

Abstract
Somatic hypermutation (SHM) underlies the generation of a diverse repertoire of high-affinity antibodies. It is effected by a two-step process: (i) DNA lesions initiated by activation-induced cytidine deaminase (AID), and (ii) lesion repair by the combined intervention of DNA replication and repair factors that include mismatch repair (MMR) proteins and translesion DNA synthesis (TLS) polymerases. AID and TLS polymerases that are crucial to SHM, namely polymerase (pol) θ, pol ζ and pol η, are induced in B cells by the stimuli that are required to trigger this process: B-cell receptor crosslinking and CD40 engagement by CD154. These polymerases, together with MMR proteins and other DNA replication and repair factors, could assemble to form a multi-molecular complex (‘mutasome’) at the site of DNA lesions. Molecular interactions in the mutasome would result in a ‘polymerase switch’, that is, the substitution of the high-fidelity replicative pol δ and pol ε with the TLS pol θ, pol η, Rev1, pol ζ and, perhaps, pol ι, which are error-prone and crucially insert mismatches or mutations while repairing DNA lesions. Here, we place these concepts in the context of the existing in vivo and in vitro findings, and discuss an integrated mechanistic model of SHM.
Features of somatic hypermutation
Antibody diversity and B-cell development are underpinned by sequential immunoglobulin (Ig) gene recombination. This assembles noncontiguous Ig variable (V), diversity (D) and joining (J) genes into a functional V(D)J DNA segment, thereby producing the diverse pre-immune repertoire of B-cell receptors (BCRs), B-cell clonotypes and corresponding antibodies. Pre-immune naive B cells express IgM with a low-to-moderate affinity for antigen. In response to an infectious agent or a self-antigen, selected IgM clones undergo somatic hypermutation (SHM), mainly in the germinal centers of secondary lymphoid organs. They mutate the V(D)J portion of their antibody-encoding genes, thus providing the basis for positive selection by antigen of higher-affinity BCR mutants and, eventually, the high-affinity antibodies characteristic of a mature immune response. During the germinal center reaction, the Ig heavy (H)-chain locus undergoes an additional change: class-switch DNA recombination (CSR). CSR replaces the constant (C)μ region of the H chain with a downstream Cγ, Cα or Cε region, thereby providing antibodies with new biological effector functions.
SHM emerged before CSR in phylogeny, being fully functional in sharks, whose antibody responses show evidence of mutational selection [1]. SHM depends on transcription and preferentially targets the RGYW/WRCY (R=A or G, Y=C or T, W=A or T) mutational hotspot [2–6]. It introduces mainly point mutations and, rarely, deletions or insertions into rearranged V(D)J gene sequences at a rate of 10−3 per base per cell generation, which is a millionfold higher than the rate of spontaneous mutation in the genome at large, but avoids C regions (Figure 1, small inset) [4–13]. SHM is restricted to the Ig locus and a few other genes because abnormal and widespread mutations in the genome are detrimental to cell homeostasis and favor the emergence of neoplasia and autoimmunity. In the Ig locus, SHM is highly regulated, starting with its specific induction. Indeed, the stimuli that induce SHM [i.e. crosslinking of the BCR, the engagement of CD40 and co-engagement of CD80 and CD86 on the B-cell surface by CD154 and CD28 expressed on the surface of activated T cells, and cytokines, such as interleukin (IL)-4] specifically upregulate activation-induced cytidine deaminase (AID) [14] and selected (lesion bypass) translesion DNA synthesis (TLS) polymerases, which are key mediators of SHM [15–19] (Figure 1). Here, we discuss the current information, derived mainly from in vivo observations, on the roles of mismatch repair (MMR) proteins and TLS polymerases, and biochemical studies of DNA repair factors, and integrate it into the context of an overall model of SHM.
Figure 1.
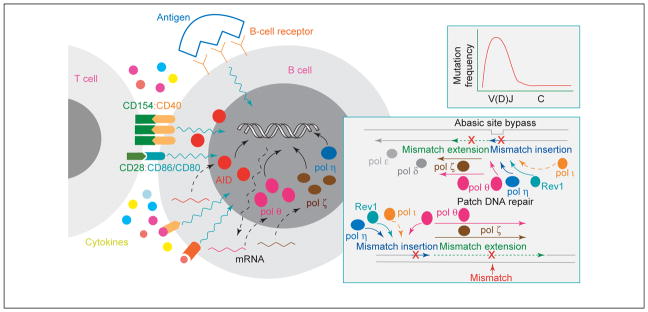
BCR cross-linking and T-cell contact through CD40:CD154 engagement and CD86/CD80:CD28 co-engagement are required for the induction of SHM. CD40:CD154 upregulates AID expression (large red circles). BCR crosslinking upregulates the error-prone TLS pol θ (large pink ovals) and pol ζ catalytic subunit Rev3 (brown ovals), which, with other TLS polymerases, namely, pol η (dark blue ovals), Rev1 (light green circles) and, perhaps, pol ι (orange ovals), are recruited into the DNA repair process that results in the insertion of mutations. The small inset depicts mutation frequency in V(D)J and C regions. The large inset depicts the polymerase switch. Somatic mutations (red crosses) are introduced by TLS polymerase(s) during DNA synthesis, while bypassing an abasic site or while copying undamaged DNA in patch DNA re-synthesis of MMR or, perhaps, a mutagenic long-patch BER. Abasic site bypass requires the sequential action of two DNA polymerases: one, such as pol θ, pol η, Rev1 or, perhaps, pol ι, that inserts a nucleotide opposite the damaged template nucleotide (inserter), and the other, such as pol θ or pol ζ, that extends from the inserted nucleotide (extender). Pol η is highly inefficient at inserting a nucleotide opposite to an abasic site, but its recruitment by PCNA greatly stimulates the ability of this TLS polymerase to insert a nucleotide opposite this lesion. Pol θ is the first DNA polymerase known to bypass abasic sites efficiently by functioning as a mispair inserter and a mispair extender.
Somatic hypermutation as a two-step process: DNA lesions and repair
SHM can be considered a two-step process involving the generation of DNA lesions and their subsequent repair. AID initiates SHM [14,20,21] by deaminating deoxycytosine (dC) directly in DNA, thereby yielding deoxyuracil (dU):deoxyguanosine (dG) mispairs [10,11,22–25]. The generation of these and other lesions, including DNA cleavage [4,6,26–29], by direct AID-mediated DNA deamination and/or by the intervention of unidentified endonuclease(s) [4,6,9,28–30], would constitute the initial step in SHM. The second step, DNA repair, would be responsible for mismatch (mutation) insertion. DNA repair involves the base-excision repair (BER) protein uracil-DNA glycosylase (Ung) [22,23], MMR proteins (e.g. Msh2 and Msh6) [5,10,31,32], DNA-break repair proteins [e.g. the Mre11–Rad50–Nbs1 (MRN) complex] [12] and, finally, error-prone TLS polymerases (Box 1) [6,7,13,17,19,33].
