

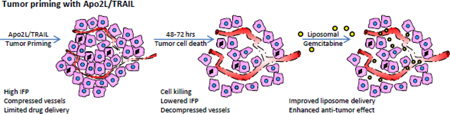
Abstract
Interstitial fluid pressure (IFP) is elevated in tumors and high IFP, a negative cancer prognosticator, is known to limit the uptake and efficacy of anti-tumor therapeutics. Approaches that alter the tumor microenvironment and enhance uptake of therapeutics are collectively referred to as tumor “priming”. Here we show that the cytotoxic biological therapy Apo2L/TRAIL can prime the tumor microenvironment and significantly lower IFP in three different human tumor xenograft models (Colo205, MiaPaca-2 and a patient gastrointestinal adenocarcinoma tumor xenograft). We found that a single dose of Apo2L/TRAIL resulted in a wave of apoptosis which reached a maximum at 8 hrs post-treatment. Apoptotic debris subsequently disappeared concurrent with an increase in macrophage infiltration. By 24 hrs post-treatment, treated tumors appeared less condensed with widening of the stromal areas which increased at 48 and 72 hrs. Analysis of tumor vasculature demonstrated a significant increase in overall vessel size at 48 and 72 hrs although the number of vessels did not change. Notably, IFP was significantly reduced in these tumors by 48 hrs after Apo2L/TRAIL treatment. Administration of gemcitabine at this time resulted in increased tumor uptake of both gemcitabine and liposomal gemcitabine and significantly improved anti-tumor efficacy of liposomal gemcitabine. These results suggest that Apo2L/TRAIL has potential as a tumor priming agent and provides a rationale for developing a sequencing schema for combination therapy such that an initial dose of Apo2L/TRAIL would precede administration of gemcitabine or other therapies.
Keywords: Apo2L/TRAIL, patient tumor xenograft, interstitial fluid pressure, gemcitabine, liposomes, drug uptake
Graphical abstract
1. Introduction
Therapeutic resistance of a tumor is often thought of as the outcome of dysregulated intrinsic signaling pathways that promote tumor cell survival or inhibit apoptosis. However, resistance can also be the result of microenvironmental factors which hinder delivery of drug (see review [1]). In part, this situation is due to immature, abnormally permeable vasculature which allows increased fluid and serum protein accumulation in the tumor microenvironment (TME) giving rise to elevated interstitial fluid pressure (IFP; [2, 3]). High IFP greatly reduces movement of therapeutics from capillaries into the tumors in comparison to normal tissues [4, 5]. This problem is compounded by the increased solid stress caused by proliferating tumor and stromal cells, as well as increased, often stiffer [6], extracellular matrix (ECM) produced by these cells [7]. Solid stress often collapses blood vessels and lymphatics, creating regions of hypoxia and preventing lymphatic drainage [8]. Overall, the reduction in functional vasculature and the increased IFP impedes transport of drugs out of the remaining perfused capillaries (reviewed in [1, 9, 10]). Additionally, the elevated IFP results in directional flow of fluid out of the tumor thereby reducing retention of drugs which are delivered [11, 12]. It has been shown that the use of some cytotoxic therapies can, by inducing apoptosis of tumor cells, lead to decompression of blood vessels (e.g. diphtheria toxin -[13] or paclitaxel [14]) and lowering of IFP (e.g. taxanes [15, 16]) as well as improved drug delivery [17]. This and other approaches to priming the abnormal tumor microenvironment for improved drug delivery, including vascular normalization [18, 19] and stromal depletion [20, 21], have recently been reviewed by Khawar et al [22].
Apo2L/TRAIL (tumor necrosis factor related apoptosis inducing ligand) is a recombinant form of TRAIL (TNF related apoptosis inducing ligand) and is a promising cytotoxic therapy that selectively targets malignant cells while sparing normal cells [23–25].TRAIL induces apoptosis through binding to the cell surface death receptors, DR4 and DR5, and activating the extrinsic apoptotic pathway. Thus it is an alternative approach to inducing apoptosis compared to traditional chemotherapies and radiation that work through the p53 dependent intrinsic pathway [25]. Many tumor cell lines [25] and patient tumor xenografts [26–28] are sensitive to killing by Apo2L/TRAIL. The mechanism(s) underlying the lack of toxicity to normal cells has still not been clarified, although multiple possibilities have been proposed including expression of decoy receptors (This, in conjunction with the observed lack of toxicity to normal cells, formed the basis for developing Apo2L/TRAIL and agonistic antibodies to DR4 and DR5 for clinical use [25, 29]). However, despite the preclinical antitumor activity of rhApo2L/TRAIL (dulanermin) and the fact that it showed no toxicity at the doses used in a Phase I trials (summarized in [30]), there was also no significant additional benefit in a randomized controlled Phase II trial comparing standard of care with and without dulanermin [30, 31]. In preclinical models Apo2L/TRAIL has shown added benefit in combination with a great variety of other therapies and a large number of tumor-specific mechanisms of these interactions have been identified (reviewed in [32]). However, whether Apo2L/TRAIL can reduce solid stress in the TME and the potential effect of Apo2L/TRAIL induced tumor cell apoptosis on tumor IFP has not been investigated. Based on the cytotoxic effects of Apo2L/TRAIL in sensitive tumors and the recent suggestion by Marcucci and Corti [33] that so-called “promoter” drugs could improve the subsequent uptake of “effector” drugs, we wondered if Apo2L/TRAIL could be used as a promoter drug to improve the efficacy of subsequently administered chemotherapy.
Here we show that a single dose of Apo2L/TRAIL induced apoptosis in both colon and pancreatic human xenograft tumors and resulted in a significant decrease in IFP by 48 hrs. In a patient gastrointestinal (GI) tumor xenograft, this was accompanied by an increase in vessel size and number of perfused tumor vessels. We administered gemcitabine (free and liposomal) at 48 hrs following Apo2L/TRAIL and observed improved uptake of both free and liposomal gemcitabine as well as a significant improvement of the anti-tumor efficacy of the liposomal formulation. These results support the conclusion that Apo2L/TRAIL can decompress the tumor and reduce IFP leading to improved uptake and anti-tumor efficacy of subsequently administered chemotherapy and thus providing a rationale for sequential administration of Apo2L/TRAIL followed by chemotherapy for improved drug delivery and therapeutic benefit.
