

Abstract
Aberrant DNA methylation is frequently observed in disease, including many cancer types, yet the underlying mechanisms remain unclear. Because germline and somatic mutations in the genes that are responsible for DNA methylation are infrequent in malignancies, additional mechanisms must be considered. Mycoplasmas spp., including Mycoplasma hyorhinis, efficiently colonize human cells and may serve as a vehicle for delivery of enzymatically active microbial proteins into the intracellular milieu. Here, we performed, for the first time, genome-wide and individual gene mapping of methylation marks generated by the M. hyorhinis CG- and GATC-specific DNA cytosine methyltransferases (MTases) in human cells. Our results demonstrated that, upon expression in human cells, MTases readily translocated to the cell nucleus. In the nucleus, MTases selectively and efficiently methylated the host genome at the DNA sequence sites free from pre-existing endogenous methylation, including those in a variety of cancer-associated genes. We also established that mycoplasma is widespread in colorectal cancers, suggesting that either the infection contributed to malignancy onset or, alternatively, that tumors provide a favorable environment for mycoplasma growth. In the human genome, ∼11% of GATC sites overlap with CGs (e.g., CGATmCG); therefore, the methylated status of these sites can be perpetuated by human DNMT1. Based on these results, we now suggest that the GATC-specific methylation represents a novel type of infection-specific epigenetic mark that originates in human cells with a previous exposure to infection. Overall, our findings unveil an entirely new panorama of interactions between the human microbiome and epigenome with a potential impact in disease etiology.
Keywords: colorectal cancer, DNA methylation, DNA methyltransferases, epigenetics, host-pathogen interactions, mycoplasma
Abbreviations
- 5mC
5-methylcytosine;
- CPGI
CpG island;
- MTase
Mycoplasma hyorhinis DNA cytosine methyltransferase
Introduction
The biology of human disease is no longer focused exclusively on human cells. A variety of microbiomes co-exist in the human body, playing fundamental roles in health and disease.1 The human microbiome contributes to cell metabolism, regulation of signaling pathways, inflammation, and immune responses. Furthermore, bacteria such as mycoplasma colonize and invade human cells, thereby reducing their susceptibility to immune defense and antibiotic treatment.2-5
Mycoplasmas (class Mollicutes) are the smallest self-replicating organisms. There are over 200 species of mycoplasma, distinguished from other bacteria by their minute size, limited genome, and lack of a cell wall. Mycoplasmas depend on host cells for survival and are parasitic microbes. In humans, mycoplasmas frequently populate mucosal surfaces of respiratory and urogenital tracts, mammary glands, and joints, often persisting as long-term asymptomatic infections3 that are likely to promote chronic aberrant states in infected tissues.
Although the role of mycoplasma infections in pathologies, including carcinogenesis, remains a matter of debate,6 evidence of mycoplasma presence in prostate, renal, gastric, colon, esophageal, lung, breast, ovarian, and melanoma tumors suggests the co-existence of mycoplasmas and tumors.7-9 Invasive bacteria, including Mycoplasmas and Mycobacteria, can induce reprogramming of somatic cells10 and oncogenic cell transformation, resulting in dysregulation of cancer-specific genes, including RAS and MYC oncogenes and p53 tumor suppressor.8,11-13 However, the molecular mechanisms that provide evidence on how mycoplasmas can modulate, genetically or epigenetically, host cell pathways remain understudied.
To this end, a common pattern observed in cancers suggests that somatic epigenetic alterations precede pro-oncogenic mutations, and that the abnormal epigenome affects the frequency of occurrence of subsequent genetic alterations that drive tumorigenesis.14-17 Recent genome-wide data also imply that epigenetic anomalies can be a key factor in cancer onset and progression. 18-22
DNA methylation, an essential element in transcriptional regulation, 23 is one of a few major epigenetic mechanisms. DNA methylation causes the conversion of cytosine to 5-methylcytosine (5mC) in the context of CG-dinucleotides. In humans, this conversion is catalyzed by DNA (cytosine-5-)-methyltransferase 1, 3A, and 3B (DNMT1, DNMT3A, and DNMT3B). CG dinucleotides are sparsely distributed in the human genome compared to other dinucleotide combinations. A higher than expected number of CGs is observed within ∼1 kb CpG islands (CPGIs), which are typically associated with the gene promoters.
Aberrant global and gene-specific DNA hypo- and hyper-methylation was reported in multiple cancer types 24-26; however, the molecular mechanisms involved in aberrant hypermethylation onset remain insufficiently understood. 27,28 Surprisingly, germline and somatic mutations in genes that are responsible for DNA methylation are infrequent in malignancies 27 [COSMIC database (http://cancer.sanger.ac.uk)]. As a result, we believe that additional mechanisms that may affect the human cell epigenome must be considered.
Here, we examined whether microbial MTases cause aberrant DNA hypermethylation in human cells. We expressed the Mycoplasma hyorhinis CG- and GATC-specific MTases in human cells and then demonstrated that these enzymes translocated to the cell nucleus, efficiently conferred a high degree of methylation on the human genome and stimulated certain pro-oncogenic and proliferation pathways in human cells. Because M. hyorhinis efficiently colonize human cells, the internalized bacteria may serve as a vehicle for delivery of enzymatically active MTases into the intracellular milieu. We also established that mycoplasma is widespread in colorectal cancers suggesting that tumors provide a favorable environment for mycoplasma growth that may facilitate further dissemination. Overall, our findings provide mechanistic clues as to how bacterial enzymes may affect the epigenetic control of human genes and, as a result, may alter cancer susceptibility in the patients with the persistent mycoplasma infections.
Materials and Methods
Reagents
All reagents were obtained from Fisher Scientific unless otherwise indicated. A murine monoclonal antibody to the V5 epitope and a secondary goat anti-mouse AlexaFluor 594 antibody were obtained from Life Technologies.
Cell culture
Cell culture media were obtained from Life Technologies unless otherwise indicated. Cell cultures were maintained at 37°C and 5% CO2. Human HT1080 fibrosarcoma was grown in DMEM supplemented with 10% fetal bovine serum (FBS). Human transformed first trimester extravillous HTR8/SVNeo trophoblasts29 were grown in RPMI-1640 supplemented with 5% FBS.
Cloning
Human codon optimized and UGA codon corrected M. hyorhinis 1191-bp Mhy1, 1239-bp Mhy2 and 1221-bp Mhy3 cDNAs were synthesized (Genewiz) based on the predicted amino acid sequence of Mhy1 (GenBank AEX13846), Mhy2 (GenBank AEX13880) and Mhy3 (GenBank AEX14156) (Table 1). cDNAs were amplified by PCR using the respective forward and reverse primers (0.2 μM each) (Table 2) and Q5 High-Fidelity DNA Polymerase (New England Biolabs). For protein expression in E. coli and human cells, the respective PCR products were inserted into the pET101/V5-His-TOPO (pET101/Mhy1, pET101/Mhy2, and pET101/Mhy3 constructs) and pEF6/V5-His-TOPO (pEF6/Mhy1, pEF6/Mhy2, and pEF6/Mhy3 constructs) vectors (Life Technologies), respectively. Plasmid DNA was purified using a HiSpeed Plasmid Maxi Column (Qiagen). All constructs were verified by DNA sequencing.
Table 1.
Amino acid sequences of M. hyorhinis MTases
| MTase | GenBank ID | Amino acid sequence |
|---|---|---|
| Mhy1 | AEX13846 | MNKKIKVFEVFAGIGSQFKALKNIEKSLDIEVESLGLIEWYLDAIISYQKIHLYKSKKV NVSIELMLSQLQSLTLSKDSKSPVSKNYFM KQNEEKLNNYYQYLLPFINTITNPNQDKKFYTDINKVQIIPKDIDIFTYSFPCQDLSQQGNQLGIQDNTRSGLLLQVKRILKQNQDRLPKTLLM ENVKSLTNKKFMSQFEDWIKFLETLGYNSSWKVLNSTDFGSSQNRERVFMVSKLNNKPFKWPLKIKHNNDLSRILESNFQPTAQILELTSK IKEKGITEFKTTTNNISKAFIKNWSNFNSENYIYNNKGFGPTLTASGANSRLKFYFKNKDIFRYINAYESFKYMGFSSKDVDKILETNLVSENKI IFMAGNSISVEVLEYIFKNLILEIKEEF |
| Mhy2 | AEX13880 | MKYYKISEIAKLESVSQKFIRKEIAKKSLISTKQNNKCLILDSHYQDWKLSYKKEASSNKKIGKIEEKVQFIDVLKEMNKLDGWNSNYKNGYKFI DLFSGAGGLSCDLVMAGFEPIASVEIMPDAVETYVYNFQNRKKKEELIETRDIRDVKVKEELYNKFKDTDIDLIVGGFPCQGFSMAGNRVV DDPRNSLYLEMLEIVKNLQPKFVLMENVQGLRTMLNGQVEQKIINDYKNIGYQINVTTLNSADYEVAQTRKRVIFIANKINKINYFPKPILQQ KEYKTLGECIEKYMYLEENKEINHIFTKHSKEMQERIKNTPEGQSVYKNYSDGWKKSPWNEPSCTIKENHGGVNIHPKLPRVLTPRELAA LQSFPDDFIFKGSKKWQLVQIGNAVPPLLAKAIGFAIKKKFGQK |
| Mhy3 | AEX14156 | MVEKQLKKIKVVELFAGVGGFRLGFERTSKLFKTIWANQWEPNKTKQWAFDCYTKHFGNSDNHVNEDIANVIDQVPEHDLLVGGFPCQD YSVARTKAEGIKGKKGVLWWSILKIIQKRHPNFILLENVDRLIKSPANQRGRDFGIMLKSLDNEGYNVEWRIIDASDYGFVQRRKRVFIFAY KKELTQIINKEQNQENILIKDGFFASEFLVESQCKKTNKSDILKNYSNLVDVSNNFSFSFYNSGSMINGNILTLDLVAKSTKKPSFLKDVIEKE EVDSKFFINDNYQKFSYLKGAKKIERTKPNGKKYTYSEGTMIFPDDLNKPARTMLTSEGSVNRSTHVIEDYVTKNLRILTPLETERINGFDDN TNTGMPERFRYFCMGNALIVPVIEKIAKQILKIWNKVN |
Table 2.
