

Abstract
A fundamental goal in catalysis is the coupling of multiple reactions to yield a desired product. Enzymes have evolved elegant approaches to address this grand challenge. A salient example is the biological conversion of methane to methanol catalyzed by soluble methane monooxygenase (sMMO), a member of the bacterial multicomponent monooxygenase (BMM) superfamily.
sMMO is a dynamic protein complex of three components: a hydroxylase, a reductase, and a regulatory protein. The active site, a carboxylate-rich non-heme diiron center, is buried inside the 251-kDa hydroxylase component. The enzyme processes four substrates: O2, protons, electrons, and methane. To couple O2 activation to methane oxidation, timely control of substrate access to the active site is critical. Recent studies of sMMO, as well as its homologs in the BMM superfamily, have begun to unravel the mechanism. The emerging and unifying picture reveals that each substrate gains access to the active site along a specific pathway through the hydroxylase. Electrons and protons are delivered via a three-amino acid pore located adjacent to the diiron center; O2 migrates via a series of hydrophobic cavities; and hydrocarbon substrates reach the active site through a channel or linked set of cavities. The gating of these pathways mediates entry of each substrate to the diiron active site in a timed sequence and is coordinated by dynamic interactions with the other component proteins. The result is coupling of dioxygen consumption with hydrocarbon oxidation, avoiding unproductive oxidation of the reductant rather than the desired hydrocarbon.
To initiate catalysis, the reductase delivers two electrons to the diiron(III) center by binding over the pore of the hydroxylase. The regulatory component then displaces the reductase, docking onto the same surface of the hydroxylase. Formation of the hydroxylase-regulatory component complex (i) induces conformational changes of pore residues that may bring protons to the active site; (ii) connects hydrophobic cavities in the hydroxylase leading from the exterior to the diiron active site, providing a pathway for O2 and methane, in the case of sMMO, to the reduced diiron center for O2 activation and substrate hydroxylation; (iii) closes the pore, as well as a channel in the case of four-component BMM enzymes, restricting proton access to the diiron center during formation of “Fe2O2” intermediates required for hydrocarbon oxidation; and (iv) inhibits undesired electron transfer to the Fe2O2 intermediates by blocking reductase binding during O2 activation. This mechanism is quite different from that adopted by cytochromes P450, a large class of heme-containing monooxygenases that catalyze very similar reactions as the BMM enzymes. Understanding the timed enzyme control of substrate access has implications for designing artificial catalysts. To achieve multiple turnovers and tight coupling, synthetic models must also control substrate access, a major challenge considering that nature requires large, multimeric, dynamic protein complexes to accomplish this feat.
Graphical Abstract
1. Introduction
How can reactions among multiple substrates be coupled to generate a desired product? This challenge is frequently seen in biocatalysis, especially in achieving the most difficult chemical transformations. One example is the biological activation of inert C–H bonds. This transformation is catalyzed by several metalloenzymes, including the heme-containing cytochromes P450,1-4 the dicopper-containing particulate methane monooxygenase,5,6 and the family of non-heme diiron-containing bacterial multicomponent monooxygenases (BMMs).7-10 These enzymes couple reactions involving four substrates (eq 1), namely, oxygen, protons, electrons, and a hydrocarbon RH.
| (1) |
Timed control of substrate addition to the active site is essential for coupling O2 activation to substrate hydroxylation. Without proper control, the following uncoupled reactions can occur (eq 2, 3):2,11,12
| (2) |
| (3) |
Such uncoupling could be detrimental to the organism, for example, by wasting reducing equivalents and by generating hydrogen peroxide, a reactive oxygen species.
Recently, we have come to understand that the desired coupling (eq 1) involves carefully programmed access of substrates to the diiron active sites in the hydroxylase components of soluble methane monooxygenase (sMMO) and its homologs in the bacterial multicomponent monooxygenase (BMM) superfamily. These enzymes are of great interest in the context of the global carbon cycle13 and bioremediation.14,15 They are complex systems typically comprising three to four protein components.8,16 The flagship enzyme, sMMO, has three components, a hydroxylase (MMOH), a reductase (MMOR), and a regulatory protein (MMOB). The 251-kDa hydroxylase is a homodimer with three subunits (α, β, and γ) in each protomer.17,18 Each α subunit hosts a diiron center, where dioxygen is activated and substrate hydroxylated. The 38-kDa reductase is responsible for providing two electrons during each round of the catalytic cycle. It transfers electrons from the reduced form of nicotinamide adenine dinucleotide (NADH) to the diiron center, via its flavin adenine dinucleotide (FAD) cofactor in the C-terminal domain and an [Fe2S2] cluster in the ferredoxin domain.12,19-22 The third component is a 16-kDa regulatory protein, which accelerates dioxygen activation and methane oxidation23 and is critical for coupling.11 Some members of the BMM superfamily, such as toluene/o-xylene monooxygenase (ToMO)24 and toluene-4-monooxygenase (T4mo),25 require an additional Rieske-type [Fe2S2] protein for reducing the hydroxylase. Dynamic interactions among these component proteins control substrate access to achieve some of the most challenging chemical conversions.
Since our previous Account published in 2007,9 we have learned that coupling in BMM enzymes relies critically upon the control of substrate access to the catalytic diiron center. Here we describe the entry pathway for each substrate as well as the molecular details of the mechanism used to control substrate access. Finally, we compare the tactics used by BMM enzymes with those adopted by P450s, a large group of related monooxygenases.