Box 1. Glossary.
- Abasic site
AP (apurinic/apyrimidinic) site. This is a site in a DNA molecule where a nucleotide base has been removed by cleavage of the glycosidic bond that links the nucleotide base to the deoxyribose-phosphate backbone.
- AID
Activation-induced cytidine deaminase. AID is a B-cell-specific enzyme that is required for SHM and CSR.
- APE
AP endonuclease. APE excises apurinic or apyrimidinic sites, thereby generating DNA nicks.
- BER
Base excision repair. BER is a repair process that recognizes and eliminates particular non-canonical bases (such as deaminated or oxidized bases) from DNA and replaces them with an appropriate base templated on the opposite strand. In BER, an altered base is removed by a DNA glycosylase, such as Ung, followed by excision of the resulting sugar phosphate. The small gap left in the DNA is filled in by the sequential actions of DNA polymerase and DNA ligase.
- Exo I
Exonuclease I, a 5′–3′ exonuclease of the Rad 2 family that can also have 3′–5′ exonuclease activity. Exo I interacts with Msh2 and Mlh1 proteins and is required for MMR that is directed by a strand break located either 3′ or 5′ to the mispair.
- MMR
Mismatch repair. MMR is a DNA repair process that recognizes and corrects mismatched nucleotide pairs by replacing the incorrect nucleotide.
- MRN
The Mre11–Rad50–Nbs1 complex, a protein complex possessing endonuclease activity. It is essential for DNA double-strand break repair, genomic stability and signaling internal surveillance mechanisms that arrest the cell cycle when there is a DNA damage (checkpoint).
- Mutasome
A putative protein complex effecting SHM. In our model, the mutasome contains TLS polymerases interacting with DNA replication and repair factors and recruits these DNA polymerases to error-prone DNA synthesis during the DNA lesion-repair process of SHM.
- Okazaki fragments
DNA replication for the lagging strand is discontinuous and away from the replication fork. The small fragments synthesized are called Okazaki fragments and are subsequently stitched together by DNA ligase.
- Patch DNA repair
The repair of a DNA lesion involves DNA excision to remove a single nucleotide (short-patch) or multiple nucleotides (long-patch) to generate a gap on the damaged strand, and subsequent re-synthesis using the other strand as a template. The repair usually occurs at a time distinct from when the genome is replicated.
- PCNA
Proliferating cell nuclear antigen. PCNA is an essential component of the DNA replication machinery, functioning as the accessory protein for DNA polymerase and required for long-stretch processive chromosomal DNA synthesis. PCNA is also required for DNA recombination and repair. In addition, it interacts with the DNA polymerases and proteins involved in DNA repair and cell-cycle regulation. It is probably involved in SHM by recruiting error-prone TLS polymerases into the process of DNA damage repair.
- Processivity
This is a measure of the length of polynucleotide that is synthesized by a DNA polymerase before this dissociates from the template. A highly processive DNA polymerase, such as pol δ or pol ε, inserts hundreds of bases before ‘falling off’ the template. A low processivity polymerase, for example, pol η, inserts only one or two nucleotides and then falls off.
- RPA
Replication protein A. RPA is the main single-stranded DNA-binding protein involved in the replication of eukaryotic DNA. RPA is also involved in DNA recombination and DNA repair. In activated B cells, RPA can associate with phosphorylated AID and enhances AID activity on transcribed double-stranded DNA.
- Translesion DNA synthesis (TLS) (also known as ‘lesion bypass’) polymerase
This is a DNA polymerase that can synthesize past a lesion (such as an abasic site) in the template strand. TLS polymerases display considerably higher rates of nucleotide misincorporation than the high-fidelity DNA replicative pol δ and pol ε, thereby generating mismatches at a high frequency opposite a lesion or when copying DNA using an undamaged DNA strand.
- Ung
Uracil-DNA glycosylases. Ung prevents mutagenesis by eliminating dU from DNA molecules by cleaving the N-glycosylic bond and initiating BER. dU residues arise from dC deamination or misincorporation of deoxyuridine monophosphate (dUMP) residues. Alternative promoter usage and splicing of this gene results in two different isoforms: the mitochondrial Ung1 and the nuclear Ung2.
AID binds to single-stranded DNA in the transcription bubble and deaminates dC to produce dU [34–36]. This activity is enhanced by the association of its phosphorylated form with single-strand-DNA-binding replication protein A (RPA) [37]. dU is not relevant to DNA, therefore the dU:dG mismatch must either be ‘replicated over’ or removed by the DNA repair machinery. Replicating over dU results in a dC→deoxythymidine (dT) transition mutation (Phase 1a) [10], whereas dU deglycosylation by Ung produces an abasic site. DNA synthesis opposite the abasic site by TLS polymerases yields dC→dT transitions, and dC→deoxyadenosine (dA) or dC→dG transversions (Figure 2, Phase 1b). The abasic site can also be excised by apurinic/apyrimidinic endonuclease (APE) and repaired by error-free short-patch BER or a mutagenic long-patch BER. In the long-patch BER, the damage-containing strand is excised by the structure-specific endonuclease Fen1, and ~2–8 nucleotides would be incorporated by TLS polymerases, thereby inserting mismatches. Alternatively, dU:dG mismatches recruit the MMR machinery, which, through the activity of an unidentified endonuclease and Exonuclease I (Exo I), creates a single-stranded DNA gap around the dU:dG mismatch. The ‘patch’ repair of such a gap by TLS polymerase(s) would result in the insertions of mismatches at dA:dT and dC:dG (Phase 2).
Figure 2.

