

Abstract
Aberrant regulation of microRNA expression in pancreatic cancers has been shown to play an important role in its inherent poor prognosis and malignant potential. MicroRNAs have also been shown to inhibit translation of genes by targeting the 3′-untranslated region (3-UTR) of mRNAs resulting in the inhibition of translation and often destruction of the mRNA. In the present study we investigated the role of the microRNA miR-202 in the apoptotic pathways of pancreatic cancer cells. The adamantyl-related molecule, 3-Cl-AHPC down-regulated expression of miR-202 and miR-578 resulting in the increased expression of mRNA and protein expression of their target genes, Max dimerization protein 1 (Mxd1/Mad1) and the Sin3A associated protein 18 (SAP18). Overexpression of pre-miR-202 led to diminished levels of Mxd1 and blocked the 3-Cl-AHPC-mediated increase in Mxd1 mRNA expression. The addition of the microRNA inhibitor 2′-O-methylated miR-202 enhanced the 3-Cl-AHPC-mediated increase of Mxd1 mRNA levels as well as 3-CI-AHPC-mediated apoptosis. We found increased Mxd1 bound to the Sin3A repressor protein complex through its increased binding with HDAC-2 and subsequently enhanced transcriptional repression in cells as evidenced by increased HDAC activity. Mxd1 also repressed human telomerase reverse transcriptase (hTERT) mRNA expression through its increased binding to the hTERT promoter site and resulted in decreased telomerase activity in cells. Our results demonstrate that down regulation of miR-202 increased the expression of its target Mxd1, followed by Mxd1 recruitment to the Sin3A repressor complex and through its dimerization with Max, and increased repression of Myc-Max target proteins.
Keywords: c-Myc, hTERT, miR-202, Mxd1/Mad1, miR-578, pancreatic cancer, Sin3A, SAP18
Abbreviations
- 3-Cl-AHPC
(E)-4-[3-(1-adamantyl)-4-hydroxyphenyl]-3-chlorocinnamic acid
- ARR
Adamantly-substituted retinoid related
- Mxd1
Max dimerization protein 1
- hTERT
human telomerase reverse transcriptase
- SAP18
Sin3A associated protein 18
- HDCA2
histone deacetylase 2
Introduction
Regulation of gene expression has been found to occur at multiple levels. In 1993, a novel class of RNAs called microRNAs (miRNAs) were found and discovered to be potent post translational regulators of gene expression in animals, plants and DNA viruses.1 MiRNAs are non-coding sequences of 18 to24 nucleotides and genetic variants in miRNA target sites are identified to be related with human diseases.2 A number of studies have indicated that miRNAs regulate mRNA translation at the post transcriptional level through their binding to the 3′-untranslated region (3′-UTR) of mRNAs. More importantly, dysregulation of miRNA expression has been found to play an important role in the inherent processes of cancer cell migration and tumor metastases.
Human miRNA genes have been frequently located at fragile sites as well as at loss of heterozygosity or common break point genomic regions linked to cancer.3 Other studies have demonstrated that global reduction in miRNA processing promotes carcinogenesis.4-7 Previous investigations have shown that aberrant expression of miR-202 was linked with diverse type of cancer such as breast6, cervical8, colorectal9, gastric tumor,10,11 follicular lymphoma;12,13 multiple myeloma.14 Depending of the cancer type, miR-202 was found to play differential roles of miR-202 as either a tumor suppressor or regulator of oncogenes.
Pancreatic cancer, the fourth leading cause of cancer of cancer-related deaths has a dismal prognosis with the rate of incidence equaling the rate of death and a 5 y survival level of 3% to 6%. It has been previously demonstrated that the miRNA profiles found in pancreatic carcinoma tissues differ significantly from those found in normal pancreatic tissue and in pancreatitis.15 It has been hypothesized that modulating the enhanced or diminished expression of specific miRNAs may be a powerful approach in the therapy of a number of malignancies including pancreatic cancer.16 A number of approaches to modulate miRNA expression have been devised.17
Adamantly-substituted retinoid related (ARR) molecules have been found to induce apoptosis in a variety of malignant cells both in vitro and in vivo.18 A number of mechanisms has been proposed by which apoptosis is achieved by the ARR class.18 We have previously demonstrated that ARRs are potent inducers of apoptosis of pancreatic cancer cells.19 We recently showed that an ARR regulated IGF-1R through miR-150 and miR-630 in apoptotic pathway in a pancreatic cancer cell line.20 Here, we demonstrate that the ARR mediated down regulation of miR-202 led to the increased expression of its target protein Mxd1 and enhanced binding of Mxd1 with the Sin3A repressor complex which resulted in the repression of c-Myc interacting proteins, and subsequently the apoptosis of pancreatic cancer cells.
Results
3-Cl-AHPC mediated down-regulation of miR-202 and miR-578
Exposure of the PANC-1 cells to 1 μM 3-Cl-AHPC resulted in the downregulation of miR-202 and miR-578 as determined by a microRNA array (Fig. 1A). Further documentation of 3-Cl-AHPC modulation of miR-202 and miR-578 was obtained by real-time-PCR using the TaqMan probe. This assay demonstrated a statistically significant decrease in the expression of both miRNAs in treated PANC-1 cells after 24 h (Fig. 1B). We also determined whether 3-Cl-AHPC modulation of expression of these microRNAs resulted in up-regulation of genes targeted by these miRNAs; which were predicted to be Mxd1 and SAP18 using the Target Scan Human 5.0 data base (Fig. 1C). 3-Cl-AHPC exposure increased Mxd1 and SAP18 mRNA levels (Fig. 1D and 1E, respectively) and protein level (Fig. 2A) in PANC-1 and MiaPaCa-2 cells within 24 h. miR-578 decreased the SAP18-3′-UTR activity in PANC-1 cells suggesting that SAP18 is target protein of miR-578 (Fig. 2B).
Figure 1.