2. Control of electron and proton transfer: gating the pore
Accurate control of electron and proton transfer is vital for tight coupling. Electron transfer initiates catalysis by reducing the diiron(III) center in the resting hydroxylase to diiron(II) (Scheme 1). In subsequent steps, however, electron transfer is undesired, because extra electrons could quench the activated oxygen species in P*, Hperoxo, and Q. Proton transfer is required for O2 activation, promoting heterolytic cleavage of the O–O bond in sMMO catalysis to generate the methane-oxidizing intermediate Q.26,27 Excess proton transfer could quench the activated oxygen species, however, leading to production of H2O and H2O2 (eqs 2, 3). Accumulating evidence indicates that electrons and protons are transmitted through the pore (Figure 1a,b),28-32 a conserved structural feature in BMM hydroxylases,29,32-34 and are gated through the interaction with the regulatory component.
Scheme 1.

The catalytic cycle of sMMO. Rred and Rox represent the reduced and oxidized reductase MMOR, respectively, and B the regulatory component MMOB.
Figure 1.

The reductase binding site on the hydroxylase. a) Structure of sMMO hydroxylase (PDB ID 1MTY), showing an area that is within 12 Å of the diiron center in one of the monomers (red) and the reductase binding site suggested by HDX-MS (orange). b) A close-up view of the pore in sMMO hydroxylase, showing the three residues that define the pore and Fe1 of the diiron center. c) Computationally docked model of the hydroxylase-reductase ferredoxin domain protein complex of sMMO. d) A close-up view of the binding interface in the docked model shown in c), viewed from the top, showing the [Fe2S2] cluster, the diiron center, and selected residues of the hydroxylase. e) Crystal structure of the hydroxylase-reductase complex of T4mo (PDB ID 4P1B). f) A close-up view of the binding interface in the docked model shown in e), viewed from the top.
2.1. The electron transfer pathway
The reduction of the diiron(III) center is mediated by the reductase component(s). To understand the mechanism of electron transfer control, we first need to identify the reductase binding site on the hydroxylase and the electron transfer pathway. As the shortest entry from the protein exterior to the diiron center, the pore is the leading candidate for the reductase binding site.16
Recent studies of sMMO, T4mo, and ToMO all confirm this proposal. In the case of sMMO, the reductase binding site on the hydroxylase was determined by hydrogen-deuterium exchange mass spectrometry (HDX-MS).35 The data indicated that the ferredoxin domain of the reductase binds to a shallow region at the dimer interface of the hydroxylase, known as the “canyon”,17 covering the pore (Figure 1a). A computational docking study supported this conclusion (Figure 1c) and revealed that the gating residue of the pore, Glu240, lies nearly on a line between the [Fe2S2] cluster of the MMOR ferredoxin domain and the diiron center of MMOH (Figure 1d).35 The importance of the pore residues Asn202 and Gln228 in ToMO (Asn214 and Glu240 in sMMO) for reductase binding was demonstrated in a biochemical study of ToMO.32
Atomic details of the hydroxylase-reductase complex were revealed in an X-ray crystallographic investigation of T4MO.36 Consistent with our findings with sMMO35 and ToMO,32,37 the reductase (T4moC) covers the pore region in the complex (Figure 1e). The crystal structure further revealed a 12-Å distance between the [Fe2S2] cluster of the reductase and the diiron center of the hydroxylase, favorable for biological electron transfer.38 The two metal clusters are linked through a hydrogen-bonding network, which connects the [Fe2S2] cluster ligand His68 with the diiron center ligand Glu231, mediated by the pore residue Gln228 (corresponding to Glu240 in MMOH) (Figure 1f).36
2.2. Control of electron transfer: binding competition at the pore
An understanding of the mechanism by which the BMM proteins control the timing of electron transfer required information about interactions between the hydroxylase, reductase, and regulatory components. Two models were considered, a non-competitive model whereby the regulatory component and the reductase bind to different sites on the hydroxylase12 and a competitive model where they compete for the same binding site on the hydroxylase. Recent structural studies strongly favor the latter. X-ray crystallographic results for the hydroxylase-regulatory protein complex revealed that the regulatory component binds to the canyon, the same region where the reductase binds, covering the pore. This conserved binding mode occurs in sMMO (Figure 2a),31 T4mo (Figure 2b),29 and PH (Figure 2c).39
Figure 2.

Crystal structures of the hydroxylase-regulatory component complexes of a) sMMO (PDB ID 4GAM), b) T4mo (PDB ID 3DHI), and c) PH (PDB ID 2INP). The hydroxylases are shown in cyan, and regulatory components in magenta.
Multiple biochemical/biophysical characterization experiments further support the binding competition. In the case of sMMO, MMOB inhibits MMOR cross-linking to MMOH in a dose-dependent manner. The MMOR ferredoxin domain protein can displace MMOB from MMOH, as observed in a titration experiment by monitoring fluorescence anisotropy of MMOB carrying a fluorescent label.35 Binding competition between the regulatory component and the reductase was also reported for T4mo.36 Consistent with the competitive model, the regulatory component retards electron transfer from the reductase to the diiron center, an effect observed in sMMO 20,35,40 and ToMO.37
Binding competition between the reductase and the regulatory components at the pore region of the hydroxylase provides a basis for electron transfer control. By fine-tuning the binding affinity, the regulatory component could inhibit reductase binding during O2 activation, blocking undesired electron transfer to the diiron center. Indeed, the regulatory component exhibited increased binding affinity to the hydroxylase after reduction of the diiron center in sMMO isolated from Methylococcus capsulatus (Bath).41
2.3. Proton and water transfer through the pore
Oxygen activation requires protons.10,26,27 As the only hydrophilic entry to the diiron center, the pore provides the route for proton transfer. Biochemical study of ToMO indicated pore residue Thr201 to be critical for proton transfer during dioxygen activation.34 Kinetic isotope effects and pH profiles suggested that another pore residue, Gln228, mediates proton ingress to, and water egress from, the active site.32
Structural studies revealed the molecular mechanism of proton transfer. In the case of sMMO, crystal structures showed Glu240 to be the gating residue in the pore, playing a key role in proton transfer.31 In the absence of other component proteins, this residue is hydrogen bonded to a water or hydronium ion on the surface of the hydroxylase.31,42 In response to the binding of the regulatory component, the carboxylic side chain of Glu240 moves inward31 in a manner suggesting a role in delivering a proton to the active site for O2 activation and, in the process, closes down the pore (Figure 3a) to block undesired water/hydronium ion ingress that would quench reactive intermediates (Scheme 1).31 A similar conformational change may occur when the reductase binds to the hydroxylase. Thus Glu240 provides the basis for proton-coupled electron transfer.35
Figure 3.

