

Abstract
Congenital heart defects represent the most common malformation at birth, occurring also in ∼50% of individuals with Down syndrome. Congenital heart defects are thought to have multifactorial etiology, but the main causes are largely unknown. We have explored the global methylation profile of fetal heart DNA in comparison to blood DNA from control subjects: an absolute correlation with the type of tissue was detected. Pathway analysis revealed a significant enrichment of differential methylation at genes related to muscle contraction and cardiomyopathies in the developing heart DNA. We have also searched for abnormal methylation profiles on developing heart-tissue DNA of syndromic and non-syndromic congenital heart defects. On average, 3 regions with aberrant methylation were detected per sample and 18 regions were found differentially methylated between groups. Several epimutations were detected in candidate genes involved in growth regulation, apoptosis and folate pathway. A likely pathogenic hypermethylation of several intragenic sites at the MSX1 gene, involved in outflow tract morphogenesis, was found in a fetus with isolated heart malformation. In addition, hypermethylation of the GATA4 gene was present in fetuses with Down syndrome with or without congenital heart defects, as well as in fetuses with isolated heart malformations. Expression deregulation of the abnormally methylated genes was detected. Our data indicate that epigenetic alterations of relevant genes are present in developing heart DNA in fetuses with both isolated and syndromic heart malformations. These epimutations likely contribute to the pathogenesis of the malformation by cis-acting effects on gene expression.
Keywords: congenital heart disease, Down syndrome, DNA methylation, epimutation, heart malformation
Abbreviations
- CHD
congenital heart defects
- DS
Down syndrome
- iCHD
isolated CHD
- DMCpGs
differentially methylated CpGs
- MS-MLPA
Methylation Specific Multiple Ligation-Dependent Probe Amplification
- TRR
transcriptional regulatory region
- qRT-PCR
quantitative real-time PCR
Introduction
Epigenetic mechanisms contribute to the regulation of multiple physiological processes during development or aging.1 Among the different epigenetic mechanisms, aberrant DNA methylation has been clearly associated to several diseases including cancer,2,3 diabetes,4,5 or psychiatric disorders.6-8 DNA methylation, by the addition of a methyl group in the 5′ carbon of cytosine, alters the structure of the DNA molecule resulting in possible modifications of gene expression patterns. Thus, aberrant DNA methylation through its transcriptional consequences may have an important role in human disease.
Congenital heart defects (CHD) represent a high percentage of clinically significant birth defects, occurring in ∼8 per 1,000 live births and being the most common malformation and an important public health burden.9 Although there is strong evidence that genetic alterations, including point mutations and chromosomal abnormalities,10–13 play a relevant role in the etiology of CHD, the molecular bases remain unclear in the majority of cases and environmental factors are also likely to contribute. Down syndrome (DS, [MIM 190685]), or trisomy 21, represents one of the most common causes of CHD, with a prevalence ranging between 43% and 58%.14,15 Given that all individuals with DS carry an identical chromosomal alteration, additional factors, genetic and/or epigenetic, must contribute to the development of CHD in this disorder.
A possible contribution of methylation abnormalities to CHD has been recently explored by studying the methylation regulatory folate-pathway.16,17 Maternal folic acid supplementation has been reported to decrease the risk of CHD associated with DS.18 Several SNPs in the folate transporter gene SLC19A1 (solute carrier family 19, [MIM 600424]) showed significant association with the incidence of CHD in DS cases. Over-transmission to DS cases with CHD of functional alleles at the MTHFR (methylenetetrahydrofolate reductase, [MIM 607093]) gene, encoding a rate-limiting enzyme in the methyl cycle, also suggested that disruption of this pathway may contribute to CHD in DS.16 Several studies and a confirmatory meta-analysis also showed association of MTHFR variants with increased risk for isolated CHD.17 In addition, an excess of de novo mutations in histone-modifying genes has been described in a cohort of patients with CHD,13 pointing out another source of epigenetic variation as a contributing factor in the etiology of heart malformations.
However, methylation profiles and other potential epigenetic abnormalities have not been explored in relation to CHD to date. Considering the relevant role of epigenetics in the regulation of gene expression in development and the increasing evidence linking epigenetic alterations with congenital malformations, we have searched for potential abnormal methylation profiles on developing heart-tissue DNA in samples with syndromic and non-syndromic CHD compared to controls. Our data indicate a relevant role of methylation abnormalities in both syndromic and non-syndromic CHD.
Results
Developing a heart methylation profile
We studied 22 DNA samples of fetal heart tissue from medically terminated pregnancies: 4 with normal development, 6 with DS without CHD, 6 with DS and CHD, and 6 with isolated CHD (iCHD) (Table 1). In order to establish the methylation profile for developing heart tissue and analyze the global performance of the assay, we first compared the methylation levels between fetal heart tissue and blood samples from a control cohort (656 subjects from 19 to 101 y old studied using Illumina Infinium HumanMethylation450 BeadChip Kit).19 This analysis revealed remarkably different profiles between tissues as shown by the well defined and tissue dependent clustering in a Principal Component Analysis with all samples (Fig. 1A). A total of 465 CpGs contiguous to 407 genes were found hypomethylated in heart tissue when compared to blood; likewise, 407 CpGs corresponding to 339 genes were identified as hypomethylated in blood. As expected, ontology and pathway-based analyses revealed strong differences in enriched sets of genes with differential methylation per tissue, further indicating the relevance of methylation patterns in the epigenetic regulation of tissue functions (Table 2).
Table 1.