2. Materials and Methods
2.1. Animal Models
CB17 SCID mice were housed in the AALAC accredited Laboratory Animal Resource and experiments carried out in accordance with RPCI Animal Care and Use Committee approved protocols. Colo205, a human colon adenocarcinoma, and MiaPaCa-2, a human pancreatic adenocarcinoma, were obtained from ATTC. An Apo2L/TRAIL resistant Colo205 cell line (Colo205 R) was generated by continuously exposing cells to 250ng/ml of Apo2L/TRAIL for several weeks (sensitivity to Apo2L/TRAIL was less than 10% compared to >80% killing of the wild type). Cell line tumors were generated by subcutaneous injection of 1 × 106 cells. The patient tumor xenograft was established by engrafting de-identified patient tumor tissue obtained through the Pathology Resource Network at RPCI subcutaneously into SCID mice. (For details, see [27]). Tumor volume was determined using the formula volume= width2 × length/2.
2.2. Experimental Design
Apo2L/TRAIL was provided by Genentech, Inc; gemcitabine (Eli Lilly) was acquired from the pharmacy at RPCI. To investigate the histology of tumors following a single dose of Apo2L/TRAIL (average tumor size approximately 200mm3), mice were injected i.p. with 1000µg Apo2L/TRAIL and tumors recovered at specified time points. Previously, we have treated tumor bearing mice with 500µg recombinant human Apo2L/TRAIL daily for two or more weeks with no signs of toxicity ([26, 27, 34]. The half-life of Apo2L/TRAIL in rodents is 3–5 min [35]. To visualize hypoxia, mice were injected, one hour prior to sacrifice, with 60mg/kg pimonidazole i.p. (Hypoxyprobe, NPI, Burlington, MA). Tumors were fixed in 10% formalin or zinc fixative and paraffin embedded. IFP was measured 48 hrs after administration of Apo2L/TRAIL or gemcitabine (75mg/kg or near MTD (maximum tolerated dose) of 140mg/kg [36]). For efficacy experiments, free gemcitabine was administered at 75 mg/kg and liposomal gemcitabine at 15 mg/kg based on preliminary experiments in which liposomal gemcitabine at 60mg/kg dose showed toxicity. Note that the dose for liposomal gemcitabine is less than that for free gemcitabine since free gemcitabine is eliminated much faster from blood as compared to liposomal formulation, the levels drop rapidly and thus overall free gemcitabine has less systemic toxicity than the liposomal formulation For drug content analysis after administration of gemcitabine, tumors were obtained from 3–5 mice at each of the indicated time points. For analysis after administration of liposomal gemcitabine, tumors were obtained from 2–3 mice/ time points.
2.3. Immunohistochemistry (IHC)
Antibodies used were: mouse anti-cleaved cytokeratin 18 (clone M30, Alexis Biochemicals), rat anti-mouse F4/80 (MCA497R, Serotec), MAb1 clone 4.3.11.31for hypoxyprobe adducts (Hypoxyprobe-1, NPI, Burlington, MA); rat anti-mouse CD31 (#553370 Pharmingen; zinc fixation) and visualized with biotinylated secondary antibodies, ABC-RTU and DAB (Vector Laboratories Inc).
2.4. Tumor interstitial fluid pressure (IFP)
IFP was measured as reported earlier [37, 38] using a wing-tip needle catheter fitted with a 23½ gauge needle is coupled to a micro pressure transducer (Millar Instruments, Model SPR-524 via a saline filled catheter). The analog output of the transducer is converted to digital signal (Data Translation Model DT9816 converter), fed to a digital amplifier (Millar Pressure Control Unit Model PCU-2000) connected to a computer. Data was acquired and plotted using a program developed in the laboratory using DT Measure Foundry (Version 4.0.7 Data Translation). Pressure data was collected at a sampling rate of 1 kHz and averaged over 1 second. IFP was measured every few millimeters along the entry path and averaged. The instrument was calibrated using a water column manometer from 0 to 30 cm of water. Several readings were obtained from each tumor.
2.5. Analysis of Blood Vessel Morphology
Four control, untreated mice and, at each time time point, 2 treated mice were sacrificed for analysis (see Fig 3I). CD31 stained slides were scanned into a ScanScope XT (Aperio) and images of areas with viable tumor were captured at 5× (2–6 images /section). CD31(+) stained vessels were identified and profiles were filled in Photoshop. Area quantification was accomplished using ImageJ: 1) each image was split into its three RGB colors and the blue was selected for quantification, 2) the threshold was adjusted to maximally colorize the filled areas and minimize background and 3) using ImageJ, we performed “Particle analysis” to enumerate the vessel sillhouettes using a minimal size of 10 px2. This was done for each of several images taken from each slide and the average vessel number and size recorded.
Fig. 3.

Priming of a patient tumor xenograft (PDX 14244) by Apo2L/TRAIL. (A–D) H&E stained sections of the periphery of tumors at selected times post-treatment. (A) Control tumor is surrounded by a connective tissue capsule (✱) and contains well-differentiated glandular structures. (B) At 4 hrs apoptotic bodies are seen in the periphery (arrow) (C) At 8 hrs the number of apoptotic cells is increased (arrow). (D) At 48 hrs cellular debris is reduced and blood vessels appear more open (arrow). Bar= 25 µm. (E–J) Analysis of vascular changes in a patient xenograft following Apo2L/TRAIL. Representative sections of (E) a control tumor and (G) a tumor 48 hrs after Apo2L/TRAIL stained with anti-CD31 to visualize blood vessels Bar= 200µm. (F, H) Profiles of vessels in E & G. (J) Vessel sizes in four control and 2 treated tumors from each timepoint were analyzed; each point is an average of pixels2/5× fields for several fields/slide
2.6. Liposomes
Liposomal gemcitabine (L-Gem) was prepared using distearolyphosphatidylcholine (DSPC), cholesterol (Chol) and PEG 5000-distearolyphosphatidylethanolamine (PEG 5000-DSPE) using a remote loading method. Briefly, the above lipids in chloroform solution were mixed in the molar ratio 1:0.5:0.05 (DSPC:Chol:PEG-DSPE molar ratio). The mixed lipid solution was dried in a rotary evaporator, taken up in a mixture of benzene and methanol (8:2 v/v), frozen in liquid nitrogen and lyophilized overnight. Residual solvents after overnight lyophilization are close to zero. The lyophilized lipid mixture was dispersed in 300mM ammonium sulfate, vortexed vigorously and subjected to three freeze/thaw cycles. The lipid dispersion was then extruded stepwise through polycarbonate membrane filters of decreasing pore sizes starting with 1µm followed by 0.8, 0.4 and finally through 0.1µm. A 10ml Thermobarrel Lipex extruder (Northern Lipids Inc. Vancouver B.C. Canada), heated to 50°C using a circulating water bath, was used for liposome extrusion. The liposomes were extruded through each of the filters between 6 to 8 times to ensure correct size. The two basic properties of the gemcitabine liposomes that we examined were size, by dynamic laser scattering (DLS), and loading efficiency. The size of the final liposome preparation was measured by dynamic laser scattering (DLS) using a Nicomp Model 370 Particle Sizer and was found to be approximately 103nm. Ammonium sulfate in the external medium of the liposome preparation was removed by overnight dialysis in HEPES buffered saline pH7.4. Dry gemcitabine was then added to the liposomes and the mixture shaken until it was completely dissolved. The liposomal gemcitabine preparation was then incubated in a water bath at 60°C for one hour and then chilled in an ice bath. The gemcitabine liposomes used in this study were not further purified after loading because the loading efficiency was close to 93–95%. The liposomes were stored at in a refrigerator 4°C until used. Fluorescent liposomes were prepared as above except that a fluorescent dye (below) was incorporated within the lipid bilayer. The membrane dye 1,1'-dioctadecyl-3,3,3',3'-tetramethylindodicarbocyanine-5,5'-disulfonic acid (DiIC18(5)-DS; Invitrogen) was added to the lipid mixture at 0.1% of the total lipid molar concentration. This dye with two covalently linked, long C18 acyl chains, the same length as the primary lipid used, incorporates in the lipid bilayer. The lipids were dispersed in HEPES buffer saline (pH 7.4) and extruded to a final size of 100nm diameter. DiIC18(5)-DS was selected based on absence of autofluorescence from biological tissue at the dye excitation and emission wavelengths of 650 and 670 nm [39].