An integrated model of SHM. This assumes that AID deaminates dC in both DNA strands. dU is not relevant to DNA and the dU:dG mismatch is ‘replicated over’ or dealt with by the DNA repair machinery. Replicating over dU results in a dC→dT transition mutation (Phase 1a), whereas dU deglycosylation by Ung gives rise to an abasic site. In the presence of PCNA (orange ring), DNA synthesis opposite the abasic site by TLS pol θ, which has both nucleotide inserter and extender activity, or by the nucleotide inserter pol η, Rev1 or, perhaps, pol ι, followed by the nucleotide extender pol θ or pol ζ, yields dC→dT transitions and dC→dA or dC→dG transversions (Phase 1b). Alternatively, the abasic site can be recognized and excised by APE or the Mre11–Rad50 lyase to create a DNA nick. This nick can be repaired by DNA pol β (light pink circles) in an error-free fashion (short-patch BER) or repaired in an error-prone fashion by a TLS polymerase through a long-patch BER also involving PCNA and Fen1. dU:dG mispairs can also be recognized by the MMR machinery, resulting in a DNA-gap formation through the intervention of an unidentified endonuclease or MRN and Exo I. Subsequently, TLS pol θ, pol η, Rev1, pol ζ and, perhaps, pol ι can effect DNA re-synthesis as part of a patch repair, thereby inserting mismatches (Phase 2). In the long-patch BER or MMR, RPA (large brown ovals) and PCNA would recruit other repair proteins to the lesion and co-ordinate their actions. MMR proteins are indicated as large green ovals. Mutated nucleotides are shown in red.
AID-resected double-stranded DNA breaks (DSBs) would constitute a proportion of the DNA lesions occurring in SHM [4,6,27–29,38,39]. However, in this review we will not address the repair of DSBs, because the involvement of these lesions in SHM is still controversial. TLS polymerases have a central role in the DNA repair of AID-induced lesions. They insert mismatches while synthesizing a DNA strand across or past a damaged nucleotide, such as in the abasic site bypass of Phase 1b, or while copying a DNA strand on an undamaged template, as in the patch repair process that is central to MMR (Phase 2) [4,6,19]. They are recruited, with MMR proteins, into a multimolecular complex or ‘mutasome’ that assembles [catalyzed by proliferating cell nuclear antigen (PCNA)] at DNA lesions. The modes by which AID-mediated DNA lesions and DNA-repair factors eventually yield mismatches are discussed here in the context of how, as we hypothesize, SHM subverts general DNA repair processes to insert mutations. Our knowledge of DNA replication and repair has been derived mainly from experiments in Escherichia coli or yeast. The precise nature of these processes in mammalian cells are as yet undefined.
Mismatch DNA repair and somatic hypermutation
The most-studied function of MMR is the correction of misincorporated nucleotides in the newly synthesized strand during DNA replication [40]. The MMR process is seemingly subverted by SHM. MMR consists of three sequential stages: (i) mismatch recognition; (ii) excision of the DNA sequence containing the mismatched nucleotide; and (iii) patch DNA re-synthesis to complete the repair. ‘SHM MMR’ is altered in the excision stage and in the DNA polymerases used during the final DNA re-synthesis stage (Table 1).
Table 1.
MMR and SHM MMR1
| MMR2 | SHM MMR | Refs3 | |
|---|---|---|---|
| Function | Correct misincorporated nucleotides during DNA replication and maintain genomic stability | Repair DNA lesions initiated by AID and introduce mutations in the Ig locus | [10,12] |
|
| |||
| Initiation stage | |||
| Mismatch sensor | Msh2–Msh6 recognizes single-nucleotide mismatches and 1–2 nucleotide insertions or deletions Msh2–Msh3 recognizes mostly more than 2-nucleotide insertions or deletions |
Msh2–Msh6 initiates SHM MMR to introduce Phase 2 mutations Msh6 also influences the AID targeting in Phase 1 of SHM Msh2–Msh3 is not involved in SHM |
[5,31,32,43, 59,84,85] |
|
| |||
| Msh2-binding enhancement | By PCNA | Not determined | |
|
| |||
| Loading of Msh2 and other proteins | Dependent on ATP hydrolysis by Msh2 | Dependent on ATP hydrolysis by Msh2 | [86] |
|
| |||
| Matchmaker protein | Mlh1 dimerizes with Pms2, Mlh3 or Pms1 Pms2 repairs a variety of mismatches Mlh3 removes frameshift intermediates |
Mlh1, Pms2 and Mlh3 all contribute to SHM Deficiency in Mlh1 or Pms1 reduces SHM and alters the mutation spectrum Deficiency in Mlh3 alters the mutation spectrum |
[5,31] [47] |
|
| |||
| Excision stage | |||
| Strand nicking | In nascent strands, the 3′-end of the leading strand and the 5′- or 3′-end of Okazaki fragments of the lagging strand provide entry points for Exo I | Template strand would be nicked by Mre11–Rad50 and APE to provide entry points for Exo I | [49] |
|
| |||
| Strand excision | By Exo I, which bidirectionally (5′→3′ and 3′→5′) cleaves the nascent strand; could extend 150 bp beyond the mismatch | By Exo I, which possibly cleaves DNA in the vicinity of dU:dG | [44] |
|
| |||
| Re-synthesis stage | |||
| DNA polymerase clamp and clamp loader | PCNA and RFC complex | Not determined | |
|
| |||
| DNA polymerase | High-fidelity pol δ and ε | Error-prone DNA pol θ, pol η, Rev1, pol ζ and, perhaps, pol ι | [17,19,59, 61,63] |
|
| |||
| DNA ligase | DNA ligase I | Not determined | |
Abbreviations: bp, base pairs; kb, kilobase; RFC, replication factor C.
MMR as associated with replication. The MMR processes associated with homologous recombination and meiosis involve different sets of MMR factors. They are not discussed here owing to space limitations.
The references listed here are mainly for SHM MMR; the reference for MMR is [40].
MMR was first characterized in Escherichia coli, in which it is mediated by Mutant S (MutS), MutL and MutH proteins. In mammals, MMR is mediated by proteins homologous to prokaryotic MMR factors, such as MutS homolog (Msh)2, Msh3 and Msh6 and the MutL homologs (Mlh)1, Mlh3, postmeiotic segregation increased (Pms)1 and Pms2. Msh2, heterodimerized with Msh6 or Msh3, is recruited by a nucleotide mispair to initiate the MMR cascade. The Msh2–Msh6 dimer (also known as MutSα) but not the Msh2–Msh3 dimer (also known as MutSβ) binds to dU:dG in vitro with an affinity comparable to its binding to dT:dG [41,42]. Consistent with the role of Msh2–Msh6 as the mismatch sensor for dU:dG in SHM, mice deficient in Msh2 and/or Msh6 but not Msh3 exhibit a decreased frequency and altered spectrum of mutations entailing decreased mutations at dA:dT with concurrent increased mutations at dC:dG [5,31,32,43–45]. During MMR, the mismatch recognition stage and the subsequent strand-excision stage are co-ordinated and regulated by the MutL heterodimer, which is formed by Mlh1 and one of its three partners, Pms1, Pms2 or Mlh3, and recruited to the lesion site by the mismatch sensor Msh2–Msh6. Mlh1 has an important role in SHM MMR. However, a compensatory MutL-independent pathway also exists, as suggested by the moderate increase in dC and dG mutations in mlh−/− mice [5,31], when compared with msh2−/− and msh6−/− mice [5,7,31,43,46]. Both Pms2 and Mlh3 have a role in SHM MMR, albeit in different ways, because mice deficient in Mlh1, Pms2 or Mlh3 display different spectra of mutations in V(D)J DNA and in the proximity of switch–switch (S–S) region junctions [45,47].