Phenotype data and most relevant alterations. List of the studied samples with information on gender, gestational age and phenotype (specifying the heart malformation) of the 16 fetuses with syndromic (DS) and isolated congenital heart defect (iCHD). The most relevant regions showing aberrant methylation per sample and the status and location of the aberrant methylation with respect to the closest gene are displayed in the 2 last columns. TOF: tetralogy of Fallot; RHH: right heart hypoplasia; HAA: hypoplasia of the ascending aorta; LHH: left heart hypoplasia; VSD: ventricular septal defect; D-TGA: dextro-transposition of the great arteries; L-TGA: levo-transposition of the great arteries; AVSD: atrioventricular septal defect; MVA: mitral valve atresia; ADA: aneurismatic dilatation of the atrium; AVA: aortic valve atresia; TAV: tricuspid aortic valve; DORV: double outlet right ventricle, CoA: coarctation of the preductal aorta; TVS: tricuspid valve stenosis; TA: truncus arteriosus; ADA: absent ductus arteriosus; PFO: patent foramen ovale
| Sample | Gender | Weeks of gestation | Phenotype | Regions with altered methylation profile | Genes included in the regions (region and status) |
|---|---|---|---|---|---|
| Control_1 | Female | 20 | Normal development | — | — |
| Control_2 | Female | 15 | Normal development | — | — |
| Control_3 | Female | 15 | Normal development | — | — |
| Control_4 | Male | 20 | Normal development | — | — |
| DS_1 | Female | 19 | DS | — | — |
| DS_2 | Female | 22 | DS | — | — |
| DS_3 | Female | 22 | DS | — | — |
| DS_4 | Female | 22 | DS | — | — |
| DS_5 | Female | 18 | DS | — | — |
| DS_6 | Male | 20 | DS | — | — |
| DS-CHD_1 | Female | 22 | DS and VSD | 5 | HTR3C (hypo), MT1X (hyper), IDH3G (hyper), GZMH (hypo), TCL1B (hyper), TCL6 (hyper), TML1 (hyper) |
| DS-CHD_2 | Female | 22 | DS and VSD | 4 | CD80 (hypo), IGDCC3 (hypo), IGDCC4 (hypo), FFAR3 (hypo), CBR1 (hyper), SETD4 (hyper) |
| DS-CHD_3 | Female | 18 | DS, AVSD and CoA | 2 | DOK7 (hyper), NOS3 (hypo) |
| DS-CHD_4 | Female | 19 | DS and TOF | 1 | SLC2A1 (hyper) |
| DS-CHD_5 | Male | 22 | DS and VSD | 3 | ZFP2 (hyper), RAET1L (hyper), KIAA0101 (hyper), |
| DS-CHD_6 | Female | 18 | DS, LHH and HAA | 5 | BIN1 (hyper), UBE2H (hyper), CHP (hyper), NXN (hyper), TCL1B (hyper), TCL6 (hyper), TML1 (hyper) |
| iCHD_1 | Male | 22 | DORV, VSD and HAA | 3 | MSX1 (hyper), CST8 (hypo), GZMH (hypo) |
| iCHD_2 | Female | 22 | TOF | 8 | RPL10A (hyper), PIM1 (hyper), C7orf25 (hyper), NPR2 (hyper), KRT71 (hypo), HRK (hyper), LZTR1 (hyper), SERHL (hyper), |
| iCHD_3 | Female | 21 | LHH, MVA, AVA and HAA | 1 | TNR (hypo) |
| iCHD_4 | Female | 22 | DORV, PFO, VSD and CoA | 2 | IGFBP3 (hyper), MTHFS (hyper) |
| iCHD_5 | Female | 22 | TVS, RHH, VSD, MVA and AA | 3 | CHST11 (hypo), MYO1E (hyper), TCL1B (hyper), TCL6 (hyper), TML1 (hyper) |
| iCHD_6 | Female | 22 | TA-type II, ADA, TAV, VSD | 0 | — |
Figure 1.
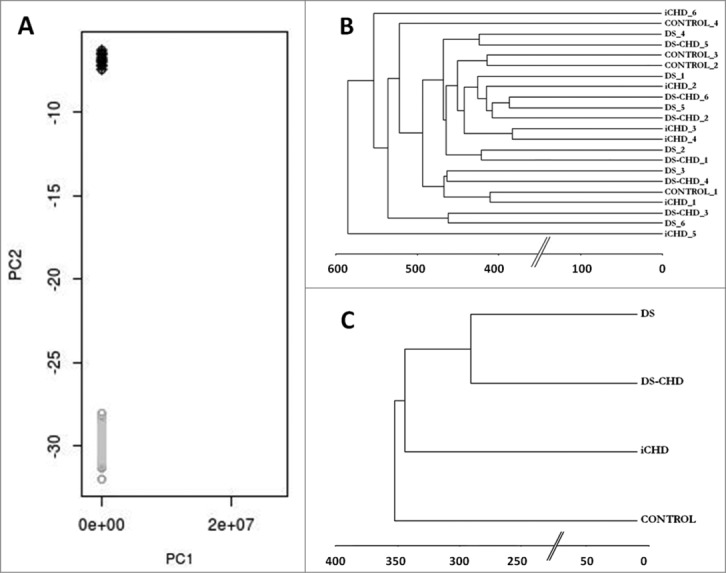
A: Principal Component Analysis revealed remarkably different methylome profiles between blood (in gray) and fetal heart DNA (in black) as shown by the well-defined clustering. B: Manhattan dendrogram with the 22 samples included in the study; samples clustered randomly considering gender and group (X chromosome was excluded). C: Manhattan dendrogram with the 4 different groups used in the study. DS samples (with and without CHD) clustered closer together compared to iCHD samples and fetuses with normal development.
Table 2.
Over-representation analyses in blood and heart tissue. The five top enriched gene ontology-based sets, and the top enriched pathways-based sets are shown, along with the P-value and Q-value for each set. Only gene-sets with a Q-value < 0.05 were considered
| Enriched gene ontology-based sets | P-value | Q-value | Enriched pathways-based sets | P-value | Q-value |
|---|---|---|---|---|---|
| Heart tissue | |||||
| Contractile fiber part | 1.62e–18 | 8.6e–17 | Striated muscle contraction | 9.73e–12 | 5.31e–09 |
| Single-organism cellular process | 1.17e–11 | 9.04e–10 | Muscle contraction | 3.58e–11 | 6.51e–09 |
| Single-multicellular organism process | 2.29e–11 | 9.04e–10 | Hypertrophic cardiomyopathy | 1.23e–06 | 0.000168 |
| Single-organism developmental process | 2.8e–08 | 7.38e–07 | Dilated cardiomyopathy | 2.79e–06 | 0.000305 |
| Anatomical structure development | 2.24e–07 | 4.42e–06 | |||
| Blood | |||||
| Immune response | 3.91e–42 | 3.33e–40 | Immune system | 7.77e–11 | 5.37e–08 |
| Leukocyte activation | 6.46e–28 | 2.75e–26 | TCR | 3.14e–09 | 7.37e–07 |
| Response to stress | 4e–26 | 1.13e–24 | Cell adhesion molecules | 3.2e–09 | 7.37e–07 |
| Cellular response to stimulus | 8.04e–19 | 1.71e–17 | Tuberculosis | 8.95e–08 | 1.27e–05 |
| Plasma membrane | 1.5e–18 | 5.07e–17 | TCR signaling in naïve CD4+ T cells | 9.19e–08 | 1.27e–05 |
Whole genome methylation pattern
Among all the 22 heart tissue DNA samples, a total of 17,877 CpGs (64.8%) were found hypomethylated, 5,370 hemimethylated (19.5%), and the remaining 4,331 hypermethylated (15.7%). A Manhattan dendrogram with all samples showed random clustering regardless of phenotype or gender, after excluding X chromosome signals (Fig. 1B). A Manhattan dendrogram per groups was also obtained. Significant differences in methylation profiles were only present between fetuses with DS and fetuses with a normal karyotype, since the two groups with DS clustered closer than those with normal development and iCHD, even when chromosome 21 data was excluded from the analysis (Fig. 1C). No clustering was detected depending on gestational age or gender. Therefore, mildly differentiated methylation patterns were identified among some of the groups analyzed, although this did not allow individual sample classification.
In order to validate the Methylation array Platform results, we used 3 additional techniques, EpiTYPER, Methylation Specific Multiple Ligation-Dependent Probe Amplification (MS-MLPA) and bisulfite sequencing, targeting a selected subset of 21 differentially methylated (DM) CpGs (DMCpGs) from 18 different regions and using the same DNAs studied by Methylation array Platform. By MS-MLPA, we validated the hypermethylation found in several intragenic CpGs of the MSX1 gene (homolog of Drosophila muscle segment homeobox 1, [MIM 142983]) in a single sample. Using EpiTYPER, 6 alterations were clearly detected and 3 additional DMCpGs gave results with the same tendency, but less strong than the Methylation array Platform results. However, it was not possible to conclude if the methylation pattern was different than in controls at 7 sites, due to either lack of validation or technical problems of EpiTYPER. Four alterations detected in several samples were analyzed by bisulfite sequencing and 2 out of 4 were confirmed. In summary, differences in methylation patterns were clearly validated at 12 of the 21 sites, with dubious results at 7 sites and apparent lack of validation at 2 of the recurrent sites. Given the lack of a consensus gold standard for methylation quantification, we consider that the global validation of the Methylation array Platform results with an additional technique (57–90%) was quite appropriate but far from perfect, indicating that a validation would be especially relevant for each potentially pathogenic aberration.