Regulatory component induced conformational changes at the pore. a) A close-up view of the pore in the hydroxylase-regulatory component complex of sMMO (PDB ID 4GAM), where the pore is closed. b) A close-up view of pore residues and ligands of the diiron(II) center in the hydroxylase-regulatory component complex of T4mo (PDB ID 3DHI).
Structures of protein complexes of T4mo revealed additional details. As observed in sMMO, the regulatory component and the reductase can both induce inward movement of the pore gating residue Gln228.29,36 A water molecule not observed in the free hydroxylase appeared in the pore in the hydroxylase-regulatory component (Figure 3b) and hydroxylase-reductase (Figure 1f) complexes, providing a putative mechanism for proton transfer. In the case of the reduced hydroxylase-regulatory component complex, this water molecule (HOH5) is close to the open coordination site for O2 binding (Figure 3b), serving as an likely proton source for dioxygen activation.29 The crystal structures also revealed an important role of the conserved Thr201 in stabilizing the hydrogen bonding network at the active site,29 providing a structural basis for the importance of this residue in proton transfer and dioxygen activation.26,34
3. Control of O2 access
After reduction of the hydroxylase, O2 needs to be activated by the reduced diiron center for substrate hydroxylation (Scheme 1). How does O2 reach the buried diiron center, and how is the access controlled?
3.1. Cavities as the O2 ingress route
Hydrophobic gas molecules, including O2 and CH4, find their path to the diiron center through a series of cavities that extend over 36-40 Å through the protein interior. Multiple lines of evidence support this pathway. Structural characterization of Xe-pressurized sMMO and PH hydroxylases revealed xenon atoms, which are similar to O2 molecules in terms of size, hydrophobicity, and polarizability,43 localized in these cavities. Halogenated alkanes, substrate analogs for sMMO bearing heavy atoms, can also occupy these cavities.44 Mutagenesis of residues in each of the three hydrophobic cavities in ToMO altered the dioxygen diffusion rate in a manner that correlated with the size of the cavities.45 These cavities are observed in all crystallographically characterized BMM hydroxylases and are likely to be conserved across the superfamily.30
3.2. Control of O2 access by the regulatory component
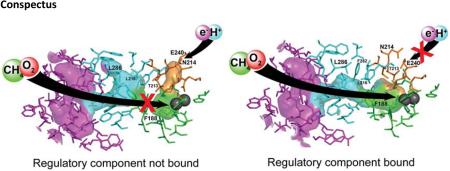
An interesting feature of these cavities was evident from the early studies. In several cases, cavities 1 and 2 are disconnected (Figure 4a),30 raising the question of how gas molecules could migrate through such a disconnected pathway. The crystal structure of the hydroxylase-regulatory component complex of sMMO provided critical insight.31 It revealed that, upon binding of the regulatory component MMOB, cavities 1 and 2 become connected (Figure 4b). MMOB residues Ser111 and Tyr8, at the interface with MMOH in the complex, trigger allosteric changes that alter the conformation of Phe188, the gating residue between the two cavities (Fig. 4).
Figure 4.

Substrate access through the cavities and the pore is triggered by binding of the regulatory component. a) In free hydroxylase of sMMO, cavities 1 and 2 are disconnected, and the pore is open (PDB ID 1MTY). Xe atoms were found in cavity 2 in MMOH crystals pressurized with Xe gas.44 b) When the regulatory component binds, the cavities are connected and the pore is closed (PDB ID 4GAM). The concerted motion of these two alterations is striking.
The crystal structure also explains the importance of the N-terminal tail of MMOB. The regulatory components of ToMO, T4mo, PH, and most other members of the BMM superfamily do not have such tail, but in the case of sMMO, enzyme activity is severely compromised when the N-terminal tail is truncated or even partially truncated.31,46,47 The N-terminal tail is disordered in free MMOB, as revealed by solution NMR spectral studies,48,49 but it adopts an unusual ring-like conformation when bound to the surface of MMOH (Fig. 2a).31 A key residue in this tail in sMMO is Tyr8, which triggers allosteric changes in MMOH that lead to the changes depicted in Figure 4, with assistance from residue Ser111, which resides in the core of the regulatory protein.
Another important finding is that the function of MMOB in facilitating O2 ingress depends on the oxidation state of the diiron center. In a study of the MMOH-MMOB complex in solution, the conformation of the MMOB N-terminal tail was assessed using nitroxide spin-labeled MMOB mutants by double electron-electron resonance spectroscopy.41 The data revealed that the N-terminal tail switches from a flexible to an ordered conformation in response to reduction of the diiron center from the diiron(III) to the diiron(II) state. The allosteric alterations observed crystallographically in the MMOH-MMOB complex are therefore not occurring for the diiron(III) form of the hydroxylase in solution, because Tyr8, a critical residue that induces allosteric changes in MMOH, is disordered and lacking a stable contact with the hydroxylase. Thus oxygen, and most likely the structurally similar hydrocarbon substrate methane, do not have access to the active site until the diiron center is reduced to diiron(II), when the N-terminal tail becomes ordered and Tyr8 forms a stable interaction with MMOH. Through such a mechanism, access of O2 and CH4 to the active site is coupled to reduction of the diiron center.