3-Cl-AHPC mediated expression of miR-202 and miR-578 in pancreatic cancer cells. (A) miR-202 and miR-578 were down regulated in presence of 3-Cl-AHPC as demonstrated in microRNA array. (B). Changes in miRNAs levels were validated by quantitative Real time PCR. Cells were treated with 3-Cl-AHPC for 24 h. (C). 3′-UTR binding sites of miR-202 and miR-578 target genes Mxd1 and SAP18, respectively. (D, E). Increased expression of mRNAs of their target genes Mxd1 and SAP18 by SYBR-Green RT-PCR. Methodologies were as described in Materials and Methods. Error bars represent the mean of 3 separate determinations ± standard deviation (SD). *, ** and *** (<0 .05, <0.01 and <0 .001) indicate significantly differences between control and treated samples using the t-Test.
Figure 2.

Overexpression of pre-miR-202 blocked the expression of its target protein Mxd1 mRNA and decreased Mxd1-3′-UTR activity. (A, B) Exposure of PANC-1 cells to 3-Cl-AHPC increased Mxd1 and SAP18 proteins levels as demonstrated by Western blots analysis and miR-578 miRNA mimic decreased SAP18-3′-UTR luciferase activity in cells (C, D). Increased expression of pre-miR-202 expression vector and pre-miR-202 reduced expression of Mxd1 mRNA in pre-miR-202 stably transfected cells. (D) Over-expression of pre-miR-202 blocked 3-Cl-AHPC mediated mRNA expression of Mxd1. (E) miR-202 decreased Mxd1-3′-UTR activity in transiently transfected cells. Error bars represent the mean of 3 separate determinations ± the standard deviation (SD). *, ** and *** (<0 .05, <0 .01 and <0 .001) were significantly differences between control and treated samples and •• was significantly different between 3-Cl-AHPC treated vector and pre-miR-202 expressing cells using the t-Test.
Over-expression of miR-202 blocked Mxd1 expression and decreased Mxd1-3′-UTR activity
In order to further demonstrate miR-202 modulation of Mxd1 in the PANC-1 cells, pre-miR-202 was stably over-expressed using a pre-miR-202 expression vector (pre-miR-202) in PANC-1 cells resulting in a 36-fold increase in pre-miR-202 expression (Fig. 2C); pre-miR-202 overexpression resulted in inhibition of Mxd1 mRNA levels (Fig. 2D) and blocked the 3-Cl-AHPC mediated increased expression of Mxd1 mRNA levels (Fig. 2D). miR-202 decreased the miR-202 binding sites containing Mxd1-3′-UTR activity in PANC-1 cells (Fig. 2E). These results supported the identification of Mxd1 as a miR-202 target protein.
Incubation of the PANC-1 cells with the inhibitor 2′-O-meyhylated miR-202 antisense (OME-miR-202) blocked miR-202 over expression (Fig. 3A). The addition of miR-202 inhibitor itself or 3-Cl-AHPC resulted in enhanced Mxd1 mRNA expression in cells (Fig. 3B). While, increased expression of pre-miR-202 by itself or in the presence of 3-Cl-AHPC significantly blocked apoptosis in the PANC-1 cells, indicating the inhibitory role of miR-202 to apoptosis induction (Fig. 3C).
Figure 3.
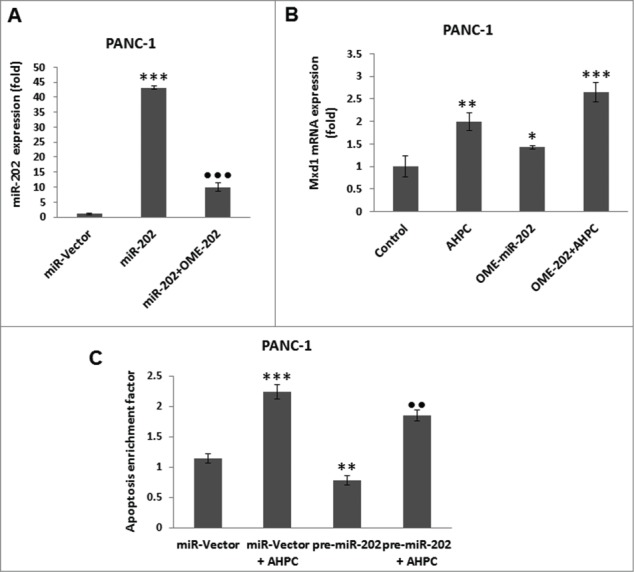
Inhibitor of miR-202 (OME-202) blocked the miR-202 mediated decrease of Mxd1 and pre-miR-202 overexpression induced apoptosis in PANC-1 cells. (A) Inhibitor 2′-O-methylated miR-202 (OME-202) inhibited miR-202 expression in transiently transfected cells. (B) OME-miR-202 alone and in the presence of 3-Cl-AHPC increased Mxd1 mRNA levels. Cells were transiently transfected with OME-miR-202 and then treated with 1 μM 3-Cl-AHPC for 24 h. (C) pre-miR-202 expression blocked the apoptosis in cells. The apoptosis induction was measured by cytoplasmic histone-associated-DNA-fragments using ELISA (enrichment factor = OD of PDCD5 expressed lysate / OD of vector lysate, OD at 405 nm-490 nm). Error bars represent the mean of 3 separate determinations ± SD. All pre-miR -202 expressed samples are significantly different from miR-vector. **, ***, and ••• (<0 .01 and <0 .001, respectively) were significantly different between control vehicle, ss-vector and ••• represented comparison between mi-202 to miR-202 + OME-202 treated samples.
