

Abstract
Fetal hemoglobin (Hb F) is an important genetic modulator of the beta-hemoglobinopathies. The regulation of Hb F levels is influenced by transcription factors. We used phylogenetic footprinting to screen transcription factors that have binding sites in HBG1 and HBG2 genes’ noncoding regions in order to know the genetic determinants of the Hb F expression. Our analysis showed 354 conserved motifs in the noncoding regions of HBG1 gene and 231 motifs in the HBG2 gene between the analyzed species. Of these motifs, 13 showed relation to Hb F regulation: cell division cycle-5 (CDC5), myelo-blastosis viral oncogene homolog (c-MYB), transcription factor CP2 (TFCP2), GATA binding protein 1 (GATA-1), GATA binding protein 2 (GATA-2), nuclear factor erythroid 2 (NF-E2), nuclear transcription factor Y (NF-Y), runt-related transcription factor 1 (RUNX-1), T-cell acute lymphocytic leukemia 1 (TAL-1), YY1 transcription factor (YY1), beta protein 1 (BP1), chicken ovalbumin upstream promoter-transcription factor II (COUP-TFII), and paired box 1 (PAX-1). The last three motifs were conserved only in the noncoding regions of the HBG1 gene. The understanding of genetic elements involved in the maintenance of high Hb F levels may provide new efficient therapeutic strategies in the beta-hemoglobinopathies treatment, promoting reduction in clinical complications of these genetic disorders.
Keywords: Hb F, beta-hemoglobinopathies, transcription factors, phylogenetic footprinting
Introduction
Fetal hemoglobin (Hb F), which consists of two alpha-globin chains and two gamma-globin chains (α2γ2), is predominantly expressed in the fetal period. Its levels decrease to less than 1% of total hemoglobin in adult life, the period in which adult hemoglobin (Hb A) (α2β2) represents most of the composition. Through their high affinity for binding to oxygen molecules, the increased Hb F concentrations act as a modulator of the phenotype of beta-hemoglobinopathies, hemolytic anemias that result from beta-globin gene mutations.1,2 In sickle cell disease, high Hb F levels may dilute the amount of Hb S (HBB:c.20 A > T), thus inhibiting or retarding the polymerization process. This change reduces the severity of the disease.3 In the thalassemia beta major, the increased production of γ-globin chains reduces the imbalance of the α chain/non-α chain and increases the total hemoglobin synthesis. Thus, the increase in γ-globin gene expression is clinically relevant in the treatment of diseases in which altered beta-globin is involved.2,4,5
The evolutionary process of the γ-globin genes differed during the divergence of mammals. Prosimian primates (suborder Strepsirhini) have a single γ-globin gene, the expression of which occurs in the embryonic stage, along with ε-globin gene expression. Simian primates (suborder Haplorhini) have two γ-globin genes, the expression of which occurs during the fetal period, a change which differed from that seen in prosimians.6,7 Molecular evidence suggests that γ-globin gene expression during the fetal period may have occurred after the divergence between prosimians and apes, ∼55 million years ago.8 The evolutionary history of these genes indicates the occurrence of a tandem duplication of 5.5-kb DNA fragment, before the divergence between Platyrrhini and Catarrhini occurred (∼35 million years ago). Based on this information, it is believed that fetal γ-globin gene recruitment and gene duplication occurred during the same period of evolutionary history.8,9
After gene duplication, the coding regions of the γG-globin (HBG2) and γA-globin (HBG1) genes differed by only one amino acid at position 136 of the polypeptide chain, while the noncoding (introns) and transcribed and untranslated (5′ and 3′-UTR) regions differ by 3% and 14%, respectively.10 Although apes present two γ-globin genes, the activation and expression periods of these genes differ. In Platyrrhini, only one of the γ-globin genes is functional; in Catarrhini, both genes are expressed.6,8
Hb F regulation is influenced by transcription factors. These genetic elements act in the globin gene regulation and in the switch from Hb F to Hb A expression. This process involves factors that are well established in the literature, including BCL11A and SOX6.11–13
Because the beta-hemoglobinopathies are considered the most common monogenic diseases in the world and because high Hb F concentrations in these conditions may result in clinical improvement of patients,14,15 knowledge of transcription factors that act in Hb F regulation can reveal an important area in which therapeutic strategies and pharmacological agents are needed in order to increase the life expectancy of patients.