Differential methylation analyses
DMCpGs in single cases
Each subject with CHD was compared to the control group as a pool, because no clustering was seen depending on gestational age or gender among the samples analyzed. For each DS-CHD subject, a comparison with the group of DS without CHD was also done. Individual findings per sample are summarized in Table 1.
A total of 24 DMCpGs corresponding to 17 regions were found in iCHD cases (70.6% hyper / 29.4% hypomethylated) with a mean of 2.8 DMCpGs per sample (Supplementary Table S1). Most DM regions (70.6%) were located in the transcriptional regulatory region (TRR, ± 500 bp from predicted transcription start sites), 17.6% in the gene body and 11.8% in intergenic regions.
The comparison between individual DS-CHD cases with 2 control groups (normal development and DS without CHD) revealed 22 DMCpGs (68.4% hyper / 31.6% hypomethylated) corresponding to 19 regions (Supplementary Table S2). On average, 3.3 DMCpGs were identified per sample. Regarding location, 79% sites lied in TRR, 15.8% in the gene body, 5.3% in intergenic regions, and 5.3% in both gene body and TRR for the nearby gene.
In case iCHD_1, a fetus presenting with double outlet right ventricle, ventricular septal defect and hypoplasia of the ascending aorta, hypermethylation of a cluster of 8 contiguous CpG sites located in the MSX1 gene body was identified (Fig. 2A). Hypermethylation was then validated by both MS-MLPA and EpiTyper. Sanger sequencing of the region and 1.2 kb flanking interval (chr4:4863673–4864873) did not reveal any genetic alteration associated to the hypermethylation state. By quantitative real-time PCR (qRT-PCR) of heart tissue mRNA, significantly increased expression of MSX1 was observed in iCHD_1 (∼3-fold) with respect to controls (Fig. 2B).
Figure 2.
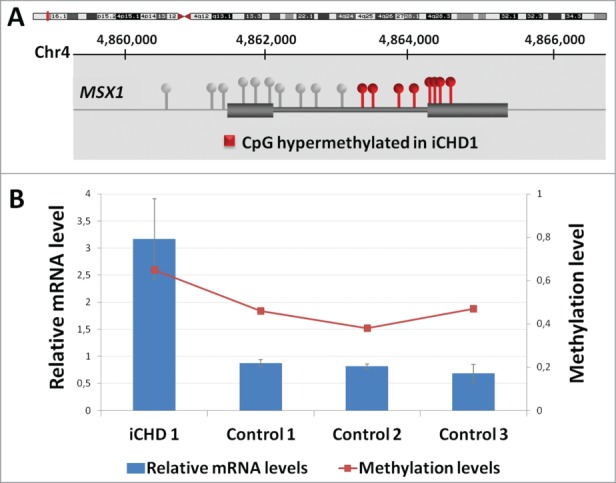
Hypermethylation of 8 CpG sites at a CpG island located within the MSX1 gene in iCHD_1. A: The position of the gene with respect to the chromosomal band 4p16.2 is shown (Hg19), as well as the location of the CpGs with respect to the 2 exons of the gene and the hypermethylated sites (in red). The MSX1 gene has been implicated in cardiac development in animal models. B: Relative MSX1 mRNA levels in heart tissue of the case iCHD_1 and controls by qRT-PCR (scale on the left) along with absolute methylation levels (β values, scale on the right). For mRNA levels, data were normalized such that the level of one control was 1.0 represented by the gray line and the results represent the mean +/− SD of 3 independent experiments conducted in triplicate.
In case iCHD_4, a fetus presenting double outlet right ventricle, patent foramen ovale, ventricular septal defect and coarctation of the preductal aorta, a hypermethylated CpG was detected in the TRR of the MTHFS gene (5,10-methenyltetrahydrofolate synthetase, [MIM 604197]), related to folate metabolism. The flanking region was sequenced by Sanger to discard a genetic alteration. By qRT-PCR of heart tissue mRNA, significantly decreased expression of MTHFS was observed in iCHD_4 with respect to 3 controls. As MTHFS has 2 different transcripts (A: ENST00000258874, protein coding; and B: ENST0000050919, putative nonsense mediated decay), a semi-quantitative analysis was done to determine whether the methylation alteration resulted in isoform-specific alterations, using RIMBP2 [MIM 611602] as a control gene considering its uniform expression in heart. A decreased expression of both A and B MTHFS transcripts was evident in sample iCHD_4, although the reduction was higher for transcript B (Fig. 3).
Figure 3.
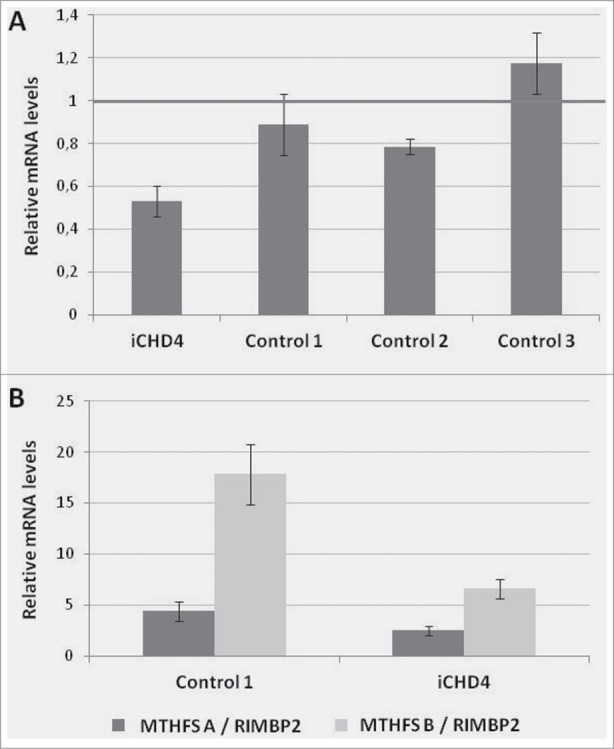
Hypermethylation at one CpG located in the TRR of MTHFS in case iCHD_4. A: Relative MTHFS mRNA in heart tissue of the case and controls by qRT-PCR. Data were normalized such that the level of another control was 1.0 represented by the gray line. The results represent the mean +/− SD of 2 independent experiments conducted in triplicate. B: Relative MTHFS mRNA in heart tissue of the case and one control by semiquantitative analysis using agarose gel. Two transcripts of MTHFS were quantified using RIMBP2 as a control gene. The results represent the mean +/− SD of 3 independent experiments.
Recurrent alterations
A total of 6 DMCpGs corresponding to 6 regions were identified in more than one sample of fetuses with CHD, after excluding those located in CpGs containing relatively common SNPs (>0.5%) that could interfere with the analysis. Two regions were discarded after Sanger sequencing, as the same single nucleotide change was found in both samples, suggesting the existence of a population-specific SNP at that position. The remaining 4 regions were analyzed by bisulfite sequencing and 2 regions were validated, next to the GZMH (Granzyme H, [MIM 116831]) and TCL1B (T-cell lymphoma/leukemia 1B, [MIM 603769]) genes (Supplementary Table S3). In GZMH, an epigenetic alteration and a genetic variant preventing the methylation of the CpG were found at the same location in 2 different samples.
