

Abstract
Here we report inherited dysregulation of protein phosphatase activity as a cause of intellectual disability (ID). De novo missense mutations in 2 subunits of serine/threonine (Ser/Thr) protein phosphatase 2A (PP2A) were identified in 16 individuals with mild to severe ID, long-lasting hypotonia, epileptic susceptibility, frontal bossing, mild hypertelorism, and downslanting palpebral fissures. PP2A comprises catalytic (C), scaffolding (A), and regulatory (B) subunits that determine subcellular anchoring, substrate specificity, and physiological function. Ten patients had mutations within a highly conserved acidic loop of the PPP2R5D-encoded B56δ regulatory subunit, with the same E198K mutation present in 6 individuals. Five patients had mutations in the PPP2R1A-encoded scaffolding Aα subunit, with the same R182W mutation in 3 individuals. Some Aα cases presented with large ventricles, causing macrocephaly and hydrocephalus suspicion, and all cases exhibited partial or complete corpus callosum agenesis. Functional evaluation revealed that mutant A and B subunits were stable and uncoupled from phosphatase activity. Mutant B56δ was A and C binding–deficient, while mutant Aα subunits bound B56δ well but were unable to bind C or bound a catalytically impaired C, suggesting a dominant-negative effect where mutant subunits hinder dephosphorylation of B56δ-anchored substrates. Moreover, mutant subunit overexpression resulted in hyperphosphorylation of GSK3β, a B56δ-regulated substrate. This effect was in line with clinical observations, supporting a correlation between the ID degree and biochemical disturbance.
Introduction
Unlike protein kinases, mutations in serine/threonine (Ser/Thr) protein phosphatases have not commonly been associated with disorders of human development. There are 2 major Ser/Thr protein phosphatase families in the cell: protein phosphatase 1 (PP1) and protein phosphatase 2A (PP2A), together accounting for more than 90% of all phospho-Ser/Thr dephosphorylations. PP2A consists of a catalytic subunit (C), a substrate binding regulatory subunit (B), and a scaffolding subunit (A) that links B and C. Unlike the generally expressed A and C subunits, there is a plethora of B subunits with different expression patterns (1). The differential substrate preferences of the nearly 100 different PP2A holoenzymes that, in theory, can be formed by 2 C isoforms, 2 A isoforms, and at least 23 types of B subunits is largely unknown (2), particularly within the context of a whole organism. Apparently, PP2A-dependent protein dephosphorylation has a potential for regulation that may be just as fine-tuned as protein phosphorylation. Unlike protein phosphorylation, associations between mutations in PP2A subunits and genetic diseases or syndromes have not been described until recently, when 4 de novo PPP2R5D and 3 de novo PPP2R1A mutations were found among the first 1,133 parent-child trios sequenced in the United Kingdom Deciphering Developmental Disorders project (3).
Here, we add clinical descriptions and functional data to the DDD findings and present 9 additional cases with de novo PP2A subunit mutations; 7 in PPP2R5D, encoding the regulatory B56δ subunit, and 2 in PPP2R1A, encoding the scaffolding Aα subunit. Taken together, of the 11 mutations in PPP2R5D, 6 mutations and 2 mutations were identical; 3 of the 5 mutations in PPP2R1A were also identical. All Aα mutations and all but one of the B56δ mutations had the potential to hinder access of catalytically competent C subunits to B56δ-regulated substrates, suggesting a common dominant-negative disease mechanism mainly affecting B56δ-regulated Ser/Thr dephosphorylation.
Results
In cases with intellectual disability (ID) of unknown etiology, parent-child trio exome sequencing was performed to find de novo and recessive mutations that could explain the condition. De novo missense mutations in 2 subunits of the Ser/Thr phosphatase PP2A were identified in 16 individuals from the United Kingdom (7 cases), the Netherlands (7 cases), Israel (1 case), and Norway (1 case).
The 7 United Kingdom cases were found among 1,133 chromosomally normal parent-child trios (3). This suggests that the prevalence of PP2A subunit mutations in the moderate-to-severe ID group without pathogenic copy number aberrations is around 0.6%. In the United Kingdom, this was part of the large DDD project (http://www.ddduk.org); in other cases, this was done as part of routine diagnostics. In 11 cases, de novo missense mutations in PPP2R5D, encoding the regulatory B56δ PP2A subunit, were found. In 5 other cases, a de novo missense mutation in PPP2R1A, encoding the scaffolding Aα subunit of PP2A, was found. Six mutations and 2 mutations in PPP2R5D were identical, and 3 PPP2R1A mutations were identical. Details on all mutations can be found in Table 1. Other trio exome sequencing results indicating a de novo change of possible relevance or a recessive condition of potential interest can be found in Supplemental Table 1 (supplemental material available online with this article; doi:10.1172/JCI79860DS1). In 10 cases, such findings were made, but based on bioinformatic evaluation of the variants and the clinical features of the patients, all but one of these findings could easily be excluded as causative factors for the phenotype. The exception was case 15, which had heterozygocity for a TMEM67 splice mutation and a few signs that were compatible with a ciliopathy (e.g., unilateral postaxial polydactyly). However, this could also be a random finding, since a second TMEM67 mutation was not found upon Sanger sequencing. In addition, detecting the same de novo missense mutations in patients with identical clinical features is, in itself, evidence in support of causality, especially when supported by functional data (see below). As a crude estimation, the likelihood of finding 10 de novo missense mutations in the same 9–amino acid stretch of B56δ by chance should be less than 10–50 (see Statistics).
Table 1. De novo mutation details and the corresponding cases.
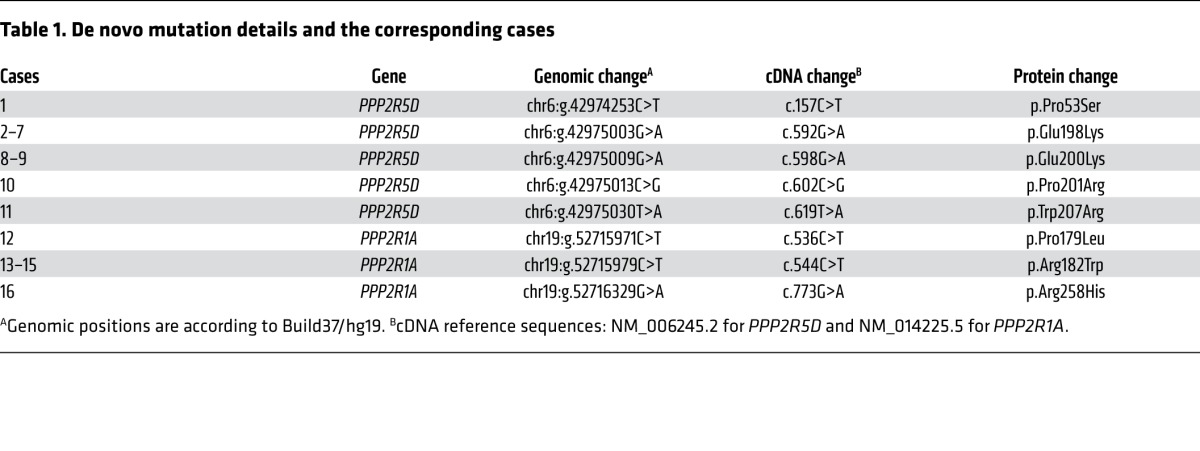
The clinical features of the 11 PPP2R5D cases and the 5 PPP2R1A cases are summarized in Tables 2 and 3, respectively. Despite mutations occurring in 2 different PP2A subunit genes with different biochemical functions (regulatory and scaffolding), there are clinical similarities between the cases. All patients were born after a normal pregnancy, and 15/16 cases had birth weights within normal range. In 2 cases, breech deliveries were reported, and in 2 other cases, emergency cesarean sections had to be performed. After birth, ID and hypotonia were common features in all cases. Despite pronounced and long-lasting hypotonia, feeding difficulties were usually not a major problem, and only one case had gastrostomy. In 12/16 cases, the degree of ID was severe, and this correlated with very late independent walking, usually around age 6–7 years. The exceptions were the 4 patients with E200K, P201R, or W207R PPP2R5D mutations (see below for functional explanation), who learned to walk between 1½ and 2¼ years of age and had mild/moderate ID (Table 2). These 4 cases were also the only ones with language development beyond a few words. Seven out of 16 patients had epilepsy, including one of the mild ID cases. Only one patient had short stature (case 1 with a P53S mutation, see Table 2), and he was the only PPP2R5D case that was microcephalic. In the other PPP2R5D cases, head circumferences were from upper-normal range to pronounced macrocephaly, and in the latter cases, hydrocephalus was suspected. In contrast, most PPP2R1A cases were normocephalic or microcephalic, and hydrocephalus was initially suspected in only one case (Table 3). In all these patients, the corpus callosum was absent or almost absent, a feature that distinguished PPP2R1A cases from PPP2R5D cases. In contrast, facial features were overlapping (Figure 1): A hypotonic and sometimes also elongated face with tented upper lip, mild hypertelorism with downslanting palpebral fissures, and frontal bossing in the PPP2R5D cases.