The aim of this study was to use phylogenetic footprinting to screen transcription factors that have binding sites in the γ-globin genes’ noncoding regions in order to understand the genetic determinants that act in the modulation of Hb F levels.
Methods
We used the VISTA bioinformatics tool (http://www-gsd.lbl.gov/vista/), which identifies conservation patterns on a genome-wide scale.16,17 Phylogenetic footprinting analysis was used to identify regulatory elements conserved between different species. The decision to use this method was based on the assumption that sequences of biological importance and noncoding regions, in particular, are conserved among related species as a result of functional pressure. Thus, the identification of conserved elements in the noncoding regions of the γ-globin genes may indicate that these elements play a functional role in gene regulation and, consequently, in the maintenance of Hb F levels.
Because they present differences in the noncoding regions, both γ-globin genes (HBG1 and HBG2) were included in the phylogenetic footprinting analysis. The sequences of the New World monkeys, popularly known as the common marmoset (Callithrix jacchus) and tufted capuchin (Cebus apella); those of Old World monkeys, known as the Sumatran orangutan (Pongo abelli), the rhesus monkey (Macaca mulatta), the chimpanzee (Pan troglodytes), and the tarsier monkey (Tarsius syrichta); and one basal primate of the Haplorhini suborder, were all compared to human (Homo sapiens) γ-globin gene sequences in order to search for transcription factors involved in the regulation of these genes. The gene sequences for each species were obtained from the National Center for Biotechnology Information database under the following gene IDs: H. sapiens (GenBank: NG_000007.3), P. troglodytes (GenBank: NW_001249430.1), M. mulatta (GenBank: NC_007871.1), P. abelli (GenBank: NC_012602.1), C. jacchus (GenBank: NC_013906.1), C. apella (GenBank: U57043.1), and T. syrichta (GenBank: M3397.1).
The computational scheme for alignment and conservation analysis was based on the local alignment program known as BLAT (BLAST-like alignment tool) in order to identify homology. The data were then processed and globally aligned using the MLAGAN (multiple alignment) program. Alignments were visualized in the VISTA Browser by defining the colors and patterns of peaks and valleys. Next, the conservation of the region was evaluated. For the conservation evaluation, we used the program’s default parameters (70% identity over 100 bp). To identify transcription factors, rVISTA was used. It associates the TRANSFAC database (913 motifs) with the comparative sequences analysis.18
Results and Discussion
The comparison of the gene sequences in the noncoding regions of the human γ-globin genes to those of P. troglodytes, M. mulatta, P. abelli, C. jacchus, C. Apella, and T. syrichta revealed conservation levels above 70% over 100 bp of the γ-globin gene, according to the criteria established using the VISTA tools. The similarity pattern observed in the coding regions was expected from the comparison of the selected species, since these sequences are responsible for specific protein coding during the development of the organism. The high degree of similarity observed in the noncoding regions suggests the existence of functionality conserved between compared species. The noncoding regions that have high rates of conservation may indicate potential binding sites for specific transcription factors.18–20
The results showed that the conservation degree observed in the γ-globin genes sequences differed between the species analyzed, and that the HBG1 gene was found to have higher levels of conservation (Fig. 1).
Figure 1.
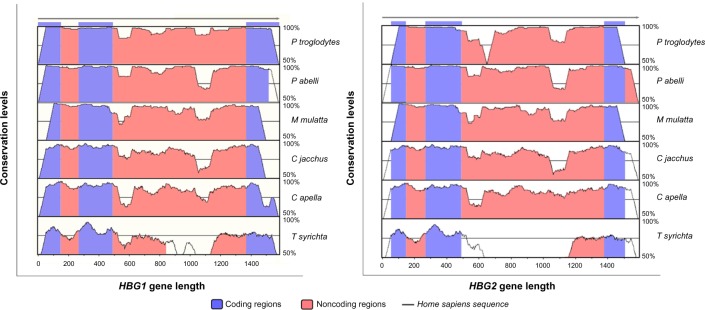
VISTA plots obtained from the comparison analysis of genomic sequences of the nonconding regions from the γ-globin genes. The plots contain genomic sequences of γA-globin and γG-globin genes from H. sapiens (gray arrow), P. troglodytes (line 1), P. abelli (line 2), M. mulatta (line 3), C. jacchus (line 4), C. apella (line 5), and T. syrichta (line 6), showing the patterns of peaks and valleys. In blue are represented γ-globin coding regions (exons) and in red are represented the noncoding regions (introns). The results indicate similarity conservation of coding and noncoding sequences greater than 70%, suggesting functionality for the noncoding regions.