Mxd1 induced repression by recruiting the Sin3A complex in response to 3-Cl-AHPC
Mxd1/Mad1 is a member of the Myc/Max/Mad network of transcriptional regulators and acts as a repressor via tethering the co-repressor complexes possessing histone deacetylase activity to Myc/Max/Mad DNA consensus sequences.21-24 Mad1 proteins are known to antagonize the function of Myc by binding to and repressing the same E-box target DNA sequences.24-27 We hypothesized that Mxd1 might recruit the Sin3A repressor complex to repress the function of Myc target genes in the presence of 3-Cl-AHPC. Immunoprecipitation studies of Mxd1 using PANC-1 cell lysates revealed its increase binding to Sin3A whereas 3-Cl-AHPC exposure decreased the level of Sin3A protein at 48 h in Western blot (Fig. 4A). Exposure of cells to 3-Cl-AHPC increased histone deacetylase 2 (HDAC2) binding with Mxd1 and also enhanced HDCA2 protein expression in cells (Fig. 4A). We also found that 3-Cl-AHPC exposure increased HDAC activity at 48 h in PANC-1 cells, (Fig. 4B). Increased HDAC binding to the Sin3A complex is secondary to increased HDAC protein in the presence of 3-Cl-AHPC.
Figure 4.

Mxd1 recruited Sin3A repressor complexes in the presence of 3-Cl-AHPC and heterodimerized with Max protein. (A) 3-Cl-AHPC increased Mxd1 protein binding with Sin3A and HDAC2 as demonstrated by co-immunoprecipitation. (B) 3-Cl-AHPC enhanced HDAC activity in cells. (C) Mxd1 increased its binding with Max protein whereas that of c-Myc with Max decreased in nuclear extracts of 3-Cl-AHPC treated cells. (D) 3-Cl-AHPC increased Mxd1 protein binding to the hTERT promoter whereas that of c-Myc to the hTERT promoter was concomitantly decreased as demonstrated by CHIP assay. (E) Mxd1 protein binding decreased in c-Myc promoter. Error bars represent the mean of 3 separate determinations ± SD. *, ** and *** (<0 .05, < 0.01 and <0 .001, respectively) were significantly between control and treated cells using the t-Test
Max is an essential dimerization partner of Myc protein to activate transcriptional binding to E-box region in target gene promoters. When max heterodimerize with Mxd1, the Max-Mxd1 complex antagonize Myc-dependent cell transformation through transcriptional repression.27,28 In order to understand the role of Mxd1, we performed co-immunoprecipitation studies. We found that on treatment of PANC-1 cells with to 3-Cl-AHPC Max protein had increased binding with Mxd1 with time, whereas binding between Max and c-Myc decreased (Fig. 4C).
Xu D et al.29 reported that during the differentiation of HL-60 leukemia cells the human telomerase reverse transcriptase (hTERT) promoter was switched to the off position by its decreased binding of the c-Myc/Max dimer and increased binding of the Mxd1/Max dimer and the associated decrease in histone deacetylation. Following 3-Cl-AHPC exposure of PANC-1 cells, we found enhanced binding of Mxd1 to the hTERT promoter with concomitantly decreased binding of cMyc to the hTERT promoter in cells using chromatin mmunoprecipitation assays (Fig. 4D). Moreover, hTERT mRNA and protein expression were reduced in PANC-1 and MiaPaCa-2 cells exposed to 3-Cl-AHPC (Fig. 5A and B). Telomerase activity was also decreased in both cells (Fig. 5C and D) after a treatment with 3-Cl-AHPC or its analog AHPN. We demonstrated previously that the effects of AHPN on cellular growth inhibition and the induction of apoptosis were identical to those of 3-Cl-AHPC.30,31 These results suggest that increased Mxd1-Max binding to the hTERT promoter repressed hTERT expression and activity in both cell lines.
Figure 5.
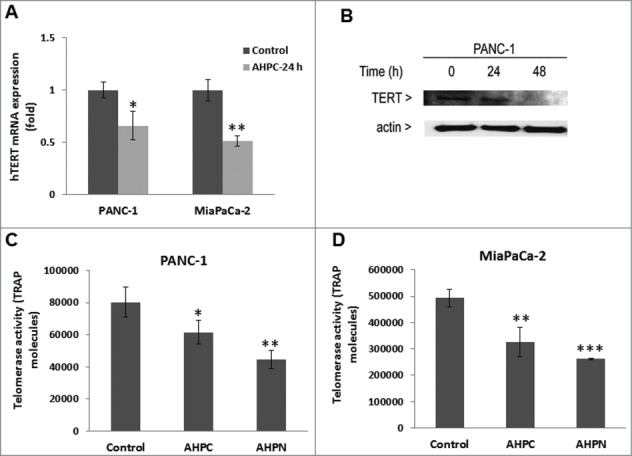
3-Cl-AHPC decreased hTERT activity in pancreatic cancer cells. (A, B) 3-Cl-AHPC decreased hTERT mRNA and protein expression. (C, D) 3-Cl-AHPC decreased telomerase activities in cells. Error bars represent the mean of 3 separate determinations ± SD. *, **, and *** (<0 .05, <0 .01 and <0 .001) were significantly different in the treated samples compared to control using the t-Test.
In order to determine the role of hTERT in apoptosis, knockdown of hTERT was accomplished by transiently transfecting sh-TERT-KD into PANC-1 cells (Fig. 6A and C). The expression of sh-RNA-TERT reduced hTERT mRNA levels (Fig. 6B) and increased apoptosis compared to the vector control transfected cells as indicated by DNA fragmentation of sh-TERT-2 transfected cells (Fig. 6A) supporting the anti-apoptotic role of hTERT in pancreatic cancer cells. We found that over-expression of hTERT inhibited 3-Cl-AHPC mediated apoptosis marginally in PANC-1 cells (Fig. 6D). Increased expression of hTERT mRNA levels was found in hTERT over–expressing cells (Fig. 6E). Inhibition of apoptosis by overexpression of hTERT and apoptosis induction by sh-TERT expressing cells occurred to a similar extent (Fig. 6C and D). This result suggested that hTERT has a minor role in modulating apoptosis in cells. Inhibition of apoptosis by overexpression of hTERT and apoptosis induction by sh-TERT expression while having a dramatic effect on hTERT mRNA levels, had a statistically significant but minimal effect on apoptosis induction. This result suggests that the presence of other agents involved in apoptosis induction.
Figure 6.

