

Abstract
Many human tumors express high levels of the p53 inhibitor Mdm2, resulting from amplification of the Mdm2 locus or aberrant post-translational regulation of the Mdm2 protein. While the importance of Mdm2 in regulating p53 is clear, Mdm2 also has p53-independent roles. For example, overexpression of Mdm2 results in genomic instability in a p53-independent manner. In addition, Mdm2 has many additional binding partners; some such as the tumor suppressor p73 have also been implicated in genomic instability. In this study, cells and tumors with Mdm2 overexpression and p73 loss exhibit increased genomic instability as compared to either alteration alone and cooperate in development of B-cell lymphomagenesis. Cytogenetic analyses of mouse embryonic fibroblasts and pre-malignant B-cells demonstrate that loss of p73 exacerbates the chromosome breaks and fusions observed in Mdm2Tg cells. B-cell lymphomas from Mdm2Tg;p73+/− mice retain the remaining p73 allele, and exhibit elevated levels of the anti-apoptotic protein Bcl2 and thus dampen apoptosis. In summary, Mdm2 overexpression and p73 loss cooperate in genomic instability and tumor development, indicating that the oncogenic functions of Mdm2 are a combined effect of inhibiting p53 and p73 functions. Given that p73 is lost or silenced in human B-cell lymphomas [1–4], the Mdm2Tg;p73+/− mouse serves as a model for human disease and may provide additional insight into the pathways that contribute to B-cell lymphomagenesis.
Keywords: Mdm2, p73, B-cell lymphoma, genomic instability
Introduction
The p53 pathway is altered in over 90% of human cancers by several mechanisms including mutation or deletion of the TP53 gene itself and overexpression of the p53 inhibitor, MDM2. Mdm2 is a RING finger-containing E3 ubiquitin ligase that targets p53 for proteasomal degradation. Loss of Mdm2 leads to an embryonic lethal phenotype that is p53-dependent [5, 6]. More relevant to human tumors is the finding that many tumors produce high levels of Mdm2 resulting from amplification of the Mdm2 locus or aberrant post-translational regulation of the MDM2 protein [3]. While the importance of Mdm2 in regulating p53 is clear, Mdm2 also has p53-independent roles. For example, overexpression of Mdm2 in one mouse model resulted in multiple rounds of DNA synthesis without cell division leading to polyploidy in a p53-independent manner in mammary epithelial cells [7] and Mdm2 overexpression results in genomic instability in a p53-independent manner in NIH3T3 cells and mouse embryonic fibroblasts (MEFs) [8]. Mdm2Tg mice that express 2–4 fold elevated Mdm2 levels are tumor prone and develop a different tumor spectrum from that of p53-null mice [9], further supporting a p53-independent function of Mdm2 during tumorigenesis. In addition, analysis of these Mdm2 transgenic mice also showed genomic instability in primary pre-B cells, splenocytes, and purified splenic B cells [10]. Mdm2 overexpression also enhances the increased genomic instability observed in aging mice, further supporting a role for Mdm2 overexpression in genomic instability [11].
In addition, Mdm2 has many other binding partners including the p53 family member, p73 [12]. The TP73 gene expresses seven alternatively spliced C-terminal isoforms (TAp73), which contain a complete amino-terminal transactivation domain and four N-terminal isoforms (ΔNp73), which lack the transactivation domain. A mouse deficient for all isoforms of p73 exhibits neurological and inflammatory defects resulting in a high incidence of mortality in p73−/− pups (80% of pups do not survive past 30 days). A p73-deficient mouse heterozygous for all isoforms is tumor prone and when combined with p53 deficiency, results in increased metastasis [13]. Mdm2 specifically binds the TAp73 isoforms with similar affinity to that of Mdm2 for p53 [14]. Although Mdm2 is an E3 ubiquitin ligase, it does not target TAp73 for degradation, but blocks its transcriptional activation [15, 16]. The TAp73 isoforms can transactivate p53 targets such as Bax, Puma, and p21 to induce apoptosis and cell cycle arrest [17, 18]. In addition, the TAp73 isoforms also have unique, p53-independent targets and cellular functions. For example, TAp73 plays a role in antioxidant metabolism through its direct activation of glucose-6-phosphate 1-dehydrogenase (G6PD), the rate limiting enzyme of a major glucose metabolic pathway (pentose phosphate pathway) [19]. Furthermore, unlike p53-deficient mice, a TAp73 specific knockout mouse exhibits genomic instability in a cell-specific manner [20]. In contrast, loss of the ΔNp73 isoforms has no effect on genomic stability, but mice exhibit neurodegeneration. In addition, ΔNp73 inhibits p53 and TAp73 activity by competitive binding to the promoters of target genes such as Puma and p21 in both untreated cells and cells treated with a range of DNA-damaging agents [21, 22]. Consequently, MEFs and thymocytes from ΔNp73−/− mice exhibit an enhanced apoptotic response. Lastly, E1A and RasV12 transformed ΔNp73−/− MEFs injected into nude mice were unable to form tumors unlike wild type E1A and RasV12 transformed MEFs, indicating the ΔNp73 isoforms are necessary for transformed cells to form tumors in vivo [21]. Given these data, the TAp73 isoforms are thought to have tumor suppressor functions and the ΔNp73 isoforms to have oncogenic functions.
