

Abstract
Background
Healthcare organizations, compendia, and drug knowledgebase vendors use varying methods to evaluate and synthesize evidence on drug-drug interactions (DDIs). This situation has a negative effect on electronic prescribing and medication information systems that warn clinicians of potentially harmful medication combinations.
Objective
To provide recommendations for systematic evaluation of evidence from the scientific literature, drug product labeling, and regulatory documents with respect to DDIs for clinical decision support.
Methods
A conference series was conducted to develop a structured process to improve the quality of DDI alerting systems. Three expert workgroups were assembled to address the goals of the conference. The Evidence Workgroup consisted of 15 individuals with expertise in pharmacology, drug information, biomedical informatics, and clinical decision support. Workgroup members met via webinar from January 2013 to February 2014. Two in-person meetings were conducted in May and September 2013 to reach consensus on recommendations.
Results
We developed expert-consensus answers to three key questions: 1) What is the best approach to evaluate DDI evidence?; 2) What evidence is required for a DDI to be applicable to an entire class of drugs?; and 3) How should a structured evaluation process be vetted and validated?
Conclusion
Evidence-based decision support for DDIs requires consistent application of transparent and systematic methods to evaluate the evidence. Drug information systems that implement these recommendations should be able to provide higher quality information about DDIs in drug compendia and clinical decision support tools.
1 Background
Exposure to potential drug-drug interactions (DDIs) is a significant source of preventable drug-related harm that requires proper management to avoid medical errors [1]. Studies indicate DDIs harm 1.9 to 5 million inpatients per year and cause 2,600 to 220,000 emergency department visits per year [2–4].
The importance of DDIs as a risk factor for patient harm led the Centers for Medicare and Medicaid Services (CMS) to include DDI clinical decision support (CDS) alerts in the agency’s guidelines for achieving meaningful use of electronic health records (i.e., CMS Meaningful Use Core Measure 2) [5]. However, evidence indicates that DDI decision support systems have not successfully reduced exposure to DDIs [6–8]. In the United States, most alerting systems rely on clinical content created, maintained, and sold by knowledgebase vendors [9]. Each organization implements their own approach to classifying DDIs with limited agreement between systems [10–12]. Additionally, CDS systems often alert for DDIs that have limited clinical relevance, which may increase alert fatigue [13] and lead to inappropriate responses [14–16].
In spite of a desire among providers of DDI decision support tools to provide clinically relevant content, improving the state-of-the art poses several challenges. High quality evidence to support the existence of many DDIs is lacking, there are few controlled clinical studies conducted in relevant populations [17–19], and individual case reports are underreported and often lack information [20]. Compendia and knowledgebase editors use differing approaches to identify and evaluate evidence [10–12]. There are no guidelines or standards for determining clinical relevance of interactions via consistent systematic evaluation or classification [9, 21]. Without such guidance, DDIs may also be inappropriately extrapolated to other drugs within the same therapeutic or pharmacologic class [22]. In an effort to reduce legal liability, system vendors might have an incentive to include almost all possible DDIs, including those that confer extremely low risk to exposed patients [9, 23].
We conducted a conference series to develop specific recommendations to improve the quality of CDS alerts for DDIs. These activities were supported in part by a conference grant from the Agency for Healthcare Research and Quality (AHRQ) and donations from health information technology (IT) vendors. Use of funds was at the sole discretion of the University of Arizona and according to Department of Health and Human Services requirements. This paper describes recommendations by the Evidence Workgroup to develop and maintain a standard set of DDIs for CDS alerts.
2 Methods
Fifteen individuals with expertise in DDIs, clinical pharmacology, drug information, evidence evaluation, biomedical informatics, and health IT were invited to participate as workgroup panelists. Members represented diverse backgrounds such as academia; journal, compendia, and knowledgebase editors; healthcare organizations; US Food and Drug Administration (FDA); and the US Office of the National Coordinator for Health IT (ONC). We met via webinar from January 2013 to February 2014, with live meetings held in Washington DC (May 2013) and Phoenix, Arizona (September 2013). To ensure that we addressed the most pressing issues we first conducted a search of the literature for papers describing methods for evaluating DDI evidence. From these articles the following questions were then developed by the conference organizers and then reviewed and agreed upon by consensus of the members: (1) What is the best approach to evaluate DDI evidence? (2) What evidence is required for a DDI to be applicable to an entire class of drugs? (3) How should a structured evaluation process be vetted and validated?