hTERT depletion is essential for induction of apoptosis in PANC-1 cells. (A) Knockdown of TERT led to nuclear fragmentation at 72 h of transfection in PANC-1 cells as determined by Phase Contrast Microscopy. (B) hTERT mRNA expression decreased in stably transfected sh-TERT knockdown cells. (C) sh-RNA-TERT knockdown cells induced apoptosis in transiently transfected cells. (D) Overexpression of hTERT blocked 3-Cl-AHPC mediated apoptosis. The apoptosis induction was measured by flow cytometry using Annexin V-FITC binding together with propidium iodide (PI) staining. Percentage of apoptotic cells corresponds to lower right (early apoptotic cells, annexin V positive, PI-negative) quadrants (right panel). (E) Increased hTERT mRNA expression after 72 h in over-expressing hTERT cells. Error bars represent the mean of 3 separate determinations ± SD. * and *** (<0 .05, and <0 .001) indicate significantly differences between vector and sh-TERT knockdown cells and ♦♦ (<0 .01) indicate significantly difference apoptosis between 3-Cl-AHPC treated and hTERT overexpressing cells using the t-Test.
Discussion
Exposure of PANC-1 pancreatic carcinoma cells to 3-Cl-AHPC resulted in the deceased expression of miR-202 followed by the increased expression of Mxd1 and the Sin3A scaffold protein SAP18 mRNA and protein. Transfection of the PANC1 cells with a miR-202 expression vector resulted in the elevation of miR-202 levels and the significant decrease in Mxd1 levels, thereby demonstrating that the elevation of Mxd1 levels following 3-Cl-AHPC exposure was directly due to the decrease in miR-202 levels. Modulation of miR-202 levels has been demonstrated in a number of malignancies. Interestingly, several of these studies have suggested that miR-202 can function as a tumor suppressor. Zhang et al.32 found that miR-202 expression is downregulated in human hepatocellular carcinoma cells while its overexpression suppressed their proliferation and tumorigenicity. Similar observations were made by Zhao et al.,11 who found decreased expression of miR-202-3p expression in gastric carcinoma cells. Increased miR-202-3p levels in these cells inhibited cell proliferation through its targeting of the transcription factor Gli1. Recently, Bisio et al.33 demonstrated that miR-202 is a p53 target protein and that the induction of p53 in cells resulted in the enhanced p53 binding to the p53 responsive element in the miR-202 promoter. Such reports led to the conclusion that the function of miR-202 as either a tumor suppressor or oncogene depending on the context in which its levels are modulated.
We found that exposure of pancreatic cancer cells to the ARR 3-Cl-AHPC resulted in the down-regulation of miR-202, increased levels of Mxd1 and the induction of apoptosis. Elevated Mxd1 levels have been found to inhibit Myc signaling. 22,34 Salehi-Tabar et al.35 demonstrated relatively modest and opposite effects on Myc and Mxd1 levels in the presence of c-Myc/Max and Mxd1/Max heterodimers bound to DNA. Binding to c-Myc target gene promoters by Mxd1/Max, resulted in repression of c-Myc target gene transcription. Since Myc-induction of malignant transformation has been shown to involve the activation of a number of pathways, inhibition of Myc-mediated steps by enhanced levels of Mxd1 could result in the inhibition of these processes.
Deregulation of Myc has been identified as the primary instigator of malignant transformation in several studies.36 Enhanced expression of Myc has been shown to occur through a number of different pathways including translocation and amplification of Myc genes.28 Myc is commonly altered through balanced translocation in Burkitts lymphoma37 and is frequently translocated in multiple myeloma cells.37,38 Myc is one of the most highly amplified oncogenes occurring in different human cancers.39 Numerous studies have demonstrated the potential of deregulated Myc to function as a major murine oncogene during tumor formation in the presence of other genetic changes.40,41 Further studies have shown that deregulated Myc expression may not only play a major role in tumor initiation but also contribute to tumor maintenance.42
Myc dimerizes with Max to bind DNA Myc response elements, enhance transcription and thus mediate many of its functions.43 The members of the Mxd1 family (Mxd1, MxI1, Mxd3 and Mxd4) induce cellular differentiation, inhibit cell proliferation and Myc-induced transformation and suppress Myc-mediated development of cancer.28 Therefore, members of the Mxd1 family may function as tumor suppressors and their loss resulted in increased susceptibility to tumorigenesis.28 Specific inactivation of Myc has been shown to reverse malignant properties of various tumors.28 In the present study, we have demonstrated that 3-Cl-AHPC through its downregulation of miR-202 led to increased mRNA and protein expression of Mxd1. Increased Mxd1 levels resulted in enhanced binding of Mxd1 to Max accompanied by decreased binding of Max to Myc. In addition, increased binding of Mxd1 to the transcriptional repressor complex Sin3A and associated increase in histone deacetylase activity and repression of hTERT mRNA expression occurred in pancreatic cancer cells. These changes were accompanied by increased binding of Mxd1 to the hTERT promoter site and resulted in decreased telomerase activity in the cells. Thus, we have demonstrated the decreased TERT activity can contribute to apoptosis induction.
The Sin3A/HDAC co-repressor complex is a multi-protein complex that regulates gene transcription through its modification of chromatin architecture resulting in compaction of chromatin structure and inhibition of the transcriptional machinery. Sin3A is capable of interacting with a number of transcriptional factors through its four paired amphipathic helices (PAH 1–4), through its histone deacetylase interaction domain (HID) it interacts with HDAC leading to chromatin deacetylation. Recent data suggests the PAH1 and 2 predominately interact with various transcription factors while PAH3, HID and PAH4 interact with other subunits of the co-repressor complex.44 Other studies have shown that the Sin3 demonstrates similarity and interacts with helix-loop-helix protein of the Myc (Mad-Max1) family transcription factors.21,23 Exposure of the PANC-1 cells to 3-Cl-AHPC resulted in the enhanced expression of the Sin3A associated protein SAP18 as well as Mxd1. This result was followed by the increased binding of Mxd1 to Sin3A and HDAC2 which is component of the Sin3A protein complex. The enhanced binding of Mxd1 to Sin3A resulted in a significant increase in HDAC activity as well as increased binding of Mxd1 and decreased Myc binding to the hTERT promoter. This switch in binding (increased Mxd1 and decreased Myc binding) was associated with a decrease in hTERT mRNA expression in the PANC-1 and MiaPACa-2 cell lines as well as decreased hTERT protein levels in PANC-1 cells. Furthermore, decreased telomerase activity was found in the PANC-1 and MiaPaCa-2 cells. We have previously demonstrated that exposure of PANC-1 and MiaPaCa-2 pancreatic cancer cells to 3-Cl-AHPC resulted in the induction of apoptosis. In order to further demonstrate a causal relationship between decreased hTERT levels and the induction of apoptosis, levels of hTERT were decreased in cells using a knockdown approach. We found that the resulting decreased hTERT levels in the knock-down cells resulted in the induction of apoptosis.
