the primary sensory neurons after peripheral nerve injury: implications in the development of neuropathic pain. Neuroscience 2013; 240:147-54; PMID:23466809; http://dx.doi.org/ 10.1016/j.neuroscience.2013.02.053 [DOI] [PubMed] [Google Scholar]
- 8. Denk F, McMahon SB. Chronic pain: emerging evidence for the involvement of epigenetics. Neuron 2012; 73:435-44; PMID:22325197; http://dx.doi.org/ 10.1016/j.neuron.2012.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Klengel T, Mehta D, Anacker C, Rex-Haffner M, Pruessner JC, Pariante CM, Pace TWW, Mercer KB, Mayberg HS, Bradley B, et al. . Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat Neurosci 2013; 16:33-41; PMID:23201972; http://dx.doi.org/ 10.1038/nn.3275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guo JU, Ma DK, Mo H, Ball MP, Jang M-H, Bonaguidi M. a, Balazer J. a, Eaves HL, Xie B, Ford E, et al. . Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat Neurosci 2011; 14:1345-51; PMID:21874013; http://dx.doi.org/ 10.1038/nn.2900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain 1992; 50:355-63; PMID:1333581; http://dx.doi.org/ 10.1016/0304-3959(92)90041-9 [DOI] [PubMed] [Google Scholar]
- 12. Meissner A, Gnirke A, Bell GW, Ramsahoye B, Lander ES, Jaenisch R. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res 2005; 33:5868-77; PMID:16224102; http://dx.doi.org/ 10.1093/nar/gki901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hartung T, Zhang L, Kanwar R, Khrebtukova I, Reinhardt M, Wang C, Therneau TM, Banck MS, Schroth GP, Beutler AS. Diametrically opposite methylome-transcriptome relationships in high- and low-CpG promoter genes in postmitotic neural rat tissue. Epigenetics 2012; 7:421-8; PMID:22415013; http://dx.doi.org/ 10.4161/epi.19565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Seijffers R, Mills CD, Woolf CJ. ATF3 increases the intrinsic growth state of DRG neurons to enhance peripheral nerve regeneration. J Neurosci 2007; 27:7911-20; PMID:17652582; http://dx.doi.org/ 10.1523/JNEUROSCI.5313-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wan G, Yang K, Lim Q’En, Zhou L, He BP, Wong HK, Too H-P. Identification and validation of reference genes for expression studies in a rat model of neuropathic pain. Biochem Biophys Res Commun 2010; 400:575-80; PMID:20804730; http://dx.doi.org/ 10.1016/j.bbrc.2010.08.106 [DOI] [PubMed] [Google Scholar]
- 16. Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci 2007; 10:1361-8; PMID:17965656; http://dx.doi.org/ 10.1038/nn1992 [DOI] [PubMed] [Google Scholar]
- 17. Ben-Yaakov K, Dagan SY, Segal-Ruder Y, Shalem O, Vuppalanchi D, Willis DE, Yudin D, Rishal I, Rother F, Bader M, et al. . Axonal transcription factors signal retrogradely in lesioned peripheral nerve. EMBO J 2012; 31:1350-63; PMID:22246183; http://dx.doi.org/ 10.1038/emboj.2011.494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jankowski MP, Cornuet PK, Mcilwrath S, Koerber HR, Albers KM. SRY-Box containing gene 11 (Sox11) transcription factor Is required for neuron survival and neurite growth. Neuroscience 2006; 143:501-14; PMID:17055661; http://dx.doi.org/ 10.1016/j.neuroscience.2006.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bird AP, Wolffe AP. Methylation-induced repression-belts, braces, and chromatin. Cell 1999; 99:451-4; PMID:10589672; http://dx.doi.org/ 10.1016/S0092-8674(00)81532-9 [DOI] [PubMed] [Google Scholar]