Oligonucleotide primers for cloning of M. hyorhinis MTases
| PCR product | Primers pair (5'-3' sequence) |
|---|---|
| Mhy1 | CACCATGAATAAGAAGATCAAGGTG |
| |
AAATTCCTCCTTAATCTCGAGAATC |
| Mhy2 | CACCATGAAGTACTATAAGATTTCCGAGATCG |
| |
CTTCTGGCCGAACTTCTTTTTG |
| Mhy3 | CACCATGGTGGAGAAGCAGCTCA |
| GTTGACCTTGTTCCAGATCTTCAG |
Cell transfection
Cells were seeded in wells of a 24 well plate and incubated for 16 h to reach 70% confluency. Cells were transfected with the linearized plasmid DNA using Lipofectamine LTX (Life Technologies). In 48 h post-transfection, cells were seeded in 10 cm dish. Blasticidin-resistant clones were selected in medium supplemented with 5 μg/ml blasticidin S (Life Technologies).
Methylation-interference assay
Digestion of genomic DNA (1 μg) with MspI, HpaII, MboI and Sau3AI was conducted in 100 μl reactions (New England Biolabs). Digested DNA was separated on 1% agarose gel and visualized by ethidium bromide.
Methylation-specific PCR
PCR was performed in 50 μl reactions consisting of Standard Taq Reaction buffer, Taq DNA polymerase (New England Biolabs), dNTPs (200 μM each), bisulfite converted DNAs (50 ng), and the forward and reverse primers (0.5 μM each) specific for either unmethylated and methylated DNA (Table 3). PCR products were separated on 2% agarose gel, visualized by ethidium bromide, extracted from a gel and sequenced.
Table 3.
Oligonucleotide primers for bisulfite sequencing
| Target | Primers pair (5′-3′ sequence) | |
|---|---|---|
| pET101 | 1 | ATTGATTGGTTTTAATTTTTGGAGT |
| AAATAACCTTCCCCATTATAATTCTTC | ||
| 2 | AATGGGGAAGGTTATTTAGTTT | |
| TAAATTAACTCACTCATTAAACACC | ||
| 3 | GTTATTTTTTGATTTTGTGGATAAT | |
| AAAAACCTCTAACACATACAACTCC | ||
| 4 | GAGTTGTATGTGTTAGAGGTTTTTAT | |
| CCCTAAATAATTTTTCTCTAATCCC | ||
| 5 | TTAGTGTAGGTAGTTTTTATAGTAATGGTA | |
| AAAAAAAATCAATTCAAAATAATAAATATA | ||
| MMP-2 | U | GGTGGTTATATGTATTGAGTTAGTGA |
| ACTCTTTATCCATTTTAAAAACAAC | ||
| M | GCGGTTATACGTATCGAGTTAGC | |
| ACTCTTTATCCGTTTTAAAAACGAC | ||
| AKT3 | TTTTGAAATTTAGTAGGTGTTTTGAG | |
| GTAAAACGACGGCCAGTTAGGTGTTTTGAGGTAATATTTTGG | ||
| AAAACTTAAAAATTCCCAACCTCTT | ||
| ID1 | AGGGATTTTTAGTTGGAGTTGAATT | |
| CTTTTTCCAAACTCCTTAAACACC | ||
Bisulfite sequencing
Genomic DNA was purified from cultured human cells using the genomic DNA purification system (Zymo Research). The bisulfite DNA conversion was performed using the EZ DNA Methylation-Lightning system (Zymo Research). DNA fragments were amplified by PCR in 50 μl reactions using EpiMark Hot Start Taq DNA Polymerase (New England Biolabs), dNTPs (200 μM each), bisulfite converted DNA template (10-50 ng), and the forward and reverse primers (0.5 μM each) (Table 3). Primers were designed using MethPrimer.30 PCR products were purified, inserted into the pMiniT vector (New England Biolabs) and transformed into 10-β E. coli cells. Individual positive E. coli clones were randomly picked and their DNA inserts were sequenced.
MTase sequence specificity
E. coli BL21(DE3) cells were transformed using the pET101/Mhy1, pET101/Mhy2 and pET101/Mhy3 constructs. Transformed cells were diluted in 10 ml Lennox broth supplemented with 100 μg/ml ampicillin and grown at 37°C on a shaker at 150 rpm. In 16 h cultures were diluted at a 1:20 ratio (v/v) in the Lennox broth-100 μg/ml ampicillin medium and grown to reach OD600 = 0.7. IPTG (1 μM) was then added and cells were grown at 18°C for an additional 12 h. Cells were collected by centrifugation, plasmid DNAs were isolated using a Zippy plasmid purification system (Zymo Research). Bisulfite sequencing was performed as described in the “Bisulfite Sequencing” subsection above using the primers specific to the pET101 plasmid (Table 3).
MeDIP-Seq and bioinformatics analysis
Genomic DNA was fragmented to an average size of 200 bp using a S-4000 sonicator (Misonix). DNA fragments were end-repaired, A-tailed and ligated with custom adapters (Illumina). Methylated DNA was immunoprecipitated using the Methylated-DNA IP system (Zymo Research). Both methylated and non-methylated DNAs were subjected to PCR enrichment using adapter-specific primers (Illumina). DNA was sequenced using a HiSeq 1000 Sequencer (Illumina) with 57 × 106–151 × 106 reads/sample. Sequence reads were trimmed. Low quality reads were filtered out. Reads were mapped to the human reference genome (assembly 37.3) and read coverage was calculated at every genomic position using our custom software (Fofanov, unpublished). Reads with low quality mapping were excluded. MeDIP-seq data were deposited to the NCBI Sequence Reads Archive (BioProject ID 233511). Genome-wide MeDIP profiles of methylation marks were generated using our custom software (Chernov, unpublished). Circular histograms of MeDIP profiles were generated using Circos. 31 MeDIP profiles at the individual gene loci were visualized using the Integrative Genomic viewer.32
In situ immunofluorescent microscopy
Cells were seeded in wells of a Lab-Tek II CC2 glass chamber (Nalge Nunc) and grown to reach 70% confluency. Cells were fixed for 10 min in 4% paraformaldehyde at 25°C, permeabilized for 5 min in 0.25% Triton X-100, and blocked for 1 h in 2% BSA at 25°C. Cells were incubated for 1 h with the V5 antibody followed by incubation for 30 min with the goat anti-mouse AlexaFluor 594 antibody at 25°C. Cells were mounted on slides using the Prolong Gold reagent (Life Technologies) containing 4′,6-diamidino-2-phenylindole (DAPI). Confocal microscopy was conducted using a LSM 710 NLO Zeiss Multiphoton Laser Point scanning confocal microscope equipped with a multi-photon Mai-Tai laser HB - DeepSee system (690–1024 nm). Images were acquired using ZEN software (Zeiss).
Transcriptome analysis
Total RNA was purified using a Direct-zol RNA purification system (Zymo Research) and quantified with a Nanodrop ND-1000 spectrophotometer (Thermo Scientific). RNA integrity was determined using an Experion electrophoresis system (Bio-Rad). RNA (500 ng) was labeled using a RNA Amplification system (Illumina). Labeled RNA (750 ng) was hybridized for 16–18 h at 58°C to HumanHT-12v4 Expression BeadChip slides (Illumina). Slides were washed, developed using Fluorolink Streptavidin-Cy3 reagent (GE Healthcare), and scanned using a BeadArray Reader (Illumina). The fluorescence intensity data were processed using Feature Extraction software (Agilent). Bioinformatics analysis was performed using GeneSpring GX (Agilent) and the data with P > 0.05 were excluded. Our transcriptome data were deposited to the Gene Expression Omnibus (accession number GSE53853). Functional clustering was conducted using Ingenuity Pathway Analyses (Qiagen). The association of genes with biological functions and canonical pathways was based on the respective p-values that were calculated using the right-tailed Fisher exact test as previously described.33 Heat map diagrams were generated using GenePattern.34
Detection of mycoplasma in colorectal cancer specimens
Unselected primary colorectal cancer and corresponding non-tumoral tissues were obtained through the Cooperative Human Tissue Network (CHTN) from 751 patients who had undergone curative surgery in various hospitals, including Philadelphia, Tennessee, Ohio and Alabama between 1985 and 2004. Clinical information included age at diagnosis, gender, race, tumor location, and surgical stage; and pathology data that included grade of differentiation. Sanford-Burnham Institutional Review Board approval was obtained for this work. The study was performed according to the ethical guidelines of the Declaration of Helsinki. Colorectal tumor and normal colon specimens were collected during the surgical procedures, immediately frozen in liquid nitrogen and stored at −80°C. Genomic DNA was isolated from the tumor and matching normal adjacent tissues as previously described. 35,36 DNA concentration and integrity were determined using a Qubit fluorometer (Life Technologies) and gel electrophoresis on 1% agarose gel, respectively. Quantitative real-time PCR was carried out in 10 μl reactions containing 200 μM forward and reverse primers specific to the rRNA gene [5′- GGGAGCAAACAGGATTAGATACCCT-3′ and 5′-TGCACCATCTGTCACTCTGTTAACCTC-3′, respectively37], 50 ng of template DNA and SYBR Green I Master Mix (Roche). Real-time PCR was performed using a LightCycler 480 PCR cycler (Roche) as follows: 10 min denaturation at 95°C, followed by 40 cycles of 10 s at 95°C and 30 s at 60°C. DNA from HepG2 cells infected with M. hyorhinis and uninfected HepG2 cells were used as the positive and negative controls, respectively. Cycle threshold (Ct) values were calculated using LightCycler 480 SW software. PCR products were separated on 2% agarose gel and visualized by ethidium bromide. PCR products were ligated into the pSC-A-amp/kan vector (Agilent) and used to transform E. coli cells. Positive clones were analyzed by DNA sequencing. The identity of mycoplasma species was determined using BLAST (NCBI). The specimens were classified as mycoplasma positive if the query DNA exhibited both over 98% identity to DNA of mycoplasma, and the corresponding Ct below 35.