In MMR, the DNA excision is targeted exclusively to the nascent strand. In Escherichia coli, only the newly synthesized strand, which is transiently unmethylated, is nicked by the MutH endonuclease [40]. The resulting nick provides the entry point for Exo I to excise the DNA segment that contains mismatches [48]. In eukaryotes, the newly synthesized strands can be distinguished from the template strand solely by the presence of gaps between Okazaki fragments on the lagging strand or the free 3′-terminus on the leading strand [40]; these are ‘entry’ sites from which the degradation of the error-containing strand by Exo I can start. However, nascent strand-specific MMR cannot explain how AID-generated dUs in the template strand are removed during SHM MMR. This process is probably independent of DNA replication. Exo I has an important role in excising DNA in SHM MMR, as indicated by the decreased mutations in Exo I-deficient mice [44], but the endonuclease that nicks the template strand containing dU is unidentified to date. Interestingly, Mre11–Rad50, part of the MRN complex, possesses an evolutionarily conserved lyase activity [49] that could nick Ung-generated abasic sites. Accordingly, Mre11 but not the ubiquitous APE is enriched on V(D)J DNA of hypermutating B cells [49], and MRN promotes SHM [44,50], suggesting that some SHM MMR is initiated by this complex.
TLS polymerases in somatic hypermutation and ‘polymerase switch’
The substitution of the high-fidelity replicative polymerases with error-prone TLS polymerases (polymerase switch) [51,52] during DNA re-synthesis has a significant role in introducing mismatches during SHM DNA repair. TLS polymerases introduce mismatches at: (i) dC:dG while bypassing abasic sites arising from Ung-deglycosylation of dU; (ii) dC:dG and dA:dT while extending past these abasic site mispairs (Phase 1b); and (iii) dC:dG and dA:dT while copying undamaged DNA template during patch DNA re-synthesis in MMR or, perhaps, a long-patch BER (Phase 2). DNA replication, BER and patch DNA repairs are performed by high-fidelity pol δ and pol ε because preserving genomic integrity is vital for the cell. Owing to the geometry constraints imposed by a nucleotide to be copied, these high-fidelity polymerases stop at a damaged base, thereby stalling the replication fork. At the stalled replication fork, they would be substituted by TLS polymerases, such as pol θ, pol η, Rev1, pol ζ, pol ι, pol κ, pol λ and pol μ [53,54]. By synthesizing a DNA strand across or past a damaged nucleotide, these TLS polymerases help the cell to tolerate DNA damage and continue dividing [55]. However, they are highly error-prone, even when copying undamaged DNA. The bypass of damaged bases or abasic sites mainly involves the intervention of two different enzymatic activities: one that incorporates a nucleotide opposite the lesion (mispair insertion, for example, by pol η, Rev1 or pol ι) and another that elongates past the mispair from the newly created primer end (mispair extension, for example, by pol ζ) (Figure 1, large inset). Following an initial mispair extension by pol ζ, further DNA extension is resumed by high-fidelity pol δ or pol ε [51,52,56].
Whereas DNA-damage-repairing pol β and TLS pol μ, pol λ and pol κ are apparently not involved in SHM [13,33], the TLS pol θ, pol η, Rev1, pol ζ and, perhaps, pol ι contribute to this process [4,7,13,17,19,57–61]. Pol η preferentially introduces mutations at dA:dT, as indicated by the dC:dG-biased mutation spectrum in Xeroderma pigmentosum variant (XP-V) patients, who are congenitally deficient in pol η, and in pol η-deficient mice [18,59,60]. In these mice, the overall frequency of mutations is normal but mutations at dA:dT are decreased by two-thirds [59,60]. Pol ι is a paralog of pol η. Pol ι-deficiency causes a slight reduction in overall mutation frequency only when combined with pol η deficiency, suggesting that pol ι has only a marginal role in SHM [62,63]. Rev1 effectively bypasses an abasic site by inserting a dC opposite the lesion, thereby introducing a dC→dG transversion at an AID-mediated abasic site, as shown in chicken DT40 B cells and Rev1-deficient mice [58,61]. Pol ζ, which consists of the catalytic Rev3 and the regulatory Rev7 subunits, effects TLS by extending past a mispair. The inhibition of rev3 expression by specific oligonucleotides inhibited damage-induced DNA mutagenesis and impaired SHM in the Ig and in bcl-6 loci in Burkitt lymphoma B cells in vitro [17]. A comparable effect was reported in vivo in transgenic mice expressing an antisense rev3 RNA that partially downregulated the expression of this gene [57].
Pol η, Rev1 and pol ζ act together to insert mismatches in SHM. The function of pol η or Rev1 is largely restricted to mispair insertion because of their low processivity. Pol ζ is inefficient at replicating through DNA lesions but efficiently extends a DNA strand past mispairs inserted by pol η or Rev1. Thus, pol ζ and pol η contribute, albeit differently, to SHM. Inhibition of rev3 expression or deficiency in pol η resulted in only partial impairment of SHM [17,57,59,60], suggesting that another TLS polymerase has a dominant role in this process. Pol θ is the first DNA polymerase known to bypass efficiently an abasic site by functioning as both a mispair inserter and a mispair extender [64]. It synthesizes through a damaged DNA template containing randomly inserted abasic sites with an efficiency comparable to that displayed on the corresponding undamaged template [64]. Although residues in the polymerase catalytic site are highly homologous to high-fidelity family A DNA polymerases, pol θ displays an extremely low fidelity [54,64]. Pol θ possibly introduces mismatches while bypassing abasic sites and/or extending past abasic sites in Phase 1b, and while copying undamaged templates in Phase 2, as indicated by the decreased overall frequency of mutations at dC:dG and dA:dT in pol θ−/− mice, in which an in-frame stop codon in exon 1 and the replacement of exons 2 to 5 with a neomycin resistance gene deleted most of the gene product [19]. The contribution of pol θ to Phase 1b and Phase 2 is further emphasized by the skewed mutation spectrum in mice expressing an altered form of this polymerase [65], and would explain the residual mutations found at dA:dT in pol η-deficient mice [59,60]. Interestingly, the mismatches introduced by TLS polymerases could trigger a new round of MutSα-initiated MMR, resulting in an amplification of mutations.