2.7. Fluorescent liposomes-quantification of perfusion and uptake
Fluorescent liposomes (200µl) were injected via the tail vein, tumors recovered 24 hrs later and embedded in OCT. Sections (0.8µm) were viewed using a Nikon eclipse Ti inverted fluorescence microscope with a Cy5 filter cube. Fluorescence micrographs were taken using a SPOT camera (Model RTke, Diagnostic Instruments, Inc.). The number of perfused blood vessels in each field of view (FOV) at 20× was determined using ImageJ with the threshold intensity set at 200 to 255 for the 8bit monochrome images to obtain binary images of the micrographs. The number of perfused vessels was then determined using particle analyses defining a particle by setting the minimum and maximum pixel counts at 5 and 5000 respectively. Total tumor uptake of liposomes was determined as the sum total of the product of pixel intensity (between 5 and 255) and the number of pixels. Between 4 and 8 FOVs per tumor section and two tumors each from a control untreated group and an Apo2L/TRAIL treated group were analyzed.
2.8. Pharmacokinetics of gemcitabine and liposomal gemcitabine uptake
Gemcitabine and its metabolite dFdU were analyzed by LC-MS/MS. Briefly, mouse tissue and plasma samples were homogenized in PBS and extracted with ice cold acetonitrile containing the internal standard colofarabine 2.5ng/ml. The extracts were dried and taken up in HPLC mobile phase A (below). LC-MS/MS analysis was performed in positive ion mode using a Thermo Scientific Surveyor MS Pump Plus and Thermo Scientific TSQ Quantum Ultra fitted with Atlantis™ dC18 5 µm, 2.1 × 150 mm column at 30°C. Mobile phase A consisted of 2% acetonitrile 0.5% formic acid and mobile phase B was 98% acetonitrile 0.5% formic acid and initial gradient was 1% B and increasing to 100% B at 7.5 minutes. Using the LC-MS/MS method used at RPCI for evaluation of clinical samples, two calibration curves were prepared with a range of 0.5ng/ml to 1000ng/ml and duplicate quality controls were run at 3 concentrations (7.5, 75ng/ml and 750ng/ml). Overall average precision of the gemcitabine calibrators was 6.89% with % accuracy ranging from 94.5% to 107.9%. Gemcitabine quality controls had an average precision of 1.36% with % accuracy ranging from 94% to 101%. Gemcitabine for calibration was obtained from Toronto Research Chemicals (Cat# G305000, Lot# 17-ANR-123-1).
2.9. Statistics
Statistical analysis was performed with the Student t test or Anova (Sigma Plot, Systat Software, or GraphPad Prism). Data is plotted with the mean and standard error. A p< 0.05 was considered significant.
3. Results
3.1. A single dose of Apo2L/TRAIL induces changes in the tumor microenvironment
To investigate the effects of a single dose of Apo2L/TRAIL, we administered 1000µg Apo2L/TRAIL to mice bearing the Apo2L/TRAIL sensitive human cell line xenograft Colo205 [23]. Tumors were recovered at 2, 8, 24 and 48 hrs after treatment and examined histologically. In untreated tumors (Fig 1A), dense clusters of tumor cells were surrounded by thin areas of compact stroma and contained many mitotic figures Apo2L/TRAIL induced apoptosis in these tumors so that by 2 hrs post-treatment (Fig 1B), apoptotic bodies were apparent in localized foci. By 8 hrs Fig 1C), the tumors contained widespread regions of apoptotic debris and appeared less compact with widened stromal regions; residual debris was still seen 24 hrs post-treatment (Fig 1D). However by 48 hrs (Fig 1E), debris was no longer apparent, tumors appeared less compact and vessels appeared enlarged. IHC confirmed that the number of apoptotic cells peaked at 8 hrs after treatment (Fig 1F–I). The number of labeled cells in 3 representative fields was counted and is shown graphically. Infiltration of the tumors by macrophages also peaked at 8 hrs after treatment (Fig 1J–M) suggesting that macrophages are responsible for clearing the debris.
Fig. 1.

Kinetics of apoptosis induced in human Colo205 xenografts by a single Apo2L/TRAIL treatment. (A–E) H&E sections post-treatment: (A) Control, tumors are compact with mitotic figures (arrows); (B) at 2 hrs foci of apoptosis are seen (arrows); (C) at 8 hrs widespread areas of apoptotic bodies and cellular debris are evident (arrows) and stromal areas are wider (arrowhead); (D) at 24 hrs debris remnants are seen (arrow) and (E) at 48 hrs post-treatment, the debris is cleared and the tumor microenvironment appears less compact (arrowhead). (F–I) IHC for apoptotic cells (cleaved cytokeratin18+ cells); bar graph indicates average number of cells in 3 fields. (J–M) IHC for macrophages (F4/80+ cells); bar graph indicates average number of cells in three fields. Bar= 50µm
3.2. A single dose of Apo2L/TRAIL reduces interstitial fluid pressure
To determine whether Apo2L/TRAIL affected IFP, we measured IFP in the tumors of control, untreated mice and compared levels to tumors of treated mice 48 hrs post-treatment when maximal changes in the TME were observed histologically. Measurements were not made on the same tumor before and after treatment because this technique is invasive and disrupts the tumor microenvironment In the Apo2L/TRAIL sensitive Colo205 tumors, we saw a significant drop in IFP (p< 0.01), Fig 2A). In contrast, tumors from a resistant clone did not undergo a similar drop in IFP (Fig 2B). An Apo2L/TRAIL sensitive pancreatic adenocarcinoma cell line, MiaPaCa-2 [40] also underwent a significant drop in IFP (p< 0.0001, Fig 2C). Previously, we have found that some patients’ GI tumor xenografts are sensitive to Apo2L/TRAIL [26, 27]. Therefore, we tested a patient GI tumor xenograft (PDX #14244) and observed a significant drop in IFP at 48 hrs after Apo2L/TRAIL in this tumor also (Fig 2D, p< 0.01).