Results
Cloning of M. hyorhinis MTases
Based on the presence of conserved motifs, we identified 3 previously uncharacterized MTase genes located at positions 42823-44016, 94325-95566, and 475074-476297 bp in M. hyorhinis GDL chromosome (Fig. 1). 38 We refer to the encoded proteins as Mhy1, Mhy2, and Mhy3 (Tables 1 and 2). To facilitate the uninterrupted translation of Mhy1, Mhy2, and Mhy3 in E. coli and human cells, we employed synthetic gene constructs that replaced amber UGA codons (Trp in Mycoplasmas) with UGG (Trp) and altered codon usage.
Figure 1.

M. hyorhinis MTases. (A) conserved motifs in M. hyorhinis MTases. The amino acid sequence alignment of M. hyorhinis Mhy1, Mhy2 and Mhy3, Mfe1 from M. fermentans, Uur1 from Ureaplasma urealyticus and Mpe1 from M. penetrans. Conserved motifs are indicated by numbers. A consensus sequence is shown at the bottom. (B) MTase loci in M. hyorhinis genomes. Positions of the Mhy1 (40.5–46.5 kb), Mhy2 (92–98 kb), and Mhy3 (472.5–480 kb) genes in M. hyorhinis chromosome. Genes are: dnaJ, chaperone protein; mnmA, tRNA-specific 2-thiouridylase; Mhy1, CG-specific MTase; MYM_0038, hypothetical protein; nox, NADH oxidase; MYM_0074, hypothetical protein; lepA, GTP-binding protein; Mhy2, CG-specific MTase; MYM_0077, hypothetical protein; MYM_0078, ATP-binding ABC transporter; MYM_0384, putative endonuclease or phosphatase; MYM_0385, glutamyl aminopeptidase; Mhy3, GATC-specific MTase; MYM_0387, hypothetical protein; hsdS-3 and hsdS-4, type I DNA MTases. Genes are shown as solid color bars. The direction of transcription is indicated by arrows. Putative MTase and endonuclease genes are red and blue, respectively. Other genes are green.
Specificity of mycoplasma MTases
To identify the sequence specificity of MTases, we employed E. coli BL21(DE3) cells, which lack the intrinsic cytosine-specific dcm methylation. Following transformation of E. coli cells with the recombinant Mhy1, Mhy2, and Mhy3, we analyzed plasmid DNA methylation (Fig. 2A). Using bisulfite sequencing (Table 3), we determined that Mhy1 incorporated 5mCs exclusively within the mCG sequence context. Mhy2 methylated DNA at the (G/A)mCATG(C/T) and mCG DNA sites and, therefore, exhibited the properties of the multi-specific DNA MTases. 39 Remarkably, Mhy2 methylated in a random manner only a fraction of the available CG sites. This is in contrast to Mhy1 that methylated all CGs in the DNA substrate. Lastly, Mhy3 incorporated 5 mC exclusively within the GATmC context. However, the GATmC-specific Mhy3 could also methylate mCGs if its GATC recognition sequence overlaps with the CG dinucleotides in the DNA substrate (e.g., CGATmCG). Thus, all 3 MTases were capable of generating methylated mCG sites that are canonical epigenetic marks in humans.
Figure 2.
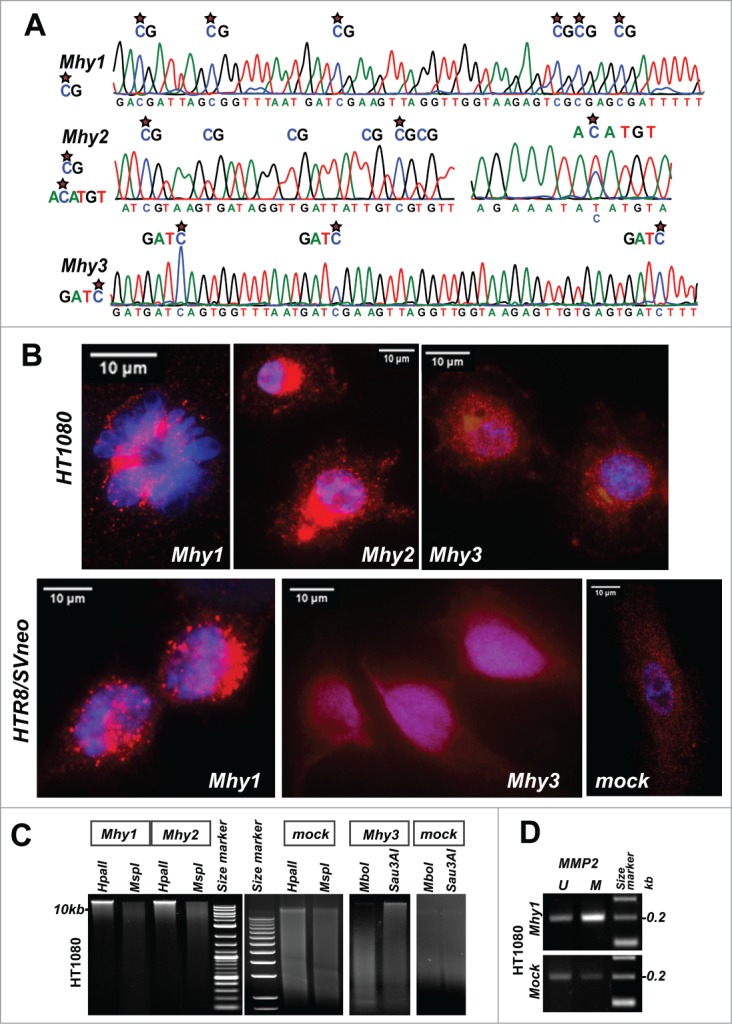
Mycoplasma MTases translocate to the human cell nuclei and methylate the human genome. (A) substrate specificity of Mhy1 (mCG), Mhy2 (mCG and AmCATGT), and Mhy3 (GATmC). Bisulfite DNA sequencing chromatograms of DNA substrates are shown. Stars indicate 5 mC. (B) confocal images of HT1080/Mhy1, HT1080/Mhy2, HT1080/Mhy3, HTR8/Mhy1, HTR8/Mhy3, and HTR8/mock cells. Upper left panel shows the condensed chromatin in a single HT1080/Mhy1 cell. Red, the V5 immunostaining. Blue, DAPI. (C) DNA Methylation-interference assay. HT1080/Mhy1, HT1080/Mhy2, HT1080/Mhy3 and HT1080/mock genomic DNA was digested using HpaII, MspI (a methylation-insensitive isoschizomer of HpaII), Sau3AI and MboI (a methylation-insensitive isoschizomer of Sau3AI). (D) methylation-specific PCR of a CPGI promoter region of the MMP2 gene in HT1080/Mhy1 and HT1080/mock cells. PCR products specific to methylated (M) and unmethylated (U) DNA are shown.
Expressed mycoplasma MTases translocated into human nuclei
To determine the effect of mycoplasma MTases in human cells, we transfected HT1080 fibrosarcoma cells and extravillous HTR8/SVNeo trophoblasts with Mhy1, Mhy2, and Mhy3, and generated HT1080/Mhy1, HT1080/Mhy2, HT1080/Mhy3, and HTR8/Mhy1, and HTR8/Mhy3 cells, respectively. As a control, we used cells transfected with β-galactosidase/lacZ (HT1080/mock and HTR8/mock cells). Strikingly, we observed that the V5-tagged Mhy1 protein was transported into the nucleus and largely associated with the condensed chromatin in HT1080/Mhy1 cells (Fig. 2B). Consistently, in HTR8/Mhy1 cells, the Mhy1-specific immunostaining was detected primarily in the nucleus. Mhy2 also accumulated in the nucleus and the perinuclear space in HT1080/Mhy2 cells. Likewise, Mhy3 was readily transported to the nucleus in both HT1080/Mhy3 and HTR8/Mhy3 cells. By contrast, the control V5-tagged β-galactosidase was localized in the cytoplasm.
Mycoplasma MTases exhibited DNA methylation activity in human cells
To determine if Mhy1 and Mhy2 methylated human DNA, we digested genomic DNA from HT1080/Mhy1, HT1080/Mhy2, and HT1080/mock cells using HpaII endonuclease. HpaII cleaves only the unmethylated 5′-CCGG-3′ sites. Genomic DNA from HT1080/Mhy1 and HT1080/Mhy2 cells was significantly protected from HpaII while HT1080/mock DNA was cleaved by HpaII (Fig. 2C). Similarly, Mhy3 protected HT1080/Mhy3 DNA from Sau3AI because Sau3AI cleaves 5’-GATC-3’ but not the 5’-GATmC-3’ sequence. In turn, HT1080/Mhy1, HT1080/Mhy2, HT1080/Mhy3 and HT1080/mock DNAs were readily cleaved by MspI and MboI, methylation-insensitive isoschizomers of HpaII and Sau3AI, respectively. These data suggested that expressed mycoplasma MTases caused methylation of the respective target sites in the human genome.