The mutasome
It has been suggested that AID mediates the assembly of a multimolecular complex involving specific DNA repair and error-prone TLS activities: the ‘mutasome’ [66]. The mutasome would actively facilitate the polymerase switch that, by effecting faulty DNA re-synthesis in MMR and long-patch BER of Phase 2, is probably central to SHM. Although there is no direct evidence for the assembly of a mutasome in SHM, biochemical studies indicate that TLS polymerases are recruited at the DNA-damage site through interactions with different DNA-repair factors as part of a complex that also comprises PCNA, RPA, MMR and BER proteins, and, possibly, MRN. The unique features of PCNA suggest that this protein has a crucial role in recruiting other components of the mutasome and in promoting the access of TLS polymerases to the DNA lesion. PCNA is possibly the ultimate multifunctional matchmaker protein for DNA transactions [67]. It forms a homotrimeric clamp around the DNA, functions as a platform for other repair proteins and is an essential component of the eukaryotic chromosomal DNA replisome [67]. PCNA is a polymerase processivity factor and is key to DNA replication and repair. It has a crucial role in promoting the access of TLS polymerases to the replication fork, where DNA synthesis is paused by a lesion [68–70]. By functioning as a ‘docking bay’ for different proteins and interacting with TLS polymerases, PCNA is crucially involved in abasic site bypass, BER and MMR, thereby modulating the fidelity of DNA synthesis and co-ordinating various aspects of DNA synthesis and repair [71].
By interacting with pol η, Rev1, pol ζ, pol ι and, possibly, pol θ, PCNA enhances the efficiency of these TLS polymerases in nucleotide incorporation opposite undamaged and damaged sites [54]. In human cells, the stalling of the replication machinery at damaged DNA results in mono-ubiquitination of PCNA. Mono-ubiquitinated PCNA recruits pol η and enhances its enzymatic activity, thereby facilitating the DNA polymerase switch [51,52,72]. Pol η is highly inefficient at inserting a nucleotide opposite an abasic site but interaction with PCNA greatly stimulates its ability to insert a nucleotide opposite this lesion [54]. PCNA also dramatically stimulates TLS synthesis of UV-damaged or abasic site-containing DNA by pol ζ [68]. It contributes significantly to MMR by forming a stable ternary complex with Msh2–Msh6, thereby enhancing the mispair-binding specificity of Mutα [73–75]. This ternary complex is transferred from PCNA to mispaired bases in an ATP-dependent fashion [76]. PCNA associates with MRN [77] to effect DNA repair, possibly as a part of the mutasome. Furthermore, it co-localizes with the MMR excision enzyme Exo I in replication foci and participates in strand excision and DNA re-synthesis [78,79]. Finally, PCNA would recruit TLS polymerases into MMR, as suggested by its ability to facilitate the interaction between pol η and Msh2–Msh6 [42].
RPA binds to single-stranded DNA with a high affinity and interacts specifically with multiple proteins. It associates with PCNA and is essential in multiple DNA-metabolism processes, including DNA replication, recombination and NER, BER and DSB repair [80]. In activated B cells, RPA associates with AID phosphorylated by protein kinase A (PKA), thereby enhancing AID activity on transcribed double-stranded DNA [37,81]. Following deamination, AID can be released from the transcribed SHM substrate, leaving RPA bound to DNA [37,81]. This DNA-bound RPA would function as a platform for the entry of PCNA and other DNA repair proteins, including Ung [82], and for the assembly of the mutasome, leading to Phase 1b and Phase 2 of SHM. By contrast, because only phosphorylated AID, which is found specifically in B cells, interacts with RPA, the overexpression of AID in nonlymphoid cells would uncouple AID from RPA, thereby aborting recruitment of the mutasome and catalyzing low-level non-specific deamination. This would leave dU:dG mismatches to be resolved only through replication-over, thereby yielding a predominance of dC or dG transitions [37]. Thus, PCNA and RPA would have a central role in SHM by: (i) recruiting and enhancing the efficiency of TLS polymerases in abasic site bypass (Phase 1b); and (ii) recruiting and co-ordinating DNA re-synthesis by such TLS polymerases in a mutagenic MMR or long-patch BER (Phase 2).
Conclusions and perspectives
Considerable progress has been made in our understanding of SHM since the identification of AID and its DNA deamination activity [21,22,83]. However, some key questions have yet to be answered. One key question is what mediates SHM target specificity? AID targeting of selected DNA sequences could depend on modifications in chromatin structure and interactions of AID with specific cofactors. However, none of the available experimental data provides convincing evidence explaining how actively transcribed non-Ig genes are protected from dC deamination, and how dC deamination is limited to a few kilobases downstream of V promoters and avoids the constant region. In addition, AID deaminates dC only in a single-stranded DNA substrate. The top (non-transcribed) but not the bottom (transcribed) strand exists in this configuration in the transcription bubble. The absence of strand-polarity in SHM indicates that AID targets the top and bottom DNA strands with comparable efficiency, perhaps implying that the top strand is also transcribed in an antisense fashion. However, active transcription of the top strand has yet to be shown.
Another key question is what triggers the polymerase switch and how is this effected? As discussed, SHM is initiated by AID-mediated DNA lesions, and most mutations are introduced by the TLS pol θ, pol η, Rev1, pol ζ and, perhaps, pol ι. Indeed, in humans and mice, pol θ and pol ζ are preferentially expressed in hypermutating B cells [17–19], and pol η is upregulated in mouse germinal center B cells [18]. The mechanisms by which the high-fidelity pol δ and pol ε are substituted with TLS pol θ, pol η, Rev1, pol ζ or, perhaps, pol ι, thereby altering the genome-guardian BER and MMR pathways to become mutagenic in SHM, have yet to be defined. The precise roles of pol θ and pol η at different stages of the SHM process will be addressed by constructing double-knockout mice generated from single-knockout pol θ−/−, polη−/−, ung−/− or msh2−/− mice. Likewise, the role of the mutasome in promoting the access of TLS polymerases to the DNA lesion(s) will be explored using mice deficient in the DNA replication and repair factors, including RPA and PCNA. Biochemical and molecular studies should elucidate the dynamic changes of the mutasome during different phases of SHM. Collectively, these studies should elucidate the relative contributions of events, such as dU:dG lesion frequency and the rate of DNA repair, that could dictate the efficiency and nature of the overall SHM process.