The phylogenetic footprinting analysis and the screening of the transcription factors based on the TRANSFAC database revealed 354 conserved motifs in the noncoding regions of the HBG1 gene and 231 motifs of the HBG2 gene among the species analyzed. After analyzing the information in the literature regarding the role and involvement of each of these transcription factors in the Hb F modulation, we identified 13 elements involved in γA-globin gene regulation and 10 elements associated with γG-globin gene regulation.
The 10 transcription factor candidates for the regulation of both γ-globin genes are cell division cycle-5 (CDC5), myeloblastosis viral oncogene homolog (c-MYB), transcription factor CP2 (TFCP2), GATA binding protein 1 (GATA-1), GATA binding protein 2 (GATA-2), nuclear factor erythroid 2 (NF-E2), nuclear tran¬scription factor Y (NF-Y), runt-related transcription factor 1 (RUNX-1), T-cell acute lymphocytic leukemia 1 (TAL-1), and YY1 transcription factor (YY1). Three other transcription factors (beta protein 1 (BP1), chicken ovalbumin upstream promoter-transcription factor II (COUP-TFII), and paired box 1 (PAX-1)) are involved in γA-globin gene regulation, but not in γG-globin gene regulation. Among the transcription factors highlighted in our analysis, some behave as transcriptional activators of the γ-globin genes and thus act as positive regulators of Hb F expression. Meanwhile, some regulatory elements are transcriptional repressors and other factors form protein complexes that act in the regulation of the globin genes. There are also transcription factors that act indirectly in γ-globin gene regulation.
GATA-2 and TAL-1 are transcriptional activators of the γ-globin genes. GATA-2 is expressed during the initial stages of the precursor hematopoietic lineage and appears to be involved in self-renewal, proliferation, and cell survival. Some reports have shown that the β-globin gene cluster has 16 binding sites preserved for the GATA transcription factor, and most of these sites are located in the locus control region (LCR). Furthermore, only the ε-globin gene and the γ-globin genes have GATA binding sites, which are absent from the δ-globin gene and the β-globin gene.21 TAL-1 participates in the chromatin looping formation between the LCR and the γ-globin gene, and it acts by recruiting required proteins in looping formation (Ldb1 and LMO2, for example).22 By promoting interaction between the gene and the regulatory region, TAL-1 acts as an activator of Hb F expression.
Some activators of the γ-globin genes are involved in hereditary persistence of fetal haemoglobin (HPFH) mutations. When combined with DNMT1, RAP74, and SNEV, CDC5 can bind to the single point mutation -198 (T > C) (British nondeletional HPFH) in the γA-globin gene promoter, the Hb F levels of which range from 1.8% to 13%.23,24 In Brazilian nondeletional HPFH (γA -195 C > G), PAX-1 is able to bind to the TTCCGC sequence in the HBG1 gene promoter only in the presence of this mutation, a finding that indicates that this transcription factor may play a role in the activation of Hb F expression.25 The binding of these transcription factors in point mutations that generates increased Hb F levels may indicate that these genetic elements play a role as regulators of Hb F concentrations.