In conclusion, miRNAs have been found to be tissue specific and their deregulation plays an important role in tumorigenesis, tumor maintenance as well as the biologic properties of tumor invasion and tumor metastases. It has been speculated that modulation of miRNA levels in malignancies may prove to be a significant therapeutic approach in the treatment of cancer. In the present study, we demonstrated that the addition of the ARR 3-Cl-AHPC to pancreatic cells resulted in the modulation of miR-202 levels as well as its target mRNA and protein levels resulting in apoptosis and thus demonstrating that molecules that target microRNAs may indeed have therapeutic potential.
Materials and methods
Reagents and Cell culture
(E)-4-[3-(1-adamantyl)-4-hydroxyphenyl]-3-chlorocinnamic acid (3-Cl-AHPC) was synthesized as previously described.45 Human pancreatic carcinoma cell lines, PANC-1 and MiaPaCa-2 were obtained from the American Type Culture Collection (ATCC, Rockville, MD) and were maintained in DMEM-F12 medium containing 10% FBS and 100 μg/ml gentamycin. DMEM-F12 medium, fetal bovine serum (FBS) and lipofectamine 2000 were purchased from Invitrogen (Carlsbad, CA). Antibodies and their sources were as follows: anti-Mxd1 (Mad1), anti-SAP18, anti-Sin3A, anti-c-Myc, anti-Max, and anti-HDAC2 antibodies (Santa Cruz Biotechnology, Santa Cruz, CA); and anti-TERT, anti-β-tubulin and actin antibodies (Oncogene Research Products, Boston, MA). Six-[3-(Adamantyl)-4-hydroxyphenyl]-2-naphthalene carboxylic acid (CD437)/AHPN and puromycin were purchased from Sigma-Aldrich (St. Louise, MO).
miRNA and mRNA quantitation
PANC-1 cells were exposed to 1 μM 3-Cl-AHPC for 30 h and then RNA samples preparation, hybridization to G4470A 15K Human miRNA Microarray (V2) (Agilent Technologies, Santa Clara, CA); as previously described.15 Total RNA was prepared from PANC-1 cell lines using TRIzol reagent (Invitrogen) as recommended by the manufacturer and purified using the Rneasy Mini Kit (Qiagen, Valencia, CA). For miRNA validation and mRNA quantitation, total RNA was purified with RNeasy Mini kit (Qiagen). A 100 ng of RNA was used to prepare cDNA using the TaqMan microRNA reverse transcription kit (Applied Biosystems, Grand Island, NY) using specific miRNA primers. The miRNA sequence specific primer and probes for miR-202, miR-578, endogenous control RNU6B and TaqMan 2X PCR Master mix for TaqMan miRNA assays were purchased from Applied Biosystems and used as described by manufacturer's instructions. Real-time qRT-PCR and analysis were performed using the Applied Biosystems 7500 Real Time PCR system. Data analysis was performed with ΔCt values of miRNAs from each sample were calculated by normalizing with internal control RNU6B and each value were mean of 3 replicates.
For real time PCR (RT-PCR), cDNA was prepared using the SuperScript III First-Strand cDNA synthesis system for RT-PCR (Invitrogen) and analyzed in triplicate using the 2 × SYBR Green PCR Master Mix (Applied Biosystem) and the ABI Prism 7500 sequence detection system. PCR consisted of 40 cycles of 95 °C for 10 min followed by 95°C for 15 sec and 60 °C for 60 sec. The oligonucleotide primer sets were synthesized by Integrated DNA technology Inc.. (Coralville, IA).
The primer set for each gene is listed : Mxd1, forward 5′-TGAACATGGTTATGCCTCCA-3′; reverse, 5′-ACTTGATTCGGGTCCAAGTG-3′ SAP18, forward 5′-CCACTGTTGCTACGGGTCTT-3’; reverse, 5′-AAGGTTGCATCCATCCAAGT-3′; hTERT, forward 5′- TCTTCCTACGCTTCATGTGC -3′; reverse 5′- ACAGCTTGTTCTCCATGTCG -3′; β-actin, forward 5′-TCCTTCCTGGGCATGGAG-3′; reverse 5′-AGGAGGGGCAATGATCTT-3′.
miR-202 expression vector and Mxd1 3′-UTR validation
For miR-202 over-expression, miRNASelect pEP-has-miR-202 expression vector was purchased from Cell Biolabs Inc.. (San Diego, CA) and stably transfected into PANC-1 cells using lipofectamine 2000 and the stable cell lines were selected with puromycin. Mxd1-3′-UTR and SAP18-3′-UTR clones containing Mxd1 and SAP18 binding sites were constructed using the pMirTarget vector (OriGene Technology, MD). Cells were transfected with Mxd1-3-UTR vector and Mxd1-3′-UTR Plus miR-202 expression vectors and harvested after 24 h post-transfection. Luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega, Madison, WI) on a BioTeK Synergy HT (BioTeK Instrument Inc., Vermont). SAP18–3′-UTR activity assays were performed as described above and miR-578 miRNA mimic (Sigma-Aldrich) has been used in transfection. For inhibition of miRNAs, O’methylated miR-202 (OME-202) sequence was used as an inhibitor for miR-202; the sequence was synthesized by Integrated DNA Technology Inc.