Acknowledgments
We thank David Schatz, Patricia Gearhart, John Schimenti and Michael Lieber for helpful discussions. Owing to space limitations, we could cite only a fraction of the papers relevant to the topic discussed in this review article. We apologize to the authors of the publications that are not cited here. This work was supported by NIH grants AR 40908, AI 45011 and AI 60573 to P.C.
References
- 1.Flajnik MF, Du Pasquier L. Evolution of innate and adaptive immunity: can we draw a line? Trends Immunol. 2004;25:640–644. doi: 10.1016/j.it.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 2.Chang B, Casali P. The CDR1 sequences of a major proportion of human germline Ig VH genes are inherently susceptible to amino acid replacement. Immunol Today. 1994;15:367–373. doi: 10.1016/0167-5699(94)90175-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Milstein C, et al. Both DNA strands of antibody genes are hypermutation targets. Proc Natl Acad Sci U S A. 1998;95:8791–8794. doi: 10.1073/pnas.95.15.8791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu X, et al. Immunoglobulin somatic hypermutation: double-strand DNA breaks, AID and error-prone DNA repair. J Clin Immunol. 2003;23:235–246. doi: 10.1023/a:1024571714867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li Z, et al. The generation of antibody diversity through somatic hypermutation and class switch recombination. Genes Dev. 2004;18:1–11. doi: 10.1101/gad.1161904. [DOI] [PubMed] [Google Scholar]
- 6.Xu Z, et al. DNA lesions and repair in immunoglobulin class switch recombination and somatic hypermutation. Ann N Y Acad Sci. 2005;1050:146–162. doi: 10.1196/annals.1313.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Diaz M, Casali P. Somatic immunoglobulin hypermutation. Curr Opin Immunol. 2002;14:235–240. doi: 10.1016/s0952-7915(02)00327-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Papavasiliou FN, Schatz DG. Somatic hypermutation of immunoglobulin genes; merging mechanisms for genetic diversity. Cell. 2002;109:S35–S44. doi: 10.1016/s0092-8674(02)00706-7. [DOI] [PubMed] [Google Scholar]
- 9.Honjo T, et al. AID: how does it aid antibody diversity? Immunity. 2004;20:659–668. doi: 10.1016/j.immuni.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 10.Neuberger MS, et al. Somatic hypermutation at A·T pairs: polymerase error versus dUTP incorporation. Nat Rev Immunol. 2005;5:171–178. doi: 10.1038/nri1553. [DOI] [PubMed] [Google Scholar]
- 11.Barreto VM, et al. Activation-induced deaminase: controversies and open questions. Trends Immunol. 2005;26:90–96. doi: 10.1016/j.it.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 12.Maizels N. Immunoglobulin gene diversification. Annu Rev Genet. 2005;39:23–46. doi: 10.1146/annurev.genet.39.073003.110544. [DOI] [PubMed] [Google Scholar]
- 13.Seki M, et al. DNA polymerases and somatic hypermutation of immunoglobulin genes. EMBO Rep. 2005;6:1143–1148. doi: 10.1038/sj.embor.7400582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muramatsu M, et al. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 15.Zan H, et al. Induction of Ig somatic hypermutation and class switching in a human monoclonal IgM+ IgD+ B cell line in vitro: definition of the requirements and modalities of hypermutation. J Immunol. 1999;162:3437–3447. [PMC free article] [PubMed] [Google Scholar]
- 16.Zan H, et al. B cell receptor engagement and T cell contact induce bcl-6 somatic hypermutation in human B cells: identity with Ig hypermutation. J Immunol. 2000;165:830–839. doi: 10.4049/jimmunol.165.2.830. [DOI] [PubMed] [Google Scholar]
- 17.Zan H, et al. The translesion DNA polymerase ζ plays a major role in Ig and bcl-6 somatic hypermutation. Immunity. 2001;14:643–653. doi: 10.1016/s1074-7613(01)00142-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zeng X, et al. DNA polymerase η is an A-T mutator in somatic hypermutation of immunoglobulin variable genes. Nat Immunol. 2001;2:537–541. doi: 10.1038/88740. [DOI] [PubMed] [Google Scholar]
- 19.Zan H, et al. The translesion DNA polymerase θ plays a dominant role in immunoglobulin gene somatic hypermutation. EMBO J. 2005;24:3757–3769. doi: 10.1038/sj.emboj.7600833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muramatsu M, Honjo T. Complex layers of genetic alteration in the generation of antibody diversity. Trends Immunol. 2001;22:66–68. doi: 10.1016/s1471-4906(00)01818-4. [DOI] [PubMed] [Google Scholar]
- 21.Longerich S, et al. AID in somatic hypermutation and class switch recombination. Curr Opin Immunol. 2006;18:164–174. doi: 10.1016/j.coi.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 22.Petersen-Mahrt SK, et al. AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature. 2002;418:99–103. doi: 10.1038/nature00862. [DOI] [PubMed] [Google Scholar]
- 23.Rada C, et al. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr Biol. 2002;12:1748–1755. doi: 10.1016/s0960-9822(02)01215-0. [DOI] [PubMed] [Google Scholar]
- 24.Di Noia J, Neuberger MS. Altering the pathway of immunoglobulin hypermutation by inhibiting uracil-DNA glycosylase. Nature. 2002;419:43–48. doi: 10.1038/nature00981. [DOI] [PubMed] [Google Scholar]
- 25.Neuberger MS, et al. Immunity through DNA deamination. Trends Biochem Sci. 2003;28:305–312. doi: 10.1016/S0968-0004(03)00111-7. [DOI] [PubMed] [Google Scholar]
- 26.Papavasiliou FN, Schatz DG. Cell-cycle-regulated DNA double-stranded breaks in somatic hypermutation of immunoglobulin genes. Nature. 2000;408:216–221. doi: 10.1038/35041599. [DOI] [PubMed] [Google Scholar]
- 27.Bross L, et al. DNA double-strand breaks: prior to but not sufficient in targeting hypermutation. J Exp Med. 2002;195:1187–1192. doi: 10.1084/jem.20011749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zan H, et al. AID-dependent generation of resected double-strand DNA breaks and recruitment of Rad52/Rad51 in somatic hypermutation. Immunity. 2003;18:727–738. doi: 10.1016/s1074-7613(03)00151-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Casali P, Zan H. Class switching and Myc translocation: how does DNA break? Nat Immunol. 2004;5:1101–1103. doi: 10.1038/ni1104-1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Honjo T, et al. AID to overcome the limitations of genomic information. Nat Immunol. 2005;6:655–661. doi: 10.1038/ni1218. [DOI] [PubMed] [Google Scholar]