BP1 and CP2 (transcription factor CP2) are indirect transcriptional activators of the γ-globin genes. BP1 reduces β-globin gene expression in cells of the erythroid lineage, both in the early stages of cell maturation and in fully mature cells. It acts as a negative regulator of adult Hb levels (Hb AA)26 and may act as an upregulation factor of Hb F. CP2 is involved in the transcriptional activation of the α-globin genes and plays an important role in globin gene switch expression through the formation of a protein complex with GATA-1.27,28 Along with nuclear factor erythroid 4 (NF-E4), CP2 forms the heterodimeric protein complex stage selector protein (SSP), which is involved in the preferential expression of the γ-globin genes.29
In contrast, c-MYB, COUP-TFII, and GATA-1 act as negative regulators of Hb F levels. c-MYB regulates erythroid progenitor differentiation.30 Literature reports have shown that elevated c-MYB levels inhibit γ-globin expression in the K562 cell line,31 and they feature this transcription factor as a negative regulator of Hb F levels. Aerbajinai et al.32 used siRNA transfection to show that the knockdown of COUP-TFII resulted in the induction of γ-globin gene expression during adult erythropoiesis. Furthermore, COUP-TFII joins to the BCL11A transcription factor through RID1 and RID2 motifs, thus forming a repressor complex that is able to bind to key regulation regions in Hb F genes.33 GATA-1 is essential for survival and terminal maturation of erythroid precursors.21 In the case of globin gene regulation, GATA-1 has been identified as a transcription factor involved in silencing the γ-globin genes due to its participation in the formation of the Hb F repressor protein complex, examples of which include the association with BCL11A, SOX6, FOG1, and NuRD13 and the association with NF-Y, COUP-TFII, and BCL11A.34 Moreover, the loss of GATA-1 binding sites in γ-globin gene promoters influences the prevention of HBG1 and HBG2 gene silencing.35
YY1 (YY1 transcription factor) acts by activating and repressing a variable number of genes through histone modifications.27 The literature reported that YY1 consists of a transcriptional repressor of ε-globin and γ-globin genes.25,36 In Brazilian nondeletional HPFH (-195 C > G), the authors reported decreased interaction between YY1 and the HBG1 gene promoter region, which thus reduced the repressional activity exerted by this transcription factor.25
RUNX-1 regulates the expression of specific genes involved in the control of hematopoiesis.37,38 The NF-E2 transcription factor is an important element that regulates the globin gene expression and acts in the formation of the components of the hemoglobin molecule.39,40
NF-Y is involved in both γ-globin gene activation and the repression processes. In general, NF-Y recruits GATA-2 and thus forms an activating complex of γ-globin gene transcription, and BCL11A, GATA-1, and COUPTFII, forming a complex that acts as a transcriptional repressor.34 Thus, the NF-Y transcription factor can act as either an activator or a repressor of γ-globin gene expression, depending on the recruited regulator elements.
Figure 2 shows the possible mechanisms of interaction exerted by the transcription factors selected from the in silico analysis in hematopoietic processes, erythropoietic processes, and the regulation of globin gene expression.
Figure 2.

Representation of the interaction between transcription factors and regulation of hematopoiesis, erythropoiesis, and the globin genes. The 13 transcription factors selected from in silico analysis are showed. The continuous arrows indicate elements that act as transcriptional activators. The dashed arrows indicate indirect activators. The bars show transcriptional repressors. Boxes without staining (NF-E4 and SSP) indicate factors that comprise protein complexes involved in globin regulation. The different colors of transcription factors represent different forms of action: dark green represents direct transcriptional activation, orange transcriptional activation mediated by the presence of specific mutations, light green indirect transcriptional activation, red transcriptional repression, and blue control of hematopoiesis and erythropoiesis and synthesis of globin chains. We used the Microsoft PowerPoint to elaborate this figure.
In addition to the 13 transcription factors in our analysis, other elements are known for regulating γ-globin gene expression. They include Krüppel-like factor family members and NF-E4. Krüppel-like factor 1 (KLF1) is an indirect repressor of γ-globin gene expression. KLF1 activates the expression of BCL11A, a transcription factor known as a key repressor of Hb F expression.41 Krüppel-like factor 11 (KLF11) is predominantly expressed in the erythroid lineage cells. Gene expression studies have revealed that KLF11 can act as a transcriptional activator of embryonic and fetal globin.42 The transcription factor known as NF-E4, which is part of the SSP complex along with CP2, can direct the LCR to the promoter regions of the γ-globin genes and thus maintain high Hb F levels.29,43 However, these factors are not conserved among species of the suborders Catarrhini and Platyrrhini. When we compared only the primates of the Old World and the New World to each other, these transcription factors were found to be conserved. Thus, we can infer that these elements do not exert significant influence on Hb F production in T. syrichta. In this species, fetal hemoglobin is expressed along with the ε-globin, unlike to happen on globin ontogeny of the other analyzed species of primates.
Our results corroborate the reports in the literature on the participation of numerous genetic factors in γ-globin gene regulation. Many studies have revealed genetic regulators that can provide promising information for the development of effective strategies for the induction of Hb F levels.1,11,44,45 Sankaran and Orkin15 performed a literature review regarding the elements that act in the switch from Hb F to Hb A expression, and they showed that most of the molecules identified as regulators in this process are transcription factors. These findings reinforce the importance of screening such regulatory elements in order to provide more accurate information on these factors’ mechanisms of action in the increase of Hb F levels. Ideally, this information will result in the clinical improvement of patients with beta-hemoglobinopathies.