Workgroup members were provided access to articles that were deemed relevant for consideration. Experts also identified relevant studies and these were obtained for workgroup members. Each key question was evaluated in light of the available evidence and the collective experience of the workgroup members. Responses to each key question were written and then modified to improve clarity or address issues or concerns. Workgroup recommendations were posted on a project Internet site and feedback was sought from other stakeholders via dissemination to professional societies and organizations.
3 Results
A summary of our recommendations when evaluating the DDI evidence is shown in Table 1. Details about these recommendations are described more fully below.
Table 1.
Summary of Evidence Workgroup Recommendations for Systematic Evaluation of DDI Evidence
| Recommendation | Comments |
|---|---|
| Recommendations to be Adopted in the Short Term | |
| Apply consistent terminology |
|
| Apply DIPS for evaluating DDI case reports [20] |
|
| A new approach for evaluating a body of evidence |
|
| Evaluate statements/evidence in FDA documents and product labeling by same criteria as published evidence |
|
| Classify DDIs by therapeutic/pharmacologic class only when the evidence applies, or can be reasonably extrapolated, to the entire class of drugs |
|
| Recommendations for Future Work | |
| Evaluate DRIVE Instrument | |
| Develop systematic DDI search criteria |
|
| Identify or develop a system to evaluate and communicate the strength of DDI evidence with graded recommendations [49] |
AHRQ = Agency for Healthcare Research and Quality; CDS = clinical decision support; DDI = drug-drug interaction; DIPS = Drug Interaction Probability Scale; DRIVE = DRug Interaction eVidence Evaluation (DRIVE); FDA = Food and Drug Administration; GRADE = Grading of Recommendations, Assessment, Development, and Evaluation; PD = pharmacodynamics; PK = pharmacokinetic; WHO = World Health Organization
3.1 Terminology
We recommend consistent use of relevant terminology for evaluation of DDI evidence. In the process of answering the Key Questions, we identified several terms requiring clarification. A complete list of definitions agreed upon by the workgroup is provided in Appendix A, with some key terms described below.
A DDI is defined a clinically meaningful alteration in the exposure and/or response to a drug (object drug) that has occurred as a result of the co-administration of another drug (precipitant drug) [24, 25]. Response can refer to either precipitating an adverse event or altering the therapeutic effect of the object drug. A potential DDI is defined as the co-prescription of two drugs known to interact, and therefore a DDI could occur in the exposed patient [25]. Although the distinction between a DDI and a potential DDI is important, we refer to both as DDI throughout this paper for simplicity. A clinically relevant DDI is defined as one associated with either toxicity or loss of efficacy that warrants the attention of healthcare professionals. We recommend use of the term seriousness, rather than severity, to describe the extent to which a DDI can or does cause harm [26].
We developed a working definition for narrow therapeutic index (NTI) because many clinically relevant DDIs involve drugs with a NTI. Similar terms include narrow therapeutic ratio and narrow therapeutic range (NTR). We considered existing NTI/NTR definitions (See Appendix B) inadequate for guiding the evaluation of DDIs [27]. Although the FDA is developing a definition and list of NTI drugs in the context of bioequivalence, these guidelines/definitions would generally be stricter than is needed for managing DDIs in clinical practice. Therefore, we define NTI drugs as those for which even a small change in drug exposure may lead to toxicity or loss of efficacy. Several scenarios describe what may constitute a “small” change in drug levels: 1) <100% (<2-fold) increase in area under the concentration-time curve (AUC) for the object drug may lead to serious adverse events; or <50% decrease in AUC for the object drug may result in a loss of efficacy with serious therapeutic consequences (e.g., failure of contraception, or virologic failure due to subtherapeutic drug levels).
3.2 Key Question 1: What is the Best Approach to Evaluate DDI Evidence?
Figure 1 illustrates a conceptual model for evaluating DDIs to guide clinical decision making. The first step relates to establishing sufficient evidence that a DDI exists, followed by questions of clinical relevance and how to present DDI recommendations to health professionals. We primarily focused on identifying the best approach to evaluate that a DDI exists, with additional consideration for how to establish clinical relevance.