Fig. 2.

IFP in xenograft tumors 48 hrs after Apo2L/TRAIL. (A) Colo205: IFP is significantly reduced, p<0.005, (B) Colo205 Apo2L/TRAIL resistant clone: IFP is not significantly different, (C) MiaPaCa-2: IFP is significantly significantly reduced, p< 0.0001, (D) Patient tumor xenograft: IFP is significantly reduced, p< 0.005.
3.3. Apo2L/TRAIL induced changes in the tumor microenvironment
The kinetics of apoptosis in patient xenograft PDX #14244 were similar to those observed in Colo205 (Fig 3). Tumors were recovered from four control mice (0 hr) and two mice each at 4, 8, 24, 48 and 72 hrs following a single dose of Apo2L/TRAIL. The untreated tumors (Fig 3A) were surrounded by a connective tissue capsule and consisted of glands separated by stroma. At 4 hrs (Fig 3B), areas of apoptotic cells were seen in the periphery and became more widespread at 8 hrs (Fig 3C). This peak of apoptosis correlated with a high degree of infiltration by macrophages (F4/80+ cells) which were especially dense in areas of obvious apoptosis (Fig S-1). Apoptotic debris was removed by 48 hrs, at which time blood vessels containing RBCs could be identified within the interior of the tumor (Fig 3D, arrow).
To quantify the changes in the vasculature, we analyzed the vessels in these tumors. Sections of zinc fixed tumors were stained with anti-CD31, photographed and the vessel outlines were filled manually using Adobe Photoshop. Using ImageJ, the images were converted to black and white profiles. Representative fields from stained sections of untreated control tumors vs. 48 hrs post-treatment tumors (Fig 3 E, G) and the vessel profiles derived from each section are shown (Fig 3 F, H). The images of vessel profiles were analyzed for total vessel count and average vessel size; multiple (3–5) different 5× fields were averaged per section. While the total number of CD31(+) vessels did not change (data not shown), the average vessel size increased. At 48 hours, 1 of 2, and by 72 hours, 2 of 2, tumors contained vessels which were increased in size over those in the four control tumors (Fig 3I). Because CD31(+) staining alone is not indicator of vessel patency, as we have previously reported (Xu eat., 2007), we next asked how Apo2L/TRAIL pretreatment would alter the distribution of NIR dye labelled liposomes. We injected mice with liposomes containing fluorescent DiIC18 (5)-DS at 48 hrs post-Apo2L/TRAIL treatment. The advantage of using this dye is the negligible autofluorescence in the near infrared (670nm). Representative images of liposomal uptake are shown (S-2-A-control, B-Apo2L/TRAIL primed). Apo2L/TRAIL primed tumors had both increased numbers of perfused vessels (S-4C) and increased total uptake of liposomes (S-4D). That this increase in average vessel size and increased patency is related to increased functionality is supported by a significant reduction in tumor hypoxia. Untreated tumors were uniformly hypoxic (Fig 4A) whereas at 48 and 72 hrs post-treatment, tumors were extremely heterogeneous with many areas which did not stain (Fig 4B, C).
Fig. 4.

Hypoxia in PDX #14244 is reduced following a single treatment with Apo2L/TRAIL. (A) Control tumors are homogeneously hypoxic (hypoxyprobe-brown). (B, C) At 48 and 72 hrs following a single dose of Apo2L/TRAIL, tumors have areas of reduced hypoxia. Bar = 200µm.
3.4. Gemcitabine uptake is increased after a single dose of Apo2L/TRAIL
To determine whether the decompression of tumor vessels and concomitant decrease in IFP could improve uptake of subsequently administered therapeutics, we compared gemcitabine uptake in control and Apo2L/TRAIL primed mice. Tumor bearing mice received gemcitabine either 30 min or 48 hrs after the priming dose of Apo2L/TRAIL. At several time intervals after gemcitabine administration (0.5 hr, 1 hr, 2 hrs and 6 hrs), tumors were recovered for pharmacological analysis. When gemcitabine was given 30 min after Apo2L/TRAIL, there was no difference in gemcitabine uptake in the tumors of untreated and pre-treated mice (Fig 5A). However, when gemcitabine was administered at 48 hrs after Apo2L/TRAIL, the uptake peaked at 1 hour and remained slightly higher through the remainder of the 6 hrs (Fig 5B). Apo2L/TRAIL priming resulted in a 35% increase in gemcitabine uptake compared to concurrent administration Fig 5C.
Fig. 5.

Tumor priming with Apo2L/TRAIL 48 hrs prior to administration of free gemcitabine increased gemcitabine uptake. Gemcitabine was administered either (A) 30 min or (B) 48 hrs after Apo2L/TRAIL. (Error bars shown in one direction; n= 3–5). (C) Comparison of gemcitabine uptake (area under the curve, AUC) in these two protocols shows a 33% enhancement when gemcitabine was administered 48 hours after tumor priming with Apo2L/TRAIL. (D) Efficacy of combination therapy: Apo2L/TRAIL (large shaded arrow) was given on day 1 and gemcitabine was started 48 hrs later and given on days 3, 5, and 7 (small hatched arrows) to groups as indicated in the legend. (PDX 14244, n= 5, average= heavy dashed line).
3.5. Apo2L/TRAIL priming enhances efficacy of gemcitabine
We next asked whether tumor priming and the increased gemcitabine uptake would result in improved efficacy. Mice with PDX #14244 were divided into four groups which received either: (1) saline, (2) a single dose of Apo2L/TRAIL on day 1, (3) 3 doses of gemcitabine (75 mg/kg) on days 3, 5, and 7, or (4) Apo2L/TRAIL on day 1 followed by 3 doses of gemcitabine on days 3, 5, and 7. Tumor growth was monitored for 10 days during which the controls, Apo2L/TRAIL alone or gemcitabine alone treated mice showed continued tumor growth. In comparison, tumors in mice which received sequential combination therapy showed a trend to slower growth, although the difference is not statistically significant. (Fig 5 D, Fig S-3). The lack of anti-tumor efficacy in the group which received a single dose of Apo2L/TRAIL underscores the observation that although PDX #14244 is Apo2L/TRAIL sensitive, daily dosing is needed to significantly control tumor growth [34]. Interestingly, gemcitabine treatment alone (either moderate-75 mg/kg or high-140 mg/kg dose) did not lower IFP (Fig S-4) providing additional rationale for initiating combination therapy with Apo2L/TRAIL.