Table 2. Clinical features in cases with de novo PPP2R5D missense mutations.
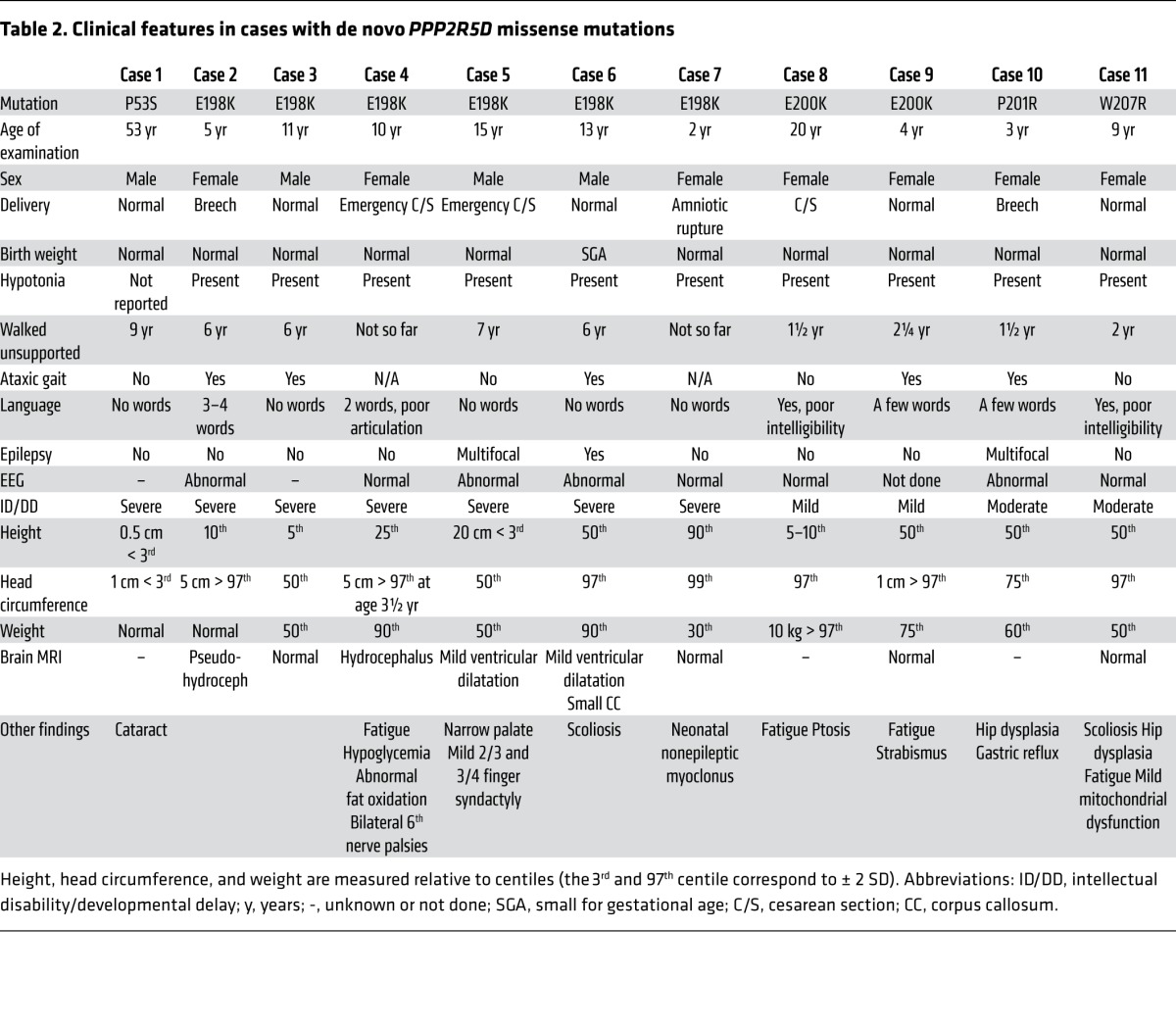
Table 3. Clinical features in cases with de novo PPP2R1A missense mutations.
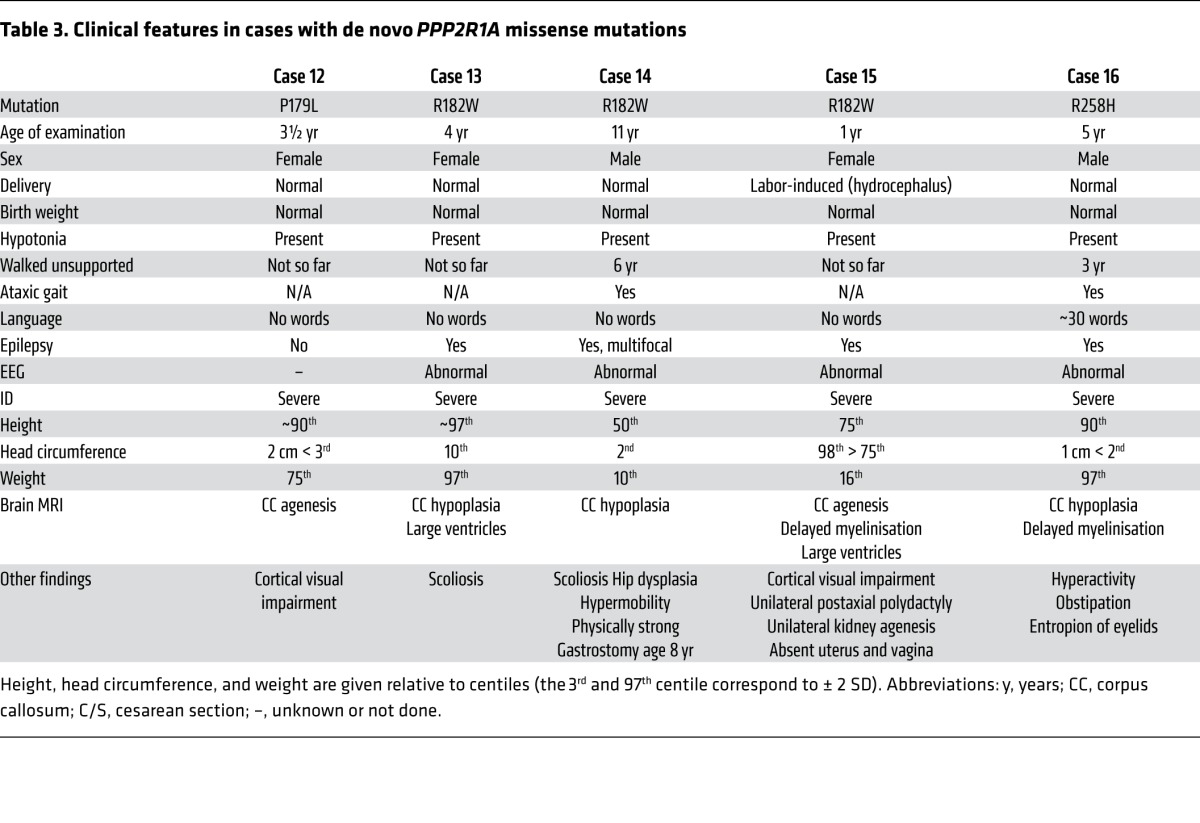
Figure 1. Facial photographs.