- 31.Martin A, Scharff MD. AID and mismatch repair in antibody diversification. Nat Rev Immunol. 2002;2:605–614. doi: 10.1038/nri858. [DOI] [PubMed] [Google Scholar]
- 32.Rada C, et al. Mismatch recognition and uracil excision provide complementary paths to both Ig switching and the A/T-focused phase of somatic mutation. Mol Cell. 2004;16:163–171. doi: 10.1016/j.molcel.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 33.Diaz M, Lawrence C. An update on the role of translesion synthesis DNA polymerases in Ig hypermutation. Trends Immunol. 2005;26:215–220. doi: 10.1016/j.it.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 34.Chaudhuri J, et al. Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature. 2003;422:726–730. doi: 10.1038/nature01574. [DOI] [PubMed] [Google Scholar]
- 35.Pham P, et al. Processive AID-catalysed cytosine deamination on single-stranded DNA simulates somatic hypermutation. Nature. 2003;424:103–107. doi: 10.1038/nature01760. [DOI] [PubMed] [Google Scholar]
- 36.Ramiro AR, et al. Transcription enhances AID-mediated cytidine deamination by exposing single-stranded DNA on the nontemplate strand. Nat Immunol. 2003;4:452–456. doi: 10.1038/ni920. [DOI] [PubMed] [Google Scholar]
- 37.Chaudhuri J, et al. Replication protein A interacts with AID to promote deamination of somatic hypermutation targets. Nature. 2004;430:992–998. doi: 10.1038/nature02821. [DOI] [PubMed] [Google Scholar]
- 38.Papavasiliou FN, Schatz DG. The activation-induced deaminase functions in a postcleavage step of the somatic hypermutation process. J Exp Med. 2002;195:1193–1198. doi: 10.1084/jem.20011858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Unniraman S, et al. Identification of an AID-independent pathway for chromosomal translocations between the Igh switch region and Myc. Nat Immunol. 2004;5:1117–1123. doi: 10.1038/ni1127. [DOI] [PubMed] [Google Scholar]
- 40.Kunkel TA, Erie DA. DNA mismatch repair. Annu Rev Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- 41.Zhang Y, et al. Reconstitution of 5′-directed human mismatch repair in a purified system. Cell. 2005;122:693–705. doi: 10.1016/j.cell.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 42.Wilson TM, et al. MSH2-MSH6 stimulates DNA polymerase η, suggesting a role for A:T mutations in antibody genes. J Exp Med. 2005;201:637–645. doi: 10.1084/jem.20042066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martomo SA, et al. A role for Msh6 but not Msh3 in somatic hypermutation and class switch recombination. J Exp Med. 2004;200:61–68. doi: 10.1084/jem.20040691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bardwell PD, et al. Altered somatic hypermutation and reduced class-switch recombination in exonuclease 1-mutant mice. Nat Immunol. 2004;5:224–229. doi: 10.1038/ni1031. [DOI] [PubMed] [Google Scholar]
- 45.Stavnezer J, Schrader CE. Mismatch repair converts AID-instigated nicks to double-strand breaks for antibody class-switch recombination. Trends Genet. 2005;22:23–28. doi: 10.1016/j.tig.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 46.Li Z, et al. Examination of Msh6- and Msh3-deficient mice in class switching reveals overlapping and distinct roles of MutS homologues in antibody diversification. J Exp Med. 2004;200:47–59. doi: 10.1084/jem.20040355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu X, et al. A role for the MutL mismatch repair Mlh3 protein in immunoglobulin class switch DNA recombination and somatic hypermutation. J Immunol. 2006;176:5426–5437. doi: 10.4049/jimmunol.176.9.5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schofield MJ, Hsieh P. DNA mismatch repair: molecular mechanisms and biological function. Annu Rev Microbiol. 2003;57:579–608. doi: 10.1146/annurev.micro.57.030502.090847. [DOI] [PubMed] [Google Scholar]
- 49.Larson ED, et al. MRE11/RAD50 cleaves DNA in the AID/UNG-dependent pathway of immunoglobulin gene diversification. Mol Cell. 2005;20:367–375. doi: 10.1016/j.molcel.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 50.Yabuki M, et al. The MRE11–RAD50–NBS1 complex accelerates somatic hypermutation and gene conversion of immunoglobulin variable regions. Nat Immunol. 2005;6:730–736. doi: 10.1038/ni1215. [DOI] [PubMed] [Google Scholar]
- 51.Fischhaber PL, Friedberg EC. How are specialized (low-fidelity) eukaryotic polymerases selected and switched with high-fidelity polymerases during translesion DNA synthesis? DNA Repair. 2005;4:279–283. doi: 10.1016/j.dnarep.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 52.Friedberg EC, et al. Trading places: how do DNA polymerases switch during translesion DNA synthesis? Mol Cell. 2005;18:499–505. doi: 10.1016/j.molcel.2005.03.032. [DOI] [PubMed] [Google Scholar]
- 53.Rattray AJ, Strathern JN. Error-prone DNA polymerases: when making a mistake is the only way to get ahead. Annu Rev Genet. 2003;37:31–66. doi: 10.1146/annurev.genet.37.042203.132748. [DOI] [PubMed] [Google Scholar]
- 54.Prakash S, et al. Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu Rev Biochem. 2005;74:317–353. doi: 10.1146/annurev.biochem.74.082803.133250. [DOI] [PubMed] [Google Scholar]
- 55.Hubscher U, et al. Eukaryotic DNA polymerases. Annu Rev Biochem. 2002;71:133–163. doi: 10.1146/annurev.biochem.71.090501.150041. [DOI] [PubMed] [Google Scholar]
- 56.McCulloch SD, et al. Enzymatic switching for efficient and accurate translesion DNA replication. Nucleic Acids Res. 2004;32:4665–4675. doi: 10.1093/nar/gkh777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Diaz M, et al. Decreased frequency of somatic hypermutation and impaired affinity maturation but intact germinal center formation in mice expressing antisense RNA to DNA polymerase ζ. J Immunol. 2001;167:327–335. doi: 10.4049/jimmunol.167.1.327. [DOI] [PubMed] [Google Scholar]