Despite the reported interaction between Mdm2 and TAp73, to date, no studies have addressed the in vivo relevance or physiological consequence of this interaction. Given that Mdm2 has p53-independent functions, we propose that the oncogenic activity of Mdm2 is a combined effect of inhibiting p53 and p73 functions. Here, we present the first evidence that Mdm2 overexpression and p73 loss cooperate to induce genomic instability, resulting in accelerated spontaneous tumorigenesis. Mdm2Tg;p73+/− mice develop a high incidence of B-cell lymphoma and analysis of pre-malignant B-cells reveals increased chromosome fusions in these mice. Furthermore, we show that B-cell lymphomas from Mdm2Tg;p73+/− mice exhibit deregulation of the B-cell anti-apoptotic protein, Bcl2 and a dampened apoptotic response. Taken together, these data indicate that Mdm2 overexpression and p73 loss cooperate in genomic instability and result in accelerated B-cell lymphomagenesis.
Results
Loss of p73 exacerbates genomic instability and results in increased growth of Mdm2Tg MEFs
Given that Mdm2 can inhibit p73 transcriptional activity and that both overexpression of Mdm2 and loss of TAp73 have been implicated in genomic instability and tumor development, we first examined whether these two alterations may have an additive effect on genomic instability by examining metaphase spreads in Mdm2Tg;p73+/− and Mdm2Tg;p73−/− mouse embryonic fibroblasts (MEFs). Mdm2Tg transgenic mice were generated using a cosmid containing the entire Mdm2 gene and these mice express 2–4 fold elevated Mdm2 levels [9]. The p73-deficient mice are either heterozygous or null for all isoforms of p73 [13]. Passage 1 mouse embryonic fibroblasts (MEFs) were examined for chromosome abnormalities such as chromosome breaks, chromatid breaks, chromosome fragments, and chromosome fusions. As previously reported, Mdm2 overexpression alone resulted in a small increase in chromosome breaks and fusions compared to wild type MEFs [8] but p73 loss does not (Fig. 1A). In contrast, Mdm2 overexpression combined with loss of one copy or both copies of p73 results in more severe abnormalities (Fig. 1A,B). Mdm2Tg;p73+/− and Mdm2Tg;p73−/− MEFs exhibited a significant (p<0.01) increase in the percentage of cells with chromosome and chromatid breaks (Fig 1A; first panel) when compared to Mdm2Tg or p73−/− MEFs. These MEFs also have other chromosomal abnormalities such as numerous chromosome fragments and fusions (Fig 1B). Although there was a statistically significant (p<0.05) increase in the percentage of cells with chromosome fusions in Mdm2Tg MEFs when compared to wild type MEFs, loss of p73 did not exacerbate this phenotype (Fig 1A; second panel). Lastly, there was a statistically significant increase (p<0.001) in the percentage of cells with four or more chromosome abnormalities per metaphase in Mdm2Tg;p73+/− and Mdm2Tg;p73−/− MEFs compared to Mdm2Tg or p73−/− alone.
Figure 1. Mdm2 overexpression and p73 loss cooperate in genomic instability.

(A) Metaphases from passage 1 day 13.5 MEFs were examined for chromosome or chromatid breaks, and fusions. The total numbers of metaphases evaluated are indicated in parentheses next to the genotypes examined. Significant differences between Mdm2Tg and Mdm2Tg;p73+(−)/− are indicated above each graph. (B) Photographs of representative metaphase spreads from wild type, Mdm2Tg and Mdm2Tg;p73+/− MEFs. Chromatid breaks are shown in panels 2–4 and chromosome fusions are indicated in panel 4 by arrow heads. Chromosome fragments are indicated by red circles.
Since genomic instability has both growth inhibiting and promoting effects, we next evaluated the effect of Mdm2 overexpression and p73 loss on MEF cell proliferation. An equal number of cells for each genotype were placed in culture; Mdm2Tg;p73+/− and Mdm2Tg;p73−/− MEFs continued to proliferate after 5 passages, whereas Mdm2Tg and p73+/− MEFs begin to senescence at passage 3, as indicated by lack of proliferation, positive SA-β-GAL staining, and morphological changes consistent with senescence (Fig. 2A,B). In addition, loss of p73 in an Mdm2Tg background resulted in an increase in S and a decrease in G2M phases of the cell cycle by passage 3 as measured by flow cytometry using propidium iodide staining (Fig. 2C).
Figure 2. Loss of p73 in an Mdm2Tg background results in a growth advantage for MEFs.
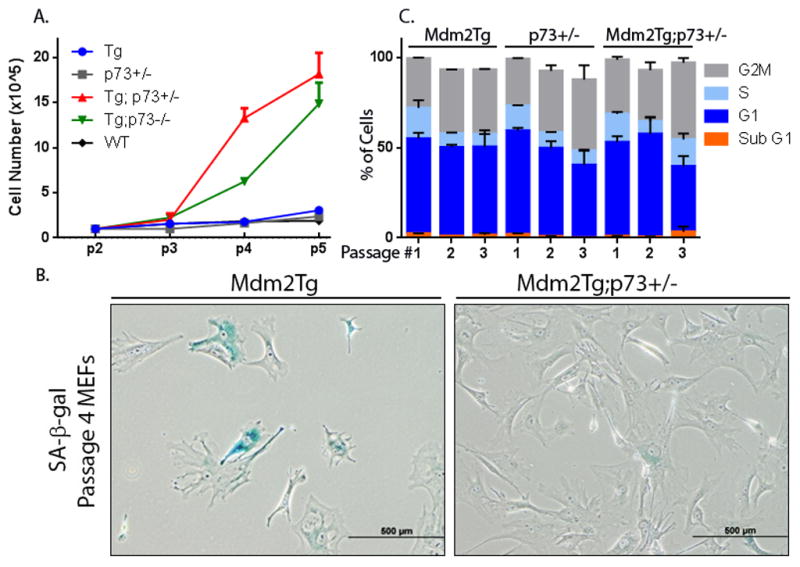
(A) Equal numbers of passage 2 MEFs from the indicated genotypes were plated in triplicate. Viability of cells was determined at each passage by Trypan Blue exclusion. (B) SA-β-gal staining was used to examine senescence in Passage 4 MEFs of the indicated genotypes. (C) Equal numbers of cells for passages 1, 3, and 4 MEFs of the indicated genotypes were plated and allowed to grow for one day. Cell-cycle profiles were used to determine the percentage of total cells in G1, S, and G2M-phases for each genotype at passages 1–3.