The display shows the facial features of the PPP2R5D cases that consented to facial photographs being shown and the PPP2R1A cases.
The finding of recurrent and clustered de novo missense mutations in 2 PP2A subunit genes (PPP2R5D and PPP2R1A) suggested a dominant-negative– or gain-of-function–related disease mechanism, rather than haploinsufficiency or loss-of-function. All but one of the PPP2R5D mutations (E198K, E200K, P201R, and W207R) clustered in a highly conserved acidic loop that faces the A and C subunits (4–6). This acidic surface corresponds to the extended loop between α-helices 3 and 4 of HEAT domain 2 in the crystal structure of the highly related B56γ isoform (Figure 2A). Only the P53S mutation (case 1 in Table 2) localized outside this loop, i.e., in the B56δ-specific N-terminal domain.
Figure 2. Binding of mutant and WT B56δ to the A or C subunits.

(A) Model of the highly conserved B56 acidic loop, harboring 4 ID-associated B56δ missense mutations, based on crystallographic data from PP2A-B56γ (PDB code: 2IAE)(5). The corresponding residues in B56γ (highlighted in white) are displayed with amino acid numbering according to B56δ. Note that the E198 residue directly contacts the C subunit. Color code: C subunit, blue; A subunit, yellow; B56γ, red. The structure was analyzed and visualized with Molsoft MolBrowser 3.7. (B) Cellular binding assays of ID-associated B56δ mutants and endogenous A and C subunits. EGFP-tagged WT B56δ, 5 ID-associated B56δ mutants (P53S, E198K, E200K, P201R, and W207R), or EGFP alone (–) were ectopically expressed in HEK293 cells. Following EGFP trapping, the presence of endogenous A and C subunits in the trapped complexes was examined by immunoblotting (IB). After quantification of the band intensities with ImageJ software, the ratios between EGFP and C signals — and between EGFP and A signals — were determined and calculated relative to B56δ WT control. Mean values and a representative image of 4 independent experiments are shown (1-way multiple-comparisons ANOVA; *P < 0.05, **P < 0.01).
To investigate if the PPP2R5D missense mutations affected subunit interactions, a human embryonic kidney cell line — HEK293 cells, a well-known model from previous PP2A subunit interaction studies (7) — was transfected with EGFP-tagged WT or mutant B56δ subunits in order to study subunit interactions. All ID-associated B56δ mutants except P53S showed deficient holoenzyme formation, i.e., A- or C-to-B56δ association (Figure 2B). To check if others had discovered missense variants in the same acidic B56δ loop, Broad institute’s ExAC browser (http://exac.broadinstitute.org/) was consulted. Only 2 other missense variants (P196L and P201S) were reported. Interestingly, the latter variant was in the same residue as the de novo mutation (P201R) in case 10, although the amino acid change was different. Unlike P201R, we found that P201S failed to show any significant A or C binding defects (Supplemental Figure 1), further strengthening our working hypothesis that a charge change in the acidic B56δ loop could be pathogenic.
Our cellular binding assays with Glutathione S-Transferase–tagged (GST-tagged) B subunits and HemAgglutinin-tagged (HA-tagged) WT or mutant Aα subunits revealed that all 3 PPP2R1A mutations also affected PP2A holoenzyme formation (Figure 3). Surprisingly, interaction with the C subunit was hindered, despite the Aα mutations being in HEAT domains predicted to interact with B (Figure 3A). The mutations’ effect on B subunit binding was complex (Figure 3B). All Aα mutants lacked significant binding to the B55 (also called B) family members tested (isoforms B55α and B55β), as well as to the B56 (also called B’ or PR61) family members tested (B56α and B56γ). On the other hand, binding to B56δ was almost entirely retained, whereas B56ε bound significantly less. For PR72, a member of the B’’ family of PP2A regulatory subunits, binding was retained to Aα-P179L but was completely lost to the Aα-R182W and Aα-R258H mutants (Figure 3B). These (mutant) Aα binding characteristics were confirmed for endogenous B55α and B56δ subunits, for which good-quality, isoform-specific antibodies are available (Figure 4A). These data could be compatible with a dominant-negative effect on, notably, B56δ for all Aα mutants, and on PR72 for Aα-P179L, provided that C binding would be lost or diminished in the B56δ-Aα mutant complex. To provide direct evidence that mutant Aα can complex with B56δ without C, we expressed HA-Aα mutants or HA-Aα (WT) in HEK293 cells stably expressing EGFP-TEV-B56δ (WT) and analyzed the presence of endogenous C in anti-HA immunoprecipitates from the tobacco etch virus–cleaved (TEV-cleaved) eluates of GFP-trapped B56δ (Figure 4B). While C subunit was clearly present in B56δ-Aα (WT) complex (as expected from normal holoenzyme formation), it was barely detectable in the B56δ-Aα-P179L complex, indicating that mutant Aα-P179L may bind B56δ but not C. In contrast, the B56δ-Aα-R182W and B56δ-Aα-R258H complexes still bound a significant amount of C (Figure 4B). However, measurements of specific PP2A activity in these samples showed a decrease in phosphatase activity for all Aα mutants compared with WT (Figure 4C), suggesting that the C subunit present in these B56δ-mutant A-C complexes is catalytically impaired.
Figure 3. Binding of mutant and WT Aα to C and B subunits.

(A) PP2A-C subunit binding assays: HA-tagged WT Aα, 3 ID-associated Aα mutants (P179L, R182W, and R258H), or an empty HA-vector (–) were transfected into HEK293 cells. Following anti-HA immunoprecipitation, the presence of endogenous C subunit in the immunoprecipitates was examined by immunoblotting (IB). After quantification of the band intensities with ImageJ software, the ratios between HA and C signals were determined and calculated relative to WT Aα control. Mean values and a representative image of 3 independent experiments are shown (1-way multiple-comparisons ANOVA, **P < 0.01). (B) PP2A B subunit binding assays: Several GST-tagged B subunits, belonging to 3 different families (B55 or B, B56 or B’, and B’’) or GST alone (–) were coexpressed in HEK293 cells with HA-tagged WT Aα, or ID-associated Aα-P179L, R182W, and R258H mutants. The presence of HA-Aα (WT or mutant) in the complete lysates and the isolated GST pulldown complexes was determined by IB. After quantification of the band intensities with ImageJ software, the ratios between GST and HA signals were determined and calculated relative to WT Aα control (which were set to 100% for each B-type subunit pulldown). Mean values and a representative image of 3 independent experiments are shown (1-way multiple-comparisons ANOVA; *P < 0.05, **P < 0.01, ***P < 0.001).