To test these observations further, we analyzed methylation of the CPGI in the MMP2 gene promoter region in HT1080/Mhy1 versus HT1080/mock cells. MMP2 is a member of the matrix metalloprotease family involved in extracellular matrix degradation in disease, especially in cancer. We previously demonstrated that MMP2 is regulated by methylation of a respective CPGI.40 The methylation-specific PCR analysis clearly indicated that in HT1080/Mhy1 cells methylation of the MMP2 CPGI was increased compared to HT1080/mock cells (Fig. 2D). These results proved that Mhy1 effectively targeted a functionally-relevant CPGI region of an epigenetically-controlled human gene.
Mycoplasma MTases altered the epigenetic landscape in human cells
Next, we examined the genome-wide distribution of methylation that was introduced by Mhy1, Mhy2, and Mhy3 in the human genome. To obtain high-resolution methylation profiles, we employed methylated DNA immunoprecipitation – sequencing (MeDIP-seq) of DNA from HT1080/Mhy1, HT1080/Mhy2, and HT1080/Mhy3 cells. The MeDIP profile of pre-existing intrinsic methylation was determined using HT1080/mock cells. The mapping of methylation incorporated by mycoplasma MTases was performed by comparing MeDIP profiles in HT1080/Mhy1, HT1080/Mhy2, and HT1080/Mhy3 cells vs. HT1080/mock cells. The results presented in a circular histogram illustrate that the CG and GATC methylation marks correlated well with the distribution of CG and GATC sites in human chromosomes (Figs. 3A and S1). The Mhy1-specific methylation was predominant in the CPGIs with a high CG frequency. An even distribution of the Mhy3-specific methylation was consistent with the uniform distribution of GATC sites in the human genome.
Figure 3.
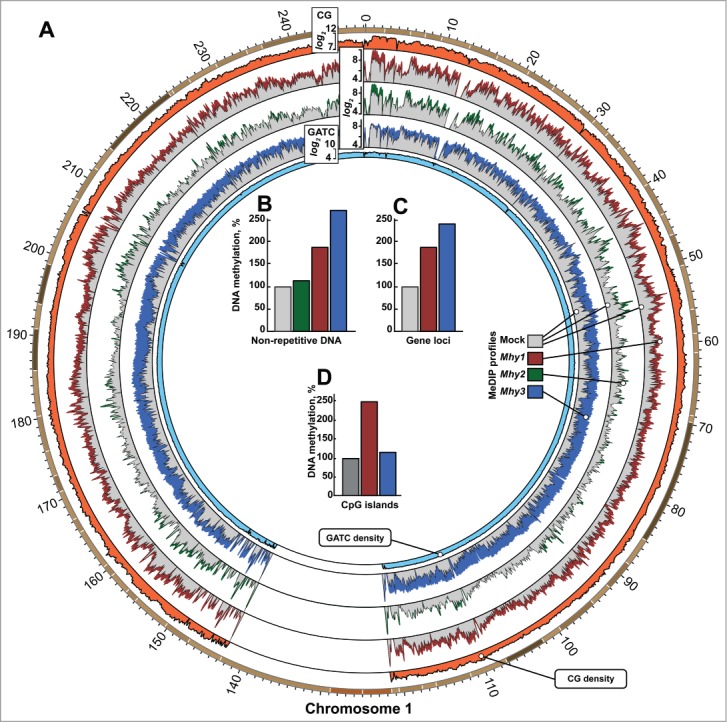
Genome-wide methylation induced by mycoplasma MTases. (A) circular histogram representation of DNA methylation induced by Mhy1 (red), Mhy2 (green), and Mhy3 (blue) in HT1080/Mhy1, HT1080/Mhy2, and HT1080/Mhy3 cells, respectively. For simplicity, the MeDIP profile of human chromosome 1 is shown. The data for human chromosomes 2–22, Y and X are in Figure S3A-O. Histogram bar heights are calculated as logarithms of a number of the individual methylation marks within a 100 kb window (see Extended Experimental Procedures). Gray histograms correspond to methylation in HT1080/mock cells. Orange and light blue histograms indicate relative density of CG and GATC sites within a 100 kb window, respectively. (B-D) bars show the methylation level in HT1080/Mhy1 (red), HT1080/Mhy2 (green), HT1080/Mhy3 (blue), and HT1080/mock cells (gray). Methylation was individually calculated for all non-repetitive genomic sequences (B), genic regions (C), and CPGI regions (D). The percentages were calculated relative to HT1080/mock cells (gray bars = 100%).
Relative to HT1080/mock cells, a total number of methylated CG and GATC sites increased >1.8-fold, >1.15-fold, and >2.7-fold in HT1080/Mhy1, HT1080/Mhy2, and HT1080/Mhy3 cells, respectively (Fig. 3B). Because methylation introduced by Mhy2 in the HT1080/Mhy2 genome was low, we analyzed further only HT1080/Mhy1 and Ht1080/Mhy3 cells. In genic regions, including exons and introns, we recorded a 1.9-fold and 2.4-fold increase in CG/Mhy1 and GATC/Mhy3 methylation, respectively (Fig. 3C). As expected, Mhy1, but not Mhy3, predominantly methylated the CG-rich genomic regions (Fig. 3D). All together, these results suggested that both CG/Mhy1- and GATC/Mhy3-specific methylation efficiently targeted the human DNA sites that were unmethylated by human DNMTs.
Epigenetic contribution of GATC-specific methylation
Approximately 7.1 × 106 GATC sites with an average frequency of ∼2.25 sites/kb are present in human non-repetitive DNA sequences. Most GATC sites are uniformly distributed. However, we identified 2,023 regions in the genome in which GATC sites clustered with a frequency of >10 sites/kb. These GATC clusters are within or in the vicinity of ∼1,000 genes, including cancer-associated TGFB1, AKT3, ID1, SMARCA4, and EPHB1. In the human genome, approximately 11% of GATC sites overlap with CGs (e.g., CGATmCG); therefore, the methylated status of the overlapping sites can be perpetuated by DNMT1. We believe that GATC sites represent an important epigenetic reservoir that can be targeted by Mhy3 in the human genome.
Gene loci methylated by Mhy1 and Mhy3
To test our MeDIP-seq results at the individual gene level, we analyzed a 2.5 kb region of the AKT3 gene. This region encompasses 9 CG and 17 GATC sites. According to MeDIP, there were one and 2 marks in HT1080/mock and HT1080/Mhy1 cells, respectively, while there were 7 methylation marks in HT1080/Mhy3 cells. Bisulfite sequencing confirmed that all 4 GATC sites existing in a 218 bp AKT3 sub-region were methylated in HT1080/Mhy3 cells while no methylation was detected in HT1080/mock and HT1080/Mhy1 cells (Fig. 4A). These results demonstrated that mycoplasma MTases were capable of altering the methylation patterns at positions that were unmethylated by human DNMTs.
Figure 4.

Methylation of AKT3, ID1, and MMP2 genes. (A) methylation of the AKT3 gene locus. Top, the AKT3 gene locus with exons indicated as vertical bars. Middle, MeDIP profiles. Solid color bars indicate the genomic position and MeDIP coverage of individual CG- and GATC-methylation marks in HT1080/mock (gray), HT1080/Mhy1 (red) and HT1080/Mhy3 (blue) in a 2.5 kb GATC cluster of the AKT3 gene (positions 243977.5–243980 kb on chromosome 1). Bottom, bisulfite sequencing chromatogram of a 218 bp sub-region with 4 GATC sites methylated in HT1080/Mhy3 cells. Arrows indicate 5 mC. (B) methylation of the ID1 gene. Top, bisulfite sequencing chromatogram of a 213 bp sub-region in the promoter of the ID1 gene locus in HT1080/Mhy1 and HT1080/Mhy3 cells. The converted and original genomic sequences are shown under chromatograms. Middle, MeDIP profiles. Solid blue bars indicate the genomic position and MeDIP coverage of the individual GATC-methylation marks in HT1080/Mhy3 cells in a 2.5 kb ID1 gene promoter (positions 30192–30194.5 kb on chromosome 20). No GATC methylation was recorded in HT1080/Mhy1 and HT1080/mock cells. Bottom, ID1 gene locus with exons (solid black bars) and CPGIs (purple bars). (C-D) methylation of the MMP2 gene. C, MeDIP profiles of a 27.5 kb MMP2 gene locus in HT1080/Mhy3, HT1080/Mhy1 and HT1080/mock cells. Solid color bars indicate the genomic position and MeDIP coverage of the individual CG- and GATC-methylation marks in HT1080/mock (gray), HT1080/Mhy1 (red) and HT1080/Mhy3 (blue) cells. CPGIs, purple rectangles. Genomic coordinates are shown at the panel bottom. (D) bisulfite sequencing diagrams of a 221 bp sub-region (indicated by a red arrow in panel C) in the CPGI promoter (purple bar) of the first exon (solid black bar) of the MMP2 gene in HT1080/Mhy1 and HT1080/mock cells. Stars and arrows indicate 5 mC.
Similarly, we analyzed a 2.5 kb region near a promoter/exon 1 of the ID1 gene. This CG- and GATC-dense region contains 157 CG and 10 GATC sites. MeDIP revealed 4 methylation marks in this 2.5 kb region in HT1080/Mhy3 cells compared to zero in HT1080/mock and HT1080/Mhy1 cells (Fig. 4B). Bisulfite sequencing of a 213 bp ID1 sub-region with 21 CG and 3 GATC sites confirmed complete GATC-specific methylation in HT1080/Mhy3 cells. All 21 CGs were unmethylated in HT1080/Mhy3 and HT1080/mock cells. Furthermore, according to MeDIP there were 47 and 71 methylation marks within the entire 27.5 kb MMP2 gene in HT1080/mock and HT1080/Mhy1 cells, respectively (Fig. 4C). Bisulfite sequencing of a 221 bp MMP2 promoter sub-region demonstrated that all 22 CG sites (100%) were methylated in HT1080/Mhy1 cells while only 14 CG sites were methylated in HT1080/mock cells (Fig. 4D). Taken together, MeDIP, bisulfite sequencing and methylation-sensitive PCR indicated that Mhy1 and, especially, Mhy3, efficiently methylated human genomic DNA.