Mdm2 overexpression and p73 loss cooperate during B-cell lymphomagenesis
To determine whether the genomic instability and proliferation changes in Mdm2Tg;p73+/− MEFs lead to spontaneous tumor formation, we monitored a cohort of wild type, Mdm2Tg, p73+/−, Mdm2Tg;p73+/−, p73−/−, and Mdm2Tg;p73−/− mutant mice in a pure C57Bl/6 background for two years or until moribund. As previously reported, complete loss of p73 results in a high incidence of mortality in the first 3 months of life due to developmental and immunological defects [23]. As a result, only a small cohort of p73−/− and Mdm2Tg;73−/− mice could be established and monitored until moribund. Fifty percent of the p73−/− and Mdm2Tg;73−/− mice were moribund and euthanized by 74 and 64 weeks of age, respectively (Fig. 3). Mdm2Tg;p73+/− mice had a significant reduction in lifespan relative to wild type, Mdm2Tg, or p73+/− littermates (Fig. 3A). By 85 weeks of age, 50% of the Mdm2Tg;p73+/− animals were moribund and had to be euthanized, whereas the median survival for Mdm2Tg mice was 104 weeks. At two years, 38 out of 41 (93%) Mdm2Tg;p73+/− mice had been sacrificed, compared to 16 out of 38 (42%) Mdm2Tg, 12 out of 40 (30%) p73+/−, 7/7 (100%) p73−/− and 4/4 (100%) Mdm2Tg;p73−/− mice. The survival time of Mdm2Tg;p73+/− mice was determined to be significantly reduced (p<0.0001) compared to the survival time of wild type, Mdm2Tg, or p73+/− mice. The reduced survival rate largely resulted from widely disseminated lymphoma involving the spleen, mesenteric lymph nodes, lung, and liver as indicated by the lymphoma free survival curve (Fig. 3B). 63% (26 of 41) of all Mdm2Tg;p73+/− mice develop lymphoma by two years of age, compared to 31.5% (12 of 38) of Mdm2Tg and 20% (8 out of 40) of p73+/− mice (Fig. 3C). Of these lymphomas, cell surface marker analysis by flow cytometry and immunohistochemistry using antibodies against B-cell markers CD19, IgM and B220 confirm that the majority of these Mdm2Tg;p73+/− (76%) and Mdm2Tg (60%) lymphomas are of B-cell origin (Fig. 3C; 4A,B).
Figure 3.

(A) Kaplan-Meier survival curves of wild type, Mdm2Tg, p73+/−, Mdm2Tg; p73+/−, p73−/−, and Mdm2Tg; p73−/− mice illustrating the percentage of mice alive up to 24 months. The median time of survival was 104 weeks for Mdm2Tg and 84 weeks for Mdm2Tg; p73+/− mice. The log-rank test was used to determine the statistical significance between the survival time of Mdm2Tg and Mdm2Tg; p73+/− mice (p<.0001). (B) Kaplan-Meier survival curves showing the fraction of mice that were lymphoma free from each indicated genotype up until 24 months. By 92 weeks of age, half of the Mdm2Tg;p73+/− mice had succumbed to lymphoma whereas more than half of the Mdm2Tg mice were disease free at the 2 year end point. The log-rank test was used to determine the statistical significance between the lymphoma-specific survival time of Mdm2Tg and Mdm2Tg; p73+/− mice (p<.0001). (C) The tumor spectrum of wild type, Mdm2Tg, p73+/−, Mdm2Tg; p73+/−, p73−/−, and Mdm2Tg; p73−/− mice presented as the percentage of each tumor type out of the total number of tumors for each genotype. The total number of tumors examined for each genotype is indicated in parentheses above each bar. B/TCL= Mixed B and T-cell lymphoma; TCL=T-cell lymphoma; BCL= B-cell lymphoma.
To determine whether the tumors in Mdm2Tg;p73+/− or p73+/− mice lose the wild type p73 allele, DNA was extracted from the spleens of mice with lymphoma and analyzed by PCR. Whereas 37.5% of p73+/− tumors (3/8) undergo loss of heterozygosity, consistent with previous reports [13] (Fig. 4C), all tumors (26/26) from Mdm2Tg;p73+/− mice retained the wild type copy of p73, indicating that either loss of one p73 allele in the Mdm2Tg background is sufficient to accelerate lymphomagenesis or that Mdm2 overexpression dampens activity of the remaining allele.
Figure 4. Loss of p73 in an Mdm2Tg background results in clonal B-cell lymphoma.