Figure 4. Analysis of Aα mutant complexes.

(A) Endogenous B subunit binding assays: HA-tagged WT Aα, Aα mutants (P179L, R182W, and R258H), or an empty HA-vector (–) were transfected into HEK293 cells. Following anti-HA immunoprecipitation, presence of endogenous B56δ and B55α subunits in the immunoprecipitates was examined by immunoblotting (IB). (B) Formation of B56δ-(mutant Aα)-C complexes: HEK293 cells stably expressing EGFP-TEV-B56δ were transfected with HA-Aα, HA-Aα mutants, or empty HA-vector (–). Following EGFP-trapping and cleavage of the trapped complexes with TEV protease, the eluates were subjected to HA immunoprecipitation and the immunoprecipitates analyzed by IB with anti-HA, anti-C, and anti-B56δ antibodies. After quantification of the band intensities (ImageJ), the ratios between C and HA signals were determined and calculated relative to WT Aα control (set to 100% in each IP-on-IP experiment). Mean values and a representative image of 4 independent experiments are shown (1-way multiple-comparisons ANOVA; *P < 0.05). (C) PP2A activity measurements in B56δ-(mutant Aα)-C complexes. The pmol number of released phosphate from K-R-pT-I-R-R phosphopeptide (350 μM) was determined by Malachite Green for each B56δ-(mutant Aα)-C complex (retrieved as in B). To obtain specific C activities, this number was divided by the amount of C in the respective samples, as determined by IB and following quantification by ImageJ software. All specific activities were eventually recalculated relative to WT Aα control (set to 100%). Mean values and one representative image of 3 independent experiments are shown (1-way multiple-comparisons ANOVA; **P < 0.01).
Because previous reports had indicated increased B56δ subunit degradation upon gradual reduction of A subunit levels (8, 9), we examined the stability of our mutant subunits, since unstable mutant proteins would preclude the interpretation of our functional data. In line with our hypothesis, we did not experience any problems expressing ID-associated B56δ subunits in cells (Figures 2–4). Furthermore, protein-synthesis–blocking experiments showed that the ID-associated B56δ (P53S, E198K) or Aα (P179L, R182W) mutants tested appeared more long-lived than the WT subunits in our assay (Supplemental Figure 2).
Taken together, for all but one (P53S in PPP2R5D) of the de novo mutations identified, our biochemical data are consistent with a common defect in PP2A-B56δ–dependent dephosphorylation. A-C binding–defective B56δ mutants, or C binding– but not B56δ binding–defective Aα-mutants, and B56δ binding Aα-mutants harboring a catalytically impaired C subunit, may all block dephosphorylation of PP2A-B56–specific substrates and interfere with phosphorylation-dephosphorylation dynamics in the brain. In line with this hypothesis, overexpression of the E198K B56δ mutant or the R182W Aα mutant in HEK293 cells resulted in increased phosphorylation of GSK-3β Ser9, an established PP2A-B56δ substrate in this cell line (Figure 5 and ref. 10).
Figure 5. Increased phosphorylation of GSK-3β Ser9 upon expression of B56δ-E198K or Aα-R182W.

GFP-tagged WT B56δ, E198K mutant B56δ, or GFP alone (–) (left); or HA-tagged WT Aα, R182W mutant Aα, or HA alone (–) (right) were expressed in HEK293 cells, and the effect on GSK-3β phosphorylation determined by immunoblotting (IB) with the indicated antibodies. Total GSK-3β and phospho-Ser9 GSK-3β signals were determined on different blots, which were both developed for vinculin to correct for loading differences. In cells expressing the ID-associated mutants, higher phospho/total GSK-3β ratios were found, relative to cells transfected with empty expression vector (in which case, this ratio was set to 100%) or cells expressing WT subunits. Mean values and one representative image of 3 independent experiments are shown (1-way multiple-comparisons ANOVA; *P < 0.05, **P < 0.01).
Discussion
The presented work demonstrates that de novo missense mutations in genes encoding PP2A subunits may cause syndromic ID — and probably also nonsyndromic ID, since the facial dysmorphism in these cases is subtle (Figure 1). The PPP2R5D and PPP2R1A mutations disrupt B56δ-dependent dephosphorylation dynamics and link PP2A dysfunction to congenital brain dysfunction.
In general, the Aα cases were more severely affected than the B56δ cases. All had severe ID, absent speech, diminished brain growth, and partial or complete agenesis of the corpus callosum (Table 3). This is in line with the expected greater difficulty to compensate for a general scaffolding (A) subunit dysfunction than a specific regulatory (B) subunit dysfunction, as reflected by our biochemical data showing additional loss or reduction of holoenzyme assembly of many different PP2A complexes (B55α, B55β, B56α, B56γ, and B56ε) for these Aα mutants (Figure 3B). The Aα scaffolding subunit is highly flexible, composed of 15 tandem repeat HEAT motifs (11) that mediate interactions with a regulatory B subunit (HEAT repeats 1–8) and the C subunit (HEAT repeats 11–15)(4, 5, 12, 13). Two ID-associated PPP2R1A mutations (P179L and R182W) cluster in HEAT domain 5 of Aα, and one (R258H) occurs in HEAT domain 7; these mutations are involved in contacts with subunits of all B families (4, 5, 12, 13). However, for these mutations, loss-of-function might be less critical than substrate protection and altered phosphorylation dynamics; hence, the retained binding to PR72 for Aα-P179L could have additional functional consequences due to sequestration of PR72 in a complex deficient in C subunit binding or with decreased specific activity of C.
Besides its scaffolding function, Aα is a major player in the biogenesis of active PP2A holoenzymes (14). This highly regulated but incompletely understood process does not only involve simple trimeric assembly of the A, B, and C subunits, but it also involves several activation steps of the C subunit, which is de novo translated as an inactive enzyme (15). It has been suggested that some of these activation steps require or are facilitated by the A subunit (16, 17), explaining why A-subunit mutations may affect the specific activity of the associated C subunit, as observed here within the B56δ-(mutant A)-C complexes (Figure 4C). Additional activity measurements performed directly in anti-HA immunoprecipitates of HA-tagged (mutated) A subunits seem to further confirm this hypothesis (Supplemental Figure 3). Hence, it can be further rationalized why mutations in the A subunit have a much more severe effect on the PP2A system as a whole, as opposed to mutations in B56δ, which affect a single PP2A holoenzyme complex.
PPP2R5D encodes the longest isoform of the B’ family of PP2A regulatory subunits and harbors unique N- and C-terminal extensions, which are predicted to be important for substrate recognition and/or subcellular targeting (18). Ten out of 11 PPP2R5D mutations were located in a conserved acidic loop of B56δ needed for holoenzyme formation (Table 1), and all mutations introduced a positively charged residue (either arginine or lysine). Only one mutation (P53S) was atypical, and this case also had a different clinical picture: it was the only PPP2R5D case with short stature and microcephaly (Table 2). In theory, P53S in the B56δ-specific N-terminal domain might change the PP2A-B56δ interaction with relevant substrates or introduce a new phosphorylation site that affects regulation by protein kinases. Such changes could easily have a gain-of-function or dominant-negative effect.
We also observed a correlation between the degree of biochemical disturbance and clinical severity. Among the PPP2R5D cases (Table 2), the 6 patients with E198K mutations were the most severely affected, in line with a near absence of A and C subunit binding. The least-affected individuals were the E200K cases, both with mild ID correlating with some residual A and C binding capability (Figure 2B). Notably, E198 is the only one of the 5 mutated residues that directly interacts with the catalytic subunit (Figure 2A and refs. 4–6). Additionally, since all B subunits, except B56δ and PR72, have been shown to make stabilizing contacts with the C subunit tail (5, 19), C-subunit binding to B56δ, and possibly PR72, may be particularly Aα dependent.