3.6. Apo2L/TRAIL priming significantly improves the efficacy of liposomal gemcitabine
We wondered whether the enhanced effect of combination therapy could be improved by using liposomal gemcitabine which has a longer half-life (reported half-life ~13 hrs; [41]) than free gemcitabine (half-life between 8.5 and 17 min; [41, 42]). We next analyzed Liposomal-gem uptake in mice bearing PDX #14244 tumors and saw increased gemcitabine uptake when liposomes were administered 48 hrs after priming (Fig 6A and Fig. 6B, calculated AUC). Finally, we treated mice with Apo2L/TRAIL alone, liposomal gemcitabine alone, or a combination strategy in which tumors were primed with Apo2L/TRAIL and then treated with liposomal gemcitabine (15 mg/kg) 48 hrs later (2 cycles given 7 days apart). As seen in Fig 6C, the combination strategy resulted in a significant improvement in tumor control in comparison to either single agent alone (p< 0.01). Again, the once weekly dosing with Apo2L/TRAIL used here is not sufficient to control tumor growth [34].
Fig. 6.

A single dose of Apo2L/TRAIL increases liposomal gemcitabine (L-gem) uptake in tumors (PDX 14244) pre-treated with Apo2L/TRAIL. (A) Graph of gemcitabine uptake (error bars one direction only, n=2–3). (B) Comparison of total L-gem uptake (AUC) by tumors in (A) shows a 45% increase in Apo2L/TRAIL treated tumors over a 24 hr period. (C) Treatment of tumor bearing mice with Apo2L/TRAIL and L-gem, either alone or in combination (Apo2L/TRAIL d1 and d7 (dark gray arrows), L-gem on d3 and d9 (light gray arrows). The tumors in the group which received the combination sequentially were significantly smaller than tumors in the other groups (n=5, ** p< 0.01).
4. Discussion
4.1 Apo2L/TRAIL as a promotor drug to prime the TME
Targeting the death receptors DR4 and DR5 with either Apo2L/TRAIL or agonistic antibodies is a promising approach for treating patients with malignant disease for several reasons: 1) these death receptors activate the extrinsic apoptotic pathway as opposed to traditional therapies which work by p53 mediated activation of the intrinsic apoptotic pathway and 2) dose-limiting toxicity to normal tissue has not occurred in initial clinical trials (e.g. [43]). However, at the doses used, neither the ligand nor the antibodies have shown significant clinical efficacy [30]. Since strategies combining Apo2L/TRAIL (or DR targeting antibodies) will likely involve combination with other reagents, understanding the mechanisms of interaction will allow us to design rationale schedules for administration of these combinations. Recently, Marcucci and Corti have discussed the concept of “promoter drugs” that are able to increase the uptake and penetration of other anti-tumor “effector drugs” [33]. There are reports that cytotoxic therapies can decompress the tumor microenvironment [13], lower IFP and improve drug delivery [15] and thus the idea of priming the tumor microenvironment with cytotoxic therapies is promising [22]. Here, we present data showing that Apo2L/TRAIL, in addition to being an effector drug, can prime a tumor, inducing apoptosis to decompress the microenvironment and resulting in lowered IFP at 48 hrs post-treatment. When gemcitabine was administered at this time, uptake of free gemcitabine was increased by approximately 35% and we observed a trend towards improved efficacy. When we repeated the experiment with a liposomal formulation of gemcitabine, not only were we able to use a substantially lower dose of gemcitabine, but uptake was improved by 45% over a 24 hr period and the antitumor efficacy was significantly improved over L-gem alone. We did not examine whether empty liposomes, i.e., without gemcitabine, reduced tumor size since it is documented in the literature that empty liposomes do not influence growth of primary tumors ([44]). This enhanced uptake of liposomal gemcitabine after tumor priming is comparable to what has been reported earlier by Lu et al 2007 ([14] in which a 43% increase in uptake of liposomal doxorubicin occurred following priming of FaDu head and neck tumors with paclitaxel. Additionally, we have shown previously that tumor priming by mild systemic heating leads to increased tumor vessel perfusion and subsequent uptake of liposomal doxorubicin [45].
In the protocol that we used for these efficacy studies, we administered Apo2L/TRAIL first to reduce solid stress and lower the IFP allowing reperfusion of compressed vessels (as shown by increased number of patent vessels and uptake of dye containing liposomes) to facilitate gemcitabine uptake. Many tumors, however, are resistant to DR4 and DR5 targeting reagents. In this regard, a variety of agents are able to sensitize many resistant tumors to Apo2L/TRAIL [25, 32]; for instance, chemotherapies (e.g. gemcitabine) can sensitize tumors by downregulation of FLIP [46] or upregulation of DR5 and/or p53 [47]. A recent pre-clinical study of combination therapy using the glioblastoma cell line D54 concluded that administering doxorubicin 18 hrs prior to an anti-DR5 agonistic antibody resulted in greater than 2-fold upregulation of DR5 on the cell surface and significantly increased apoptosis. In the glioblastoma experiments, however, increased apoptosis was not seen when anti-DR5 antibody was administered first, or simultaneously [48]. Thus, it might be possible in the case of an Apo2L/TRAIL resistant tumor to first sensitize the tumor to Apo2L/TRAIL. Additionally, we found that it takes 48–72 hrs to see the reduction in IFP which follows. This reduced IFP may be a predictive biomarker for the activity of Apo2L/TRAIL which facilitates gemcitabine uptake. Optimizing the interval between Apo2L/TRAIL and administration of the second therapy seems to be important in terms of allowing sufficient time for clearance of the apoptotic debris induced by the priming agent. Therefore, we expect that a treatment protocol could be optimized to take advantage of the priming action in which an initial dose of Apo2L/TRAIL could improve chemotherapy uptake, which in turn could increase DR5 expression and increase the sensitivity of the tumor to the next dose of Apo2L/TRAIL. Since Apo2L/TRAIL did not show systemic toxicity in clinical trials, it would be logical to begin combination therapy with a single dose of Apo2L/TRAIL. If a tumor is sensitive to Apo2L/TRAIL, this would result in better accumulation of the second therapeutic directly in the tumor and improved efficacy. As treatment progressed, the sensitizing effects of the second therapeutic would therefore be enhanced. It might also be possible to combine Apo2L/TRAIL with other approaches to reducing IFP, for example enzymatic digestion of hyaluronan which caused a reduction in IFP [49] leading to increased vessel functionality, uptake of chemotherapy and improved anti-tumor efficacy [21] or normalization of vessels with anti-angiogenic therapies [2, 50]. We have also shown that IFP can be reduced by mild hyperthermia treatment 4–6 hrs after exposure and this is maintained for at least 24 hrs resulting in decreased hypoxia and significantly improved response to radiation therapy [37] when radiation is administered 24h post-heating. Optimal interdose interval has been reported using PBPK/PD modelling for combined paclitaxel and liposomal doxorubicin [51] and this interval was identified as 24h post-paclitaxel dosing. The interdose interval of 48h in this study was based upon analyses of tumor histology and IFP at different times after Apo2L/TRAIL dosing. It is possible that PBPK/PD modeling for Apo2L/TRAIL and liposomal gemcitabine may suggest an optimal interval that is different from the one we report here and this possibility is worth pursuing in the future.