Figure 1.
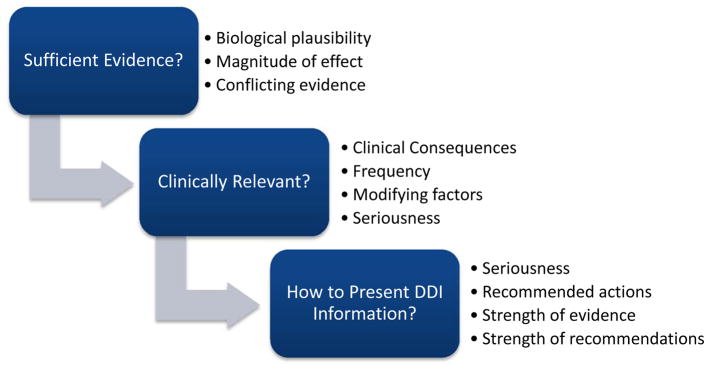
Conceptual Model for Evaluating Drug-Drug Interactions to Guide Clinical Decision-Making
When publishing a recommendation about the risk of a DDI, it is essential first to assess the quality of individual studies to prevent drawing erroneous conclusions about the entire body of evidence. Evaluation of medical treatments commonly involves hierarchical rating schemes such as those used in evidence-based medicine. However, a unique approach is needed to summarize a body of DDI evidence, which often consists of case reports, retrospective reviews, and extrapolation from in vitro studies, with few controlled clinical studies conducted in relevant populations [17, 19, 28]. Some DDIs do not require randomized controlled trials to confirm their existence. It is possible to reasonably extrapolate many interactions based on pharmacokinetic (PK) and/or pharmacodynamic (PD) properties of a drug without placing patients at unnecessary or unethical risk. High-quality observational studies and evidence from real-world use can be applied to confirm the association with adverse clinical outcomes and to evaluate the magnitude of harm and relevant risk factors. Therefore, evidence supporting a DDI may be derived from what would be regarded as less rigorous study designs for other research questions.
3.2.1 Existing DDI Evidence Evaluation Methods
We conducted a search for published methods for evaluating DDI case reports using Medline and also queried panel members for relevant studies. Only one instrument, the Drug Interaction Probability Scale (DIPS) [20], was found to be specifically developed to evaluate individual case reports for DDIs. This 10-item scale was designed to assess an adverse event for causality by a DDI. DIPS was developed to address the limitations of previous assessment instruments, such as the Naranjo scale [29], that failed to evaluate causality associated with concomitant medications. DIPS takes into consideration previous credible reports, consistency with known interactive properties, time course of the interaction, results of de-challenge and re-challenge, and alternative explanations. DIPS also meets published criteria for assessing causality in terms of guiding users to conduct an explicit, transparent, complete, and balanced assessment of the attributes important to causality assessment of whether an adverse drug interaction occurred and exists [30]. A more complete discussion of available instruments is available in Appendix C.
We also searched for published literature of methods that evaluate a collection of evidence relevant to establishing that a DDI exists (see Appendix C) and found two systematic approaches [31, 32]. The first is the approach for developing a DDI knowledgebase in Swedish and Finnish computerized CDS systems (SFINX) [31]. This system categorizes level of documentation (0–4) and clinical relevance (A–D). A “0” level of documentation reflects potentially dangerous interactions that have not been, and probably never will be, documented in clinical studies. The second approach described in the literature is a systematic assessment of DDIs for CDS systems in the Netherlands [32]. Four core parameters are used to assess each DDI: evidence supporting the interaction; clinical relevance of the potential adverse reaction; risk factors related to patient, drug, or disease characteristics; and incidence of the adverse reaction. A 5-item scale is used to assess the quality of evidence for a DDI. The approach requires the existence of a reasonable mechanistic explanation in order to establish a DDI based solely on PK or PD properties.
While the DIPS approach to case report evaluation was considered acceptable, we considered the existing methods for evaluating a body of evidence more complex than necessary because they combine DDI evidence assessment with questions of clinical relevance. Additionally, existing methods do not explicitly address reasonable extrapolation of DDIs from in vitro findings, nor do they explicitly address study quality and interpretation in the context of DDIs.