Conclusion
Based on the results provided via phylogenetic footprinting analysis and a subsequent overview of the literature, the noncoding regions of γA-globin gene have been found to have binding sites for 13 conserved transcription factor motifs involved in Hb F regulation between Old World monkeys, New World monkeys, and Tarsiidae, while the noncoding regions of γG-globin gene have 10 elements. Knowledge of the genetic elements involved in γ-globin gene regulation forms the basis of new molecular strategies that can be used in the treatment of diseases in which elevated Hb F levels can reduce clinical manifestations and provide longer life expectancies for individuals with these disorders.
Footnotes
ACADEMIC EDITOR: Jike Cui, Associate Editor
PEER REVIEW: Two peer reviewers contributed to the peer review report. Reviewers’ reports totaled 844 words, excluding any confidential comments to the academic editor.
FUNDING: The authors acknowledge Fundação de Amparo à Pesquisa do Estado de São Paulo (Fapesp), grants 2011/15570-1 and 2015/09456-2, for financial support. The authors confirm that the funder had no influence over the study design, content of the article, or selection of this journal.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
Authors’ Contributions
GCSC participated in the in silico analysis, the search for literature information of transcription factors, and the interpretation of data and article writing. LPRV participated in in silico analysis and interpretation of data. CRB-D participated in the interpretation of results. All authors read and approved the article.
REFERENCES
- 1.Menzel S, Garner C, Gut I, et al. A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nat Genet. 2007;39(10):1197–9. doi: 10.1038/ng2108. [DOI] [PubMed] [Google Scholar]
- 2.Xu XS, Hong X, Wang G. Induction of endogenous γ-globin gene expression with decoy oligonucleotide targeting Oct-1 transcription factor consensus sequence. J Hematol Oncol. 2009;2(15):1–11. doi: 10.1186/1756-8722-2-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Steinberg MH. Genetic etiologies for phenotypic diversity in sickle cell anemia. Scientific World Journal. 2009;9:46–67. doi: 10.1100/tsw.2009.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galanello R, Cao A. Relationship between genotype and phenotype. Ann N Y Acad Sci. 1998;850:325–33. doi: 10.1111/j.1749-6632.1998.tb10489.x. [DOI] [PubMed] [Google Scholar]
- 5.Thein SL. Genetic modifiers of β-thalassemia. Haematologica. 2005;90:649–60. [PubMed] [Google Scholar]
- 6.Chiu CH, Schneider H, Schneider MP, et al. Reduction of two functional y-globin genes to one: An evolutionary trend in New World monkeys (infraorder Platyrrhini) Proc Natl Acad Sci U S A. 1996;93:6510–5. doi: 10.1073/pnas.93.13.6510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson RM, Buck S, Chiu C, et al. Fetal globin expression in new world monkeys. J Biol Chem. 1996;271(25):14684–91. doi: 10.1074/jbc.271.25.14684. [DOI] [PubMed] [Google Scholar]
- 8.Meireles CM, Schneider MP, Sampaio MI, et al. Fate of a redundant y-globin gene in the atelid clade of New World monkeys: implications concerning fetal globin gene expression. Proc Natl Acad Sci U S A. 1995;92:2607–11. doi: 10.1073/pnas.92.7.2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson RM, Gumucio D, Goodman M. Globin gene switching in primates. Comp Biochem Physiol A Mol Integr Physiol. 2002;133:877–83. doi: 10.1016/s1095-6433(02)00205-2. [DOI] [PubMed] [Google Scholar]
- 10.Manca L, Masala B. Disorders of the synthesis of human fetal hemoglobin. IUBMB Life. 2008;60(2):94–111. doi: 10.1002/iub.4. [DOI] [PubMed] [Google Scholar]
- 11.Sankaran VG, Menne TF, Xu J, et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science. 2008;322:1839–42. doi: 10.1126/science.1165409. [DOI] [PubMed] [Google Scholar]