The further observation that the PDX #14244 was essentially completely hypoxic before treatment as shown by Hypoxyprobe localization by IHC is striking. In addition to improving delivery of chemotherapy, therefore, Apo2L/TRAIL treatment can also lead to reduced hypoxia in these tumors concomitant with reducing IFP. It is known that tumors develop mechanisms to survive hypoxic microenvironments and these mechanisms contribute to the ability of tumor cells to survive chemotherapy (see reviews [52, 53]) so this reduction in hypoxia may very well be an additional factor contributing to increased response to chemotherapy.
In summary, our results demonstrate a previously unappreciated aspect of the response of tumors to Apo2L/TRAIL treatment which, by direct killing of tumor cells, can reduce solid stress, reduce IFP and condition a tumor prior to administration of chemotherapy. These results support the further development of combination therapy and provide a rationale for a dosing strategy in which Apo2L/TRAIL is given initially and after a 48–72 hr interval to allow clearance of debris, followed by chemotherapy. Further studies examining any changes to liposomal gemcitabine biodistribution following Apo2L/TRAIL pretreatment will be needed before adopting this strategy for tumor priming.
Supplementary Material
Acknowledgments
We appreciate the contributions of Jeanne Prendergast and the technical assistance of Cory Mavis and Steve Turowski.
Grant Support
This work was supported by the National Institutes of Health R01 CA108888 (EAR) and utilized Shared Resources supported by the RPCI’s Comprehensive Cancer Center Support grant (NIH/NCI P30CA016056).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Authors’ Contributions:
Conception and design: E.A. Repasky, B.L. Hylander, A. Sen, S.H. Beachy,
Development of methodology: B.L. Hylander, A. Sen, S.H. Beachy, S. Ullas, R. Pitoniak, J. Qiu, J. Prey, G. Fetterly, E.A. Repasky
Acquisition of data: B.L. Hylander, A. Sen, S.H. Beachy, S. Ullas, R. Pitoniak, J. Qiu, J.D. Prey, G.J. Fetterly, E.A. Repask
Analysis and interpretation of data: B.L. Hylander, A. Sen, S.H. Beachy, S. Ullas, J. Qiu, J.D. Prey, G.J. Fetterly, E.A. Repasky
Writing, review and/or revision of manuscript: B.L. Hylander, A. Sen, S.H. Beachy, R. Pitoniak, J.F. Gibbs, J. Qiu, J.D. Prey, G.J. Fetterly, E.A. Repasky
Administrative, technical, or material support: B.L. Hylander, A Sen, E.A. Repasky
Study supervision: B.L. Hylander, A. Sen, E.A. Repasky
REFERENCES
- 1.Heldin CH, Rubin K, Pietras K, Ostman A. High interstitial fluid pressure - an obstacle in cancer therapy. Nat Rev Cancer. 2004;4:806–813. doi: 10.1038/nrc1456. [DOI] [PubMed] [Google Scholar]
- 2.Fukumura D, Jain RK. Tumor microvasculature and microenvironment: targets for anti-angiogenesis and normalization. Microvasc Res. 2007;74:72–84. doi: 10.1016/j.mvr.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stohrer M, Boucher Y, Stangassinger M, Jain RK. Oncotic pressure in solid tumors is elevated. Cancer Res. 2000;60:4251–4255. [PubMed] [Google Scholar]
- 4.Jain RK. Normalizing tumor microenvironment to treat cancer: bench to bedside to biomarkers. J Clin Oncol. 2013;31:2205–2218. doi: 10.1200/JCO.2012.46.3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Netti PA, Hamberg LM, Babich JW, Kierstead D, Graham W, Hunter GJ, Wolf GL, Fischman A, Boucher Y, Jain RK. Enhancement of fluid filtration across tumor vessels: implication for delivery of macromolecules. Proc Natl Acad Sci U S A. 1999;96:3137–3142. doi: 10.1073/pnas.96.6.3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Netti PA, Berk DA, Swartz MA, Grodzinsky AJ, Jain RK. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res. 2000;60:2497–2503. [PubMed] [Google Scholar]
- 7.Stylianopoulos T, Martin JD, Chauhan VP, Jain SR, Diop-Frimpong B, Bardeesy N, Smith BL, Ferrone CR, Hornicek FJ, Boucher Y, Munn LL, Jain RK. Causes, consequences, and remedies for growth-induced solid stress in murine and human tumors. Proc Natl Acad Sci U S A. 2012;109:15101–15108. doi: 10.1073/pnas.1213353109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stylianopoulos T, Martin JD, Snuderl M, Mpekris F, Jain SR, Jain RK. Co-evolution of solid stress and interstitial fluid pressure in tumors during progression: Implications for vascular collapse. Cancer Res. 2013 doi: 10.1158/0008-5472.CAN-12-4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov. 2011;10:417–427. doi: 10.1038/nrd3455. [DOI] [PubMed] [Google Scholar]
- 10.Minchinton AI, Tannock IF. Drug penetration in solid tumours. Nat Rev Cancer. 2006;6:583–592. doi: 10.1038/nrc1893. [DOI] [PubMed] [Google Scholar]
- 11.Swartz MA, Lund AW. Lymphatic and interstitial flow in the tumour microenvironment: linking mechanobiology with immunity. Nat Rev Cancer. 2012;12:210–219. doi: 10.1038/nrc3186. [DOI] [PubMed] [Google Scholar]
- 12.Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7:653–664. doi: 10.1038/nrclinonc.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Padera TP, Stoll BR, Tooredman JB, Capen D, di Tomaso E, Jain RK. Pathology: cancer cells compress intratumour vessels. Nature. 2004;427:695. doi: 10.1038/427695a. [DOI] [PubMed] [Google Scholar]
- 14.Lu D, Wientjes MG, Lu Z, Au JL. Tumor priming enhances delivery and efficacy of nanomedicines. J Pharmacol Exp Ther. 2007;322:80–88. doi: 10.1124/jpet.107.121632. [DOI] [PubMed] [Google Scholar]