Comparisons between groups
Only a few DMCpGs were identified when comparing the different groups of cases with controls. For some comparisons no DMCpGs were found. A total of 8 DMCpGs were identified between DS (with or without CHD) and controls, corresponding to 8 different regions in chromosomes other than 21: 7 CpGs were located in TRR and 1 in the gene body. In 7 of these 8 loci, case samples showed relative hypermethylation with respect to controls (Supplementary Table S4).
Comparing all cases (DS, DS-CHD and CHD) with controls, 13 DMCpGs corresponding to 8 different regions were identified, being 6 in TRR, 5 in gene bodies and 2 in intergenic regions. At 75% of them, cases presented hypermethylation when compared to controls (Supplementary Table S5).
Hypermethylation of multiple CpGs in the GATA4 (GATA binding protein 4, [MIM 600576]) gene body was seen in the entire group of cases compared to controls (Fig. 4A and B). In order to explore the functional consequences of GATA4 hypermethylation, we quantified GATA4 mRNA levels in heart tissue samples of 5 cases and 2 controls by qRT-PCR. Significantly higher expression of GATA4 transcript was evident in cases, with exception of a single DS sample (Fig. 4C and D).
Figure 4.
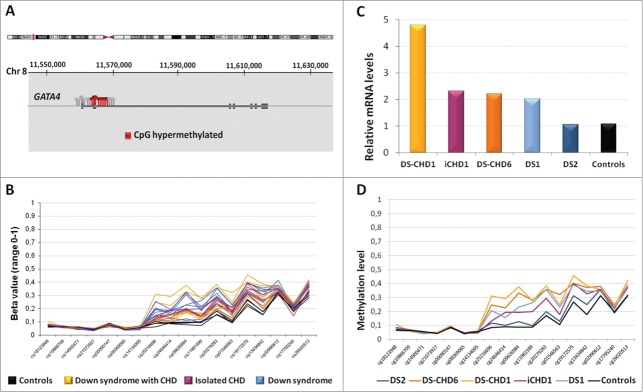
Hypermethylation at several CpGs at GATA4 locus in cases with respect to controls. A: The location of the GATA4 gene with respect to the chromosomal band 8p23.1 is shown (Hg19), as well as the CpGs analyzed. In gray, GATA4 CpGs analyzed in this study; in red, the CpGs significantly altered in patients. B: Beta value of the CpGs in controls (black) with respect to the 3 groups of patient samples (colored lines, one per sample). C: Relative GATA4 mRNA in heart tissue of several patients and controls by qRT-PCR. Data were normalized such that the level of another control was 1.0 represented by the gray line. The results represent the mean ± SD of 2–4 independent experiments conducted in triplicate. D: Methylation levels (in β values) at all the GATA4 CpG sites of the same samples studied by qRT-PCR are shown. Cases with abnormal expression and higher methylation (cases DS_1, DS-CHD_1, DS-CHD_6, iCHD_1) have been grouped in both panels.
Enrichment analyses
Two different overrepresentation analyses (pathways and ontology terms) were done using all genes located next to DMCpGs per group of patients. In the group of iCHD, a significant enrichment of the anatomical structure morphogenesis and postembryonic development ontology terms as well as the insulin-like growth factor regulating pathway was observed. In DS-CHD, the prostaglandin formation and the doxorubicin pathways were found enriched, while the Parkinson disease pathway was enriched in the entire group of DS cases. The gene ontology term or pathway, the genes contained in the set and the P- and q-values are detailed in Table 3.
Table 3.
Over-representation analyses of differentially methylated genes. Enriched pathways, genes included in the pathway, P-value and Q-value (corrected for multiple testing using the false discovery rate method) for each pathway are shown. Only gene-sets with a Q-value lower than 0.05 were considered
| Enriched gene ontology-based sets | Genes | P-value | Q-value |
|---|---|---|---|
| iCHD | |||
| Anatomical structure morphogenesis | CHST11; KRT71; TNR; MYO1E; RPL10A; MSX1; LZTR1 | 0.0010 | 0.022 |
| Cell death | CHST11; PIM1; GZMH; IGFBP3; HRK; MSX1 | 0.0022 | 0.022 |
| Anatomical structure development | CHST11; NPR2; KRT71; TNR; MYO1E; IGFBP3; RPL10A; MSX1; LZTR1 | 0.0023 | 0.022 |
| Post-embryonic development | CHST11; MYO1E | 0.0028 | 0.022 |
| Cell growth | IGFBP3; TNR; MSX1 | 0.0036 | 0.022 |
| Enriched pathway-based sets |
Genes |
P-value |
Q-value |
| iCHD | |||
| Regulation of Insulin-like Growth Factor (IGF) Transport and Uptake by Insulin-like Growth Factor Binding Proteins (IGFBPs) | IGFBP3, GZMH | 0.0002 | 0.0010 |
| DS-CHD | |||
| Doxorubicin Pathway (Pharmacokinetics and dynamics, Cardiomyocytes) | NOS3; CBR1 | 0.0006 | 0.0040 |
| Anti-Inflammatory metabolism (Prostaglandin, Arachidonic acid, Thromboxanes)* | CBR1; SETD4 | 0.0031 | 0.0093 |
| DS, DS-CHD | |||
| Parkinson disease pathway* | SEPT5; UBA7 | 0.0022 | 0.0022 |
More than one pathway source reported the same enriched pathway.
Discussion
We have explored for the first time the global methylation profile of fetal heart DNA in comparison to blood DNA from control subjects. Two clearly distinguished groups of DM regions and genes were evident with an absolute correlation with the type of tissue, allowing the perfect classification of individual samples. While in blood DNA the most significantly enriched pathways were related to the immune response, pathways related to muscle contraction and cardiomyopathies were identified as DM in the developing heart tissue DNA. These data highlight the relevant methylation differences between tissues and the strengths of analyzing the tissue affected.