4. Hydrocarbon substrate ingress and product egress
The small hydrocarbon CH4 finds its route to the active site through cavities in sMMO. Some BMM enzymes, however, process much larger, and in some case polar, hydrocarbons. In addition, the hydroxylation products are all more hydrophilic than the original substrate. What are the pathways for trafficking these substrates and products to and from the active site?
4.1. ToMO/T4MO: through the channel and the cavities
A prominent feature of the four-component toluene monooxygenases ToMO and T4mo is a 6 – 10 Å-wide, 35 Å-long channel in their hydroxylase components (Figures 5a, b). It extends from the protein surface to the active site and merges with cavities 1 and 2 at their junction.28,29 The channel was proposed as the substrate and product migration pathway in toluene monooxygenases (TMOs), because the product analog bromophenol occupied the channel in a crystal structure of ToMO.28 The possibility that this channel might also serve as the O2 access pathway was unsupported by a mutagenesis study of ToMO, where variants with narrowed or widened channels did not affect the rate of O2 diffusion to the diiron center.45 The cavities, which exist in TMOs as the O2 migration pathway, can also provide aromatic substrates access to the active site.50,51 Additional studies indicated the cavities to be preferred as the substrate ingress pathway, whereas the more hydrophilic channel was the preferred product egress pathway.51
Figure 5.

Channels in TMO. a) The channel in free hydroxylase of ToMO (PDB ID 2INC) and a magnified view of the channel in the inset. b) The channel in free hydroxylase of T4mo (PDB ID 3DHG). c) The channel in the hydroxylase-regulatory component complex of T4mo (PDB ID 3DHH). Channel in one of the hydroxylase monomer are shown; b) and c) only show one of the hydroxylase monomer. The channel and pore are shown in green and orange, respectively.
The opening of the channel is also controlled by the regulatory component. Upon formation of the hydroxylase-regulatory component complex in T4mo, the channel collapses, making it inaccessible to small molecules (Figure 5c).29 This effect blocks solvent access to the active site during dioxygen activation, thereby increasing the coupling efficiency. Binding of the reductase to the hydroxylase leaves the channel open.36
4.2. PH: through the pore or the cavities?
Similar channels occur in the hydroxylase components of PH and sMMO as revealed by computational analysis, but they are more constricted and less likely to be functional.30 In the case of PH, the migration pathways for the phenolic substrate and product are unclear. One possibility is through the cavities, because halogenated alcohols of different sizes could be located by X-ray diffraction in the cavities of the sMMO hydroxylase co-crystals,52 but direct evidence for such is lacking in the case of PH. The pore is an alternative option, considering its hydrophilicity and the larger opening (6 Å diameter) than the pores that occur in other BMM enzymes.39
5. Similar function, different strategies: a comparison with cytochromes P450
The heme-containing cytochrome P450 monooxygenases catalyze the same overall reaction as BMM enzymes (eq 1), but are distinct in many respects. P450s are 40-55 kDa single-polypeptide proteins. Most of them do not require a regulatory component, also referred to as an effector protein, except for a few cases including P450cam, the reductase of which also serves as the effector.53 Moreover, P450s adopt very different tactical approach to controlling substrate access to the catalytic heme active site.
In the electron transfer step, delivery of the first electron to the heme iron center is gated by substrate binding, which prevents wasteful discharge of the reductant in the absence of substrate.1,2,4 Such a gating effect is achieved by displacing a water ligand from the heme iron upon substrate binding; as a result, the heme iron switches from low-spin to high-spin with a concomitant increase in the redox potential, which favors reduction.54-57 This control mechanism is not observed in BMM enzymes. In the subsequent O2 activation step for P450s, an Fe(IV)=O species known as Compound I2,58 forms by oxygenation of the reduced heme site and subsequent e−/2H+ transfer. Quenching of Compound I (eq 2) by further electron transfer is disfavored over substrate oxidation as a result of the close proximity of substrate to this oxidizing species.2,59
Control of proton and water access to the active site is also important for P450s. Without proper control, the peroxo intermediate that precedes formation of Compound I is quenched, 2 releasing H2O2 (eq 3). The hydrophobicity of the active site2,59 as well as the conserved alcohol-acid pair, residues Thr252 and Asp251 in P450cam, 2,4,60 are essential for proper proton transfer. In addition, substrate binding expels undesired water molecules from the active site61 and, in many cases, induces a closed conformation that shields the active site from exposure to exterior solvent. This conformational change has been observed in P450cam61-63 and in P450s processing specific substrates,64-67 but may not be relevant to P450s processing more diverse substrates.68 Such global conformational dynamics are also critical for substrate specificity.62 In contrast, BMM enzymes use the regulatory component to block undesired proton and water access during O2 activation, by closing the pore as well as the channel in the case of TMO. In addition, the hydroxylases of BMM enzymes do not undergo any global conformational changes.