(A.) Representative histological sections stained with H&E or subject to immunohistochemistry on spleen and lung tissue samples from an Mdm2Tg; p73+/− mouse with B-cell lymphoma. B220/CDR45 is a marker of B-cells and CD3 is a marker of T-cells. (B) Representative FACS analysis on Mdm2Tg; p73+/− tumors, using antibodies against B220 and IgM or CD19 as indicated. (C). Representative examples of LOH analysis on tumor and normal tissue samples from Mdm2Tg and Mdm2Tg; p73+/− mice. PCR for p73 from B-cell lymphomas (T1, T2) and respective normal kidney tissue (N1,N2) from Mdm2Tg; p73+/− mice and T-cell lymphomas (T3,T4) and respective normal kidney (N3,4) from p73+/− mice. All tumors from Mdm2Tg;p73+/− mice retained the wild type copy of p73 whereas ≈38% of tumors from p73+/− mice underwent LOH. (D) Southern blot analysis of DNA from indicated Mdm2Tg, Mdm2Tg; p73+/−, and p73−/− B-cell lymphoma samples with a JH probe to detect IgH loci rearrangements. The position of the germ-line (gl) band is shown in the wild type sample. DNA from a normal spleen was used as control. When available, DNA from spleen (S) and lymph node (LN) from the same mouse was used. White asterisks denote samples with two or more rearranged bands in addition to the germline band.
To further characterize these B-cell lymphomas, we analyzed IgH rearrangements in tumor DNA by southern blotting. Compared to wild type spleen DNA, which contains a single germline band, all Mdm2Tg, Mdm2Tg;p73+/− and p73−/− B-cell lymphoma samples contained 1–2 rearranged bands (Fig. 4D). When possible, a peripheral lymph node from the same mouse was analyzed and found to contain identical IgH rearrangements as splenic tumors (lanes 6–7 and 11–12). These data indicate that the B-cell lymphomas in Mdm2Tg and Mdm2Tg;p73+/− mice are clonal, likely derived from single cells that had undergone recombination at the IgH locus.
Mdm2 overexpression and p73 loss increase genomic instability in B cells
Since p73 loss exacerbates genomic instability in Mdm2Tg MEFs, we hypothesized that pre-malignant B cells from Mdm2Tg;p73+/− mice may have increased genomic instability making them more susceptible to development of B-cell lymphomas. In order to analyze metaphases in pre-malignant B cells, we purified B cells from total splenocyte populations of 9 month old mice using negative selection, resulting in the depletion of unwanted cells and enrichment of CD19+ B-cells. A total of 136 metaphases from four independent Mdm2Tg;p73+/− purified splenic B cell cultures, each with an Mdm2Tg littermate control, were evaluated for chromosomal aberrations. Pre-malignant B cells from Mdm2Tg;p73+/− mice had a significantly greater percentage of cells with fusions than those from Mdm2Tg mice (8.5% vs 1.5%, respectively, p<0.05) and a significantly higher percentage of cells with aberrant metaphases (>2 abnormalities per cell) (19% vs 3.2%, respectively, p<0.05) (Figure 5A). Examples of metaphases from purified B cells are shown in Figure 5B.
Figure 5. Mdm2Tg; p73+/− mice exhibit genomic instability in pre-malignant B-cells.
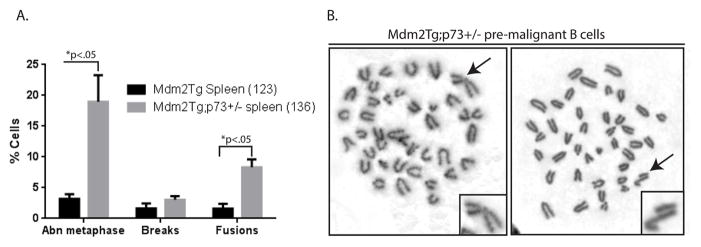
Metaphases of pre-malignant (9 months of age) purified splenic B-cells from Mdm2Tg;p73+/− and Mdm2Tg littermate controls were examined for chromosomal abnormalities. (A) The percentages of cells with abnormal metaphases, breaks, and fusions were determined. The total number of metaphases examined is indicated in parentheses. The Mdm2Tg bar represents 3 mice and the Mdm2Tg;p73+/− bar represents 4 mice. Abnormal metaphases (Abn) were characterized by 2 or more aberrations. P values (t-tests) are shown above each category. (B) Representative metaphase spreads from pre-malignant B-cells. The arrow and magnified chromosome in the left panel depicts a chromosome fusion and in the right panel depicts a chromatid break.
Mdm2Tg mice with p73 loss exhibit increased Bcl2 protein expression
Given the proliferative advantage in Mdm2Tg;p73+/− and Mdm2Tg;p73−/− MEFs, we hypothesized that one or more apoptotic programs may be dampened in these animals contributing to development of B cell lymphomagenesis. To investigate the potential mechanism(s) contributing to this phenotype, we considered apoptotic genes that are regulated by both p53 and p73. One candidate gene is the anti-apoptotic oncogene Bcl2. Bcl2 is negatively regulated by p53 through direct binding of wild type p53 to the Bcl-2 promoter, preventing Bcl2 binding to and inhibition of pro-apoptotic Bcl2 family members Bad, Bak, and Bax, resulting in apoptosis [24]. Treatment of wild type and p53 null lymphoblastic leukemia lines with IR demonstrated that down regulation of Bcl2 and subsequent apoptosis are p53-dependent [25]. p73, on the other hand, induces apoptosis by regulating the interaction between Bcl2 and Bax [26]. Although both Mdm2Tg and Mdm2Tg;p73+/− mice predominately develop B-cell lymphoma, the latency in tumor development is significantly shorter for Mdm2Tg;p73+/− mice. In order to evaluate if the combined effect of Mdm2 overexpression and p73 loss on Bcl2 expression may be contributing to lymphomagenesis, we measured protein expression of Bcl2 by western blot analysis and immunohistochemistry. Mdm2Tg;p73+/− and Mdm2Tg;p73−/− tumors exhibited a drastic increase in Bcl2 compared to wild type control (10.8 and 10.6 fold respectively), whereas Mdm2Tg exhibited a 6.3 fold increase compared to wild type control spleen (Fig 6A,B). Immunohistochemistry using an antibody against Bcl2 confirms the up regulation of Bcl2 in B cell lymphomas arising in Mdm2Tg;p73+/− mice (Fig. 6C). To determine whether this difference in Bcl2 expression affects apoptosis, we used a TdT-mediated dUTP nick end labeling (TUNEL) assay to identify apoptotic cells in B cell lymphoma sections from Mdm2Tg and Mdm2Tg;p73+/− mice. TUNEL-positive cells were present in the tumors of both genotypes, but the amount of apoptosis is greatly enhanced in the Mdm2Tg tumors with low expression of Bcl2 (Fig. 6C). These data indicate that loss of p73 in an Mdm2Tg background results in increased Bcl2 expression and dampened apoptosis in B-cell lymphomas. Taken together, our data support a model by which Mdm2 inhibition of p53 and p73 cooperate in lymphomagenesis via inhibition of apoptosis.