We have also addressed the possible contribution of epimutations altering DNA methylation in fetal heart cells to the development of CHD, either idiopathic or associated with trisomy 21. Although no clearly differentiated general patterns of abnormal methylation were identified in any group of CHD, a few remarkable changes were defined mostly in individual cases. In order to minimize false positive signals and other bias secondary to the complexity of Methylation array Platform data analysis,20 we used very stringent filtering and threshold criteria for the final definition of DMCpG sites, along with validation by additional techniques in the relevant cases. Interestingly, most DMCpG sites in CHD cases were hypermethylated (75.4%) with respect to controls, and were located in the TRR (70.2%). We also documented the functional correlate of several methylation aberrations in the same tissue. In all tested cases, intragenic hypermethylation was associated with overexpression of the affected gene, reassuring the presence of the aberrant epigenetic mark and indicating its functional relevance on gene expression, most likely by cis-acting regulation. The association of intronic hypermethylation with overexpression in cis is not that surprising. While promoter hypermethylation is generally related to repressed expression of the downstream gene,21,22 methylation alterations outside promoters may have different regulatory impacts on gene expression.23
When comparing the methylation patterns between groups, the only highly relevant difference was at several sites of the GATA4 gene, hypermethylated in cases with respect to controls. This hypermethylation, present in all fetuses with DS (with or without CHD) and iCHD, was also associated with increased GATA4 expression in the affected tissue. GATA4 encodes a member of the GATA-binding protein family expressed in yolk sac endoderm and embryonic heart that regulates downstream genes critical for myocardial differentiation and function. Deletions and point mutations of GATA4, as well as gene duplications, have been associated with CHD, including septal and conotruncal defects.24-27 Given the consistently abnormal methylation in all DS samples, deregulation of GATA4 methylation is likely a consequence of trisomy 21 that might contribute to the high risk of CHD in DS. However, methylation or expression levels were not different between the groups of DS with and without CHD, implying other still unknown modifiers for the penetrance of this phenotype in DS. In addition, abnormal GATA4 hypermethylation and overexpression were also found in fetuses with iCHD, presumably caused by heterogeneous etiologies. Thus, GATA4 hypermethylation could be a consequence of expression deregulation in cardiac tissue secondary to several underlying causes of CHD. Interestingly, increased rate of cardiac defects and alteration in the methylation pattern of the GATA4 gene promoter in the heart has also been demonstrated in the offspring of vitamin A-deficient weaning rats.28 Our data reinforce the idea that GATA4 may be a dosage-sensitive major player in heart development, as different genetic and genomic mutations as well as primary or secondary epigenetic marks appear to contribute to the risk of CHD.
Some of the detected methylation abnormalities represent clear candidate epimutations for direct involvement in the pathogenesis of the disease. Hypermethylation of a CpG island in the MSX1 gene with associated 3-fold increase in MSX1 expression was documented in a single fetus with double outlet right ventricle, ventricular septal defect and hypoplasia of the ascending aorta. MSX1 encodes a member of the muscle segment homeobox gene family and its expression is associated with epithelio-mesenchymal interactions at many locations during embryogenesis in vertebrates.29,30 Heterozygous mutations in MSX1 have been associated with non-syndromic cleft lip and/or palate [MIM 608874], Witkop-type (tooth and nail) ectodermal dysplasia 3 [MIM 189500], and autosomal dominant hypodontia [MIM 106600], while its haploinsufficiency is thought to contribute to the developmental anomalies of the Wolf-Hirschhorn syndrome [MIM 194190] caused by terminal 4p16 microdeletions.31,32 A relevant role of MSX1 and its paralog MSX2 (Muscle segment homeobox 2, [MIM 123101]) in cardiac development has been demonstrated in mice.33 Both genes regulate outflow tract morphogenesis by protecting secondary heart field precursors against apoptosis and by inhibiting excessive proliferation of cardiac neural crest, endothelial and myocardial cells in the conotruncal cushions.33 Disruption of Msx1 and Msx2 in mice causes outflow tract alignment defects including double-outlet right ventricle, overriding aorta and pulmonary stenosis, being also associated with reduced expression of Hand1 (Heart and neural crest derivatives-expressed 1, [MIM 602406]) and Hand2 (Heart and neural crest derivatives-expressed 2, [MIM 602407]).34 Additional evidence implicating MSX1 in human CHD has been recently reported. A genome-wide association study for atrial septal defects revealed a strong signal on chromosome 4p16, next to the MSX1 gene.35 A 3.8 Mb de novo duplication of the region containing MSX1 was also described in a woman with aortic valve dysplasia as the only phenotype.12 Exome sequencing in individuals with CHD revealed a high de novo mutation load in histone-modifying genes,13 altering proteins such as H1B that cooperate with MSX1 to control transcription and myogenesis.36 Therefore, we propose that the single-gene epimutation of unknown cause identified in case iCHD_1 heart DNA could be causative of the abnormal cardiac development through impaired MSX1-dependent mesenchymal embryogenesis.
In case iCHD_4, an epimutation in the folate metabolic pathway gene MTHFS associated with reduced mRNA levels in heart tissue was detected. This pathway has been previously explored in relation to different malformations including CHD and DS. Maternal folate deficiency and hypomorphic alleles in the folate synthetic pathway37,38 have been reported in association with increased risk in the offspring of both iCHD and trisomy 21.16,18,37,38 The findings on iCHD_4 further reinforce the hypothesis that deregulation of the folate pathway, either by genetic mutations or epigenetic mechanisms, can contribute to CHD.
Remarkably, two DMCpGs were recurrently identified in more than one case of CHD. A hypermethylation at the TCL1B gene was detected in three samples, two DS with CHD and one iCHD. TCL1B enhances the phosphorylation and activation of AKT1 (V-akt murine thymoma viral oncogene homolog 1) and AKT2 (V-akt murine thymoma viral oncogene homolog 2), two proteins involved in heart function and heart development. Disruption of Akt1 in mice causes CHD with a reduction in cell proliferation.39 Mutations in other genes related to cell growth such as Hes1, or apoptosis such as Alk5, have already been associated to CHD in animal models. Hes1 knockout mouse embryos display defects in proliferation at early developmental stages, which induce a reduction in cardiac neural crest cells and failure to completely extend the outflow tract.40 Increased post-migratory neural crest cell apoptosis in mice defective for Alk5 leads to severe outflow tract defects.41 Deregulation of TCL1B expression by epimutation could therefore contribute to CHD by secondary disruption of AKT signaling. The unavailability of additional tissue from these cases with TCL1B epimutations precluded further functional analyses.
Interestingly, an epigenetic alteration and a single nucleotide change that modify the methylation of a CpG were found at the same location in two different samples. In case iCHD_1, hypomethylation with no sequence change at a CpG in the promoter region of GZMH was validated by bisulfite sequencing; in case DS-CHD_1, a homozygous SNP changing the G nucleotide and preventing methylation of CpG was also found (rs66505306). The frequency of this SNP considering data from the 1000 Genomes Project is 6.46%, with estimated homozygosity of 0.42%. The occurrence in two different samples of an epigenetic event and a rare genetic variant altering methylation at the same CpG exemplifies the relevance of both types of alterations in expression regulation and suggests GZMH as another candidate gene to contribute to CHD susceptibility.
Overrepresentation analysis with the genes located next to the DMCpG identified cell growth and cell death pathways significantly enriched in iCHD samples. The regulation of both processes is essential for morphogenesis in development, as the formation of several structures requires a precise control of cell and tissue growth. At early stages in development, cardiomyocytes have a high proliferation rate, which decreases progressively in late gestation.24 As an example, a proliferating-center in the caudal coelomic wall has been proved to be crucial in the elongation of the heart tube at both its venous and arterial pole, providing a morphological mechanism for early heart formation. When this process fails, the formation of the atria and the right ventricle results impaired.42 Therefore, the misregulation of pathways related to cell growth and death at early stages in development could lead to an abnormal heart development.
In summary, our data strongly indicate that some epigenetic alterations are present in the DNA of developing heart tissue of fetuses with CHD, both isolated and syndromic. These methylation aberrations contribute to the etiology and/or pathogenesis of the malformation through deregulation of gene expression, providing evidences of novel genes and pathways relevant for heart development. Given that lineage commitment of stem or progenitor cells is tightly regulated by epigenetic mechanisms further work is granted to better define the role of epigenetic aberrations in CHD and their relationship with genetic sequence and environment.