A brain-restricted phenotype is not unexpected for B56δ mutations because PPP2R5D is expressed mainly in the brain, particularly in the striatum (18, 20, 21). The brain-restricted phenotype of the Aα mutations (P179L, R182W, and R258H) is more unexpected, since PPP2R1A is generally expressed as a common scaffolding subunit for many different PP2A holoenzymes (22). Nevertheless, other malformations than severe corpus callosum hypogenesis were not found in the 5 PPP2R1A cases (Table 3). The restricted phenotype may be related to our finding that mutated A subunits bound most tightly to B56δ and (for P179L) also PR72 (Figure 3B), both of which are expressed in the brain, notably in the striatum where both B56δ and PR72 (encoded by PPP2R3A) control the dephosphorylation of the neural dopamine-regulated inhibitor of PP1 (DARPP-32) (23–25).
Despite the severe intellectual dysfunction in most patients, B56δ does not appear essential for mammalian brain development, since Ppp2r5d knockout mice have intact learning and memory despite ataxia and tauopathy (21). This also suggests that our patients’ ID was not caused by haploinsufficiency or a mere loss of function. It is therefore tempting to speculate that the mutated B56δ subunits may not only interfere in a dominant manner with dephosphorylation of B56δ binding PP2A substrates, as shown for GSK-3β in HEK293 cells (Figure 5), but also with subcellular anchoring of PP2A via B56δ, and thereby with control of localized signaling. Thus, A-C binding–deficient B56δ mutants may still form complexes with B56δ partners, but without promoting dephosphorylation. Such dysphosphorylation may have far-reaching consequences for regulation of localized signaling. One example could be the signaling complex scaffolded by the neural variant of the cAMP-dependent PKA anchoring protein mAKAP, that binds B56δ and several other phosphatases (PP1, PP2B) and kinases (PKA, PDK1, RSK3, ERK5) (26, 27). Dephosphorylation of PP2A-B56 substrates may also be hindered, e.g., the transcription factor and PKA-substrate HAND (28), the neural cyclin-dependent kinase 5 (CDK5) activator CDK5R1 (21), and DARPP-32 (24, 25). PKA activates DARPP-32 directly by Thr-34 phosphorylation and indirectly by PP2A-B56δ–dependent activation through Thr-75 dephosphorylation (24). B56δ has several sites for PKA phosphorylation that activate PP2A-B56δ phosphatase activity (24, 29). Moreover, PKA-activated PP2A-B56δ–dependent dephosphorylation of another DARPP-32 phosphorylation site (Ser-97) induces nuclear import — mediating dopamine-dependent epigenetic functions (25) — and lack of nuclear PP2A-B56δ targeting has been associated with juvenile myoclonic epilepsy (30). There is also evidence that PP2A-B56δ regulates both expression (30) and activity (31) of tyrosine hydroxylase, the rate-limiting enzyme in catecholamine synthesis. Which of these candidate substrates will eventually be of pathologic relevance in our ID patients remains to be further determined in appropriate cellular and animal models.
A model for a common dominant-negative effect of mutant B56δ and Aα subunits is depicted in Figure 6. This model explains why all our biochemical findings are compatible with B56δ-dependent PP2A dysregulation. Our model is also supported by a recent paper showing that Aα can form a tight complex in vitro with members of the B’’ subunit family, including PR72, in the absence of the C subunit (32). It may be of interest to study if fingolimod (FTY720), a PP2A activator and immunosuppressant that is licensed for treatment of multiple sclerosis (33–35), or FDA-approved compounds of the phenothiazine family that were recently discovered as PP2A activators (36) may improve brain function in these patients.
Figure 6. Mechanistic model.

Top panels model the physiological situation in which the B-type subunit dictates subcellular targeting, substrate specificity, and substrate dephosphorylation by the C subunit. Conceivably, certain B subunits (like B56δ) or A-B dimers could dock to substrates independent of holoenzymes (alternatives labeled with question marks). Bottom panel displays the pathological situation in which (i) a B subunit mutation hindering interaction of the A and C subunit, (ii) an A subunit mutation hindering interaction of the C subunit but not the B subunit, or (iii) an A subunit mutation resulting in the incorporation of a catalytically impaired C subunit into the trimeric complex result in protection of B subunit–directed substrate dephosphorylation by a competition-based, dominant-negative mechanism. The common feature in all ID cases described here is hindered access of the PP2A activity to B56δ-specific PP2A substrates. S, substrate; P, phosphate.
For future evaluation of de novo mutation origin (paternal or maternal)(37), it is of interest that all 3 PPP2R1A mutations also are found in the Sanger Institute’s Catalogue of Somatic Mutations in Cancer (the COSMIC database; http://cancer.sanger.ac.uk/cosmic), mainly in endometrial and ovarian cancers (38, 39). Aα P179L/P179R, R182W, and R258H are by far the most prevalent mutations. A growth advantage may also explain mutation recurrence if these de novo mutations turn out to be solely paternal (40). Since 88% of the cancer-associated Aα mutations are of the missense-variant, a dominant-negative effect also in cancer promotion is likely. None of our patients have been diagnosed with or treated for cancer. The cancer risk might not be increased, in line with what is usually the case for congenital gain-of-function mutations in other cancer-related pathways like the RAS/MAPK pathway or the PI3K/AKT cascade. Only further patients and patient follow-ups will answer this question, but a major cancer risk seems unlikely.
The tumor-suppressor effect of PP2A may operate by KRAS/MAPK cascade inhibition, KRAS/ARF/TP53 cascade inhibition, or PI3K/AKT/TP53 cascade inhibition (33). Somatic mosaic activation of the PI3K/AKT cascade causes the megalencephaly-capillary malformation-polymicrogyria (MCAP) and megalencephaly-polymicrogyria-polydactyly-hydrocephalus (MPPH) group of overgrowth syndromes (41). These patients have variable ID, a tendency to develop hydrocephalus and epilepsy, and dysmorphic facial features, including frontal bossing with hypotonia, tented upper lip, and deep-set eyes. The latter features are shared with several of our patients (Figure 1). It is therefore relevant to consider B56δ-dependent PP2A dysregulation syndrome (which we propose to be designated B56deltopathies) among the differential diagnoses to the MCAP/MPPH group of syndromes, at least in some cases. It is conceivable that the B56δ mutations may affect only a subgroup of PP2A substrates located distally in the PIK3 signaling cascade — such as GSK-3β Ser9, a well-established Akt phosphorylation site — since the proximal steps do not appear to be subject to B56δ-dependent dephosphorylation (42).
In summary, we have demonstrated that de novo missense mutations in the PPP2R5D and PPP2R1A genes encoding PP2A subunits represent a new mechanism for ID, due to disrupted B56δ-dependent dephosphorylation dynamics and PP2A dysfunction.
Methods
Case detection.
There were 7 Dutch patients (from RUMC: cases 1, 8–9, and 11; from UMC Utrecht: cases 7, 15, and 16). Six of these cases were identified through routine diagnostic exome sequencing of patient-parent trios. These patients showed no pathogenic copy number changes (high-resolution copy number array). Putative de novo variants were validated by Sanger sequencing of blood DNA. In 4 of 7 cases, other de novo variants were identified, but none of these were likely to cause the phenotype (Supplemental Table 1). A possible exception was case 16, in which heterozygosity for a likely pathogenic mutation in the TMEM67 was identified. The patient had clinical features partly consistent with a ciliopathy (unilateral postaxial polydactyly, unilateral kidney agenesis, and absent uterus). However, a second TMEM67 mutation was not identified after Sanger sequencing. Cases 1 and 11 have been previously published as part of studies showing the power and impact of next-generation sequencing–based (NGS-based) technologies in a clinical diagnostic setting without clinical details on the patients’ phenotypes or functional evaluation of the mutations (43, 44). Case 8 was identified as part of large-scale resequencing study of candidate ID genes using molecular inversion probes (MIPs). PPP2R5D was one of 42 candidate ID genes tested in 1,300 cases with a clinical diagnosis of ID and in whom previous molecular diagnostic tests were negative.