- 15.Griffon-Etienne G, Boucher Y, Brekken C, Suit HD, Jain RK. Taxane-induced apoptosis decompresses blood vessels and lowers interstitial fluid pressure in solid tumors: clinical implications. Cancer Res. 1999;59:3776–3782. [PubMed] [Google Scholar]
- 16.Taghian AG, Abi-Raad R, Assaad SI, Casty A, Ancukiewicz M, Yeh E, Molokhia P, Attia K, Sullivan T, Kuter I, Boucher Y, Powell SN. Paclitaxel decreases the interstitial fluid pressure and improves oxygenation in breast cancers in patients treated with neoadjuvant chemotherapy: clinical implications. J Clin Oncol. 2005;23:1951–1961. doi: 10.1200/JCO.2005.08.119. [DOI] [PubMed] [Google Scholar]
- 17.Jang SH, Wientjes MG, Au JL. Enhancement of paclitaxel delivery to solid tumors by apoptosis-inducing pretreatment: effect of treatment schedule. J Pharmacol Exp Ther. 2001;296:1035–1042. [PubMed] [Google Scholar]
- 18.Tong RT, Boucher Y, Kozin SV, Winkler F, Hicklin DJ, Jain RK. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res. 2004;64:3731–3736. doi: 10.1158/0008-5472.CAN-04-0074. [DOI] [PubMed] [Google Scholar]
- 19.Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 20.Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, Feig C, Nakagawa T, Caldwell ME, Zecchini HI, Lolkema MP, Jiang P, Kultti A, Thompson CB, Maneval DC, Jodrell DI, Frost GI, Shepard HM, Skepper JN, Tuveson DA. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut. 2013;62:112–120. doi: 10.1136/gutjnl-2012-302529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:418–429. doi: 10.1016/j.ccr.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khawar IA, Kim JH, Kuh HJ. Improving drug delivery to solid tumors: Priming the tumor microenvironment. Journal of controlled release : official journal of the Controlled Release Society. 2015;201c:78–89. doi: 10.1016/j.jconrel.2014.12.018. [DOI] [PubMed] [Google Scholar]
- 23.Ashkenazi A, Pai RC, Fong S, Leung S, Lawrence DA, Marsters SA, Blackie C, Chang L, McMurtrey AE, Hebert A, DeForge L, Koumenis IL, Lewis D, Harris L, Bussiere J, Koeppen H, Shahrokh Z, Schwall RH. Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest. 1999;104:155–162. doi: 10.1172/JCI6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ashkenazi A, Herbst RS. To kill a tumor cell: the potential of proapoptotic receptor agonists. J Clin Invest. 2008;118:1979–1990. doi: 10.1172/JCI34359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ashkenazi A, Holland P, Eckhardt SG. Ligand-based targeting of apoptosis in cancer: the potential of recombinant human apoptosis ligand 2/Tumor necrosis factor-related apoptosis-inducing ligand (rhApo2L/TRAIL) J Clin Oncol. 2008;26:3621–3630. doi: 10.1200/JCO.2007.15.7198. [DOI] [PubMed] [Google Scholar]
- 26.Naka T, Sugamura K, Hylander BL, Widmer MB, Rustum YM, Repasky EA. Effects of tumor necrosis factor-related apoptosis-inducing ligand alone and in combination with chemotherapeutic agents on patients' colon tumors grown in SCID mice. Cancer Res. 2002;62:5800–5806. [PubMed] [Google Scholar]
- 27.Hylander BL, Pitoniak R, Penetrante RB, Gibbs JF, Oktay D, Cheng J, Repasky EA. The anti-tumor effect of Apo2L/TRAIL on patient pancreatic adenocarcinomas grown as xenografts in SCID mice. J Transl Med. 2005;3:22. doi: 10.1186/1479-5876-3-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sugamura K, Gibbs JF, Belicha-Villanueva A, Andrews C, Repasky EA, Hylander BL. Synergism of CPT-11 and Apo2L/TRAIL against two differentially sensitive human colon tumor xenografts. Oncology. 2008;74:188–197. doi: 10.1159/000151366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holland PM. Targeting Apo2L/TRAIL receptors by soluble Apo2L/TRAIL. Cancer Lett. 2013;332:156–162. doi: 10.1016/j.canlet.2010.11.001. [DOI] [PubMed] [Google Scholar]
- 30.Lemke J, von Karstedt S, Zinngrebe J, Walczak H. Getting TRAIL back on track for cancer therapy. Cell death and differentiation. 2014;21:1350–1364. doi: 10.1038/cdd.2014.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Soria JC, Mark Z, Zatloukal P, Szima B, Albert I, Juhasz E, Pujol JL, Kozielski J, Baker N, Smethurst D, Hei YJ, Ashkenazi A, Stern H, Amler L, Pan Y, Blackhall F. Randomized phase II study of dulanermin in combination with paclitaxel, carboplatin, and bevacizumab in advanced non-small-cell lung cancer. J Clin Oncol. 2011;29:4442–4451. doi: 10.1200/JCO.2011.37.2623. [DOI] [PubMed] [Google Scholar]
- 32.Amm HM, Oliver PG, Lee CH, Li Y, Buchsbaum DJ. Combined modality therapy with TRAIL or agonistic death receptor antibodies. Cancer Biol Ther. 2011;11:431–449. doi: 10.4161/cbt.11.5.14671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marcucci F, Corti A. How to improve exposure of tumor cells to drugs: promoter drugs increase tumor uptake and penetration of effector drugs. Adv Drug Deliv Rev. 2012;64:53–68. doi: 10.1016/j.addr.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 34.Sharma R, Buitrago S, Pitoniak R, Gibbs JF, Curtin L, Seshadri M, Repasky EA, Hylander BL. Influence of the implantation site on the sensitivity of patient pancreatic tumor xenografts to Apo2L/TRAIL therapy. Pancreas. 2014;43:298–305. doi: 10.1097/MPA.0000000000000099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kelley SK, Harris LA, Xie D, Deforge L, Totpal K, Bussiere J, Fox JA. Preclinical studies to predict the disposition of Apo2L/tumor necrosis factor-related apoptosis-inducing ligand in humans: characterization of in vivo efficacy, pharmacokinetics, and safety. J Pharmacol Exp Ther. 2001;299:31–38. [PubMed] [Google Scholar]