Hypermethylation of cancer-specific genes
To determine whether cancer-specific genes were affected by Mhy1 and Mhy3, we analyzed methylation of genes frequently mutated in cancers (www.broadinstitute.org/ccle). Based on MeDIP, Mhy1 and, especially, Mhy3, increased DNA methylation in 76 cancer-specific genes (Fig. 5A). More genes were targeted by Mhy3 as compared to Mhy1. This can be explained by the absence of the pre-existing GATC methylation in HT1080 fibrosarcoma. In agreement, we identified a significant increase in GATC-specific methylation in important cancer-specific genes, including ADAMTS2, APC, BRCA2, KRAS, MSH3, PTEN, RAD50, SMAD4, TGFBR2, TP53, and VEGFC (Fig. 5A). We estimated, for example, that methylation of the APC tumor suppressor increased ∼1.4-fold and ∼2.8-fold in HT1080/Mhy1 and HT1080/Mhy3 cells, respectively (Fig. 5B). Methylation of the SMAD4 tumor suppressor gene increased ∼1.9-fold and ∼4.4-fold in HT1080/Mhy1 and HT1080/Mhy3 cells, respectively (Fig. 5B). However, we also identified that promoter regions of certain cancer-specific genes, including MYC, JUN, and TGFB1, were not efficiently targeted by Mhy1 and Mhy3 (Fig. 5B).
Figure 5.
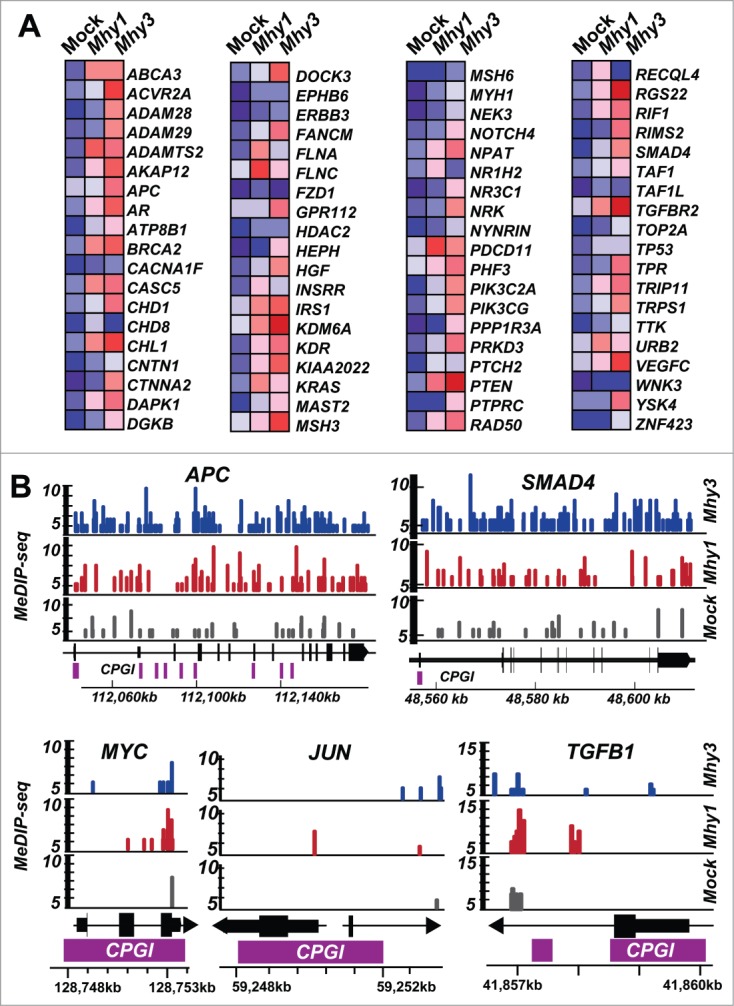
Methylation of cancer-specific genes. (A) heatmap of the CG- and GATC-methylation in 76 cancer-specific genes in HT1080/Mhy1, HT1080/Mhy3 and HT1080/mock cells. Red and blue correspond to the high and the low methylation levels according to MeDIP, respectively. (B) MeDIP profiles of the selected gene loci. Solid color bars indicate the genomic position and MeDIP coverage of the individual CG- and GATC-methylation marks in HT1080/mock (gray), HT1080/Mhy1 (red) and HT1080/Mhy3 (blue) cells in the APC, SMAD4, MYC, JUN, and TGFB1 gene loci. Solid bars indicate exons (black) and CPGIs (purple). Chromosomal coordinates are shown at the bottom of the diagrams.
Because we did not observe any significant effect of Mhy1 and Mhy3 on the proliferation of HT1080/Mhy1 and HT1080/Mhy3 cells, respectively, we suspected that the actively transcribed genes were less susceptible to methylation by mycoplasma MTases. In agreement, expression levels of only a limited number of genes were affected in HT1080/Mhy1 and HT1080/Mhy3 cells relative to HT1080/mock cells. Thus, only 7 and 9 genes were up- and down-regulated in HT1080/Mhy1 cells versus HT1080/mock cells, respectively (Fig. 6A). In HT1080/Mhy3 cells, 13 genes were upregulated and 2 genes were downregulated vs. HT1080/mock cells. Functional clustering of the differentially expressed genes revealed activation of the cellular immune response and inflammatory pathways via upregulation of IL-17, CCL2, and IL8 genes (Fig. 6B).
Figure 6.

Transcriptome analysis in fibrosarcoma and trophoblasts. (A) top regulated genes in HT1080/Mhy1, HT1080/Mhy3 relative to HT1080/mock cells. Up- and downregulated genes are in red and green, respectively. (B) affected functional (blue) and canonical pathways (orange) in HT1080/Mhy1 and HT1080/Mhy3 cells identified by the functional clustering analysis. The pathways with P ≤ 0.05 are shown. (C) top up- (red) and down- (green) regulated genes in HRT8/Mhy1 cells relative to HRT8/mock cells. (D) affected disease (blue) and canonical pathways (orange) in HRT8/Mhy1 cells. Only pathways with P ≤ 0.05 are shown.
Activation of proliferation-specific pathways in GATC-methylated trophoblasts
We focused our further analysis on non-cancerous first trimester extravillous HTR8/SVneo trophoblasts. These non-malignant cells exhibit progenitor-like plasticity. 41 Expressed Mhy1 and Mhy3 were both trafficked into the trophoblast nucleus (Fig. 2B) and introduced genome-wide CG- and GATC-specific methylation in multiple gene loci (confirmed by bisulfite sequencing; data not shown).
The transcriptome analysis demonstrated that the expression levels of 250 genes (109 upregulated and 141 downregulated) and 143 genes (85 upregulated and 58 downregulated) were affected in HTR8/Mhy1 and HTR8/Mhy3 cells, respectively, versus HTR8/mock cells (Figs. 6C and 7A). Functional clustering analysis identified that cellular development, growth, proliferation, survival, and mobility, and inflammatory and immune response pathways were affected in HTR8/Mhy1 cells (Fig. 6C,D). Upregulation of S100A10, DKK1, CXCL12, and ITGB4 and downregulation of ABTB1, CEBPB, and HES1 contributed to the activation of proliferation-specific pathways in HTR8/Mhy1 cells.
Figure 7.
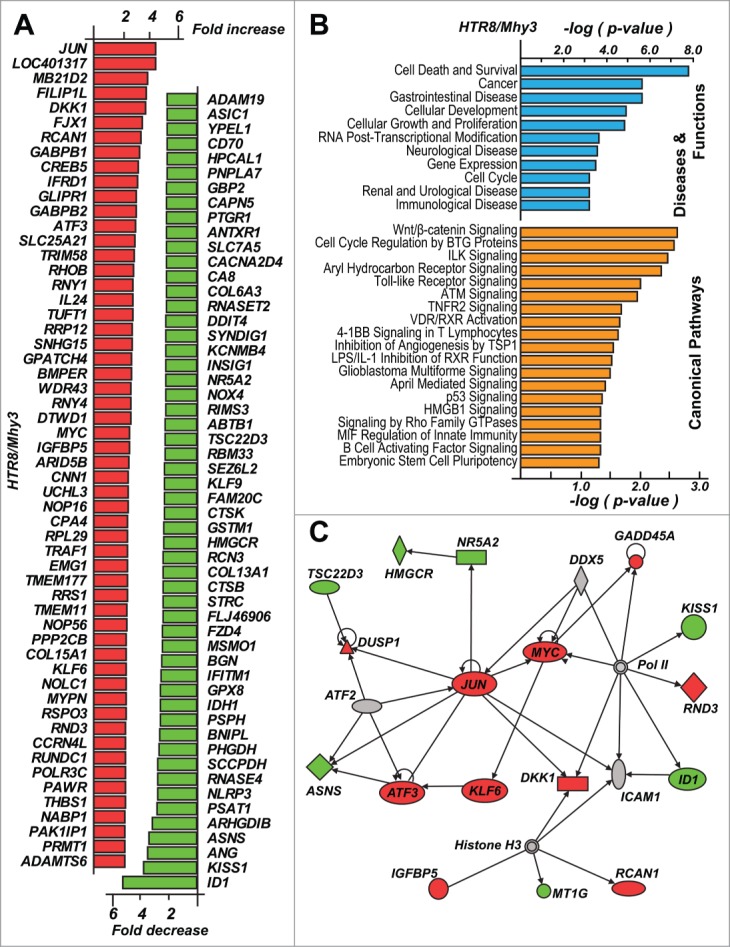
Activation of the cancer-specific pathways in trophoblasts. (A) top up- and down-regulated genes in HRT8/Mhy3 cells relative to HTR8/mock cells. (B) affected disease (blue) and canonical pathways (orange) in HRT8/Mhy3 cells. The pathways with P ≤ 0.05 are shown. (C) proliferation specific regulatory network in HTR8/Mhy3 cells. Directly interacting molecules are connected by arrows. Up- and downregulated genes are in red and green, respectively.