The 7 United Kingdom patients (cases 3–5, 10, and 12–14 in Tables 2 and 3) were recruited to the DDD study by the United Kingdom National Health Service or the Republic of Ireland Regional Genetics Service (3). Recruitment criteria were patients with neurodevelopmental disorders and/or congenital anomalies, abnormal growth parameters, dysmorphic features, and unusual behavior. DNA samples from patients and parents were analyzed by the Wellcome Trust Sanger Institute using high-resolution microarray analysis (array-CGH and SNP-genotyping) to investigate copy number variations (CNVs) in the child, and exome sequencing to investigate single nucleotide polymorphisms (SNPs) and small insertions/deletions (indels). Putative de novo sequence variants were validated using targeted Sanger sequencing of blood-sample DNA. All genomic variants were annotated with the most severe consequence predicted by Ensembl Variant Effect Predictor (VEP) (45) and their minor allele frequencies observed in diverse population samples. Likely, diagnostic variants were fed back to referring clinical geneticists for validation and discussion with the family via the patient’s record in Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources (DECIPHER; Ensembl) (46), where they can be viewed in an interactive genome browser. Full genomic datasets were also deposited under accession number EGAS00001000775 in the European Genome-Phenome Archive (www.ebi.ac.uk/ega).
The de novo PPP2R5D mutations in the Norwegian (case 2) and Israeli (case 6) patients were identified by exome sequencing of parent-child trios in a diagnostic setting. Only the PPP2R5D variant remained as true de novo after filtering and verification by Sanger sequencing of blood DNA, and no pathogenic copy number changes were detected by a high-resolution copy number array.
Biochemical investigations.
To study the functional consequences of the de novo missense mutations, WT Aα and B56δ (isoform 1) cDNAs were cloned into HA-tag (pMB001) and EGFP-tag (pEGFP-C1) eukaryotic expression vectors, respectively. The different PP2A B-subunit cDNAs were in a GST-tag eukaryotic expression vector, as described (19). PCR-based site-directed mutagenesis (Stratagene) was performed directly in the pMB001 or pEGFP vectors with proofreading Pwo polymerase (Roche Applied Science) and complementary DNA oligonucleotide primers (Sigma-Aldrich) containing the desired point mutations (primer sequences in Supplemental Table 2). All mutations were confirmed by sequencing (LGC Limited). Thereafter, HEK293 cells (ATCC) were transfected with PEI transfection reagents according to standard protocol. Forty-eight hours after transfection, cells were rinsed with PBS, lysed in 200 μl NET buffer (50 mM Tris pH 7.4, 150 mM NaCl, 15 mM EDTA, and 1% Nonidet P-40) containing protease and phosphatase inhibitor cocktail (Roche Applied Science), and centrifuged for 15 minutes at 13,000 g. In case phosphatase activity needed to be measured, phosphatase inhibitors were omitted from the lysis buffer.
For EGFP trapping, cell lysates were incubated at 4°C for 1 hour with wash buffer (10 mM Tris-HCl pH 7.5, 0.5 mM EDTA, and 150 mM NaCl) and 15 μl GFP-trap-A beads (ChromoTek GmbH) on a rotating wheel. The beads were washed 4 times with 0.3 ml of wash buffer.
For GST pulldown, cell lysates were incubated at 4°C for 1 hour with NENT100 buffer (20 mM Tris-HCl pH 7.4, 1 mM EDTA, 0.1% Nonidet P-40, 25% glycerol, and 100 mM NaCl) containing 1 mg/ml bovine serum albumin and 25 μl glutathione-Sepharose beads (GE Healthcare) on a rotating wheel. The beads were washed 2 times with 0.3 ml of NENT100 containing 1 mg/ml bovine serum albumin, and 2 times with 0.3 ml of NENT300 containing 300 mM NaCl.
For HA immunoprecipitation, the lysates were precleared with Protein A-Sepharose beads (GE Healthcare) for 1 hour, and incubated on a rotating wheel at 4°C for 2 hours with 1 μg HA antibody (Sigma-Aldrich) in TBS/1% Nonidet P-40. Protein A-Sepharose beads were added for 1 hour, and beads were washed 2 times in NENT300 and 2 times in TBS/0.1% Nonidet P-40. Alternatively, 25 μl HA-agarose beads (Sigma-Aldrich) were directly added to the lysates and incubated on a rotating wheel in 500 μl TBS/0.1% Nonidet P-40 for 1.5 hours at 4°C. Beads were washed 4 times in TBS/0.1% Nonidet P-40.
In all cases, bound proteins were eluted by the addition of NuPAGE sample buffer (Invitrogen) and boiling. The eluted proteins were subsequently analyzed by SDS-PAGE on 4%–12% (wt/vol) Bis-Tris gels (Bio-Rad) and Western blotting. The membranes were blocked in 5% milk solution in TBS/0.1% Tween 20 for 1 hour at room temperature and subsequently incubated with the primary antibody overnight at 4°C. The following primary antibodies were used: mouse monoclonal anti-GST (Sigma-Aldrich), anti-HA (Sigma-Aldrich), anti-GFP (Corning), anti–PP2A-A subunit (supplied by S. Dilworth, Middlesex University, London, United Kingdom), anti–PP2A-C subunit (BD Biosciences); and rabbit polyclonal anti-B55α (Cell Signaling Technology), and anti-B56δ (20). After washing in TBS/0.1% Tween 20, the membranes were incubated at room temperature for 1 hour with horseradish peroxidase–conjugated secondary antibodies (Dako) and developed using a Pierce enhanced chemiluminescence detection system (Thermo Fisher Scientific). All densitometric quantifications were done with ImageJ software.
For the IP-on-IP approach, a polyclonal population of HEK293 cells stably expressing EGFP-TEV-B56δ was used (selected with 2 μg/ml puromycin). The EGFP-TEV expression vector was a gift of E. Heroes (KU Leuven, Leuven, Belgium). Forty-eight hours after transfection with HA-Aα (pMB001) or HA-mutant Aα (pMB001), EGFP trapping was performed, and the trapped complexes were incubated overnight at 4°C with 0.2 μg/μl of recombinant TEV protease in TEV cleavage buffer (TBS, 1 mM DTT, 0.5 mM EDTA). Following addition of EDTA (1 mM), PMSF (1 mM), and TLCK (1 mM), the TEV eluates were subjected to HA immunoprecipitation with HA-agarose beads and the washed immunoprecipitates were analyzed by immunoblotting with anti-HA, anti–PP2A-C, and anti-B56δ antibodies (20, 21).
For PP2A activity measurements, the HA-agarose beads were washed once more with 20 mM Tris HCl pH 7.4 plus 1 mM DTT, and finally resuspended in 60 μl enzyme dilution buffer (catalog 20-169, Millipore). All assays were performed with 20 μl of this phosphatase suspension and 4.5 μl of 2 mM stock of K-R-pT-I-R-R phosphopeptide (catalog 12-219, Millipore) for 10–60 minutes at 30°C (still in the linear range of the assay). The released free phosphate was determined by the addition of malachite green solution (10/1 mix of solution A [catalog 20-105, Millipore] and solution B [catalog 20-104, Millipore]). After 15 min incubation at room temperature, absorbance at 630 nm was measured in a multi-channel spectrophotometer. Picomolar amounts of phosphate released were calculated by comparison with a standard curve of known KH2PO4 concentrations, as outlined in the manufacturer’s instructions (Ser/Thr Phosphatase Assay Kit 1, Millipore). Specific phosphatase activity was obtained by correcting these absolute values for amount of C present in the samples, as determined by immunoblotting with anti-C antibodies and quantification of the signals by ImageJ software.