Materials and Methods
Samples
We selected heart tissue samples from four different groups of fetuses from medically terminated pregnancies: with normal development (n = 4), with DS without CHD (n = 6), with DS and CHD (n = 6) and with CHD not presenting DS (n = 6). In addition to fetal ultrasounds (including echocardiography), a detailed necropsy report was available for all samples included in the study (Table 2) (Supplementary material). To minimize the potential bias introduced by other variables known to influence methylation profiles, case and control samples were matched by gender and gestational age.43,44 As dendrogram showed no clustering depending on gestational age and gender in our cohort, all samples were pooled together. Matching for the type of CHD between DS and no-DS samples was not possible. All samples of fetuses with CHD also underwent chromosomal microarray analysis to discard copy number changes potentially related to the CHD.10
Whole heart tissue was used in all cases for nucleic acid isolation (Supplementary material).
Methylation array
All 22 samples were studied by using the Illumina Infinium HumanMethylation27 array Platform, which covers 27,578 cytosine positions located in genes, intergenic regions and miRNA. Samples from different groups were randomly distributed in the chip to avoid position bias. Biological and technical replicas were also included in order to determine the quality of the experiment (Supplementary material).
Bioinformatics and statistical analyses
Two different types of data analysis were performed in order to detect methylation alterations between groups and also in individual samples, with and without previous data normalization (Supplementary material). The results of both analyses were similar.
Developing a heart methylation profile
In order to explore the methylation profile in developing heart, methylation data from fetal heart tissue DNA was compared to data obtained from blood DNA. The entire dataset of 22 fetuses was used to establish the methylation profile in developing heart in comparison with a data set from 656 blood DNA samples from adult control subjects (age range, 19–101 y old). The control dataset was generated with the Illumina Infinium HumanMethylation450 BeadChip Kit.19 Only CpGs present in both platforms were identified and used for the comparative analyses (Supplementary material).
Differential methylation analyses
DMCpGs in single cases
Considering the probable heterogeneity of causes underlying CHD, we compared the methylomic data of individual samples with respect to controls. Each sample of iCHD was compared with the control group (n = 4); each sample of DS with CHD was compared with the control group and with the group of DS without CHD. A CpG was considered to be DM when the β value difference was greater than ±0.25 in not normalized data. In the analyses with normalized data, DM was considered significant if the log fold change was greater than ±1.8 and with an adjusted P-value <0.05. All CpGs that could be altered by described SNPs (dbSNP138) were discarded.
Recurrent alterations
A recurrent alteration was considered when two samples with CHD harbored the same aberration (hyper- or hypo-methylation) at the same CpG. Recurrent CpGs were selected if at least one sample presented a log fold change grater that ±1.8 (M-value) and an additional sample presented the same type of aberration even with a log fold change lower than ±1.8 (at least higher than 0.9).
For recurrent abnormalities, the presence of a SNP at the CpG was also explored. CpGs that could be altered by SNPs with population frequency higher than 0.5% were discarded. For recurrent alterations in positions with low frequency SNPs or without reported SNPs, Sanger sequencing was used in order to find or exclude genetic changes.
Comparisons between groups
Comparisons between groups were also done with the Genome Studio and R packages considering different group combinations (patients vs. controls, all patients with CHD vs. controls, all patients with DS vs. controls, patients with DS and CHD vs. patients with DS without CHD, patients with iCHD vs. controls, patients with iCHD vs. patients with DS-CHD). Since no DMCpG between groups were found using Genome Studio, comparisons were performed using normalized data. Only DMCpGs with a P-value lower than 0.05 and a log Fold change greater than ±0.7 were considered.
Validations
To validate the alterations detected by the Illumina Infinium HumanMethylation27 array Platform, three different techniques were used: Methylation Specific Multiple Ligation-Dependent Probe Amplification (MS-MLPA), EpiTYPER analysis and bisulfite sequencing (Supplementary material). We decided which technique to use depending on technical requirements. We also explored by Sanger sequencing the presence of genetic alterations as potential causes of methylation aberrations in cases with relevant findings and in recurrent alterations, as it was mentioned above.
Quantification of mRNA
The expression of three genes (GATA4, MSX1 and MTHFS) was evaluated by quantitative real-time PCR (qRT-PCR) (Supplementary material). Amplification of the AGPAT1 transcript was used as mRNA control for relative quantification.
A semiquantitative approach was used for MTHFS in order to detect the relative expression of two different transcripts, using RIMBP2 as a control gene.
Enrichment analyses
The closest genes to the DMCpGs were analyzed using a computational resource, Consensus Path DB,45 to obtain an overview of the pathways or the ontology terms which could be altered, with the aim of identifying overrepresented sets. Genes nearby tissue-specific DMCpGs (either blood or heart) were analyzed to define enriched pathways and ontology-based sets in each tissue. Genes close to DMCpGs identified in individual heart sample analysis (Supplementary material, Tables 1 and 2), as well as the ones detected in the group comparisons (Supplementary material, Tables 4 and 5), were also used for the pathway enrichment analysis. The P-value and Q-value of each enriched set was considered.
Ethics statement
All studies were performed after obtaining written informed consent from the family. The project was approved by the Medical Ethical Committee of the Vall d’Hebron Hospital and performed conform the declaration of Helsinki.
Disclosure of Potential Conflicts of Interest
Luis A. Pérez-Jurado is scientific advisor of qGenomics, a privately held company that provides genomic services to the scientific and medical community.
Acknowledgments
We would like to thank Verena Terrado for technical assistance, Gabriela Palacios for critical comments and suggestions and the Spanish National Genotyping Center (CeGen) where the Methylation array Platform was performed. We also would like to thank all participant families for their consent.