Importantly, we observed upregulation of the multiple cancer-specific genes, including the MYC and JUN proto-oncogenes and the ATF3, KLF6, DKK1, IGFBP5, and RCAN1 transcription factors, in HTR8/Mhy3 cells (Fig. 7A). As a result, major cellular functions including cell death, survival, growth, and proliferation appeared to be impacted (Fig. 7B,C). In addition, cancer-specific pathways, such as WNT/β-catenin, toll-like receptor, TNFR2, glioblastoma multiforme, p53, and stem cell pluripotency, apparently were affected because of GATC/Mhy3 methylation in trophoblasts (Fig. 7B).
Mycoplasma infections are widespread in human colorectal cancers
To support a putative association of mycoplasma with cancer, we analyzed 71 paired colorectal tumor and matching normal tissue specimens for the presence of mycoplasma DNA. We employed quantitative real-time PCR using primers specific to conserved regions of the rRNA gene. The resulting 270 bp PCR product was sequenced to determine the taxonomic identity of the species (Fig. 8). We detected mycoplasma DNA in 50 cases (70.4%). Only 21 cases were negative for mycoplasma in our CRC cohort. Notably, ∼20% of all detected mycoplasma species were identified as M. hyorhinis. Most frequently, we detected mycoplasma in both the tumor and matching normal specimens (Table 4). In several cases, however, mycoplasma was recorded in normal tissue, but not in tumors. It is possible that mycoplasma infections arise in normal colonic epithelium. Because of the aberrant CG and especially GATC methylation marks inserted by mycoplasma methyltransferases in the normal cell genome, a subset of normal epithelium may then be driven toward oncogenic transformation. Our data also indicate that the mycoplasma GATC-specific Mhy3 is fully functional in the mycoplasma-infected human cells, thus supporting a potential impact of the bacterial enzymes on the epigenetic landscape in humans and, ultimately, tumor development (Chernov et al., submitted for publication elsewhere).
Figure 8.
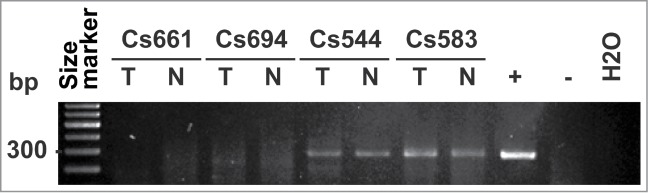
Detection of mycoplasma in colorectal cancer specimens. A 270 bp PCR product specific to the mycoplasma rRNA gene was amplified from the total DNA isolated from colorectal cancer tumor (T) and matching normal (N) specimens (negative cases Cs661 and Cs694, and positive cases Cs544 and Cs583). DNA size markers and the mycoplasma-positive (HepG2 cells infected with M. hyorhinis) and negative (uninfected HepG2 cells and H2O) controls are shown.
Table 4.
Mycoplasma infection in colorectal tumors and matching normal tissue specimens. The presence (+) or absence (−) of mycoplasma was in the samples was analyzed by quantitative real-time PCR and then confirmed by DNA sequencing. Gender: female, F; male, M. Race: Asian, A; African-American, B; Caucasians, W. Tumor localization: proximal (p) or distal (d) colon. Tumor differentiation: poor, P; moderate, M; well-differentiated, W. Tumor staging is based on the Dukes classification system. ND, not determined; UN, unknown
| Case | Normal | Tumor | Gender | Age at diagnosis | Race | Location | Differentiation | Stage |
|---|---|---|---|---|---|---|---|---|
| 38 | + | + | F | 72 | B | p | WM | C |
| 54 | + | + | F | 34 | W | p | P | D |
| 62 | + | + | M | 62 | W | p | M | B |
| 92 | + | + | M | 37 | W | d | M | B |
| 193 | ND | + | F | 68 | W | d | M | B |
| 336 | + | + | F | 75 | B | p | WM | B |
| 402 | + | + | M | 50 | UN | d | M | C |
| 407 | + | + | M | 68 | UN | d | M | C |
| 408 | + | + | F | 78 | UN | p | M | B |
| 414 | + | + | M | 77 | B | d | M | A |
| 418 | + | + | F | 70 | UN | p | M | C |
| 446 | + | + | F | 87 | UN | p | M | B |
| 458 | ND | + | M | 54 | W | p | W | B |
| 480 | ND | + | M | 74 | W | p | W | B |
| 482 | + | + | F | 46 | B | p | W | C |
| 491 | ND | + | F | 67 | UN | d | M | B |
| 498 | + | + | F | 92 | A | p | M | D |
| 504 | ND | + | F | 76 | W | p | P | B |
| 506 | ND | + | M | 77 | W | p | M | C |
| 522 | + | + | F | 66 | W | p | M | D |
| 526 | − | + | M | 73 | W | p | M | D |
| 527 | + | + | M | 49 | B | d | M | B |
| 536 | + | + | F | 61 | B | p | UN | O |
| 538 | + | + | F | 88 | W | p | M | C |
| 544 | + | + | F | 66 | W | p | M | B |
| 557 | + | + | M | 65 | W | p | UN | D |
| 583 | ND | + | M | 51 | W | p | M | B |
| 584 | + | + | M | 69 | W | d | UN | C |
| 590 | + | + | F | 76 | W | p | M | C |
| 595 | + | + | M | 84 | W | p | UN | B |
| 603 | − | + | M | 37 | UN | p | M | B |
| 605 | − | + | M | 39 | W | d | M | D |
| 608 | + | + | M | 57 | W | d | M | B |
| 609 | + | + | M | 56 | W | p | M | C |
| 610 | − | + | M | 60 | W | d | M | B |
| 612 | + | + | M | 79 | W | p | UN | C |
| 613 | − | + | F | 45 | W | p | P | A |
| 634 | + | + | M | 85 | W | p | M/P | B |
| 656 | + | + | F | 64 | W | p | M | C |
| 662 | + | + | M | 92 | W | p | M | C |
| 664 | ND | + | F | 88 | W | d | M | C |
| 672 | + | + | F | 60 | W | p | M | B |
| 692 | ND | + | M | 78 | B | p | MP | B |
| 694 | + | + | M | 62 | W | d | M | C |
| 700 | + | + | F | 76 | W | p | M | B |
| 703 | + | + | F | 67 | W | p | P | C |
| 707 | + | + | F | 75 | W | p | UN | B |
| 726 | + | + | M | 73 | W | d | M | D |
| 729 | + | + | M | 74 | W | UN | UN | M |
| 733 | + | + | M | 64 | W | d/p | W/M | C |
| 55 | ND | − | M | 68 | B | d | WM | B |
| 89 | + | − | M | 39 | W | p | UN | A |
| 199 | − | − | M | 61 | UN | d | M | A |
| 267 | − | − | F | 68 | B | UN | UN | M |
| 413 | ND | − | F | 61 | UN | p | M | B |
| 447 | ND | − | M | 72 | W | d | M | D |
| 455 | + | − | M | 37 | B | p | MP | B |
| 485 | ND | − | F | 81 | B | p | W | B |
| 486 | + | − | M | 75 | UN | p | M | B |
| 492 | ND | − | F | 45 | W | d | WM | B |
| 514 | + | − | F | 72 | W | p | M | D |
| 523 | ND | − | F | 80 | W | d | M | B |
| 525 | − | − | M | 60 | B | p | M | B |
| 535 | ND | − | M | 69 | W | d | M | D |
| 611 | − | − | F | 86 | W | p | P | C |
| 661 | − | − | M | 67 | B | d | M | A |
| 677 | − | − | F | 71 | W | p | P | B |
| 691 | − | − | F | 53 | W | p | MP | C |
| 728 | + | − | F | 84 | UN | p | M | B |
| 730 | − | − | F | 56 | UN | p | M | D |
| 739 | − | − | F | 75 | W | d | M | C |
Alternatively, mycoplasmas adapted for survival in tumor cells may spread to neighboring “normal” adjacent tissue. Our data do not exclude that secondary mycoplasma infections appeared after tumors have already progressed. A high percentage of mycoplasma-positive tumors together with our demonstration of the potential impact by mycoplasma MTases on the epigenetic landscape support the potential role of mycoplasma in cancer etiology.
Discussion
Bacteria of the endogenous flora in the human body outnumber human cells by at least one order of magnitude. A plethora of symbiotic and pathogenic bacteria in different body habitats form the microbiome, which is now considered an essential component of human biology, evolution, health, and disease. 1 Host cells can potentially affect the microbiome via genetic variations, immune-mediated stress, or accessibility of essential metabolites. Conversely, host adaptation of the intracellular pathogenic microorganisms can adversely influence normal cell functions. This may be greater likelihood for species that engage in chronic infections by invading cells and avoidance of immune recognition and activation. The diminutive size of Mycoplasma genomes (0.58 Mb-1.2 Mb) suggests that parasitism of host cells is commonplace as compensation for lost metabolic potential.
Previous studies that have expressed bacterial DNA methyltransferases in eukaryotic cells, e.g., the dam adenine DNA methyltransferase gene from E. coli in Drosophila42 and Plasmodium falciparum,43 roughly probed chromatin structure in vivo. In contrast to these earlier studies with E. coli dam methyltransferases, we generated the first evidence that the mycoplasma CG-specific Mhy1 and Mhy2, and GATC-specific Mhy3 MTases established aberrant genome-wide methylation and significantly altered the epigenetic landscape in human cells. We demonstrated that MTases from the intracellular parasite, such as M. hyorhinis, readily translocated to the human cell nucleus. In the nucleus, mycobacterial enzymes selectively and effectively methylated cytosines within the respective CG and GATC sites in human genomic DNA. We yet lack direct experimental evidence that MTase nuclear trafficking also takes place in human cells naturally infected by M. hyorhinis. However, reports by others indicated that M. genitalium proteins, including cytadhesins P140 and P110, and EF-Tu elongation factor, were readily trafficked into the human cell nucleus after only a few hours post-infection, 5 thus supporting the potential role of MTases in the host cell epigenome aberrations.