Figure 6. Combined Mdm2 overexpression and p73 loss result in increased Bcl-2 expression and reduced apoptosis.
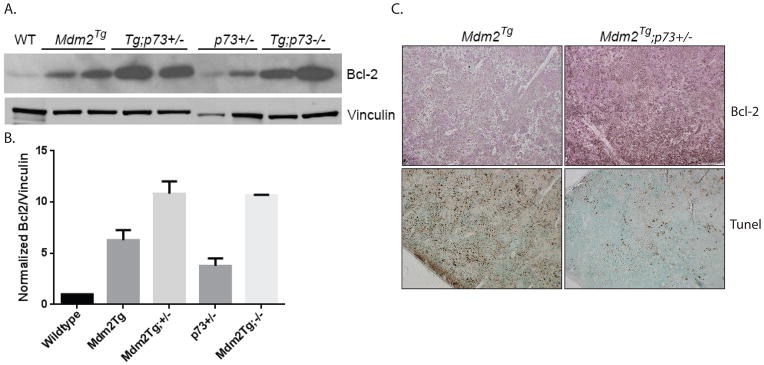
(A) Analysis of Bcl-2 protein expression in Mdm2Tg;p73+/− B-cell lymphomas. Protein lysates (50ug) from B-cell lymphomas from Mdm2Tg, Mdm2Tg;p73+/−, p73+/−, and Mdm2Tg;p73−/− mice were western blotted for Bcl-2 and the loading control Vinculin (2 tumor samples from each genotype). Lysate from a wild type spleen was used as a control (Lane 1). (B) Quantification of Bcl-2 protein expression as shown in the western blot in Figure 6A. Quantification was performed using ImageJ software. Statistical significance between genotypes was calculated by averaging two biological replicates from each genotype followed by two-tailed unpaired student t-tests. (C). Representative histological sections of B-cell lymphomas subjected to immunohistochemistry using an antibody against mouse Bcl-2 (Top 2 panels) and TUNEL staining (Bottom 2 panels). Of the 12 lymphomas that arose in Mdm2Tg mice, 7 were examined by immunohistochemistry to identify the cell of origin and apoptosis. Of the 26 lymphomas that arose in Mdm2Tg;p73+/− mice, 25 were examined by immunohistochemistry (Note: the remaining tumors were evaluated using flow cytometry; See Figure 4B).
Discussion
Despite the reported physical interaction between Mdm2 and TAp73, no studies to date have addressed the physiological consequences of this interaction. Our data demonstrate the predominant tumor type found in both Mdm2Tg and Mdm2Tg;p73+/− mice is B-cell lymphoma. However, Mdm2Tg;p73+/− mice develop a higher incidence of B-cell lymphoma and the average time to onset is significantly shorter than observed in Mdm2Tg and p73+/− mice. These data indicate that Mdm2 overexpression alone sensitizes mice to B-cell lymphomagenesis (Figure 3) [10] and that loss of p73 represents an additional event in the pathogenesis, resulting in accelerated tumor development. In order to determine the mechanism underlying the accelerated tumorigenesis in Mdm2Tg;p73+/− mice, we investigated genomic instability, which is a hallmark of human cancers.
Mdm2 overexpression results in genomic instability in a p53-independent manner, and loss of TAp73 is also linked to genomic instability. Therefore, we investigated and found that loss of even a single copy of p73 in an Mdm2Tg background results in exacerbated genomic instability. Since both Mdm2Tg;p73+/− and Mdm2Tg;p73−/− MEFS exhibit similar chromosome defects, these data indicate that loss of one copy is sufficient for the phenotype or that the remaining p73 allele is somehow silenced. Previous work [27] and our data (Fig.1) demonstrate that MEFs with complete loss of both p73 isoforms are phenotypically normal. However, in the absence of functional p53, p73 is essential for maintaining genome integrity, as evidenced by severe ploidy defects in p53−/−;p73−/− MEFs as compared to p53−/− MEFs [27]. Since Mdm2 overexpression results in dampened p53 activity, [27], it is possible that the genomic instability phenotype of Mdm2Tg;p73+/− MEFs results from inactivation of p53 coupled with p73 loss. If this were true, p53−/−;p73−/− and Mdm2Tg;p73+/− MEFS would have a similar phenotype, which is not the case. Whereas p53−/−;p73−/− MEFs exhibit severe polyploid, Mdm2Tg;p73+/− MEFs primarily have chromosome and chromatid breaks and fusions. Given that less than 1% of all metaphases analyzed from Mdm2Tg;p73+/− MEFs were polyploidy in contrast to p53−/−;p73−/− MEFs, it is likely the observed genomic instability results from a distinct mechanism. Although the exact mechanism of the increased genomic instability is still unknown, these data indicate that the phenotype observed in Mdm2Tg;p73+/− MEFs likely results from a baseline level of Mdm2 induced genomic instability, which is exacerbated by loss of p73 in a p53 compromised background.