Funding
This work was supported by grants of the Spanish Fondo de Investigación Sanitaria of the Ministry of Economy and Competitiveness and FEDER funds [FIS PI10/2512 and PI13/2812], an intramural project of the CIBERER, the Catalan Government [SGR2009/1274, SGR2014/1468 and ICREA Acadèmia] and a predoctoral fellowship of the Fondo de Investigación Sanitaria [FIS FI08/00365] to CS-J.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1. Heyn H, Esteller M. DNA methylation profiling in the clinic: applications and challenges. Nat Rev Genet 2012; 13:679-92; PMID:22945394; http://dx.doi.org/ 10.1038/nrg3270 [DOI] [PubMed] [Google Scholar]
- 2. Hansen KD, Timp W, Bravo HC, Sabunciyan S, Langmead B, McDonald OG, Wen B, Wu H, Liu Y, Diep D, et al. . Increased methylation variation in epigenetic domains across cancer types. Nat Genet 2011; 43:768-75; PMID:21706001; http://dx.doi.org/ 10.1038/ng.865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Das PM, Singal R. DNA methylation and cancer. J Clin Oncol 2004; 22:4632-42; PMID:15542813; http://dx.doi.org/ 10.1200/JCO.2004.07.151 [DOI] [PubMed] [Google Scholar]
- 4. Fradin D, Le Fur S, Mille C, Naoui N, Groves C, Zelenika D, McCarthy MI, Lathrop M, Bougnères P. Association of the CpG methylation pattern of the proximal insulin gene promoter with type 1 diabetes. PLoS One 2012; 7:e36278; PMID:22567146; http://dx.doi.org/ 10.1371/journal.pone.0036278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Volkmar M, Dedeurwaerder S, Cunha DA, Ndlovu MN, Defrance M, Deplus R, Calonne E, Volkmar U, Igoillo-Esteve M, Naamane N, et al. . DNA methylation profiling identifies epigenetic dysregulation in pancreatic islets from type 2 diabetic patients. EMBO J 2012; 31:1405-26; PMID:22293752; http://dx.doi.org/ 10.1038/emboj.2011.503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rao JS, Keleshian VL, Klein S, Rapoport SI. Epigenetic modifications in frontal cortex from Alzheimer's disease and bipolar disorder patients. Transl Psychiatry 2012; 2:e132; PMID:22760556; http://dx.doi.org/ 10.1038/tp.2012.55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Connor CM, Akbarian S. DNA methylation changes in schizophrenia and bipolar disorder. Epigenetics 2008; 3:55-8; PMID:18398310; http://dx.doi.org/ 10.4161/epi.3.2.5938 [DOI] [PubMed] [Google Scholar]
- 8. Abdolmaleky HM, Yaqubi S, Papageorgis P, Lambert AW, Ozturk S, Sivaraman V, Thiagalingam S. Epigenetic dysregulation of HTR2A in the brain of patients with schizophrenia and bipolar disorder. Schizophr Res 2011; 129:183-90; PMID:21550210; http://dx.doi.org/ 10.1016/j.schres.2011.04.007 [DOI] [PubMed] [Google Scholar]
- 9. Kalter H, Warkany J. Medical progress. Congenital malformations: etiologic factors and their role in prevention (first of two parts). N Engl J Med 1983; 308:424-31; PMID:6337330; http://dx.doi.org/ 10.1056/NEJM198302243080804 [DOI] [PubMed] [Google Scholar]
- 10. Serra-Juhé C, Rodríguez-Santiago B, Cuscó I, Vendrell T, Camats N, Torán N, Pérez-Jurado LA. Contribution of rare copy number variants to isolated human malformations. PLoS One 2012; 7:e45530; PMID:23056206; http://dx.doi.org/ 10.1371/journal.pone.0045530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Soemedi R, Wilson IJ, Bentham J, Darlay R, Töpf A, Zelenika D, Cosgrove C, Setchfield K, Thornborough C, Granados-Riveron J, et al. . Contribution of global rare copy-number variants to the risk of sporadic congenital heart disease. Am J Hum Genet 2012; 91:489-501; PMID:22939634; http://dx.doi.org/ 10.1016/j.ajhg.2012.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hitz MP, Lemieux-Perreault LP, Marshall C, Feroz-Zada Y, Davies R, Yang SW, Lionel AC, D’Amours G, Lemyre E, Cullum R, et al. . Rare copy number variants contribute to congenital left-sided heart disease. PLoS Genet 2012; 8:e1002903; PMID:22969434; http://dx.doi.org/ 10.1371/journal.pgen.1002903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zaidi S, Choi M, Wakimoto H, Ma L, Jiang J, Overton JD, Romano-Adesman A, Bjornson RD, Breitbart RE, Brown KK, et al. . De novo mutations in histone-modifying genes in congenital heart disease. Nature 2013; 498:220-3; PMID:23665959; http://dx.doi.org/ 10.1038/nature12141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Weijerman ME, van Furth AM, van der Mooren MD, van Weissenbruch MM, Rammeloo L, Broers CJM, Gemke RJBJ. Prevalence of congenital heart defects and persistent pulmonary hypertension of the neonate with Down syndrome. Eur J Pediatr 2010; 169:1195-9; PMID:20411274; http://dx.doi.org/ 10.1007/s00431-010-1200-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. De Rubens Figueroa J, del Pozzo Magaña B, Pablos Hach JL, Calderón Jiménez C, Castrejón Urbina R. Heart malformations in children with Down syndrome. Rev Esp Cardiol 2003; 56:894-9; PMID:14519277; http://dx.doi.org/ 10.1016/S0300-8932(03)76978-4 [DOI] [PubMed] [Google Scholar]
- 16. Locke AE, Dooley KJ, Tinker SW, Cheong SY, Feingold E, Allen EG, Freeman SB, Torfs CP, Cua CL, Epstein MP, et al. . Variation in folate pathway genes contributes to risk of congenital heart defects among individuals with Down syndrome. Genet Epidemiol 2010; 34:613-23; PMID:20718043; http://dx.doi.org/ 10.1002/gepi.20518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yin M, Dong L, Zheng J, Zhang H, Liu J, Xu Z. Meta analysis of the association between MTHFR C677T polymorphism and the risk of congenital heart defects. Ann Hum Genet 2012; 76:9-16; PMID:22175539; http://dx.doi.org/ 10.1111/j.1469-1809.2011.00687.x [DOI] [PubMed] [Google Scholar]
- 18. Bean LJH, Allen EG, Tinker SW, Hollis ND, Locke AE, Druschel C, Hobbs CA, Leary O, Romitti PA, Royle MH, et al. . Lack of maternal folic acid supplementation is associated with heart defects in down syndrome: a report from the national Down syndrome project. Birth Defects Res A Clin Mol Teratol 2011; 91:885-93; PMID:21987466; http://dx.doi.org/ 10.1002/bdra.22848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, Klotzle B, Bibikova M, Fan JB, Gao Y, et al. . Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell 2013; 49:359-67; PMID:23177740; http://dx.doi.org/ 10.1016/j.molcel.2012.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Blair JD, Price EM. Illuminating potential technical artifacts of DNA-methylation array probes. Am J Hum Genet 2012; 91:760-2; PMID:23040498; http://dx.doi.org/ 10.1016/j.ajhg.2012.05.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Karpf AR. Epigenomic reactivation screening to identify genes silenced by DNA hypermethylation in human cancer. Curr Opin Mol Ther 2007; 9:231-41; PMID:17608021 [PubMed] [Google Scholar]
- 22. Baylin SB. DNA methylation and gene silencing in cancer. Nat Clin Pract Oncol 2005; 2 1:S4-11; PMID:16341240; http://dx.doi.org/ 10.1038/ncponc0354 [DOI] [PubMed] [Google Scholar]