Our MeDIP profiles established that in agreement with an average frequency of ∼2.25 GATC sites/kb in humans the Mhy3-induced GATC-specific hypermethylation was evenly distributed in human genome. GATC/Mhy3-specific hypermethylation was readily detected in the cancer-specific genes such as APC, TP53, BRCA2, KRAS, PTEN, SMAD4, and VEGFC. Despite generally even distribution of GATC in the human genome, we identified over 1000 clusters with a high GATC density (≥10 GATC sites/kb). These clusters are localized in the vicinity of multiple genes, including ID1. In trophoblasts, ID1 is a transcriptionally active gene characterized by the absence of the CG methylation in its promoter-associated CPGI. Upon expression of Mhy3, we recorded methylation of all available GATC sites, but not the CG sites, in the ID1 gene promoter. The GATC methylation correlated with the transcriptional downregulation of ID1. Therefore, we propose that methylation of GATC sites and, especially, GATC clusters, represents a novel type of infection-dependent epigenetic mark.
Normally, methylation at GATC sites does not occur in human cells. It is likely that transient GATC methylation in human cells emerges in the course of M. hyorhinis infection. However, at GATC sites overlapping with CGs (e.g., 5’-CGATmCG-3’), methylation will likely be perpetuated by the CG-specific human DNMT1 independently from Mhy3. We estimate that ∼7.8 × 105 GATC sites overlapping with CGs exist in human non-repetitive DNA. These overlapping sites represent a significant reservoir for long-term aberrant methylation that can persist in the human genome following exposure to M. hyorhinis.
Based on our transcriptome data, the upregulation of ATF3, KLF6, DKK1, RCAN1, IGFBP5, RND3, DUSP1, JUN, and MYC, and the downregulation of the ID1 gene was observed in trophoblasts expressing Mhy3. Taken together, these affected genes regulate cell signaling pathways, including WNT/β-catenin, toll-like receptor, TNFR2, glioblastoma multiforme, stem cell pluripotency, and p53. Activation of these pathways is related to pre-malignancy. Our results agree well with the existing reports by others who studied the infected human cell transcriptome. 8,10-13,44,45
In contrast with non-cancerous cells, in fibrosarcoma cells aberrant CG/GATC methylation affected a limited number of genes, predominantly the immune response genes. Presumably, pre-existing epigenetic and genetic abnormalities mitigated the effect of methylation by Mhy1 and Mhy3 in cancerous HT1080 cells. It is also tempting to hypothesize that relative to normal cells cancer cells better tolerate epigenetic aberrations, and, therefore, tumors may represent a reservoir for long-term survival and propagation of mycoplasmas. In agreement, we detected the presence of mycoplasma DNAs in 70.4% of the 71 colorectal cancer cases we examined. M. hyorhinis was detected in ∼20% of the mycoplasma-positive tumors. Additionally, there is growing evidence that mycoplasma is also present in other cancer types.7-9 Thus, it is possible to envision scenarios in which mycoplasma infections play an important etiological role in cancer or other diseases, in which epigenetic alterations take place. Alternatively, but not exclusively, the infection-driven epigenetic alterations potentially contribute to the disease progression, especially if such exogenous microbiome-related methylation is better tolerated by a pathological cell phenotype.
Strikingly, both M. hyorhinis Mhy1 and Mhy2 recognized CG sites and, thus, contributed to the human intrinsic CG methylation that is normally mediated by DNMTs. Other mycoplasma strains also possess CG-specific MTases.46 This suggests that the CG-specific methylation may be a significant factor for bacterial survival and host adaptation. Importantly, in normal human cells, the methylation of CG sites is strictly controlled in a developmental manner and unregulated CG hypermethylation is deleterious to the cell.47 We estimated that in Mhy1-expressing fibrosarcoma cells a number of methylation marks in the non-repetitive DNA regions increased >1.8-fold and that these marks were predominant in the CPGI regions. Evidence based on both MeDIP profiles and bisulfite sequencing suggested that partially methylated CPGIs of multiple genes became fully methylated in fibrosarcoma cells. Complete methylation of all available CGs in a partially methylated MMP2 CPGI promoter region confirmed this conclusion. However, we also observed that certain actively transcribed genes, including ID1, MYC, JUN, and TGFB1, escaped CG-specific hypermethylation by Mhy1 in fibrosarcoma. These findings suggested that efficiently transcribed genes were largely protected from hypermethylation by Mhy1.
We acknowledge that methylation we observed in the cells with the enforced expression of mycoplasma MTases represents a non-physiologic scenario. We anticipate that a considerably lower methylation level may exist in human cells infected by mycoplasma naturally. Also, because Mhy1 and Mhy2 target CG sites, distinguishing such sites from the intrinsic methylation by DNMTs impose a challenge, especially in highly heterogeneous patient DNA specimens.
We anticipate that cytosines aberrantly methylated by M. hyorhinis MTases could be identified in tumors exposed to mycoplasma infection. However, long-term maintenance of these methylated cytosines will be compromised if the infection was cleared. An overlap of GATC with CGs permits the long-term maintenance of aberrant methylation by endogenous DNMT1. We hypothesize that this “molecular archeology” detection of putative methylation marks in human DNA specimens could serve as an indicator of current or pre-existing mycoplasma exposures and serve for predictive, diagnostic, and therapeutic purposes. Because we did not expect the phenomenon to be restricted to the 3 mycoplasma MTases studied here, our findings unveil a potential new panorama of crosstalk between the human microbiome and epigenome.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary Material
Acknowledgment
This manuscript is dedicated to the memory of Professor N. I. Matvienko.
Funding
This work was supported by a Research Fellowship to AVC from the Institute of Predictive and Personalized Medicine of Cancer, Barcelona, Spain; by NIH Grants CA077470, CA083017 and CA157328 (to AYS) and by a Grant from Fondo Investigaciones Sanitarias of the Instituto de Salud Carlos III (to MP).
References
- 1. Pflughoeft KJ, Versalovic J. Human microbiome in health and disease. Annu Rev Pathol 2012; 7:99-122; PMID:21910623; http://dx.doi.org/ 10.1146/annurev-pathol-011811-132421 [DOI] [PubMed] [Google Scholar]
- 2. Falkow S. Bacterial entry into eukaryotic cells. Cell 1991; 65:1099-102; PMID:1905978; http://dx.doi.org/ 10.1016/0092-8674(91)90003-H [DOI] [PubMed] [Google Scholar]
- 3. Razin S, Yogev D, Naot Y. Molecular biology and pathogenicity of mycoplasmas. Microbiol Mol Biol Rev 1998; 62:1094-156; PMID:9841667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kornspan JD, Tarshis M, Rottem S. Invasion of melanoma cells by Mycoplasma hyorhinis: enhancement by protease treatment. Infect Immun 2010; 78:611-7; PMID:19917715; http://dx.doi.org/ 10.1128/IAI.01017-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ueno PM, Timenetsky J, Centonze VE, Wewer JJ, Cagle M, Stein MA, Krishnan M, Baseman JB. Interaction of Mycoplasma genitalium with host cells: evidence for nuclear localization. Microbiology 2008; 154:3033-41; PMID:18832309; http://dx.doi.org/ 10.1099/mic.0.2008/020735-0 [DOI] [PubMed] [Google Scholar]
- 6. Cimolai N. Do mycoplasmas cause human cancer? Canadian journal of microbiology 2001; 47:691-7; PMID:11575494; http://dx.doi.org/ 10.1139/w01-053 [DOI] [PubMed] [Google Scholar]
- 7. Barykova YA, Logunov DY, Shmarov MM, Vinarov AZ, Fiev DN, Vinarova NA, Rakovskaya IV, Baker PS, Shyshynova I, Stephenson AJ, et al. . Association of Mycoplasma hominis infection with prostate cancer. Oncotarget 2011; 2:289-97; PMID:21471611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Namiki K, Goodison S, Porvasnik S, Allan RW, Iczkowski KA, Urbanek C, Reyes L, Sakamoto N, Rosser CJ. Persistent exposure to Mycoplasma induces malignant transformation of human prostate cells. PloS one 2009; 4:e6872; PMID:19721714; http://dx.doi.org/ 10.1371/journal.pone.0006872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yang H, Qu L, Ma H, Chen L, Liu W, Liu C, Meng L, Wu J, Shou C. Mycoplasma hyorhinis infection in gastric carcinoma and its effects on the malignant phenotypes of gastric cancer cells. BMC Gastroenterol 2010; 10:132; PMID:21062494; http://dx.doi.org/ 10.1186/1471-230X-10-132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Masaki T, Qu J, Cholewa-Waclaw J, Burr K, Raaum R, Rambukkana A. Reprogramming adult Schwann cells to stem cell-like cells by leprosy bacilli promotes dissemination of infection. Cell 2013; 152:51-67; PMID:23332746; http://dx.doi.org/ 10.1016/j.cell.2012.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Logunov DY, Scheblyakov DV, Zubkova OV, Shmarov MM, Rakovskaya IV, Gurova KV, Tararova ND, Burdelya LG, Naroditsky BS, Ginzburg AL, et al. . Mycoplasma infection suppresses p53, activates NF-kappaB and cooperates with oncogenic Ras in rodent fibroblast transformation. Oncogene 2008; 27:4521-31; PMID:18408766; http://dx.doi.org/ 10.1038/onc.2008.103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tsai S, Wear DJ, Shih JW, Lo SC. Mycoplasmas and oncogenesis: persistent infection and multistage malignant transformation. Proc Natil Acad Sci U S A 1995; 92:10197-201; PMID:7479753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gong M, Meng L, Jiang B, Zhang J, Yang H, Wu J, Shou C. p37 from Mycoplasma hyorhinis promotes cancer cell invasiveness and metastasis through activation of MMP-2 and followed by phosphorylation of EGFR. Mol Cancer Ther 2008; 7:530-7; PMID:18347140; http://dx.doi.org/ 10.1158/1535-7163.MCT-07-2191 [DOI] [PubMed] [Google Scholar]