- 20. Saxonov S, Berg P, Brutlag DL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc Natl Acad Sci U S A 2006; 103:1412-7; PMID:16432200; http://dx.doi.org/ 10.1073/pnas.0510310103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ng CW, Yildirim F, Yap YS, Dalin S, Matthews BJ, Velez PJ, Labadorf A, Housman DE, Fraenkel E. Extensive changes in DNA methylation are associated with expression of mutant huntingtin. Proc Natl Acad Sci U S A 2013; 110:2354-9; PMID:23341638; http://dx.doi.org/ 10.1073/pnas.1221292110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ball MP, Li JB, Gao Y, Lee J-H, LeProust EM, Park I-H, Xie B, Daley GQ, Church GM. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat Biotechnol 2009; 27:361-8; PMID:19329998; http://dx.doi.org/ 10.1038/nbt.1533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kulis M, Heath S, Bibikova M, Queirós AC, Navarro A, Clot G, Martínez-Trillos A, Castellano G, Brun-Heath I, Pinyol M, et al. . Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat Genet 2012; 44:1236-42; PMID:23064414; http://dx.doi.org/ 10.1038/ng.2443 [DOI] [PubMed] [Google Scholar]
- 24. Bacon A, Kerr NCH, Holmes FE, Gaston K, Wynick D. Characterization of an enhancer region of the galanin gene that directs expression to the dorsal root ganglion and confers responsiveness to axotomy. J Neurosci 2007; 27:6573-80; PMID:17567818; http://dx.doi.org/ 10.1523/JNEUROSCI.1596-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen C-L, Broom DC, Liu Y, de Nooij JC, Li Z, Cen C, Samad OA, Jessell TM, Woolf CJ, Ma Q. Runx1 determines nociceptive sensory neuron phenotype and is required for thermal and neuropathic pain. Neuron 2006; 49:365-77; PMID:16446141; http://dx.doi.org/ 10.1016/j.neuron.2005.10.036 [DOI] [PubMed] [Google Scholar]
- 26. Inoue K, Ozaki S, Shiga T, Ito K, Masuda T, Okado N, Iseda T, Kawaguchi S, Ogawa M, Bae S-C, et al. . Runx3 controls the axonal projection of proprioceptive dorsal root ganglion neurons. Nat Neurosci 2002; 5:946-54; PMID:12352981; http://dx.doi.org/ 10.1038/nn925 [DOI] [PubMed] [Google Scholar]
- 27. Bergsland M, Ramsköld D, Zaouter C, Klum S, Sandberg R, Muhr J. Sequentially acting Sox transcription factors in neural lineage development. Genes Dev 2011; 25:2453-64; PMID:22085726; http://dx.doi.org/ 10.1101/gad.176008.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jankowski MP, McIlwrath SL, Jing X, Cornuet PK, Salerno KM, Koerber HR, Albers KM. Sox11 transcription factor modulates peripheral nerve regeneration in adult mice. Brain Res 2009; 1256:43-54; PMID:19133245; http://dx.doi.org/ 10.1016/j.brainres.2008.12.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tajerian M, Alvarado S, Millecamps M, Vachon P, Crosby C, Bushnell MC, Szyf M, Stone LS. Peripheral nerve injury is associated with chronic, reversible changes in global DNA methylation in the mouse prefrontal cortex. PLoS One 2013; 8:e55259; PMID:23383129; http://dx.doi.org/ 10.1371/journal.pone.0055259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Viet CT, Ye Y, Dang D, Lam DK, Achdjian S, Zhang J, Schmidt BL. Re-expression of the methylated EDNRB gene in oral squamous cell carcinoma attenuates cancer-induced pain. Pain 2011; 152:2323-32; PMID:21782343; http://dx.doi.org/ 10.1016/j.pain.2011.06.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hanani M. Satellite glial cells in sensory ganglia: from form to function. Brain Res Brain Res Rev 2005; 48:457-76; PMID:15914252; http://dx.doi.org/ 10.1016/j.brainresrev.2004.09.001 [DOI] [PubMed] [Google Scholar]