- 36.Braakhuis BJ, van Dongen GA, Vermorken JB, Snow GB. Preclinical in vivo activity of 2',2'-difluorodeoxycytidine (Gemcitabine) against human head and neck cancer. Cancer Res. 1991;51:211–214. [PubMed] [Google Scholar]
- 37.Sen A, Capitano ML, Spernyak JA, Schueckler JT, Thomas S, Singh AK, Evans SS, Hylander BL, Repasky EA. Mild elevation of body temperature reduces tumor interstitial fluid pressure and hypoxia and enhances efficacy of radiotherapy in murine tumor models. Cancer Res. 2011;71:3872–3880. doi: 10.1158/0008-5472.CAN-10-4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Winslow T, Eranki E, Ullas S, Singh AK, Repasky EA, Sen A. International journal of hyperthermia : the official journal of European Society for Hyperthermic Oncology. North American Hyperthermia Group; 2015. A pilot study of the effects of mild systemic heating on human head and neck tumor xenografts: analysis of tumor perfusion, interstitial fluid pressure, hypoxia and efficacy of radiation therapy. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luo S, Zhang E, Su Y, Cheng T, Shi C. A review of NIR dyes in cancer targeting and imaging. Biomaterials. 2011;32:7127–7138. doi: 10.1016/j.biomaterials.2011.06.024. [DOI] [PubMed] [Google Scholar]
- 40.Vogler M, Durr K, Jovanovic M, Debatin KM, Fulda S. Regulation of TRAIL-induced apoptosis by XIAP in pancreatic carcinoma cells. Oncogene. 2007;26:248–257. doi: 10.1038/sj.onc.1209776. [DOI] [PubMed] [Google Scholar]
- 41.Moog R, Burger AM, Brandl M, Schuler J, Schubert R, Unger C, Fiebig HH, Massing U. Change in pharmacokinetic and pharmacodynamic behavior of gemcitabine in human tumor xenografts upon entrapment in vesicular phospholipid gels. Cancer Chemother Pharmacol. 2002;49:356–366. doi: 10.1007/s00280-002-0428-4. [DOI] [PubMed] [Google Scholar]
- 42.Shipley LA, Brown TJ, Cornpropst JD, Hamilton M, Daniels WD, Culp HW. Metabolism and disposition of gemcitabine, and oncolytic deoxycytidine analog, in mice, rats, and dogs. Drug Metab Dispos. 1992;20:849–855. [PubMed] [Google Scholar]
- 43.Herbst RS, Eckhardt SG, Kurzrock R, Ebbinghaus S, O'Dwyer PJ, Gordon MS, Novotny W, Goldwasser MA, Tohnya TM, Lum BL, Ashkenazi A, Jubb AM, Mendelson DS. Phase I dose-escalation study of recombinant human Apo2L/TRAIL, a dual proapoptotic receptor agonist, in patients with advanced cancer. J Clin Oncol. 2010;28:2839–2846. doi: 10.1200/JCO.2009.25.1991. [DOI] [PubMed] [Google Scholar]
- 44.Graeser R, Bornmann C, Esser N, Ziroli V, Jantscheff P, Unger C, Hopt UT, Schaechtele C, von Dobschuetz E, Massing U. Antimetastatic effects of liposomal gemcitabine and empty liposomes in an orthotopic mouse model of pancreatic cancer. Pancreas. 2009;38:330–337. doi: 10.1097/MPA.0b013e31819436e6. [DOI] [PubMed] [Google Scholar]
- 45.Xu Y, Choi J, Hylander B, Sen A, Evans SS, Kraybill WG, Repasky EA. International journal of hyperthermia : the official journal of European Society for Hyperthermic Oncology. Vol. 23. North American Hyperthermia Group; 2007. Fever-range whole body hyperthermia increases the number of perfused tumor blood vessels and therapeutic efficacy of liposomally encapsulated doxorubicin; pp. 513–527. [DOI] [PubMed] [Google Scholar]
- 46.Haag C, Stadel D, Zhou S, Bachem MG, Moller P, Debatin KM, Fulda S. Identification of c-FLIP(L) and c-FLIP(S) as critical regulators of death receptor-induced apoptosis in pancreatic cancer cells. Gut. 2011;60:225–237. doi: 10.1136/gut.2009.202325. [DOI] [PubMed] [Google Scholar]
- 47.Seol JW, Chaudhari AA, Lee YJ, Kang HS, Kim IS, Kim NS, Park JH, Kim TH, Seol DW, Park SY. Regulation of DR-5 protein and mitochondrial transmembrane potential by gemcitabine, a possible mechanism of gemcitabine-enhanced TRAIL-induced apoptosis. Oncol Rep. 2007;18:523–529. [PubMed] [Google Scholar]
- 48.Weber TG, Poschinger T, Galban S, Rehemtulla A, Scheuer W. Noninvasive monitoring of pharmacodynamics and kinetics of a death receptor 5 antibody and its enhanced apoptosis induction in sequential application with Doxorubicin. Neoplasia. 2013;15:863–874. doi: 10.1593/neo.13932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thompson CB, Shepard HM, O'Connor PM, Kadhim S, Jiang P, Osgood RJ, Bookbinder LH, Li X, Sugarman BJ, Connor RJ, Nadjsombati S, Frost GI. Enzymatic depletion of tumor hyaluronan induces antitumor responses in preclinical animal models. Mol Cancer Ther. 2010;9:3052–3064. doi: 10.1158/1535-7163.MCT-10-0470. [DOI] [PubMed] [Google Scholar]
- 50.Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, Jain RK. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev. 2011;91:1071–1121. doi: 10.1152/physrev.00038.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ait-Oudhia S, Straubinger RM, Mager DE. Systems pharmacological analysis of paclitaxel-mediated tumor priming that enhances nanocarrier deposition and efficacy. J Pharmacol Exp Ther. 2013;344:103–112. doi: 10.1124/jpet.112.199109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cosse JP, Michiels C. Tumour hypoxia affects the responsiveness of cancer cells to chemotherapy and promotes cancer progression. Anticancer Agents Med Chem. 2008;8:790–797. doi: 10.2174/187152008785914798. [DOI] [PubMed] [Google Scholar]
- 53.Rohwer N, Cramer T. Hypoxia-mediated drug resistance: novel insights on the functional interaction of HIFs and cell death pathways. Drug Resist Updat. 2011;14:191–201. doi: 10.1016/j.drup.2011.03.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.