- 58.Simpson LJ, Sale JE. Rev1 is essential for DNA damage tolerance and non-templated immunoglobulin gene mutation in a vertebrate cell line. EMBO J. 2003;22:1654–1664. doi: 10.1093/emboj/cdg161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Delbos F, et al. Contribution of DNA polymerase η to immunoglobulin gene hypermutation in the mouse. J Exp Med. 2005;201:1191–1196. doi: 10.1084/jem.20050292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Martomo SA, et al. Different mutation signatures in DNA polymerase η- and MSH6-deficient mice suggest separate roles in antibody diversification. Proc Natl Acad Sci U S A. 2005;102:8656–8661. doi: 10.1073/pnas.0501852102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jansen JG, et al. Strand-biased defect in C/G transversions in hypermutating immunoglobulin genes in Rev1-deficient mice. J Exp Med. 2006;203:319–323. doi: 10.1084/jem.20052227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McDonald JP, et al. 129-derived strains of mice are deficient in DNA polymerase ι and have normal immunoglobulin hypermutation. J Exp Med. 2003;198:635–643. doi: 10.1084/jem.20030767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Martomo SA, et al. Normal hypermutation in antibody genes from congenic mice defective for DNA polymerase ι. DNA Repair. 2006;5:392–398. doi: 10.1016/j.dnarep.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 64.Seki M, et al. High-efficiency bypass of DNA damage by human DNA polymerase Q. EMBO J. 2004;23:4484–4494. doi: 10.1038/sj.emboj.7600424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Masuda K, et al. DNA polymerase θ contributes to the generation of C/G mutations during somatic hypermutation of Ig genes. Proc Natl Acad Sci U S A. 2005;102:13986–13991. doi: 10.1073/pnas.0505636102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Reynaud CA, et al. What role for AID: mutator, or assembler of the immunoglobulin mutasome? Nat Immunol. 2003;4:631–638. doi: 10.1038/ni0703-631. [DOI] [PubMed] [Google Scholar]
- 67.Maga G, Hübscher U. Proliferating cell nuclear antigen (PCNA): a dancer with many partners. J Cell Sci. 2003;116:3051–3060. doi: 10.1242/jcs.00653. [DOI] [PubMed] [Google Scholar]
- 68.Garg P, et al. Proliferating cell nuclear antigen promotes translesion synthesis by DNA polymerase ζ. J Biol Chem. 2005;280:23446–23450. doi: 10.1074/jbc.C500173200. [DOI] [PubMed] [Google Scholar]
- 69.Garg P, Burgers PM. Ubiquitinated proliferating cell nuclear antigen activates translesion DNA polymerases η and REV1. Proc Natl Acad Sci U S A. 2005;102:18361–18366. doi: 10.1073/pnas.0505949102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lehmann AR. Replication of damaged DNA by translesion synthesis in human cells. FEBS Lett. 2005;579:873–876. doi: 10.1016/j.febslet.2004.11.029. [DOI] [PubMed] [Google Scholar]
- 71.Warbrick E. The puzzle of PCNA’s many partners. Bioessays. 2000;22:997–1006. doi: 10.1002/1521-1878(200011)22:11<997::AID-BIES6>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 72.Kannouche PL, et al. Interaction of human DNA polymerase η with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol Cell. 2004;14:491–500. doi: 10.1016/s1097-2765(04)00259-x. [DOI] [PubMed] [Google Scholar]
- 73.Lee SD, Alani E. Analysis of interactions between mismatch repair initiation factors and the replication processivity factor PCNA. J Mol Biol. 2006;355:175–184. doi: 10.1016/j.jmb.2005.10.059. [DOI] [PubMed] [Google Scholar]
- 74.Clark AB, et al. Functional interaction of proliferating cell nuclear antigen with MSH2–MSH6 and MSH2–MSH3 complexes. J Biol Chem. 2000;275:36498–36501. doi: 10.1074/jbc.C000513200. [DOI] [PubMed] [Google Scholar]
- 75.Flores-Rozas H, et al. Proliferating cell nuclear antigen and Msh2p–Msh6p interact to form an active mispair recognition complex. Nat Genet. 2000;26:375–378. doi: 10.1038/81708. [DOI] [PubMed] [Google Scholar]
- 76.Lau PJ, Kolodner RD. Transfer of the MSH2·MSH6 complex from proliferating cell nuclear antigen to mispaired bases in DNA. J Biol Chem. 2003;278:14–17. doi: 10.1074/jbc.C200627200. [DOI] [PubMed] [Google Scholar]
- 77.Maser RS, et al. Mre11 complex and DNA replication: linkage to E2F and sites of DNA synthesis. Mol Cell Biol. 2001;21:6006–6016. doi: 10.1128/MCB.21.17.6006-6016.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dzantiev L, et al. A defined human system that supports bidirectional mismatch-provoked excision. Mol Cell. 2004;15:31–41. doi: 10.1016/j.molcel.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 79.Nielsen FC, et al. Characterization of human exonuclease 1 in complex with mismatch repair proteins, subcellular localization and association with PCNA. Oncogene. 2004;23:1457–1468. doi: 10.1038/sj.onc.1207265. [DOI] [PubMed] [Google Scholar]
- 80.Binz SK, et al. Replication protein A phosphorylation and the cellular response to DNA damage. DNA Repair. 2004;3:1015–1024. doi: 10.1016/j.dnarep.2004.03.028. [DOI] [PubMed] [Google Scholar]
- 81.Basu U, et al. The AID antibody diversification enzyme is regulated by protein kinase A phosphorylation. Nature. 2005;438:508–511. doi: 10.1038/nature04255. [DOI] [PubMed] [Google Scholar]
- 82.Matsumoto Y. Molecular mechanism of PCNA-dependent base excision repair. Prog Nucleic Acid Res Mol Biol. 2001;68:129–138. doi: 10.1016/s0079-6603(01)68095-4. [DOI] [PubMed] [Google Scholar]
- 83.Muramatsu M, et al. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J Biol Chem. 1999;274:18470–18476. doi: 10.1074/jbc.274.26.18470. [DOI] [PubMed] [Google Scholar]
- 84.Li Z, et al. The mismatch repair protein Msh6 influences the in vivo AID targeting to the Ig locus. Immunity. 2006;24:393–403. doi: 10.1016/j.immuni.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 85.Li Z, et al. A role for Mlh3 in somatic hypermutation. DNA Repair. doi: 10.1016/j.dnarep.2006.02.003. in press. [DOI] [PubMed] [Google Scholar]
- 86.Martin A, et al. Msh2 ATPase activity is essential for somatic hypermutation at A-T basepairs and for efficient class switch recombination. J Exp Med. 2003;198:1171–1178. doi: 10.1084/jem.20030880. [DOI] [PMC free article] [PubMed] [Google Scholar]