3.2.2 The Need for a New DDI Evidence Evaluation Instrument
Given the limitations of the available tools, we agreed that a new assessment instrument was needed to objectively evaluate a body of evidence to establish the existence of a DDI. It was further agreed that this instrument should include concepts from previously published DDI evidence rating instruments [31, 32] but with fewer categories based on the presence or absence of specific, clearly defined, types of evidence.
Specific guidance is needed for reasonably extrapolating DDIs that are unlikely to be evaluated in clinical trials. Reasonable extrapolation refers to using the knowledge of the mechanism of a DDI to predict the risk of a DDI from one pair of drugs to multiple pairs with similar pharmacologic properties. Extrapolation of PD interactions is commonplace. For example, not every possible combination of a benzodiazepine and ethanol has been studied. Yet, all benzodiazepines are assumed to interact in a similar manner with ethanol. DDIs based on PK mechanisms present more of a challenge to extrapolation because numerous elimination pathways may be involved and it is difficult to predict the magnitude of the interaction without additional data.
Based on the needs described above for a new method to establish the existence of a DDI, we developed the DRug Interaction eVidence Evaluation (DRIVE) Instrument.. The rationale behind the DRIVE Instrument is to: (1) use simple evidence categories; (2) include causality assessment with DDI case reports (via DIPS); (3) apply reasonable extrapolation, including from in vitro studies; and (4) address evidence/statements provided in product labeling; and (5) describe study quality criteria and interpretation in the context of DDIs. Systematic evidence review should include a thorough search for relevant published and unpublished literature and, therefore, we also recognized that future work should seek to develop systematic methods for conducting literature searches for assembling DDI evidence [33–37].
FDA documents, such as product labeling, preapproval reviews and post-marketing analyses are essential sources when evaluating DDI evidence [38]. In our collective experience and opinion, the majority of pre-marketing DDI studies conducted and described in product labeling are well designed and executed. However, we agree that DDI content in drug information systems does not always need to align with product labeling, even when that information is listed as a contraindication or boxed warning. This opinion is based upon examples in which the labeling is not consistent with existing evidence [39] and significant variation occurs in the DDIs listed in product labeling compared with published information [34, 35, 40–42]. We acknowledge that the purpose and guidance of labeling is unique, and also that FDA has taken important steps to improve the quality and usefulness of DDI information in product labeling [43–45]. We recommend continued effort to improve the consistency and timeliness of DDI information in product labeling, particularly for older nonproprietary drugs.
3.2.3 Assessing Clinical Relevance
If there is sufficient evidence that a DDI might require clinical management, further evaluation is needed to establish the clinical relevance. Clinical outcomes associated with the DDI must be determined, including the magnitude, variability and frequency of effects (if known), and modifying factors that may increase or decrease the risk of patient harm (see Figure 1). Depending on the context, exposure to a clinically relevant DDI might warrant a change in therapy, increased monitoring, and/or patient education. Assessing the clinical relevance of a DDI is an estimate, at best, because inter-patient variability is often unknown, and for PK DDIs, changes in the object drug can vary 4–6-fold [46, 47]. For some DDIs, it is reasonable to assign a general risk rating based on the properties of the object drug, such as those with a NTI.
We recommend providing estimates of the frequency (incidence) of adverse outcomes from DDIs when available. However, assessing these frequencies is difficult because data are largely limited to observational studies, which are susceptible to confounding. When available, the definition of the adverse outcome and the applicable population should be clearly specified. For example, we can inform patients that the risk of upper gastrointestinal (GI) bleeding is estimated to increase by 19% with combined use of selective serotonin reuptake inhibitors (SSRIs) and nonsteroidal anti-inflammatory drugs (NSAIDs) beyond the effects of each individual drug [48]. We can also estimate that 179 (95% CI = 107,319) high-risk patients (e.g., elderly, previous GI bleed) and 645 (95% CI = 387, 1,152) low-risk patients need to be treated with the SSRI and NSAID combination to cause one upper GI bleed [48]. But for most DDIs—even those that are well-documented and potentially dangerous—only rough estimates of the incidence of adverse outcomes are known.