Since the dominant tumor type in both Mdm2Tg and Mdm2Tg;p73+/− mice is B-cell lymphoma, we performed cytogenetic analysis on pre-malignant B-cells from these mice. Consistent with the MEF data, Mdm2Tg;p73+/− mice display increased genomic instability in pre-malignant B-cells when compared to Mdm2Tg mice. We observe a significant increase in the percentage of B cells with fusions as well as the percentage of B cells with abnormal metaphases (>2 abnormalities) in Mdm2Tg;p73+/− mice as compared to Mdm2Tg mice. To determine whether any common translocations occur in Mdm2Tg;p73+/− B-cell lymphomas, Giemsa-banding was performed on B-cells purified from three B-cell lymphomas, but no common alterations were identified (data not shown). Given that pre-malignant B cells from Mdm2Tg;p73+/− mice exhibit an increase in abnormal metaphases when compared to pre-malignant Mdm2Tg B cells, it is possible that this genomic instability sensitizes B cells to lymphomagenesis, resulting in accelerated tumor development and increased tumor incidence.
Another hallmark of human cancers is deregulation of apoptosis by inactivation of pro-apoptotic tumor suppressors such as p53 or overexpression of anti-apoptotic oncogenes such as Bcl-2. Bcl-2 regulates apoptosis in hematological cells, specifically in B-cells. As these tumors retain wild type p53, it is likely that Mdm2 contributes to tumorigenesis in both p53-dependent and independent mechanisms; specifically it is possible that the accelerated B-cell lymphoma phenotype results from combined loss of p53 and p73 activity and subsequent de-regulation of apoptosis. There is some precedent for this as loss of one copy of p73 in a p53+/− background results in accelerated lymphoma development and an increase in metastasis [13]. Although these mice predominately develop T-cell lymphoma, it is possible that the early onset of T-cell lymphoma precludes drawing any conclusion as to whether combined p73 and p53 loss results in B-cell lymphoma. To test this hypothesis, we examined protein expression of Bcl-2 in Mdm2Tg and Mdm2Tg;p73+/− tumor samples. We chose to examine Bcl2 for several reasons. First, the Bcl2 promoter is down-regulated by p53 in human B-cell lymphoma cell lines [24] and mice that lack functional p53 exhibit increased expression of Bcl2 in lymphocytes [28, 29]. Second, the functional domains of p53 that are required for Bcl2 repression are conserved in p73 [30], indicating Bcl2 may be a common target for p53 and p73. Third, overexpression of Mdm2 has been reported in human B-cell lymphomas [31, 32]. Lastly, in the absence of p53, p73 induces apoptosis via regulation of Bax/Bcl2 protein expression [33]. Given that overexpression of Mdm2 dampens p53 activity [10], it is possible that our mouse model mimics this scenario. Our western blot data indicate that Bcl2 expression is up-regulated upon Mdm2 overexpression alone, likely due to Mdm2-mediated p53 degradation and transcriptional inhibition. However, upon p73 loss, Bcl2 expression is further up-regulated, indicating that Bcl2 is also elevated downstream of p73 in B-cells, although this relationship in B-cells warrants further investigation. We performed reverse phase protein analysis (RPPA) of a small number of lymphomas from Mdm2Tg and Mdm2Tg;p73+/− mice and did not see a difference in expression of the Bcl2 family members, BAD, BAK1, Bax, or Bcl2L1 (Supplementary Figure 1), although the sample size was too small for statistical analysis. Given that Bcl2 binds to Bad, Bak, and Bax thus inhibiting apoptosis, expression of these family members in Mdm2Tg;p73 deficient B-cell lymphomas should be investigated in more detail. Taken together, these findings address for the first time the physiological consequence of Mdm2 overexpression coupled with p73 loss, which are common event in tumorigenesis. Based upon these data, we conclude that Mdm2 overexpression and p73 loss cooperate in B-cell lymphomagenesis through de-regulation of genomic stability and apoptosis.
Materials and Methods
Mice
p73+/− mice [13] were backcrossed to C57Bl/6 mice for four generations making them 99% C57Bl/6. Previously backcrossed Mdm2 transgenics (Mdm2Tg; pure C57Bl/6) [9] were mated to p73+/− mice [13] to generate Mdm2Tg;p73+/− mice and non-transgenic littermate controls. At indication of disease, tissues/tumors were collected and frozen, fixed in 10% formalin for paraffin embedding, and a small portion were analyzed by flow cytometry.
Chromosome stability analysis
Murine embryonic fibroblasts (MEFs) were isolated as previously described [8], cultured for one passage, treated for four hours with colcemid, harvested, and processed for metaphase analysis according to standard protocols at the Molecular Cytogenetics Core Facility at MD Anderson. In brief, metaphase preparations were stained with PI (Sigma) and DAPI (Sigma) and mounted with Vectashield (Vector Laboratories) prior to analysis by fluorescent microscopy. Each metaphase was examined for chromosomal aberrations (chromosomal breaks, chromatid breaks, and fusions). The total number of chromosomes in each metaphase was counted with computer software. Pure populations of splenic B cells were isolated from pre-malignant (9 months of age) and malignant spleens using the EasySep Mouse B Cell Isolation Kit and EasySep Magnet (StemCell Technologies). Purified splenic B cells were placed into tissue culture and stimulated with lipopolysaccharide (LPS) for 48 hours prior to harvesting and processing as described above. A portion of the purified cells were used for flow cytometry (See below).