6. Conclusions
BMM enzyme systems form dynamic complexes among their protein components to couple reactions of multiple substrates by timely control of their access to the active site. To achieve such control, different reactants are routed through different pathways in the hydroxylase. Electrons and protons traverse the pore; O2 diffuses through the cavities; and hydrocarbon substrates use either the cavities or a special channel. The regulatory component gates access to these pathways by reversible binding to the canyon region, which includes the pore, in a manner that can be regulated by the oxidation state of the diiron center. In a catalytic cycle, the reductase transmits two electrons to the diiron(III) center by docking into the canyon. Next, the regulatory component displaces the reductase and inhibits excess electron transfer during O2 activation by physically occluding the reductase. Through binding to the sMMO hydroxylase, the regulatory component restricts water and proton access by closing the pore, and, in the case of T4mo, it collapses a channel accessed by the aromatic substrates. Within all BMM hydroxylases, allosteric changes induced by binding of the regulatory proteins connect the cavities, facilitating O2 (and CH4 in the case of sMMO) access to the diiron center. At the conclusion of a full catalytic cycle, the enzyme returns to the diiron(III) state, which lowers the binding affinity of the regulatory component to the hydroxylase. The reductase is then able to compete with the regulatory component for binding to the canyon, reduce the diiron center, and initiate the next round of catalysis.
Although a unified mechanism is emerging, there are some important outstanding issues. For instance, to fully support the proposed mechanism of electron transfer control, it would be valuable to have direct evidence that activated oxygen species can be quenched in the absence of the regulatory component or, by excess reductase that could displace the regulatory component. In addition, each BMM family member has uniquely characterstic properties. An example is the unique ability of sMMO to oxidize methane, which may reflect the apparent inability to generate intermediate Q in any other enzyme. This property may derive from an as yet unknown distinctive strategy involving control of substrate access to the diiron center.
The present understanding of these control mechanisms has the potential to guide the engineering of improved biocatalysts for hydrocarbon oxidation using atmospheric oxygen. It also poses a great challenge for synthetic modeling of enzymes – how do we achieve substrate access control in synthetic models, something that nature accomplishes with the invention of large and complex proteins and protein complexes?
ACKNOWLEDGEMENTS
This work was generously supported by NIH Grant GM032134 from the National Institute of General Medical Sciences.
Biography
Stephen J. Lippard is the Arthur Amos Noyes Professor of Chemistry at the Massachusetts Institute of Technology. His research areas lie at the interface of inorganic chemistry and biology.
Weixue Wang received his PhD in Biophysics and Computational Biology from the University of Illinois at Urbana-Champaign under the direction of Eric Oldfield. He was a postdoctoral associate with Stephen J. Lippard at the Massachusetts Institute of Technology. This year he assumed a position at the Janssen Pharmaceutical Companies of Johnson & Johnson.
Alexandria Deliz Liang received her Bachelor of Arts in chemistry from New College of Florida in 2010, where she worked in the laboratory of Suzanne E. Sherman. In 2010, Alexandria matriculated to the Massachusetts Institute of technology, where she recently completed her PhD, investigating the reactivity of toluene/o-xylene monooxygenase in the laboratory of Professor Lippard.
Footnotes
The authors declare no competing financial interest.
Reference
- 1.Sono M, Roach MP, Coulter ED, Dawson JH. Heme-Containing Oxygenases. Chem. Rev. 1996;96:2841–2888. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- 2.Denisov IG, Makris TM, Sligar SG, Schlichting I. Structure and chemistry of cytochrome P450. Chem. Rev. 2005;105:2253–2277. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- 3.Shaik S, Kumar D, de Visser SP, Altun A, Thiel W. Theoretical perspective on the structure and mechanism of cytochrome P450 enzymes. Chem. Rev. 2005;105:2279–2328. doi: 10.1021/cr030722j. [DOI] [PubMed] [Google Scholar]
- 4.Poulos TL. Heme enzyme structure and function. Chem. Rev. 2014;114:3919–3962. doi: 10.1021/cr400415k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Balasubramanian R, Rosenzweig AC. Structural and mechanistic insights into methane oxidation by particulate methane monooxygenase. Acc. Chem. Res. 2007;40:573–580. doi: 10.1021/ar700004s. [DOI] [PubMed] [Google Scholar]
- 6.Balasubramanian R, Smith SM, Rawat S, Yatsunyk LA, Stemmler TL, Rosenzweig AC. Oxidation of methane by a biological dicopper centre. Nature. 2010;465:115–119. doi: 10.1038/nature08992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wallar BJ, Lipscomb JD. Dioxygen Activation by Enzymes Containing Binuclear Non-Heme Iron Clusters. Chem. Rev. 1996;96:2625–2658. doi: 10.1021/cr9500489. [DOI] [PubMed] [Google Scholar]