Thorough evidence evaluation of DDI literature should include documented methods to mitigate harm (e.g., dosage adjustment, monitoring strategies, and therapeutic alternatives) [22, 49]. Reasonable therapeutic alternatives may include DDIs ruled out by mechanistic principles, preferably with one or more negative studies that appear robust to bias and confounding.
We also recommend that modifying factors that may increase (risk factors) or decrease (mitigating factors) susceptibility to DDIs should be considered when evaluating and reporting DDI evidence. Drug-related modifying factors may include dose, duration, route of administration, order of administration, timing of dose, and comedications. Patient-related modifying factors may include age, sex, pharmacogenomics [50], comorbidity, clinical status, vital signs, laboratory values, and indication for the drug. Identifying modifying factors is essential because research shows that providing patient-specific risk factors in CDS improves the specificity of alerts [51, 52]. There are many situations where a particular DDI may not be clinically relevant to a specific patient due to mitigating factors that result in a negligible risk of adverse outcomes. For example, a precipitant drug is unlikely to produce a clinically relevant DDI for a patient with a genetic variant producing a nonfunctional CYP enzyme (i.e., poor metabolizer) [53]. However, information on factors that alter patient susceptibility to DDIs is not yet systematically reviewed in DDI guidelines [52]. In general, more research is needed to identify modifying factors to inform CDS algorithms and clinical decision-making.
More work is also needed to identify the most appropriate process to rate the quality of DDI evidence and provide graded recommendations to reduce the risk of adverse consequences [49]. We recommend considering the Grading of Recommendations, Assessment, Development, and Evaluation (GRADE) system, a well-accepted standard to indicate quality of evidence and strength of recommendations [54–57].
3.3 Key Question 2: What evidence is required for a DDI to be applicable to an entire class of medications?
CDS systems can generate nuisance alerts when they inappropriately define or represent a DDI as a “class” effect. Knowledge of the mechanism of interaction is crucial to determining whether there is basis for a class effect. Most class-based DDIs are of a PD nature, with additive (e.g., angiotensin-converting enzyme (ACE) inhibitors + angiotensin II receptor blockers (ARBs)) or opposing (e.g., beta blocker + beta agonist) pharmacologic effects. In contrast, PK interactions are rarely generalizable to all agents within a class [22, 58]. Even when there is seemingly a class effect, the magnitude of effect can vary, which often makes it necessary to consider each drug in the class individually. For example, azole antifungal agents can inhibit cytochrome P450 (CYP) 3A4. However, itraconazole and ketoconazole are much more potent inhibitors than fluconazole, so the magnitude of interaction with drugs that are primarily metabolized by CYP3A4 may differ significantly, which would impact the clinical relevance of the interaction. This can be shown by their effect on triazolam levels: itraconazole and ketoconazole increase the AUC of triazolam by 27- and 22-fold, respectively [59], whereas fluconazole causes a 4.4-fold increase in AUC [60].
The overwhelming majority of extrapolated DDIs are PD, because few studies are conducted to investigate this type of interaction. In the absence of drug-specific data, a class-based interaction may be reasonably assumed if the purported mechanism of interaction is biologically plausible and consistent with known pharmacology of one or both classes of drugs involved. Class examples include SSRIs + other serotonergic drugs related to serotonin syndrome and anticoagulation + antiplatelet agents related to bleeding.
Occasionally, PK interaction data may be extrapolated from one agent to other agents in the class if the purported mechanism of interaction involves common pharmacologic effects. For instance, NSAIDs may reduce the renal excretion of lithium and therefore increase the risk of toxicity [61]. The proposed mechanism of interaction is inhibition of renal prostaglandin synthesis by NSAIDs, which leads to reduced renal blood flow. Although lithium toxicity has not been reported with all NSAIDs, the interaction is probably applicable to the entire class based on their common ability to inhibit prostaglandin synthesis.
We recommend that DDIs should be class-based only when the evidence (or reasonable extrapolation) applies to the entire pharmacological class of drugs.
3.4 Key Question 3: How should a structured evaluation process be vetted and validated?
In Key Question 1, we recommended use of a new instrument as a standard to evaluate DDI evidence. However, any new DDI evidence evaluation instrument should undergo a rigorous evaluation. The evaluation should ensure that the instrument is easy to apply by end users and produces results that are generally concordant with other DDI evidence rating systems, except where differences are expected.