Flow Cytometry
Single-cell suspensions from normal and malignant spleen samples were fixed in ethanol and stained with APC-labeled Anti-Mouse CD45R/B220 (BD Pharmingen) and FITC labeled Anti-Mouse CD19 (BD Pharmingen) or PE labeled anti-mouse IgM (BD Pharmingen) to determine the abundant cell type present. Data acquisition was performed at the Flow Cytometry and Cellular Imaging Core Facility at MD Anderson on a FACSCalibur flow cytometer and analysis was performed with FlowJo Ssoftware.
Histological Analysis and Immunohistochemistry
Sections from paraffin-embedded tissue were dewaxed and rehydrated following standard protocols. Slides were incubated with primary antibody (Rat Anti-mouse CD45R; BD Pharmingen) (Rabbit Anti-mouse CD3; Abcam)(Rabbit Anti-mouse Bcl-2; Abcam) at a dilution of 1:100 overnight at 4 °C. For detection, the Vectastain ABC Kit was used followed by the Vector Peroxidase Substrate Kit (Vector Labs). Terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick-end labelling (TUNEL) assays were carried out on paraffin-embedded sections according to the manufacturer’s specifications (Calbiochem, Darmstadt, Germany). Senescence associated β-Galactosidase (SAβGal) assay was performed essentially as described [34].
Western and Southern blot analysis
Spleens or B-cell lymphomas were lysed in RIPA buffer and proteins were western blotted using primary antibodies against mouse Bcl2 (BD Pharmingen), and mouse vinculin (Abcam) at dilutions of 1:1000. Two tumors for each indicated genotype were evaluated for Bcl2 expression as shown in Figure 6A. The analysis was semi-quantitative using film with multiple exposures, which were in the linear range. The expression of Bcl2 and Vinculin from these films were quantified using ImageJ. Genomic DNA was isolated from normal control spleen or malignant spleens and lymph nodes and separated on a 0.8% agarose gel and transferred to Hybond-N+ nylon membrane (GE Life sciences). Hybridization was performed in 50% (vol/vol) formamide/SSC at 42 °C. The JH4-3 probe was a gift from FW Alt [35].
Analysis for loss of heterozygosity
DNA was extracted from frozen tumor tissue and adjacent normal kidney tissue. Genomic DNA was extracted from the tissue with DNAzol reagent (Invitrogen). For detection of p73, PCR was performed using the following primers: Primer 1: GGG CCA TGC CTG TCT ACA AAG AA, Primer 2: CCT TCT ACA CGG ATG AGG TG, Primer 3: GAA AGC GAA GGA GCA AAG CTG 3. The wild type band is .650 kb and the mutant band is .400kb.
Reverse Phase Protein Array (RPPA)
Tumor samples were submitted to the RPPA Core Facility at MD Anderson for analysis using 135 different antibodies. The results were analyzed by the RPPA Core Facility and are provided as Supplemental Figure 1.
Supplementary Material
Acknowledgments
We thank Elsa Flores for the p73+/− mice, Stephen Jones for the Mdm2Tg mice, and Frederick W. Alt for the JH plasmid. This work was supported by a National Institutes of Health grant (CA47296 to GL), and a T32 Training Grant (CA009299) and an American Cancer Society postdoctoral fellowships to MFR. MJY is supported in part by NIH/NCI R01 CA164346, and the Center for Genetics and Genomics and the Sister Institution Network fund of MD Anderson Cancer Center.
Footnotes
Conflict of interest. The authors declare no conflict of interest.
The authors have no competing financial interest in relation to the work described.
References
- 1.Martinez-Delgado B, et al. Frequent inactivation of the p73 gene by abnormal methylation or LOH in non-Hodgkin’s lymphomas. Int J Cancer. 2002;102(1):15–9. doi: 10.1002/ijc.10618. [DOI] [PubMed] [Google Scholar]
- 2.Corn PG, et al. Transcriptional silencing of the p73 gene in acute lymphoblastic leukemia and Burkitt’s lymphoma is associated with 5′ CpG island methylation. Cancer Res. 1999;59(14):3352–6. [PubMed] [Google Scholar]
- 3.Kawano S, et al. Loss of p73 gene expression in leukemias/lymphomas due to hypermethylation. Blood. 1999;94(3):1113–20. [PubMed] [Google Scholar]
- 4.Pluta A, et al. The role of p73 in hematological malignancies. Leukemia. 2006;20(5):757–66. doi: 10.1038/sj.leu.2404166. [DOI] [PubMed] [Google Scholar]
- 5.Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature. 1995;378(6553):203–6. doi: 10.1038/378203a0. [DOI] [PubMed] [Google Scholar]
- 6.Jones SN, et al. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature. 1995;378(6553):206–8. doi: 10.1038/378206a0. [DOI] [PubMed] [Google Scholar]
- 7.Lundgren K, et al. Targeted expression of MDM2 uncouples S phase from mitosis and inhibits mammary gland development independent of p53. Genes Dev. 1997;11(6):714–25. doi: 10.1101/gad.11.6.714. [DOI] [PubMed] [Google Scholar]
- 8.Bouska A, et al. Mdm2 promotes genetic instability and transformation independent of p53. Mol Cell Biol. 2008;28(15):4862–74. doi: 10.1128/MCB.01584-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones SN, et al. Overexpression of Mdm2 in mice reveals a p53-independent role for Mdm2 in tumorigenesis. Proc Natl Acad Sci U S A. 1998;95(26):15608–12. doi: 10.1073/pnas.95.26.15608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang P, et al. Elevated Mdm2 expression induces chromosomal instability and confers a survival and growth advantage to B cells. Oncogene. 2008;27(11):1590–8. doi: 10.1038/sj.onc.1210788. [DOI] [PubMed] [Google Scholar]