- 14. Kamiyama H, Suzuki K, Maeda T, Koizumi K, Miyaki Y, Okada S, Kawamura YJ, Samuelsson JK, Alonso S, Konishi F, et al. . DNA demethylation in normal colon tissue predicts predisposition to multiple cancers. Oncogene 2012; 31:5029-37; PMID:22310288; http://dx.doi.org/ 10.1038/onc.2011.652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Suzuki K, Suzuki I, Leodolter A, Alonso S, Horiuchi S, Yamashita K, Perucho M. Global DNA demethylation in gastrointestinal cancer is age dependent and precedes genomic damage. Cancer Cell 2006; 9:199-207; PMID:16530704; http://dx.doi.org/ 10.1016/j.ccr.2006.02.016 [DOI] [PubMed] [Google Scholar]
- 16. Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natil Acad Sci U S A 1999; 96:8681-6; PMID:10411935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yamashita K, Dai T, Dai Y, Yamamoto F, Perucho M. Genetics supersedes epigenetics in colon cancer phenotype. Cancer Cell 2003; 4:121-31; PMID:12957287; http://dx.doi.org/ 10.1016/S1535-6108(03)00190-9 [DOI] [PubMed] [Google Scholar]
- 18. De Carvalho DD, Sharma S, You JS, Su SF, Taberlay PC, Kelly TK, Yang X, Liang G, Jones PA. DNA methylation screening identifies driver epigenetic events of cancer cell survival. Cancer Cell 2012; 21:655-67; PMID:22624715; http://dx.doi.org/ 10.1016/j.ccr.2012.03.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sandoval J, Esteller M. Cancer epigenomics: beyond genomics. Curr Opin Genet Dev 2012; 22:50-5; PMID:22402447; http://dx.doi.org/ 10.1016/j.gde.2012.02.008 [DOI] [PubMed] [Google Scholar]
- 20. Bibikova M, Fan JB. Genome-wide DNA methylation profiling. Wiley Interdiscip Rev Syst Biol Med 2010; 2:210-23; PMID:20836023; http://dx.doi.org/ 10.1002/wsbm.35 [DOI] [PubMed] [Google Scholar]
- 21. Paz MF, Fraga MF, Avila S, Guo M, Pollan M, Herman JG, Esteller M. A systematic profile of DNA methylation in human cancer cell lines. Cancer Res 2003; 63:1114-21; PMID:12615730 [PubMed] [Google Scholar]
- 22. Varley KE, Gertz J, Bowling KM, Parker SL, Reddy TE, Pauli-Behn F, Cross MK, Williams BA, Stamatoyannopoulos JA, Crawford GE, et al. . Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res 2013; 23:555-67; PMID:23325432; http://dx.doi.org/ 10.1101/gr.147942.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev 2011; 25:1010-22; PMID:21576262; http://dx.doi.org/ 10.1101/gad.2037511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer 2004; 4:143-53; PMID:14732866; http://dx.doi.org/ 10.1038/nrc1279 [DOI] [PubMed] [Google Scholar]
- 25. Laird PW, Jaenisch R. The role of DNA methylation in cancer genetic and epigenetics. Annu Rev Genet 1996; 30:441-64; PMID:8982461; http://dx.doi.org/ 10.1146/annurev.genet.30.1.441 [DOI] [PubMed] [Google Scholar]
- 26. Vertino PM, Spillare EA, Harris CC, Baylin SB. Altered chromosomal methylation patterns accompany oncogene-induced transformation of human bronchial epithelial cells. Cancer Res 1993; 53:1684-9; PMID:8453642 [PubMed] [Google Scholar]
- 27. Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer 2011; 11:726-34; PMID:21941284; http://dx.doi.org/ 10.1038/nrc3130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, Marth C, Weisenberger DJ, Campan M, Young J, Jacobs I, et al. . Epigenetic stem cell signature in cancer. Nat Genet 2007; 39:157-8; PMID:17200673; http://dx.doi.org/ 10.1038/ng1941 [DOI] [PubMed] [Google Scholar]
- 29. Graham CH, Hawley TS, Hawley RG, MacDougall JR, Kerbel RS, Khoo N, Lala PK. Establishment and characterization of first trimester human trophoblast cells with extended lifespan. Exp Cell Res 1993; 206:204-11; PMID:7684692; http://dx.doi.org/ 10.1006/excr.1993.1139 [DOI] [PubMed] [Google Scholar]
- 30. Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics 2002; 18:1427-31; PMID:12424112; http://dx.doi.org/ 10.1093/bioinformatics/18.11.1427 [DOI] [PubMed] [Google Scholar]
- 31. Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. Circos: an information aesthetic for comparative genomics. Genome Res 2009; 19:1639-45; PMID:19541911; http://dx.doi.org/ 10.1101/gr.092759.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol 2011; 29:24-6; PMID:21221095; http://dx.doi.org/ 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chernov AV, Baranovskaya S, Golubkov VS, Wakeman DR, Snyder EY, Williams R, Strongin AY. Microarray-based transcriptional and epigenetic profiling of matrix metalloproteinases, collagens, and related genes in cancer. J Biol Chem 2010; 285:19647-59; PMID:20404328; http://dx.doi.org/ 10.1074/jbc.M109.088153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP. GenePattern 2.0. Nat Genet 2006; 38:500-1; PMID:16642009; http://dx.doi.org/ 10.1038/ng0506-500 [DOI] [PubMed] [Google Scholar]
- 35. Perucho M, Goldfarb M, Shimizu K, Lama C, Fogh J, Wigler M. Human-tumor-derived cell lines contain common and different transforming genes. Cell 1981; 27:467-76; PMID:6101201; http://dx.doi.org/ 10.1016/0092-8674(81)90388-3 [DOI] [PubMed] [Google Scholar]
- 36. Nakano H, Yamamoto F, Neville C, Evans D, Mizuno T, Perucho M. Isolation of transforming sequences of two human lung carcinomas: structural and functional analysis of the activated c-K-ras oncogenes. Proc Natil Acad Sci U S A 1984; 81:71-5; PMID:6320174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Young L, Sung J, Stacey G, Masters JR. Detection of Mycoplasma in cell cultures. Nature protocols 2010; 5:929-34; PMID:20431538; http://dx.doi.org/ 10.1038/nprot.2010.43 [DOI] [PubMed] [Google Scholar]
- 38. Calcutt MJ, Foecking MF, Rosales RS, Ellis RJ, Nicholas RA. Genome sequence of Mycoplasma hyorhinis strain GDL-1. JBacteriol 2012; 194:1848; PMID:22408248; http://dx.doi.org/ 10.1128/JB.00033-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lange C, Jugel A, Walter J, Noyer-Weidner M, Trautner TA. 'Pseudo' domains in phage-encoded DNA methyltransferases. Nature 1991; 352:645-8; PMID:1865925; http://dx.doi.org/ 10.1038/352645a0 [DOI] [PubMed] [Google Scholar]
- 40. Chernov AV, Sounni NE, Remacle AG, Strongin AY. Epigenetic control of the invasion-promoting MT1-MMP/MMP-2/TIMP-2 axis in cancer cells. J Biol Chem 2009; 284:12727-34; PMID:19286653; http://dx.doi.org/ 10.1074/jbc.M900273200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Weber M, Knoefler I, Schleussner E, Markert UR, Fitzgerald JS. HTR8/SVneo cells display trophoblast progenitor cell-like characteristics indicative of self-renewal, repopulation activity, and expression of "stemness-" associated transcription factors. Biomed Res Int 2013; 2013:243649; PMID:23586024; http://dx.doi.org/ 10.1155/2013/243649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wines DR, Talbert PB, Clark DV, Henikoff S. Introduction of a DNA methyltransferase into Drosophila to probe chromatin structure in vivo. Chromosoma 1996; 104:332-40; PMID:8575244; http://dx.doi.org/ 10.1007/BF00337221 [DOI] [PubMed] [Google Scholar]
- 43. Coleman BI, Ribacke U, Manary M, Bei AK, Winzeler EA, Wirth DF, Duraisingh MT. Nuclear repositioning precedes promoter accessibility and is linked to the switching frequency of a Plasmodium falciparum invasion gene. Cell Host Microb 2012; 12:739-50; PMID:23245319; http://dx.doi.org/ 10.1016/j.chom.2012.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang S, Tsai S, Lo SC. Induction of Constitutive High-Level Expression of c-Myc in 32D Cells by Mycoplasmas is Associated with their Ability to Prevent Apoptosis and Induce Malignant Transformation. Int J Biomed Sci 2006; 2:324-32; PMID:23675000 [PMC free article] [PubMed] [Google Scholar]
- 45. Hopfe M, Deenen R, Degrandi D, Kohrer K, Henrich B. Host cell responses to persistent mycoplasmas–different stages in infection of HeLa cells with Mycoplasma hominis. PloS one 2013; 8:e54219; PMID:23326599; http://dx.doi.org/ 10.1371/journal.pone.0054219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wojciechowski M, Czapinska H, Bochtler M. CpG underrepresentation and the bacterial CpG-specific DNA methyltransferase M.MpeI. Proc Natil Acad Sci U S A 2013; 110:105-10; PMID:23248272; http://dx.doi.org/ 10.1073/pnas.1207986110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Suva ML, Riggi N, Bernstein BE. Epigenetic reprogramming in cancer. Science 2013; 339:1567-70; PMID:23539597; http://dx.doi.org/ 10.1126/science.1230184 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.