We recommend evaluating the new DRIVE Instrument using a subset of 15 “high-priority” DDIs (drug pairs which should always generate an alert) approved by an ONC-commissioned panel of experts [62]. There are also several existing studies that have systematically collected evidence for a set of DDIs and then examined concordance on DDIs mentioned in drug information sources [12, 35, 40, 42, 63]. These studies can provide DDIs for which there are varying degrees of agreement across drug information sources (e.g., some DDIs that all sources mention and others that only one source mentions).
4 Discussion
This expert panel was convened to recommend an approach for evaluating DDIs in order to provide consistent, evidence-based CDS systems for healthcare providers. Because of the numerous challenges to evaluating evidence for DDIs, we propose a systematic and transparent process to evaluate evidence that supports the existence of clinically relevant DDIs. Furthermore, the use of a standardized evidence-based approach to evaluate DDI evidence will eliminate combinations with a low probability of harm and minimize legal liability for knowledgebase vendors and healthcare systems [23].
Our search for relevant tools to evaluate the DDI evidence identified no instruments that possessed all of the attributes believed to be important. Consequently, we developed DRIVE for evaluating the body of evidence for DDIs using important concepts from existing evidence evaluation methods with a focus on simplicity and explicit criteria for certain types of evidence [31, 32]. In this process, we defined several terms for use when evaluating DDIs because consistent application of terminology is requisite for systematic evaluation.
We recommend that any systematic approach to evaluating DDI evidence be validated to ensure the method is worthwhile. To that end, further evaluation of the DRIVE approach, including explicit criteria for what constitutes a well-designed and executed study should be developed. We recommend that DDI evidence reported in product labeling should be evaluated by the same criteria as published studies to establish sufficiency of DDI evidence.
For case reports, we judged the DIPS to be the most appropriate published method to evaluate whether a DDI occurred [20]. Case reports may provide the first evidence of DDIs; however, using these reports as the sole evidence source has several disadvantages. They are often poorly described, leading to speculation and potentially inaccurate inferences about causal relationships. Use of case reports that are later found to be incorrect results in erroneous data listed in prescription product labeling and/or drug information compendia that are very difficult to correct. Therefore, careful evaluation of case reports is needed to establish the existence of a DDI.
We accomplished our goal of identifying principles for establishing a systematic process for evaluating evidence for DDIs; however, more work remains in certain areas. Although the DRIVE instrument may be used in the future to affirm that a DDI exists, further evaluation is needed to establish criteria for assessing clinical relevance. This entails identifying the associated clinical effects and their magnitude, variability, and estimated frequency. Modifying factors that may increase or decrease the risk of patient harm should also be identified. Pharmacogenetic research can further improve the precision of DDI evidence and CDS by identifying patient-specific predisposing factors. More work is also needed to identify the most appropriate process to rate the quality of DDI evidence and provide graded recommendations to reduce the risk of adverse consequences [49]. We recommend considering the Grading of Recommendations, Assessment, Development, and Evaluation (GRADE) system, a well-accepted standard to indicate quality of evidence and strength of recommendations [54–57].
5. Conclusion
Stakeholders—such as editors, developers, and end users of CDS software and compendia—should require consistent application of transparent, systematic methods to assess DDI evidence. We envision these recommendations improving the evaluation of DDI evidence for CDS.
Supplementary Material
KEY POINTS.
Evidence-based clinical decision support for drug-drug interactions (DDIs) requires consistent application of transparent and systematic methods to evaluate the evidence.
An expert workgroup developed recommendations by consensus for systematic evaluation of evidence for DDIs from the scientific literature, drug product labeling, and regulatory documents.
Workgroup members expect that drug information systems will be able to provide higher quality information about DDIs in drug compendia and clinical decision support tools.