- 8.Merkx M, Kopp DA, Sazinsky MH, Blazyk JL, Müller J, Lippard SJ. Dioxygen Activation and Methane Hydroxylation by Soluble Methane Monooxygenase: A Tale of Two Irons and Three Proteins. Angew. Chem. Int. Ed. 2001;40:2782–2807. doi: 10.1002/1521-3773(20010803)40:15<2782::AID-ANIE2782>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 9.Murray LJ, Lippard SJ. Substrate Trafficking and Dioxygen Activation in Bacterial Multicomponent Monooxygenases. Acc. Chem. Res. 2007;40:466–474. doi: 10.1021/ar600040e. [DOI] [PubMed] [Google Scholar]
- 10.Tinberg CE, Lippard SJ. Dioxygen activation in soluble methane monooxygenase. Acc. Chem. Res. 2011;44:280–288. doi: 10.1021/ar1001473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Green J, Dalton H. Protein B of soluble methane monooxygenase from Methylococcus capsulatus (Bath). A novel regulatory protein of enzyme activity. J. Biol. Chem. 1985;260:15795–15801. [PubMed] [Google Scholar]
- 12.Gassner GT, Lippard SJ. Component interactions in the soluble methane monooxygenase system from Methylococcus capsulatus (Bath). Biochemistry. 1999;38:12768–12785. doi: 10.1021/bi990841m. [DOI] [PubMed] [Google Scholar]
- 13.Hanson RS, Hanson TE. Methanotrophic bacteria. Microbiol. Rev. 1996;60:439–471. doi: 10.1128/mr.60.2.439-471.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chauhan S, Barbieri P, Wood TK. Oxidation of trichloroethylene, 1,1-dichloroethylene, and chloroform by toluene/o-xylene monooxygenase from Pseudomonas stutzeri OX1. Appl. Environ. Microbiol. 1998;64:3023–3024. doi: 10.1128/aem.64.8.3023-3024.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sullivan JP, Dickinson D, Chase HA. Methanotrophs, Methylosinus trichosporium OB3b, sMMO, and their application to bioremediation. Crit. Rev. Microbiol. 1998;24:335–373. doi: 10.1080/10408419891294217. [DOI] [PubMed] [Google Scholar]
- 16.Sazinsky MH, Lippard SJ. Correlating Structure with Function in Bacterial Multicomponent Monooxygenases and Related Diiron Proteins. Acc. Chem. Res. 2006;39:558–566. doi: 10.1021/ar030204v. [DOI] [PubMed] [Google Scholar]
- 17.Rosenzweig AC, Frederick CA, Lippard SJ, Nordlund P. Crystal structure of a bacterial non-haem iron hydroxylase that catalyses the biological oxidation of methane. Nature. 1993;366:537–543. doi: 10.1038/366537a0. [DOI] [PubMed] [Google Scholar]
- 18.Elango NA, Radhakrishnan R, Froland WA, Wallar BJ, Earhart CA, Lipscomb JD, Ohlendorf DH. Crystal structure of the hydroxylase component of methane monooxygenase from Methylosinus trichosporium OB3b. Protein Sci. 1997;6:556–568. doi: 10.1002/pro.5560060305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lund J, Dalton H. Further characterisation of the FAD and Fe2S2 redox centres of component C, the NADH:acceptor reductase of the soluble methane monooxygenase of Methylococcus capsulatus (Bath). Eur. J. Biochem. 1985;147:291–296. doi: 10.1111/j.1432-1033.1985.tb08749.x. [DOI] [PubMed] [Google Scholar]
- 20.Lund J, Woodland MP, Dalton H. Electron transfer reactions in the soluble methane monooxygenase of Methylococcus capsulatus (Bath). Eur. J. Biochem. 1985;147:297–305. doi: 10.1111/j.1432-1033.1985.tb08750.x. [DOI] [PubMed] [Google Scholar]
- 21.Liu Y, Nesheim JC, Paulsen KE, Stankovich MT, Lipscomb JD. Roles of the methane monooxygenase reductase component in the regulation of catalysis. Biochemistry. 1997;36:5223–5233. doi: 10.1021/bi962743w. [DOI] [PubMed] [Google Scholar]
- 22.Kopp DA, Gassner GT, Blazyk JL, Lippard SJ. Electron-transfer reactions of the reductase component of soluble methane monooxygenase from Methylococcus capsulatus (Bath). Biochemistry. 2001;40:14932–14941. doi: 10.1021/bi015556t. [DOI] [PubMed] [Google Scholar]
- 23.Liu Y, Nesheim JC, Lee SK, Lipscomb JD. Gating effects of component B on oxygen activation by the methane monooxygenase hydroxylase component. J. Biol. Chem. 1995;270:24662–24665. doi: 10.1074/jbc.270.42.24662. [DOI] [PubMed] [Google Scholar]
- 24.Cafaro V, Scognamiglio R, Viggiani A, Izzo V, Passaro I, Notomista E, Piaz FD, Amoresano A, Casbarra A, Pucci P, Di Donato A. Expression and purification of the recombinant subunits of toluene/o-xylene monooxygenase and reconstitution of the active complex. Eur. J. Biochem. 2002;269:5689–5699. doi: 10.1046/j.1432-1033.2002.03281.x. [DOI] [PubMed] [Google Scholar]
- 25.Pikus JD, Studts JM, Achim C, Kauffmann KE, Munck E, Steffan RJ, McClay K, Fox BG. Recombinant toluene-4-monooxygenase: catalytic and Mossbauer studies of the purified diiron and rieske components of a four-protein complex. Biochemistry. 1996;35:9106–9119. doi: 10.1021/bi960456m. [DOI] [PubMed] [Google Scholar]
- 26.Lee SK, Lipscomb JD. Oxygen activation catalyzed by methane monooxygenase hydroxylase component: proton delivery during the O-O bond cleavage steps. Biochemistry. 1999;38:4423–4432. doi: 10.1021/bi982712w. [DOI] [PubMed] [Google Scholar]
- 27.Tinberg CE, Lippard SJ. Revisiting the mechanism of dioxygen activation in soluble methane monooxygenase from M. capsulatus (Bath): evidence for a multi-step, proton-dependent reaction pathway. Biochemistry. 2009;48:12145–12158. doi: 10.1021/bi901672n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sazinsky MH, Bard J, Di Donato A, Lippard SJ. Crystal Structure of the Toluene/o-Xylene Monooxygenase Hydroxylase from Pseudomonas stutzeri OX1: insight into the substrate specificity, substrate channeling, and active site tuning of multicomponent monooxygenases. J. Biol. Chem. 2004;279:30600–30610. doi: 10.1074/jbc.M400710200. [DOI] [PubMed] [Google Scholar]