- 12.Chen Z, Luo HY, Steinberg MH, Chui DH. BCL11A represses HBG transcription in K562 cells. Blood Cells Mol Dis. 2009;42:144–9. doi: 10.1016/j.bcmd.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 13.Xu J, Sankaran VG, Ni M, et al. Transcriptional silencing of γ-globin by BCL11A involves long-range interactions and cooperation with SOX6. Genes Dev. 2010;24:783–98. doi: 10.1101/gad.1897310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weatherall D, Akinyanju O, Fucharoen S, et al. Inherited disorders of hemoglobin. In: Jamison Dean T, Breman Joel G, Measham Anthony R, Alleyne George, Claeson Mariam, Evans David B, Jha Prabhat, Mills Anne, Musgrove Philip., editors. Disease Control Priorities in Developing Countries. 2nd ed. Oxford University Pressl; New York: 2006. pp. 663–80. [Google Scholar]
- 15.Sankaran VG, Orkin SH. The Switch from fetal to adult hemoglobin. Cold Spring Harb Perspect Med. 2013;3(1):a011643. doi: 10.1101/cshperspect.a011643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cooper GM, Brudno M, Stone EA, Dubchak I, Batzoglou S, Sidow A. Characterization of evolutionary rates and constraints in three mammalian genomes. Genome Res. 2004;14(4):539–48. doi: 10.1101/gr.2034704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gibbs RA, Weinstock GM, Metzker ML, et al. Rat Genome Sequencing Project Consortium. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature. 2004;428(6982):493–521. doi: 10.1038/nature02426. [DOI] [PubMed] [Google Scholar]
- 18.Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I. VISTA: computational tools for comparative genomics. Nucleic Acids Res. 2004;32:273–9. doi: 10.1093/nar/gkh458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Touchman JW, Dehejia A, Chiba-Falek O, et al. Human and mouse alpha-synuclein genes: comparative genomic sequence analysis and identification of a novel gene regulatory element. Genome Res. 2001;11(1):78–86. doi: 10.1101/gr.165801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bofelli D, Nobrega MA, Rubin EM. Comparative genomics at the vertebrate extremes. Nat Rev Genet. 2004;5(6):456–65. doi: 10.1038/nrg1350. [DOI] [PubMed] [Google Scholar]
- 21.Ikonomi P, Noguchi CT, Miller W, Kassahun H, Hardison R, Schechter AN. Levels of GATA1/GATA2 transcription factors modulate expression of embryonic and fetal hemoglobins. Gene. 2000;261(2):277–87. doi: 10.1016/s0378-1119(00)00510-2. [DOI] [PubMed] [Google Scholar]
- 22.Yun WJ, Kim YW, Kang Y, Lee J, Dean A, Kim A. The hematopoietic regulator TAL1 is required for chromatin looping between the β-globin LCR and human γ-globin genes to activate transcription. Nucleic Acids Res. 2014;42(7):4283–93. doi: 10.1093/nar/gku072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olave IA, Doneanu C, Fang X, Stamatoyannopoulos G, Li Q. Purification and identification of proteins that bind to the hereditary persistence of fetal hemoglobin −198 mutation in the γ-globin gene promoter. J Biol Chem. 2007;282(2):853–62. doi: 10.1074/jbc.M610404200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huisman THJ, Carver MFH, Baysal E, Efremov GD. A database of human hemoglobin variants and Thalassemias Summaries of mutation categories. Pennsylvania University USA and McMaster University in Canada; 1996. [Internet]. [cited Nov 20, 2013]. Available at: http://globin.cse.psu.edu/>. [Google Scholar]
- 25.Roversi FM, Cunha AF, Lanaro C, et al. The –195 C > G substitution in Brazilian hereditary persistence of fetal hemoglobin decreases NF-E1/YY1 binding and increases PAXI binding to the Aγ globin promoter region. Proceedings of the 52nd Annual American Society of Hematology Meeting, Orlando, FL, USA, December 4–7,2010. Blood. 2010;116(21):857. [Google Scholar]
- 26.Mpollo MS, Beaudoin M, Berg PE, Beauchemin H, D’Agati V, Trudel M. BP1 is a negative modulator of definitive erythropoiesis. Nucleic Acids Res. 2006;34(18):5232–7. doi: 10.1093/nar/gkl680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.National Center for Biotechnology Information [cited Sep 25, 2013]. Available from: http://http://www.ncbi.nlm.nih.gov/ [PubMed]
- 28.Bosè F, Fugazza C, Casalgrandi M, et al. Functional interaction of CP2 with GATA-1 in the regulation of erythroid promoters. Mol Cell Biol. 2006;26(10):3942–54. doi: 10.1128/MCB.26.10.3942-3954.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou W, Zhao Q, Sutton R, et al. The role of p22 NF-E4 in human globin gene switching. J Biol Chem. 2004;79(25):26227–39. doi: 10.1074/jbc.M402191200. [DOI] [PubMed] [Google Scholar]