- 23. Shenker N, Flanagan JM. Intragenic DNA methylation: implications of this epigenetic mechanism for cancer research. Br J Cancer 2012; 106:248-53; PMID:22166804; http://dx.doi.org/ 10.1038/bjc.2011.550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rojas A, Kong SW, Agarwal P, Gilliss B, Pu WT, Black BL. GATA4 is a direct transcriptional activator of cyclin D2 and cdk4 and is required for cardiomyocyte proliferation in anterior heart field-derived myocardium. Mol Cell Biol 2008; 28:5420-31; PMID:18591257; http://dx.doi.org/ 10.1128/MCB.00717-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Garg V, Kathiriya IS, Barnes R, Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS, Hirayama-Yamada K, Joo K, et al. . GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature 2003; 424:443-7; PMID:12845333; http://dx.doi.org/ 10.1038/nature01827 [DOI] [PubMed] [Google Scholar]
- 26. Pehlivan T, Pober BR, Brueckner M, Garrett S, Slaugh R, Van Rheeden R, Wilson DB, Watson MS, Hing AV. GATA4 haploinsufficiency in patients with interstitial deletion of chromosome region 8p23.1 and congenital heart disease. Am J Med Genet 1999; 83(3):201-6; PMID:10096597; http://dx.doi.org/ 10.1002/(SICI)1096-8628(19990319)83:3%3c201::AID-AJMG11%3e3.0.CO;2-V [DOI] [PubMed] [Google Scholar]
- 27. Joyce JM, Fiedler SD, Brawner SJ, Liu H-Y, Zhou X-G, Yu S. Cardiac defects are infrequent findings in individuals with 8p23.1 genomic duplications containing GATA4. Circ Cardiovasc Genet 2011; 4:620-5; PMID:21933911; http://dx.doi.org/ 10.1161/CIRCGENETICS.111.960302 [DOI] [PubMed] [Google Scholar]
- 28. Feng Y, Zhao L-Z, Hong L, Shan C, Shi W, Cai W. Alteration in methylation pattern of GATA-4 promoter region in vitamin a-deficient offspring's heart. J Nutr Biochem 2013; 24:1373-80; PMID:23333085; http://dx.doi.org/ 10.1016/j.jnutbio.2012.11.005 [DOI] [PubMed] [Google Scholar]
- 29. Hill RE, Jones PF, Rees AR, Sime CM, Justice MJ, Copeland NG, Jenkins NA, Graham E, Davidson DR. A new family of mouse homeo box-containing genes: molecular structure, chromosomal location, and developmental expression of hox-7.1. Genes Dev 1989; 3:26-37; PMID:2565278; http://dx.doi.org/ 10.1101/gad.3.1.26 [DOI] [PubMed] [Google Scholar]
- 30. Robert B, Sassoon D, Jacq B, Gehring W, Buckingham M. Hox-7, a mouse homeobox gene with a novel pattern of expression during embryogenesis. EMBO J 1989; 8:91-100; PMID:2565810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wieczorek D, Krause M, Majewski F, Albrecht B, Horn D, Riess O, Gillessen-Kaesbach G. Effect of the size of the deletion and clinical manifestation in wolf-hirschhorn syndrome: analysis of 13 patients with a de novo deletion. Eur J Hum Genet 2000; 8:519-26; PMID:10909852; http://dx.doi.org/ 10.1038/sj.ejhg.5200498 [DOI] [PubMed] [Google Scholar]
- 32. Zollino M, Di Stefano C, Zampino G, Mastroiacovo P, Wright TJ, Sorge G, Selicorni A, Tenconi R, Zappalà A, Battaglia A, et al. . Genotype-phenotype correlations and clinical diagnostic criteria in Wolf-Hirschhorn syndrome. Am J Med Genet 2000; 94:254-61; PMID:10995514; http://dx.doi.org/ 10.1002/1096-8628(20000918)94:3%3c254::AID-AJMG13%3e3.0.CO;2-7 [DOI] [PubMed] [Google Scholar]
- 33. Chen Y-H, Ishii M, Sun J, Sucov HM, Maxson RE. Msx1 and Msx2 regulate survival of secondary heart field precursors and post-migratory proliferation of cardiac neural crest in the outflow tract. Dev Biol 2007; 308:421-37; PMID:17601530; http://dx.doi.org/ 10.1016/j.ydbio.2007.05.037 [DOI] [PubMed] [Google Scholar]
- 34. Chen Y-H, Ishii M, Sucov HM, Maxson RE. Msx1 and Msx2 are required for endothelial-mesenchymal transformation of the atrioventricular cushions and patterning of the atrioventricular myocardium. BMC Dev Biol 2008; 8:75; PMID:18667074; http://dx.doi.org/ 10.1186/1471-213X-8-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cordell HJ, Bentham J, Topf A, Zelenika D, Heath S, Mamasoula C, Cosgrove C, Blue G, Granados-Riveron J, Setchfield K, et al. . Genome-wide association study of multiple congenital heart disease phenotypes identifies a susceptibility locus for atrial septal defect at chromosome 4p16. Nat Genet 2013; 45:822-4; PMID:23708191; http://dx.doi.org/ 10.1038/ng.2637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee H, Habas R, Abate-Shen C. MSX1 cooperates with histone H1b for inhibition of transcription and myogenesis. Science 2004; 304:1675-8; PMID:15192231; http://dx.doi.org/ 10.1126/science.1098096 [DOI] [PubMed] [Google Scholar]
- 37. Junker R, Kotthoff S, Vielhaber H, Halimeh S, Kosch A, Koch HG, Kassenböhmer R, Heineking B, Nowak-Göttl U. Infant methylenetetrahydrofolate reductase 677TT genotype is a risk factor for congenital heart disease. Cardiovasc Res 2001; 51:251-4; PMID:11470464; http://dx.doi.org/ 10.1016/S0008-6363(01)00286-3 [DOI] [PubMed] [Google Scholar]
- 38. Van Beynum IM, Kapusta L, den Heijer M, Vermeulen SHHM, Kouwenberg M, Daniëls O, Blom HJ. Maternal MTHFR 677C>T is a risk factor for congenital heart defects: effect modification by periconceptional folate supplementation. Eur Heart J 2006; 27:981-7; PMID:16524890 [DOI] [PubMed] [Google Scholar]
- 39. Chang Z, Zhang Q, Feng Q, Xu J, Teng T, Luan Q, Shan C, Hu Y, Hemmings BA, Gao X, et al. . Deletion of akt1 causes heart defects and abnormal cardiomyocyte proliferation. Dev Biol 2010; 347:384-91; PMID:20816796; http://dx.doi.org/ 10.1016/j.ydbio.2010.08.033 [DOI] [PubMed] [Google Scholar]
- 40. Rochais F, Dandonneau M, Mesbah K, Jarry T, Mattei M-G, Kelly RG. Hes1 is expressed in the second heart field and is required for outflow tract development. PLoS One 2009; 4:e6267; PMID:19609448; http://dx.doi.org/ 10.1371/journal.pone.0006267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang J, Nagy A, Larsson J, Dudas M, Sucov HM, Kaartinen V. Defective ALK5 signaling in the neural crest leads to increased postmigratory neural crest cell apoptosis and severe outflow tract defects. BMC Dev Biol 2006; 6:51; PMID:17078885; http://dx.doi.org/ 10.1186/1471-213X-6-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Van den Berg G, Abu-Issa R, de Boer BA, Hutson MR, de Boer PAJ, Soufan AT, Ruijter JM, Kirby ML, van den Hoff MJB, Moorman AFM. A caudal proliferating growth center contributes to both poles of the forming heart tube. Circ Res 2009; 104:179-88; PMID:19059840; http://dx.doi.org/ 10.1161/CIRCRESAHA.108.185843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. El-Maarri O, Becker T, Junen J, Manzoor SS, Diaz-Lacava A, Schwaab R, Wienker T, Oldenburg J. Gender specific differences in levels of DNA methylation at selected loci from human total blood: a tendency toward higher methylation levels in males. Hum Genet 2007; 122:505-14; PMID:17851693; http://dx.doi.org/ 10.1007/s00439-007-0430-3 [DOI] [PubMed] [Google Scholar]
- 44. Christensen BC, Houseman EA, Marsit CJ, Zheng S, Wrensch MR, Wiemels JL, Nelson HH, Karagas MR, Padbury JF, Bueno R, et al. . Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet 2009; 5:e1000602; PMID:19680444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kamburov A, Wierling C, Lehrach H, Herwig R. ConsensusPathDB–a database for integrating human functional interaction networks. Nucleic Acids Res 2009; 37:D623-8; PMID:18940869; http://dx.doi.org/ 10.1093/nar/gkn698 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.