For protein-stability analysis, HEK293 cells were transfected with EGFP-B56δ (WT); with EGFP-B56δ-P53S or EGFP-B56δ-E198K mutants (pEGFP-C1); or with HA-Aα (WT), HA-Aα-R182W, or HA-Aα-P179L mutants (pMB001), one 10 cm plate per plasmid. Twenty-four hours after transfection, each 10-cm plate was split over 6 wells on a 6-well plate, in which eventually 50 μM cycloheximide (CHX, Sigma-Aldrich) was added per well to block translation. Following incubation with CHX for different time points (0, 10, and 24 hours), whole-cell lysates were prepared in NET lysis buffer and further analyzed by immunoblotting with anti-vinculin mouse monoclonals (Sigma-Aldrich), anti-HA, or anti-GFP antibodies. Band intensities were quantified using ImageJ software.
Statistics.
Statistical analysis of biochemical data was done with 1-way multiple-comparisons ANOVA, and P < 0.05 was considered to be significant.
The calculation of the chance likelihood for finding 10 de novo mutations in the same 9–amino acid stretch of B56δ was based on the following assumptions: The target size is <10–6 of the total ORF size, the number of random missense changes per generation is on average 1, and the index is removed from the equation. This gives a chance likelihood of less than (10–6)9, or <10–54, not taking the lack of similar variants in ExAC into account. The chance of finding a similar variant in the whole ID dataset by chance is about 2 × 10–3, assuming that about 2,000 ID cases were tested. In that case, the phenotype should also be random, and this was not the case.
Study approval.
The DDD study has UK Research Ethics Committee (REC) approval (10/H0305/83, granted by the Cambridge South REC, and GEN/284/12 granted by the Republic of Ireland REC). In other cases, ascertainment of patients was part of the clinical routine. All patients’ families have consented to publication of clinical findings. Written informed consent was also obtained for publication of all facial photographs presented in Figure 1.
Supplementary Material
Acknowledgments
The DDD study presents independent research commissioned by the Health Innovation Challenge Fund (HICF-1009-003), a parallel funding partnership between the Wellcome Trust and the Department of Health, and the Wellcome Trust Sanger Institute (WT098051). For details on the members of the study, see Supplemental Table 3. The views expressed in this publication are those of the author(s) and not necessarily those of the Wellcome Trust or the Department of Health. The research team acknowledges the support of the NIH Research, through the Comprehensive Clinical Research Network. Concerning the biochemical studies, we thank S. Dilworth for the gift of monoclonal PP2A-A subunit antibody and E. Heroes for the TEV-EGFP eukaryotic expression vector. Funding was provided by the KU Leuven Research Fund (OT/13/094 to V. Janssens), the Research Foundation Flanders (G.0582.11 to V. Janssens), and the IAP program of the Belgian federal government (P7/13 to V. Janssens). D. Haesen received a fellowship of the Flemish Agency for Innovation by Science and Technology (IWT). A. Hoischen was supported by the Netherlands Organization for Health Research and Development (ZonMW 916-12-095). G. Houge was supported by HelseVest grant 911744.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2015;125(8):3051–3062. doi:10.1172/JCI79860.
References
- 1.Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J. 2001;353(pt 3):417–439. doi: 10.1042/0264-6021:3530417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang J, Phiel C. Functions of B56-containing PP2As in major developmental and cancer signaling pathways. Life Sci. 2010;87(23):659–666. doi: 10.1016/j.lfs.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deciphering Developmental Disorders Study Large-scale discovery of novel genetic causes of developmental disorders. Nature. 2015;519(7542):223–228. doi: 10.1038/nature14135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu Y, et al. Structure of the protein phosphatase 2A holoenzyme. Cell. 2006;127(6):1239–1251. doi: 10.1016/j.cell.2006.11.033. [DOI] [PubMed] [Google Scholar]
- 5.Cho US, Xu W. Crystal structure of a protein phosphatase 2A heterotrimeric holoenzyme. Nature. 2007;445(7123):53–57. doi: 10.1038/nature05351. [DOI] [PubMed] [Google Scholar]
- 6.Xu Z, et al. Structure and function of the PP2A-shugoshin interaction. Mol Cell. 2009;35(4):426–441. doi: 10.1016/j.molcel.2009.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goudreault M, et al. A PP2A phosphatase high density interaction network identifies a novel striatin-interacting phosphatase and kinase complex linked to the cerebral cavernous malformation 3 (CCM3) protein. Mol Cell Proteomics. 2009;8(1):157–171. doi: 10.1074/mcp.M800266-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Strack S, Cribbs JT, Gomez L. Critical role for protein phosphatase 2A heterotrimers in mammalian cell survival. J Biol Chem. 2004;279(46):47732–47739. doi: 10.1074/jbc.M408015200. [DOI] [PubMed] [Google Scholar]
- 9.Chen W, Arroyo JD, Timmons JC, Possemato R, Hahn WC. Cancer-associated PP2A Aalpha subunits induce functional haploinsufficiency and tumorigenicity. Cancer Res. 2005;65(18):8183–8192. doi: 10.1158/0008-5472.CAN-05-1103. [DOI] [PubMed] [Google Scholar]
- 10.Liu L, Eisenman RN. Regulation of c-myc protein abundance by a protein phosphatase 2A-glycogen synthase kinase 3β-negative feedback pathway. Genes Cancer. 2012;3(1):23–36. doi: 10.1177/1947601912448067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Groves MR, Hanlon N, Turowski P, Hemmings BA, Barford D. The structure of the protein phosphatase 2A PR65/A subunit reveals the conformation of its 15 tandemly repeated HEAT motifs. Cell. 1999;96(1):99–110. doi: 10.1016/S0092-8674(00)80963-0. [DOI] [PubMed] [Google Scholar]
- 12.Xu Y, Chen Y, Zhang P, Jeffrey PD, Shi Y. Structure of a protein phosphatase 2A holoenzyme: insights into B55-mediated Tau dephosphorylation. Mol Cell. 2008;31(6):873–885. doi: 10.1016/j.molcel.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wlodarchak N, et al. Structure of the Ca2+-dependent PP2A heterotrimer and insights into Cdc6 dephosphorylation. Cell Res. 2013;23(7):931–946. doi: 10.1038/cr.2013.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sents W, Ivanova E, Lambrecht C, Haesen D, Janssens V. The biogenesis of active protein phosphatase 2A holoenzymes: a tightly regulated process creating phosphatase specificity. FEBS J. 2013;280(2):644–661. doi: 10.1111/j.1742-4658.2012.08579.x. [DOI] [PubMed] [Google Scholar]
- 15.Fellner T, et al. A novel and essential mechanism determining specificity and activity of protein phosphatase 2A (PP2A) in vivo. Genes Dev. 2003;17(17):2138–2150. doi: 10.1101/gad.259903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hombauer H, et al. Generation of active protein phosphatase 2A is coupled to holoenzyme assembly. PLoS Biol. 2007;5(6):e155. doi: 10.1371/journal.pbio.0050155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stanevich V, Zheng A, Guo F, Jiang L, Wlodarchak N, Xing Y. Mechanisms of the scaffold subunit in facilitating protein phosphatase 2A methylation. PLoS One. 2014;9(1):e86955. doi: 10.1371/journal.pone.0086955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCright B, Rivers AM, Audlin S, Virshup DM. The B56 family of protein phosphatase 2A (PP2A) regulatory subunits encodes differentiation-induced phosphoproteins that target PP2A to both nucleus and cytoplasm. J Biol Chem. 1996;271(36):22081–22089. doi: 10.1074/jbc.271.36.22081. [DOI] [PubMed] [Google Scholar]