- 11.Lushnikova T, et al. Aging mice have increased chromosome instability that is exacerbated by elevated Mdm2 expression. Oncogene. 2011;30(46):4622–31. doi: 10.1038/onc.2011.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Riley MF, Lozano G. The Many Faces of MDM2 Binding Partners. Genes Cancer. 2012;3(3–4):226–39. doi: 10.1177/1947601912455322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flores ER, et al. Tumor predisposition in mice mutant for p63 and p73: evidence for broader tumor suppressor functions for the p53 family. Cancer Cell. 2005;7(4):363–73. doi: 10.1016/j.ccr.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 14.Zdzalik M, et al. Interaction of regulators Mdm2 and Mdmx with transcription factors p53, p63 and p73. Cell Cycle. 2010;9(22):4584–91. doi: 10.4161/cc.9.22.13871. [DOI] [PubMed] [Google Scholar]
- 15.Zeng X, et al. MDM2 suppresses p73 function without promoting p73 degradation. Mol Cell Biol. 1999;19(5):3257–66. doi: 10.1128/mcb.19.5.3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Balint E, Bates S, Vousden KH. Mdm2 binds p73 alpha without targeting degradation. Oncogene. 1999;18(27):3923–9. doi: 10.1038/sj.onc.1202781. [DOI] [PubMed] [Google Scholar]
- 17.Irwin MS, Kaelin WG. p53 family update: p73 and p63 develop their own identities. Cell Growth Differ. 2001;12(7):337–49. [PubMed] [Google Scholar]
- 18.Jost CA, Marin MC, Kaelin WG., Jr p73 is a simian [correction of human] p53-related protein that can induce apoptosis. Nature. 1997;389(6647):191–4. doi: 10.1038/38298. [DOI] [PubMed] [Google Scholar]
- 19.Du W, et al. TAp73 enhances the pentose phosphate pathway and supports cell proliferation. Nat Cell Biol. 2013;15(8):991–1000. doi: 10.1038/ncb2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tomasini R, et al. TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev. 2008;22(19):2677–91. doi: 10.1101/gad.1695308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilhelm MT, et al. Isoform-specific p73 knockout mice reveal a novel role for delta Np73 in the DNA damage response pathway. Genes Dev. 2010;24(6):549–60. doi: 10.1101/gad.1873910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grob TJ, et al. Human delta Np73 regulates a dominant negative feedback loop for TAp73 and p53. Cell Death Differ. 2001;8(12):1213–23. doi: 10.1038/sj.cdd.4400962. [DOI] [PubMed] [Google Scholar]
- 23.Yang A, et al. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature. 2000;404(6773):99–103. doi: 10.1038/35003607. [DOI] [PubMed] [Google Scholar]
- 24.Wu Y, et al. Negative regulation of bcl-2 expression by p53 in hematopoietic cells. Oncogene. 2001;20(2):240–51. doi: 10.1038/sj.onc.1204067. [DOI] [PubMed] [Google Scholar]
- 25.Findley HW, et al. Expression and regulation of Bcl-2, Bcl-xl, and Bax correlate with p53 status and sensitivity to apoptosis in childhood acute lymphoblastic leukemia. Blood. 1997;89(8):2986–93. [PubMed] [Google Scholar]
- 26.Melino G, et al. p73 Induces apoptosis via PUMA transactivation and Bax mitochondrial translocation. J Biol Chem. 2004;279(9):8076–83. doi: 10.1074/jbc.M307469200. [DOI] [PubMed] [Google Scholar]
- 27.Talos F, et al. p73 suppresses polyploidy and aneuploidy in the absence of functional p53. Mol Cell. 2007;27(4):647–59. doi: 10.1016/j.molcel.2007.06.036. [DOI] [PubMed] [Google Scholar]
- 28.Miyashita T, et al. Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene. 1994;9(6):1799–805. [PubMed] [Google Scholar]
- 29.Wang TT, et al. Effects of dehydroepiandrosterone and calorie restriction on the Bcl-2/Bax-mediated apoptotic pathway in p53-deficient mice. Cancer Lett. 1997;116(1):61–9. doi: 10.1016/s0304-3835(97)00175-4. [DOI] [PubMed] [Google Scholar]
- 30.Levrero M, et al. The p53/p63/p73 family of transcription factors: overlapping and distinct functions. J Cell Sci. 2000;113(Pt 10):1661–70. doi: 10.1242/jcs.113.10.1661. [DOI] [PubMed] [Google Scholar]
- 31.Moller MB, Nielsen O, Pedersen NT. Oncoprotein MDM2 overexpression is associated with poor prognosis in distinct non-Hodgkin’s lymphoma entities. Mod Pathol. 1999;12(11):1010–6. [PubMed] [Google Scholar]
- 32.Watanabe T, et al. Overexpression of the MDM2 oncogene in leukemia and lymphoma. Leuk Lymphoma. 1996;21(5–6):391–7. doi: 10.3109/10428199609093436. color plates XVI following 5. [DOI] [PubMed] [Google Scholar]
- 33.Amin AR, et al. A novel role for p73 in the regulation of Akt-Foxo1a-Bim signaling and apoptosis induced by the plant lectin, Concanavalin A. Cancer Res. 2007;67(12):5617–21. doi: 10.1158/0008-5472.CAN-07-0655. [DOI] [PubMed] [Google Scholar]
- 34.Dimri GP, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92(20):9363–7. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gostissa M, et al. Conditional inactivation of p53 in mature B cells promotes generation of nongerminal center-derived B-cell lymphomas. Proc Natl Acad Sci U S A. 2013;110(8):2934–9. doi: 10.1073/pnas.1222570110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.