- 30.Soza-Ried C, Hess I, Netuschil N, Schorpp M, Boehm T. Essential role of c-myb in definitive hematopoiesis is evolutionarily conserved. Proc Natl Acad Sci U S A. 2010;107(40):17304–8. doi: 10.1073/pnas.1004640107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bauer DE, Kamran SC, Orkin SH. Reawakening fetal hemoglobin: prospects for new therapies for the β-globin disorders. Blood. 2012;120(15):2945–53. doi: 10.1182/blood-2012-06-292078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aerbajinai W, Zhu J, Kumkhaek C, Chin K, Rodgers GP. SCF induces γ-globin gene expression by regulating downstream transcription factor COUP-TFII. Blood. 2009;114(1):187–94. doi: 10.1182/blood-2008-07-170712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan CM, Fulton J, Montiel-Duarte C, et al. A signature motif mediating selective interations of BCL11A with the NR2E/F subfamily of orphan nuclear receptors. Nucleic Acids Res. 2013;41(21):9663–79. doi: 10.1093/nar/gkt761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu X, Wang Y, Pi W, Liu H, Wickrema A, Tuan D. NF-Y recruits both transcription activator and repressor to modulate tissue- and developmental stage-specific expression of human γ-globin gene. PLoS One. 2012;7(10):e47175. doi: 10.1371/journal.pone.0047175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen Z, Luo HY, Basran RK, et al. A T-to-G transversion at nucleotide -567 upstream of HBG2 in a GATA-1 binding motif is associated with elevated hemoglobin F. Mol Cell Biol. 2008;28:4386–93. doi: 10.1128/MCB.00071-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raich N, Clegg CH, Grofti J, Roméo PH, Stamatoyannopoulos G. GATA1 and YY1 are developmental repressors of the human ε-globin gene. EMBO J. 1995;14(4):801–9. doi: 10.1002/j.1460-2075.1995.tb07058.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roumier C, Fenaux P, Lafage M, Imbert M, Eclache V, Preudhomme C. New mechanisms of AML1 gene alteration in hematological malignancies. Leukemia. 2003;17(1):9–16. doi: 10.1038/sj.leu.2402766. [DOI] [PubMed] [Google Scholar]
- 38.Kurokawa M, Hirai H. Role of RUNX1/Runx1 in the pathogenesis of hematological malignancies. Cancer Sci. 2003;94:841–6. doi: 10.1111/j.1349-7006.2003.tb01364.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blank V, Kim MJ, Andrews NC. Human MafG is a functional partner for p45 NF-E2 in activating globin gene expression. Blood. 1997;89:3925–35. [PubMed] [Google Scholar]
- 40.Andrews NC. Molecules in focus: the NF-E2 transcription factor. Int J Biochem Cell Biol. 1998;30:429–32. doi: 10.1016/s1357-2725(97)00135-0. [DOI] [PubMed] [Google Scholar]
- 41.Tallack MR, Perkins AC. KLF1 directly coordinates almost all aspects of terminal erythroid differentiation. Life. 2010;62(12):886–90. doi: 10.1002/iub.404. [DOI] [PubMed] [Google Scholar]
- 42.Asano H, Li XS, Stamatouyannopoulos G. FKLF, a novel Krüppel-like factor that activates human embryonic and fetal β-like globin genes. Mol Cell Biol. 1999;19(5):3571–9. doi: 10.1128/mcb.19.5.3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou W, Clouston DR, Wang X, Cerruti L, Cunningham JM, Jane SM. Induction of human fetal globin gene expression by a novel erythroid factor, NF-E4. Mol Cell Biol. 2000;20(20):7662–72. doi: 10.1128/mcb.20.20.7662-7672.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thein SL, Menzel S, Peng X, et al. Intergenic variants of HBS1 L-MYB are responsible for a major quantitative trait locus on chromosome 6q23 influencing fetal hemoglobin levels in adults. Proc Natl Acad Sci U S A. 2007;104(27):11346–51. doi: 10.1073/pnas.0611393104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lettre G, Sankaran VG, Bezerra MA, et al. DNA polymorphisms at the BCL11 A, HBS1 L-MYB and β-globin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease. Proc Natl Acad Sci U S A. 2008;105(33):11869–74. doi: 10.1073/pnas.0804799105. [DOI] [PMC free article] [PubMed] [Google Scholar]