- 29.Bailey LJ, McCoy JG, Phillips GN, Fox BG. Structural consequences of effector protein complex formation in a diiron hydroxylase. Proc. Natl. Acad. Sci. U. S. A. 2008;105:19194–19198. doi: 10.1073/pnas.0807948105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCormick MS, Lippard SJ. Analysis of Substrate Access to Active Sites in Bacterial Multicomponent Monooxygenase Hydroxylases: X-ray Crystal Structure of Xenon-Pressurized Phenol Hydroxylase from Pseudomonas sp. OX1. Biochemistry. 2011;50:11058–11069. doi: 10.1021/bi201248b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee SJ, McCormick MS, Lippard SJ, Cho US. Control of substrate access to the active site in methane monooxygenase. Nature. 2013;494:380–384. doi: 10.1038/nature11880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liang AD, Wrobel AT, Lippard SJ. A flexible glutamine regulates the catalytic activity of toluene o-xylene monooxygenase. Biochemistry. 2014;53:3585–3592. doi: 10.1021/bi500387y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Elsen NL, Bailey LJ, Hauser AD, Fox BG. Role for threonine 201 in the catalytic cycle of the soluble diiron hydroxylase toluene 4-monooxygenase. Biochemistry. 2009;48:3838–3846. doi: 10.1021/bi900144a. [DOI] [PubMed] [Google Scholar]
- 34.Song WJ, McCormick MS, Behan RK, Sazinsky MH, Jiang W, Lin J, Krebs C, Lippard SJ. Active site threonine facilitates proton transfer during dioxygen activation at the diiron center of toluene/o-xylene monooxygenase hydroxylase. J. Am. Chem. Soc. 2010;132:13582–13585. doi: 10.1021/ja1063795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang W, Iacob RE, Luoh RP, Engen JR, Lippard SJ. Electron transfer control in soluble methane monooxygenase. J. Am. Chem. Soc. 2014;136:9754–9762. doi: 10.1021/ja504688z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Acheson JF, Bailey LJ, Elsen NL, Fox BG. Structural basis for biomolecular recognition in overlapping binding sites in a diiron enzyme system. Nat. Commun. 2014;5:5009. doi: 10.1038/ncomms6009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liang AD, Lippard SJ. Component interactions and electron transfer in toluene/o-xylene monooxygenase. Biochemistry. 2014;53:7368–7375. doi: 10.1021/bi500892n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Page CC, Moser CC, Chen X, Dutton PL. Natural engineering principles of electron tunnelling in biological oxidation-reduction. Nature. 1999;402:47–52. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- 39.Sazinsky MH, Dunten PW, McCormick MS, DiDonato A, Lippard SJ. X-ray Structure of a Hydroxylase–Regulatory Protein Complex from a Hydrocarbon-Oxidizing Multicomponent Monooxygenase, Pseudomonas sp. OX1 Phenol Hydroxylase. Biochemistry. 2006;45:15392–15404. doi: 10.1021/bi0618969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blazyk JL, Gassner GT, Lippard SJ. Intermolecular electron-transfer reactions in soluble methane monooxygenase: a role for hysteresis in protein function. J. Am. Chem. Soc. 2005;127:17364–17376. doi: 10.1021/ja0554054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang W, Lippard SJ. Diiron Oxidation State Control of Substrate Access to the Active Site of Soluble Methane Monooxygenase Mediated by the Regulatory Component. J. Am. Chem. Soc. 2014;136:2244. doi: 10.1021/ja412351b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rosenzweig AC, Brandstetter H, Whittington DA, Nordlund P, Lippard SJ, Frederick CA. Crystal structures of the methane monooxygenase hydroxylase from Methylococcus capsulatus (Bath): Implications for substrate gating and component interactions. Proteins. 1997;29:141–152. [PubMed] [Google Scholar]
- 43.Schoenborn BP, Watson HC, Kendrew JC. Binding of xenon to sperm whale myoglobin. Nature. 1965;207:28–30. doi: 10.1038/207028a0. [DOI] [PubMed] [Google Scholar]
- 44.Whittington DA, Rosenzweig AC, Frederick CA, Lippard SJ. Xenon and halogenated alkanes track putative substrate binding cavities in the soluble methane monooxygenase hydroxylase. Biochemistry. 2001;40:3476–3482. doi: 10.1021/bi0022487. [DOI] [PubMed] [Google Scholar]
- 45.Song WJ, Gucinski G, Sazinsky MH, Lippard SJ. Tracking a defined route for O2 migration in a dioxygen-activating diiron enzyme. Proc. Natl. Acad. Sci. U. S. A. 2011;108:14795–14800. doi: 10.1073/pnas.1106514108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brandstetter H, Whittington DA, Lippard SJ, Frederick CA. Mutational and structural analyses of the regulatory protein B of soluble methane monooxygenase from Methylococcus capsulatus (Bath). Chem. Biol. 1999;6:441–449. doi: 10.1016/s1074-5521(99)80062-3. [DOI] [PubMed] [Google Scholar]
- 47.Chang SL, Wallar BJ, Lipscomb JD, Mayo KH. Residues in Methylosinus trichosporium OB3b methane monooxygenase component B involved in molecular interactions with reduced- and oxidized-hydroxylase component: a role for the N-terminus. Biochemistry. 2001;40:9539–9551. doi: 10.1021/bi0103462. [DOI] [PubMed] [Google Scholar]