Apoptosis
Pancreatic cancer cell lines were treated with 1 μM 3-Cl-AHPC throughout all the experiments for indicated times. Apoptosis of cells was analyzed by flow cytometry using Annexin V-FITC binding together with propidium iodide (PI) staining (Annexin V-FITC apoptosis Detection Kit 1, BD Biosciences, San Diego, CA). Data acquisition was done using a FACS Calibur flow cytometer (BD) and analyzed with Cell Quest software (BD Biosciences). Induction of cell death of miR-202 transiently transfected PANC-1 cells was assessed using the Cell Death Detection ELISAplus kit (Roche Applied Science, Indianapolis, IN).
Immunoprecipitation and Western blots
For immunoprecipitation studies, nuclear extract or whole cell lysates (300 or 1000 μg) and 1 μg of each respective antibody were used in each sample. Western blots were performed as we previously described.46
Chromatin immunoprecipitation (ChIP) assay
PANC-1 control cells were treated with 1 μM 3-Cl-AHPC for 24 h and the formaldehyde cross-linking and immunoprecipitation analysis were performed using a described method.46 Chromatin lysate was incubated with 1 μg of anti-Mxd1 and anti-c-Myc antibodies overnight at 4°C and immunoprecipitate complexes were collected with Dynal magntic beads (Invitrogen). The following primers were used in the ChIP assay samples to analyze the hTERT promoter region, forward, 5′-GGCTC-CCAGTGGATTCGC-3′; reverse, 5′-ACCAGCGCGCGGAAAGC-3′.
RT-PCR was carried out as described in the mRNA quantitation section. Data analysis was performed with ΔCt values from each Mxd1 and c-Myc bound control and 3-Cl-AHPC treated sample calculated by normalizing with the input control, and input and treated values were expressed as mean of 3 replicates.
Telomerase activity and shRNA-TERT plasmid and exogenous hTERT expression
Telomerase activities in cells were measured by fluorometric detection and real time quantification of telomerase activity using the TRAPEZE RT Telomerase Detection Kit (Millipore, Billerica, MA) and the procedure in the manufacturer's instructions.
TERT, Human 4 unique 29mer shRNA constructed in the retroviral GFP vector was purchased from OriGene Technology. The scrambled sequence shRNA-vector (ss) was used as a control. shRNA-TERT expression vectors were transiently transfected into PANC-1 cell lines using Fugene (Promega) for 48 h. The scrambled sequence shRNA-vector was used as a control. In assessing apoptosis in over-expressing hTERT cells, we utilized the lenti-hTERT-GFP (Biogenova, Rockville, Mayland). Cells were transduced for 72 h and then 3-Cl-AHPC added. After 24 h 3-Cl-AHPC treatments, cells were harvested for apoptosis assays determined by flow cytometry.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by Veterans Affairs Merit Review Grants (JAF, MID) and NCI grants (JAF, MID).
References
- 1. Lee YS, Dutta A. MicroRNAs in cancer. Annu Rev Pathol 2009; 4:199-227; PMID:18817506; http://dx.doi.org/ 10.1146/annurev.pathol.4.110807.092222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Srinivasan S, Selvan ST, Archunan G, Gulyas B, Padmanabhan P. MicroRNAs -the next generation therapeutic targets in human diseases. Theranostics 2013; 3:930-42; PMID:24396504; http://dx.doi.org/ 10.7150/thno.7026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, Shimizu M, Rattan S, Bullrich F, Negrini M, et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A 2004; 101:2999-3004; PMID:14973191; http://dx.doi.org/ 10.1073/pnas.0307323101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kumar MS, Lu J, Mercer KL, Golub TR, Jacks T. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat Genet 2007; 39:673-7; PMID:17401365; http://dx.doi.org/ 10.1038/ng2003 [DOI] [PubMed] [Google Scholar]
- 5. Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A 2006; 103:2257-61; PMID:16461460; http://dx.doi.org/ 10.1073/pnas.0510565103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, Magri E, Pedriali M, Fabbri M, Campiglio M, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res 2005; 65:7065-70; PMID:16103053; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-1783 [DOI] [PubMed] [Google Scholar]
- 7. Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, et al. MicroRNA expression profiles classify human cancers. Nature 2005; 435:834-8; PMID:15944708; http://dx.doi.org/ 10.1038/nature03702 [DOI] [PubMed] [Google Scholar]
- 8. Zhang Y, Dai Y, Huang Y, Ma L, Yin Y, Tang M, Hu C. Microarray profile of micro-ribonucleic acid in tumor tissue from cervical squamous cell carcinoma without human papillomavirus. J Obstet Gynaecol Res 2009; 35: 842-9; PMID:20149030; http://dx.doi.org/ 10.1111/j.1447-0756.2009.01055.x [DOI] [PubMed] [Google Scholar]
- 9. Ng EK, Chong WW, Jin H, Lam EK, Shin VY, Yu J, Poon TC, Ng SS, Sung JJ. Differential expression of microRNAs in plasma of patients with colorectal cancer: a potential marker for colorectal cancer screening. Gut 2009; 58:1375-81; PMID:19201770; http://dx.doi.org/ 10.1136/gut.2008.167817 [DOI] [PubMed] [Google Scholar]
- 10. Jiang Z, Guo J, Xiao B, Miao Y, Huang R, Li D, Zhang Y. Increased expression of miR-421 in human gastric carcinoma and its clinical association. J Gastroenterol 2010; 45:17-23; PMID:19802518; http://dx.doi.org/ 10.1007/s00535-009-0135-6 [DOI] [PubMed] [Google Scholar]