- 19.Longin S, Zwaenepoel K, Louis JV, Dilworth S, Goris J, Janssens V. Selection of protein phosphatase 2A regulatory subunits is mediated by the C terminus of the catalytic Subunit. J Biol Chem. 2007;282(37):26971–26980. doi: 10.1074/jbc.M704059200. [DOI] [PubMed] [Google Scholar]
- 20.Martens E, et al. Genomic organisation, chromosomal localisation tissue distribution and developmental regulation of the PR61/B’ regulatory subunits of protein phosphatase 2A in mice. J Mol Biol. 2004;336(4):971–986. doi: 10.1016/j.jmb.2003.12.047. [DOI] [PubMed] [Google Scholar]
- 21.Louis JV, et al. Mice lacking phosphatase PP2A subunit PR61/B’Δ (Ppp2r5d) develop spatially restricted tauopathy by deregulation of CDK5 and GSK3bβ. Proc Natl Acad Sci U S A. 2011;108(17):6957–6962. doi: 10.1073/pnas.1018777108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lohmann C, Kessels HW. The developmental stages of synaptic plasticity. J Physiol. 2014;592(pt 1):13–31. doi: 10.1113/jphysiol.2012.235119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ahn JH, et al. The B’’/PR72 subunit mediates Ca2+-dependent dephosphorylation of DARPP-32 by protein phosphatase 2A. Proc Natl Acad Sci U S A. 2007;104(23):9876–9881. doi: 10.1073/pnas.0703589104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahn JH, McAvoy T, Rakhilin SV, Nishi A, Greengard P, Nairn AC. Protein kinase A activates protein phosphatase 2A by phosphorylation of the B56Δ subunit. Proc Natl Acad Sci U S A. 2007;104(8):2979–2984. doi: 10.1073/pnas.0611532104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stipanovich A, et al. A phosphatase cascade by which rewarding stimuli control nucleosomal response. Nature. 2008;453(7197):879–884. doi: 10.1038/nature06994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Michel JJ, Townley IK, Dodge-Kafka KL, Zhang F, Kapiloff MS, Scott JD. Spatial restriction of PDK1 activation cascades by anchoring to mAKAPα. Mol Cell. 2005;20(5):661–672. doi: 10.1016/j.molcel.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 27.Dodge-Kafka KL, et al. cAMP-stimulated protein phosphatase 2A activity associated with muscle A kinase-anchoring protein (mAKAP) signaling complexes inhibits the phosphorylation and activity of the cAMP-specific phosphodiesterase PDE4D3. J Biol Chem. 2010;285(15):11078–11086. doi: 10.1074/jbc.M109.034868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Firulli BA, et al. PKA, PKC, and the protein phosphatase 2A influence HAND factor function: a mechanism for tissue-specific transcriptional regulation. Mol Cell. 2003;12(5):1225–1237. doi: 10.1016/S1097-2765(03)00425-8. [DOI] [PubMed] [Google Scholar]
- 29.Yu UY, Ahn JH. Phosphorylation on the PPP2R5D B regulatory subunit modulates the biochemical properties of protein phosphatase 2A. BMB Rep. 2010;43(4):263–267. doi: 10.5483/BMBRep.2010.43.4.263. [DOI] [PubMed] [Google Scholar]
- 30.Tadmouri A, et al. Cacnb4 directly couples electrical activity to gene expression, a process defective in juvenile epilepsy. EMBO J. 2012;31(18):3730–3744. doi: 10.1038/emboj.2012.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahn JH, Kim Y, Kim HS, Greengard P, Nairn AC. Protein kinase C-dependent dephosphorylation of tyrosine hydroxylase requires the B56Δ heterotrimeric form of protein phosphatase 2A. PLoS One. 2011;6(10):e26292. doi: 10.1371/journal.pone.0026292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dovega R, Tsutakawa S, Quistgaard EM, Anandapadamanaban M, Low C, Nordlund P. Structural and biochemical characterization of human PR70 in isolation and in complex with the scaffolding subunit of protein phosphatase 2A. PLoS One. 2014;9(7):e101846. doi: 10.1371/journal.pone.0101846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walter G, Ruediger R. Mouse model for probing tumor suppressor activity of protein phosphatase 2A in diverse signaling pathways. Cell Cycle. 2012;11(3):451–459. doi: 10.4161/cc.11.3.19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brinkmann V, et al. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov. 2010;9(11):883–897. doi: 10.1038/nrd3248. [DOI] [PubMed] [Google Scholar]
- 35.Perrotti D, Neviani P. Protein phosphatase 2A: a target for anticancer therapy. Lancet Oncol. 2013;14(6):e229–238. doi: 10.1016/S1470-2045(12)70558-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gutierrez A, et al. Phenothiazines induce PP2A-mediated apoptosis in T cell acute lymphoblastic leukemia. J Clin Invest. 2014;124(2):644–655. doi: 10.1172/JCI65093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Giannoulatou E, et al. Contributions of intrinsic mutation rate and selfish selection to levels of de novo HRAS mutations in the paternal germline. Proc Natl Acad Sci U S A. 2013;110(50):20152–20157. doi: 10.1073/pnas.1311381110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McConechy MK, et al. Subtype-specific mutation of PPP2R1A in endometrial and ovarian carcinomas. J Pathol. 2011;223(5):567–573. doi: 10.1002/path.2848. [DOI] [PubMed] [Google Scholar]
- 39.Shih I-M, et al. Somatic mutations of PPP2R1A in ovarian and uterine carcinomas. Am J Pathol. 2011;178(4):1442–1447. doi: 10.1016/j.ajpath.2011.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goriely A, McGrath JJ, Hultman CM, Wilkie AO, Malaspina D. “Selfish spermatogonial selection”: a novel mechanism for the association between advanced paternal age and neurodevelopmental disorders. Am J Psychiatry. 2013;170(6):599–608. doi: 10.1176/appi.ajp.2013.12101352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mirzaa GM, Riviere JB, Dobyns WB. Megalencephaly syndromes and activating mutations in the PI3K-AKT pathway: MPPH and MCAP. Am J Med Genet C Semin Med Genet. 2013;163C(2):122–130. doi: 10.1002/ajmg.c.31361. [DOI] [PubMed] [Google Scholar]
- 42.Janssens V, Rebollo A. The role and therapeutic potential of Ser/Thr phosphatase PP2A in apoptotic signalling networks in human cancer cells. Curr Mol Med. 2012;12(3):268–287. doi: 10.2174/156652412799218930. [DOI] [PubMed] [Google Scholar]
- 43.de Ligt J, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012;367(20):1921–1929. doi: 10.1056/NEJMoa1206524. [DOI] [PubMed] [Google Scholar]
- 44.Gilissen C, et al. Genome sequencing identifies major causes of severe intellectual disability. Nature. 2014;511(7509):344–347. doi: 10.1038/nature13394. [DOI] [PubMed] [Google Scholar]
- 45.McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26(16):2069–2070. doi: 10.1093/bioinformatics/btq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bragin E, et al. DECIPHER: database for the interpretation of phenotype-linked plausibly pathogenic sequence and copy-number variation. Nucleic Acids Res. 2014;42:D993–D1000. doi: 10.1093/nar/gkt937. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.