- 32. Hu P, Mclachlan EM. Macrophage and lymphocyte invasion of dorsal root ganglia after peripheral nerve lesions in the rat. Neuroscience 2002; 112:23-38; PMID:12044469; http://dx.doi.org/ 10.1016/S0306-4522(02)00065-9 [DOI] [PubMed] [Google Scholar]
- 33. Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, et al. . A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci 2008; 28:264-78; PMID:18171944; http://dx.doi.org/ 10.1523/JNEUROSCI.4178-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Emery EC, Young GT, Berrocoso EM, Chen L, McNaughton PA. HCN2 ion channels play a central role in inflammatory and neuropathic pain. Science 2011; 333:1462-6; PMID:21903816; http://dx.doi.org/ 10.1126/science.1206243 [DOI] [PubMed] [Google Scholar]
- 35. Ziller MJ, Gu H, Müller F, Donaghey J, Tsai LT-Y, Kohlbacher O, De Jager PL, Rosen ED, Bennett DA, Bernstein BE, et al. . Charting a dynamic DNA methylation landscape of the human genome. Nature 2013; 500:477-81; PMID:23925113; http://dx.doi.org/ 10.1038/nature12433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lam LL, Emberly E, Fraser HB, Neumann SM, Chen E, Miller GE, Kobor MS. Factors underlying variable DNA methylation in a human community cohort. Proc Natl Acad Sci 2012; 109:1-8; PMID:23045638; http://dx.doi.org/ 10.1073/pnas.1121249109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zouridis H, Deng N, Ivanova T, Zhu Y, Wong B, Huang D, Wu YH, Wu Y, Tan IB, Liem N, et al. . Methylation subtypes and large-scale epigenetic alterations in gastric cancer. Sci Transl Med 2012; 4:156ra140; PMID:23076357; http://dx.doi.org/ 10.1126/scitranslmed.3004504 [DOI] [PubMed] [Google Scholar]
- 38. Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Schöler A, Van Nimwegen E, Wirbelauer C, Oakeley EJ, Gaidatzis D, et al. . DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature 2011; 480:490-5; PMID:22170606 [DOI] [PubMed] [Google Scholar]
- 39. Kulis M, Queirós AC, Beekman R, Martín-Subero JI. Intragenic DNA methylation in transcriptional regulation, normal differentiation and cancer. Biochim Biophys Acta 2013; 1829:1161-74; PMID:23938249; http://dx.doi.org/ 10.1016/j.bbagrm.2013.08.001 [DOI] [PubMed] [Google Scholar]
- 40. Benner CW. Identification of a conserved periodic promoter structure in metazoans. 2009; https://escholarship.org/uc/item/5nh266bv#page-5 [Google Scholar]
- 41. Chung JM, Kim HK, Chung K. Segmental spinal nerve ligation model of neuropathic pain. Methods in Molecular Medicine. Luo Z. editor. Totowa, New Jersey: Humana Press; 2004. page 35-45. [DOI] [PubMed] [Google Scholar]
- 42. Storek B, Reinhardt M, Wang C, Janssen WGM, Harder NM, Banck MS, Morrison JH, Beutler AS. Sensory neuron targeting by self-complementary AAV8 via lumbar puncture for chronic pain. Proc Natl Acad Sci U S A 2008; 105:1055-60; PMID:18215993; http://dx.doi.org/ 10.1073/pnas.0708003105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo Q-M, et al. . Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009; 462:315-22; PMID:19829295; http://dx.doi.org/ 10.1038/nature08514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009; 10:R25; PMID:19261174; http://dx.doi.org/ 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mortazavi A, Williams BA, Mccue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 2008; 5:1-8; PMID:18175409; http://dx.doi.org/ 10.1038/nmeth.1226 [DOI] [PubMed] [Google Scholar]
- 46. Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 2010; 38:576-89; PMID:20513432; http://dx.doi.org/ 10.1016/j.molcel.2010.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.