- 11. Zhao Y, Li C, Wang M, Su L, Qu Y, Li J, Yu B, Yan M, Yu Y, Liu B, et al. Decrease of miR-202-3p expression, a novel tumor suppressor, in gastric cancer. PLoS One 2013; 8:e69756; PMID:23936094; http://dx.doi.org/ 10.1371/journal.pone.0069756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang W, Corrigan-Cummins M, Hudson J, Maric I, Simakova O, Neelapu SS, Kwak LW, Janik JE, Gause B, Jaffe ES, et al. MicroRNA profiling of follicular lymphoma identifies microRNAs related to cell proliferation and tumor response. Haematologica 2012; 97:586-94; PMID:22102710; http://dx.doi.org/ 10.3324/haematol.2011.048132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hoffman AE, Zheng T, Yi C, Leaderer D, Weidhaas J, Slack F, Zhang Y, Paranjape T, Zhu Y. microRNA miR-196a-2 and breast cancer: a genetic and epigenetic association study and functional analysis. Cancer Res 2009; 69: 5970-7; PMID:19567675; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-0236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yu J, Qiu X, Shen X, Shi W, Wu X, Gu G, Zhu B, Ju S. miR-202 expression concentration and its clinical significance in the serum of multiple myeloma patients. Ann Clin Biochem 2013; http://dx.doi.org/ 10.1177/0004563213501155; Epub 2013 Sep [DOI] [PubMed] [Google Scholar]
- 15. Park JY, Helm J, Coppola D, Kim D, Malafa M, Kim SJ. MicroRNAs in pancreatic ductal adenocarcinoma. World J Gastroenterol 2011; 17: 817-27; PMID:21412491; http://dx.doi.org/ 10.3748/wjg.v17.i7.817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell 2009; 136: 215-33; PMID:19167326; http://dx.doi.org/ 10.1016/j.cell.2009.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Costa PM, Pedroso de Lima MC. MicroRNAs as Molecular Targets for Cancer Therapy: On the Modulation of MicroRNA Expression. Pharmaceuticals (Basel) 2013; 6:1195-220; PMID:24275848; http://dx.doi.org/ 10.3390/ph6101195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dawson MI, Fontana JA. The peptidomimetic, 1-adamantyl-substituted, and flex-het classes of retinoid-derived molecules: structure-activity relationships and retinoid receptor-independent anticancer activities. Mini Rev Med Chem 2010; 10:455-91; PMID:20370709; http://dx.doi.org/ 10.2174/138955710791384045 [DOI] [PubMed] [Google Scholar]
- 19. Farhana L, Dawson MI, Das JK, Murshed F, Xia Z, Hadden TJ, Hatfield J, Fontana JA. Adamantyl Retinoid-Related Molecules Induce Apoptosis in Pancreatic Cancer Cells by Inhibiting IGF-1R and Wnt/beta-Catenin Pathways. J Oncol 2012; 2012:796729; PMID:22570653; http://dx.doi.org/ 10.1155/2012/796729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Farhana L, Dawson MI, Murshed F, Das JK, Rishi AK, Fontana JA. Upregulation of miR-150* and miR-630 induces apoptosis in pancreatic cancer cells by targeting IGF-1R. PLoS One 2013; 8:e61015; PMID:23675407; http://dx.doi.org/ 10.1371/journal.pone.0061015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ayer DE, Lawrence QA, Eisenman RN. Mad-Max transcriptional repression is mediated by ternary complex formation with mammalian homologs of yeast repressor Sin3. Cell 1995; 80:767-76; PMID:7889570; http://dx.doi.org/ 10.1016/0092-8674(95)90355-0 [DOI] [PubMed] [Google Scholar]
- 22. Ayer DE, Laherty CD, Lawrence QA, Armstrong AP, Eisenman RN. Mad proteins contain a dominant transcription repression domain. Mol Cell Biol 1996; 16:5772-81; PMID:8816491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schreiber-Agus N, Chin L, Chen K, Torres R, Rao G, Guida P, Skoultchi AI, DePinho RA. An amino-terminal domain of Mxi1 mediates anti-Myc oncogenic activity and interacts with a homolog of the yeast transcriptional repressor SIN3. Cell 1995; 80:777-86; PMID:7889571; http://dx.doi.org/ 10.1016/0092-8674(95)90356-9 [DOI] [PubMed] [Google Scholar]
- 24. Gehring S, Rottmann S, Menkel AR, Mertsching J, Krippner-Heidenreich A, Luscher B. Inhibition of proliferation and apoptosis by the transcriptional repressor Mad1. Repression of Fas-induced caspase-8 activation. J Biol Chem 2000; 275:10413-20; PMID:10744730; http://dx.doi.org/ 10.1074/jbc.275.14.10413 [DOI] [PubMed] [Google Scholar]
- 25. Hurlin PJ, Foley KP, Ayer DE, Eisenman RN, Hanahan D, Arbeit JM. Regulation of Myc and Mad during epidermal differentiation and HPV-associated tumorigenesis. Oncogene 1995; 11:2487-501; PMID:8545105 [PubMed] [Google Scholar]
- 26. Grandori C, Cowley SM, James LP, Eisenman RN. The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu Rev Cell Dev Biol 2000; 16:653-99; PMID:11031250; http://dx.doi.org/ 10.1146/annurev.cellbio.16.1.653 [DOI] [PubMed] [Google Scholar]
- 27. Luscher B. MAD1 and its life as a MYC antagonist: an update. Eur J Cell Biol 2012; 91:506-14; PMID:21917351; http://dx.doi.org/ 10.1016/j.ejcb.2011.07.005 [DOI] [PubMed] [Google Scholar]
- 28. Cascon A, Robledo M. MAX and MYC: a heritable breakup. Cancer Res 2012; 72:3119-24; PMID:22706201; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-3891 [DOI] [PubMed] [Google Scholar]