Acknowledgments
This project was funded by the Agency for Healthcare Research and Quality (AHRQ) Grant No. 1R13HS021826-01 (Malone DC-PI). RDB received support to contribute to this project from AHRQ Grant No. K12 HS019461-01, National Library of Medicine Grant No. R01 LM011838-01, and National Institute of Aging Grant No. K01 AG044433-01. Additional support was provided by Cerner, Elsevier Clinical Solutions, Epocrates, FDB (First Databank, Inc.), Truven Health Analytics, and Wolters Kluwer. The authors would like to thank Loretta Peters for her editorial assistance in preparing this manuscript. We would like to acknowledge that John Horn developed and published the Drug Interaction Probability Scale that we recommend for use in evaluating drug interaction case reports.
References
- 1.Aspden P, Institute of Medicine (U.S.). Committee on Identifying and Preventing Medication Errors. Preventing medication errors. Washington, DC: National Academies Press; 2007. [Google Scholar]
- 2.Magro L, Moretti U, Leone R. Epidemiology and characteristics of adverse drug reactions caused by drug-drug interactions. Expert Opin Drug Saf. 2012 Jan;11(1):83–94. doi: 10.1517/14740338.2012.631910. [DOI] [PubMed] [Google Scholar]
- 3.CDC. FASTSTATS - Emergency Department Visits. 2012 [cited 12/06/2013]; Available from: http://www.cdc.gov/nchs/fastats/ervisits.htm.
- 4.CDC. FASTSTATS - Hospital Utilization. 2010 [cited 12/06/2013]; Available from: http://www.cdc.gov/nchs/fastats/hospital.htm.
- 5.CMS. Eligible Professional Meaningful Use Core Measures: Measure 2 of 15. Centers for Medicare and Medicaid Services; 2010. [cited 01/09/2013]; Available from: http://www.cms.gov/Regulations-and-Guidance/Legislation/EHRIncentivePrograms/downloads/2_Drug_Interaction_ChecksEP.pdf. [Google Scholar]
- 6.Hatton RC, Rosenberg AF, Morris CT, McKelvey RP, Lewis JR. Evaluation of contraindicated drug-drug interaction alerts in a hospital setting. Ann Pharmacother. 2011 Mar;45(3):297–308. doi: 10.1345/aph.1P533. [DOI] [PubMed] [Google Scholar]
- 7.Shah VS, Weber RJ, Nahata MC. Contradictions in contraindications for drug-drug interactions. Ann Pharmacother. 2011 Mar;45(3):409–11. doi: 10.1345/aph.1P792. [DOI] [PubMed] [Google Scholar]
- 8.Horn JR, Hansten PD. Comment: evaluation of contraindicated drug-drug interaction alerts in a hospital setting. Ann Pharmacother. 2011 Jun;45(6):826. doi: 10.1345/aph.1P533a. author reply -7. [DOI] [PubMed] [Google Scholar]
- 9.Kesselheim AS, Cresswell K, Phansalkar S, Bates DW, Sheikh A. Clinical decision support systems could be modified to reduce ‘alert fatigue’ while still minimizing the risk of litigation. Health Aff (Millwood) 2011 Dec;30(12):2310–7. doi: 10.1377/hlthaff.2010.1111. [DOI] [PubMed] [Google Scholar]
- 10.Saverno KR, Hines LE, Warholak TL, Grizzle AJ, Babits L, Clark C, et al. Ability of pharmacy clinical decision-support software to alert users about clinically important drug-drug interactions. J Am Med Inform Assoc. 2011 Jan-Feb;18(1):32–7. doi: 10.1136/jamia.2010.007609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Metzger J, Welebob E, Bates DW, Lipsitz S, Classen DC. Mixed results in the safety performance of computerized physician order entry. Health Aff (Millwood) 2010 Apr;29(4):655–63. doi: 10.1377/hlthaff.2010.0160. [DOI] [PubMed] [Google Scholar]