- 48.Chang SL, Wallar BJ, Lipscomb JD, Mayo KH. Solution structure of component B from methane monooxygenase derived through heteronuclear NMR and molecular modeling. Biochemistry. 1999;38:5799–5812. doi: 10.1021/bi982992f. [DOI] [PubMed] [Google Scholar]
- 49.Walters KJ, Gassner GT, Lippard SJ, Wagner G. Structure of the soluble methane monooxygenase regulatory protein B. Proc. Natl. Acad. Sci. U. S. A. 1999;96:7877–7882. doi: 10.1073/pnas.96.14.7877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brouk M, Fishman A. Protein engineering of toluene monooxygenases for synthesis of hydroxytyrosol. Food Chem. 2009;116:114. [Google Scholar]
- 51.Hosseini A, Brouk M, Lucas MF, Glaser F, Fishman A, Guallar V. Atomic picture of ligand migration in toluene 4-monooxygenase. J. Phys. Chem. B. 2015;119:671–678. doi: 10.1021/jp502509a. [DOI] [PubMed] [Google Scholar]
- 52.Sazinsky MH, Lippard SJ. Product bound structures of the soluble methane monooxygenase hydroxylase from Methylococcus capsulatus (Bath): protein motion in the alpha-subunit. J. Am. Chem. Soc. 2005;127:5814–5825. doi: 10.1021/ja044099b. [DOI] [PubMed] [Google Scholar]
- 53.Tyson CA, Lipscomb JD, Gunsalus IC. The Roles of Putidaredoxin and P450cam in Methylene Hydroxylation. J. Biol. Chem. 1972;247:5777–5784. [PubMed] [Google Scholar]
- 54.Sligar SG. Coupling of spin, substrate, and redox equilibriums in cytochrome P450. Biochemistry. 1976;15:5399–5406. doi: 10.1021/bi00669a029. [DOI] [PubMed] [Google Scholar]
- 55.Raag R, Poulos TL. The structural basis for substrate-induced changes in redox potential and spin equilibrium in cytochrome P-450CAM. Biochemistry. 1989;28:917–922. doi: 10.1021/bi00428a077. [DOI] [PubMed] [Google Scholar]
- 56.Haines DC, Tomchick DR, Machius M, Peterson JA. Pivotal role of water in the mechanism of P450BM-3. Biochemistry. 2001;40:13456–13465. doi: 10.1021/bi011197q. [DOI] [PubMed] [Google Scholar]
- 57.Pylypenko O, Schlichting I. Structural aspects of ligand binding to and electron transfer in bacterial and fungal P450s. Annu. Rev. Biochem. 2004;73:991–1018. doi: 10.1146/annurev.biochem.73.011303.073711. [DOI] [PubMed] [Google Scholar]
- 58.Rittle J, Green MT. Cytochrome P450 compound I: capture, characterization, and C-H bond activation kinetics. Science. 2010;330:933–937. doi: 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]
- 59.Loida PJ, Sligar SG. Molecular recognition in cytochrome P-450: mechanism for the control of uncoupling reactions. Biochemistry. 1993;32:11530–11538. doi: 10.1021/bi00094a009. [DOI] [PubMed] [Google Scholar]
- 60.Schlichting I, Berendzen J, Chu K, Stock AM, Maves SA, Benson DE, Sweet RM, Ringe D, Petsko GA, Sligar SG. The catalytic pathway of cytochrome p450cam at atomic resolution. Science. 2000;287:1615–1622. doi: 10.1126/science.287.5458.1615. [DOI] [PubMed] [Google Scholar]
- 61.Lee YT, Wilson RF, Rupniewski I, Goodin DB. P450cam visits an open conformation in the absence of substrate. Biochemistry. 2010;49:3412–3419. doi: 10.1021/bi100183g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee YT, Glazer EC, Wilson RF, Stout CD, Goodin DB. Three clusters of conformational states in p450cam reveal a multistep pathway for closing of the substrate access channel. Biochemistry. 2011;50:693–703. doi: 10.1021/bi101726d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stoll S, Lee YT, Zhang M, Wilson RF, Britt RD, Goodin DB. Double electron-electron resonance shows cytochrome P450cam undergoes a conformational change in solution upon binding substrate. Proc. Natl. Acad. Sci. U. S. A. 2012;109:12888–12893. doi: 10.1073/pnas.1207123109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhao B, Guengerich FP, Bellamine A, Lamb DC, Izumikawa M, Lei L, Podust LM, Sundaramoorthy M, Kalaitzis JA, Reddy LM, Kelly SL, Moore BS, Stec D, Voehler M, Falck JR, Shimada T, Waterman MR. Binding of two flaviolin substrate molecules, oxidative coupling, and crystal structure of Streptomyces coelicolor A3(2) cytochrome P450 158A2. J. Biol. Chem. 2005;280:11599–11607. doi: 10.1074/jbc.M410933200. [DOI] [PubMed] [Google Scholar]
- 65.Sherman DH, Li S, Yermalitskaya LV, Kim Y, Smith JA, Waterman MR, Podust LM. The structural basis for substrate anchoring, active site selectivity, and product formation by P450 PikC from Streptomyces venezuelae. J. Biol. Chem. 2006;281:26289–26297. doi: 10.1074/jbc.M605478200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Muralidhara BK, Sun L, Negi S, Halpert JR. Thermodynamic fidelity of the mammalian cytochrome P450 2B4 active site in binding substrates and inhibitors. J. Mol. Biol. 2008;377:232–245. doi: 10.1016/j.jmb.2007.12.068. [DOI] [PubMed] [Google Scholar]
- 67.Savino C, Montemiglio LC, Sciara G, Miele AE, Kendrew SG, Jemth P, Gianni S, Vallone B. Investigating the structural plasticity of a cytochrome P450: three-dimensional structures of P450 EryK and binding to its physiological substrate. J. Biol. Chem. 2009;284:29170–29179. doi: 10.1074/jbc.M109.003590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Williams PA, Cosme J, Vinkovic DM, Ward A, Angove HC, Day PJ, Vonrhein C, Tickle IJ, Jhoti H. Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone. Science. 2004;305:683–686. doi: 10.1126/science.1099736. [DOI] [PubMed] [Google Scholar]