- 29. Xu D, Popov N, Hou M, Wang Q, Bjorkholm M, Gruber A, Menkel AR, Henriksson M. Switch from Myc/Max to Mad1/Max binding and decrease in histone acetylation at the telomerase reverse transcriptase promoter during differentiation of HL60 cells. Proc Natl Acad Sci U S A 2001; 98:3826-31; PMID:11274400; http://dx.doi.org/ 10.1073/pnas.071043198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang Y, Dawson MI, Ning Y, Polin L, Parchment RE, Corbett T, Mohamed AN, Feng KC, Farhana L, Rishi AK, et al. Induction of apoptosis in retinoid-refractory acute myelogenous leukemia by a novel AHPN analog. Blood 2003; 102:3743-52; PMID:12893763; http://dx.doi.org/ 10.1182/blood-2003-01-0108 [DOI] [PubMed] [Google Scholar]
- 31. Dawson MI, Harris DL, Liu G, Hobbs PD, Lange CW, Jong L, Bruey-Sedano N, James SY, Zhang XK, Peterson VJ, et al. Antagonist analogue of 6-[3′-(1-adamantyl)-4′-hydroxyphenyl]-2-naphthalenecarboxylic acid (AHPN) family of apoptosis inducers that effectively blocks AHPN-induced apoptosis but not cell-cycle arrest. J Med Chem 2004; 47:3518-36; PMID:15214780; http://dx.doi.org/ 10.1021/jm030524k [DOI] [PubMed] [Google Scholar]
- 32. Zhang Y, Zheng D, Xiong Y, Xue C, Chen G, Yan B, Ye Q. miR-202 suppresses cell proliferation in human hepatocellular carcinoma by downregulating LRP6 post-transcriptionally. FEBS Lett 2014; 588:1913-20; PMID:24704686; http://dx.doi.org/ 10.1016/j.febslet.2014.03.030 [DOI] [PubMed] [Google Scholar]
- 33. Bisio A, De Sanctis V, Del Vescovo V, Denti MA, Jegga AG, Inga A, Ciribilli Y. Identification of new p53 target microRNAs by bioinformatics and functional analysis. BMC Cancer 2013; 13:552; PMID:24256616; http://dx.doi.org/ 10.1186/1471-2407-13-552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wu Q, Yang Z, An Y, Hu H, Yin J, Zhang P, Nie Y, Wu K, Shi Y, Fan D. MiR-19a/b modulate the metastasis of gastric cancer cells by targeting the tumour suppressor MXD1. Cell Death Dis 2014; 5:e1144; PMID:24675462; http://dx.doi.org/ 10.1038/cddis.2014.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Salehi-Tabar R, Nguyen-Yamamoto L, Tavera-Mendoza LE, Quail T, Dimitrov V, An BS, Glass L, Goltzman D, White JH. Vitamin D receptor as a master regulator of the c-MYC/MXD1 network. Proc Natl Acad Sci U S A 2012; 109:18827-32; PMID:23112173; http://dx.doi.org/ 10.1073/pnas.1210037109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dang CV. MYC on the path to cancer. Cell 2012; 149:22-35; PMID:22464321; http://dx.doi.org/ 10.1016/j.cell.2012.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dalla-Favera R, Bregni M, Erikson J, Patterson D, Gallo RC, Croce CM. Human c-myc oncogene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc Natl Acad Sci U S A 1982; 79:7824-7; PMID:6961453; http://dx.doi.org/ 10.1073/pnas.79.24.7824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shou Y, Martelli ML, Gabrea A, Qi Y, Brents LA, Roschke A, Dewald G, Kirsch IR, Bergsagel PL, Kuehl WM. Diverse karyotypic abnormalities of the c-myc locus associated with c-myc dysregulation and tumor progression in multiple myeloma. Proc Natl Acad Sci U S A 2000; 97:228-33; PMID:10618400; http://dx.doi.org/ 10.1073/pnas.97.1.228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010; 463:899-905; PMID:20164920; http://dx.doi.org/ 10.1038/nature08822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Akagi K, Suzuki T, Stephens RM, Jenkins NA, Copeland NG. RTCGD: retroviral tagged cancer gene database. Nucleic Acids Res 2004; 32:D523-7; PMID:14681473; http://dx.doi.org/ 10.1093/nar/gkh013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Beer S, Zetterberg A, Ihrie RA, McTaggart RA, Yang Q, Bradon N, Arvanitis C, Attardi LD, Feng S, Ruebner B, et al. Developmental context determines latency of MYC-induced tumorigenesis. PLoS Biol 2004; 2:e332; PMID:15455033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Arvanitis C, Felsher DW. Conditional transgenic models define how MYC initiates and maintains tumorigenesis. Semin Cancer Biol 2006; 16:313-7; PMID:16935001; http://dx.doi.org/ 10.1016/j.semcancer.2006.07.012 [DOI] [PubMed] [Google Scholar]
- 43. Amati B, Brooks MW, Levy N, Littlewood TD, Evan GI, Land H. Oncogenic activity of the c-Myc protein requires dimerization with Max. Cell 1993; 72:233-45; PMID:8425220; http://dx.doi.org/ 10.1016/0092-8674(93)90663-B [DOI] [PubMed] [Google Scholar]
- 44. Grzenda A, Lomberk G, Zhang JS, Urrutia R. Sin3: master scaffold and transcriptional corepressor. Biochim Biophys Acta 2009; 1789:443-50; PMID:19505602; http://dx.doi.org/ 10.1016/j.bbagrm.2009.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fontana JA, Dawson MI, Leid M, Rishi AK, Zhang Y, Hsu CA, Lu JS, Peterson VJ, Jong L, Hobbs P, et al. Identification of a unique binding protein specific for a novel retinoid inducing cellular apoptosis. Int J Cancer 2000; 86:474-9; PMID:10797258; http://dx.doi.org/ 10.1002/(SICI)1097-0215(20000515)86:4%3c474::AID-IJC5%3e3.0.CO;2-Z [DOI] [PubMed] [Google Scholar]
- 46. Farhana L, Dawson MI, Fontana JA. Apoptosis induction by a novel retinoid-related molecule requires nuclear factor-kappaB activation. Cancer Res 2005; 65:4909-17; PMID:15930313; http://dx.doi.org/ 10.1158/0008-5472.CAN-04-4124 [DOI] [PubMed] [Google Scholar]