- 12.Abarca J, Colon LR, Wang VS, Malone DC, Murphy JE, Armstrong EP. Evaluation of the performance of drug-drug interaction screening software in community and hospital pharmacies. J Manag Care Pharm. 2006 Jun;12(5):383–9. doi: 10.18553/jmcp.2006.12.5.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van der Sijs H, Aarts J, Vulto A, Berg M. Overriding of drug safety alerts in computerized physician order entry. J Am Med Inform Assoc. 2006 Mar-Apr;13(2):138–47. doi: 10.1197/jamia.M1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller AM, Boro MS, Korman NE, Davoren JB. Provider and pharmacist responses to warfarin drug-drug interaction alerts: a study of healthcare downstream of CPOE alerts. J Am Med Inform Assoc. 2011 Dec;18( Suppl 1):i45–50. doi: 10.1136/amiajnl-2011-000262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weingart SN, Toth M, Sands DZ, Aronson MD, Davis RB, Phillips RS. Physicians’ decisions to override computerized drug alerts in primary care. Arch Intern Med. 2003 Nov;163(21):2625–31. doi: 10.1001/archinte.163.21.2625. [DOI] [PubMed] [Google Scholar]
- 16.Grizzle AJ, Mahmood MH, Ko Y, Murphy JE, Armstrong EP, Skrepnek GH, et al. Reasons provided by prescribers when overriding drug-drug interaction alerts. Am J Manag Care. United States. 2007:573–8. [PubMed] [Google Scholar]
- 17.Caccia S, Garattini S, Pasina L, Nobili A. Predicting the clinical relevance of drug interactions from pre-approval studies. Drug Saf. 2009;32(11):1017–39. doi: 10.2165/11316630-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 18.Holbrook A, Grootendorst P, Willison D, Goldsmith C, Sebaldt R, Keshavjee K. Can current electronic systems meet drug safety and effectiveness requirements? AMIA Annu Symp Proc. 2005:335–9. [PMC free article] [PubMed] [Google Scholar]
- 19.Huang SM, Strong JM, Zhang L, Reynolds KS, Nallani S, Temple R, et al. New era in drug interaction evaluation: US Food and Drug Administration update on CYP enzymes, transporters, and the guidance process. J Clin Pharmacol. 2008 Jun;48(6):662–70. doi: 10.1177/0091270007312153. [DOI] [PubMed] [Google Scholar]
- 20.Horn JR, Hansten PD, Chan LN. Proposal for a new tool to evaluate drug interaction cases. Ann Pharmacother. 2007 Apr;41(4):674–80. doi: 10.1345/aph.1H423. [DOI] [PubMed] [Google Scholar]
- 21.Hines LE, Malone DC, Murphy JE. Recommendations for generating, evaluating, and implementing drug-drug interaction evidence. Pharmacotherapy. 2012 Apr;32(4):304–13. doi: 10.1002/j.1875-9114.2012.01024.x. [DOI] [PubMed] [Google Scholar]
- 22.Hansten PD, Horn JR, Hazlet TK. ORCA: OpeRational ClassificAtion of drug interactions. J Am Pharm Assoc (Wash) 2001 Mar-Apr;41(2):161–5. doi: 10.1016/s1086-5802(16)31244-x. [DOI] [PubMed] [Google Scholar]
- 23.Ridgeley MS, Greenberg MD. Too many alerts, too much liability: Sorting through the malpractice implications of drug-drug interaction clinical decision support. Saint Louis University Journal of Health Law and Policy. 2012;5(257):257–96. [Google Scholar]
- 24.Oates JA. Chapter 5. The science of drug therapy. In: Brunton LL, editor. Goodman & Gilman’s The Pharmacological Basis of Therapeutics. 11. McGraw-Hill, Medical Publishing Division; 2006. [Google Scholar]
- 25.Hines LE, Murphy JE. Potentially harmful drug-drug interactions in the elderly: a review. Am J Geriatr Pharmacother. 2011 Dec;9(6):364–77. doi: 10.1016/j.amjopharm.2011.10.004. [DOI] [PubMed] [Google Scholar]
- 26.Talbot JCC, Aronson JK, Stephens MDB. Stephens’ detection and evaluation of adverse drug reactions : principles and practice. 6. Chichester, West Sussex, UK: John Wiley & Sons; 2011. [Google Scholar]
- 27.US Food and Drug Administration. Title 21--Food and Drugs Chapter I--Food and Drug Administration Department of Health and Human Services Subchapter D--Drugs for Human Use. 2013 [cited; Available from: http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=320.33.
- 28.Holbrook AM, Pereira JA, Labiris R, McDonald H, Douketis JD, Crowther M, et al. Systematic overview of warfarin and its drug and food interactions. Arch Intern Med. 2005 May;165(10):1095–106. doi: 10.1001/archinte.165.10.1095. [DOI] [PubMed] [Google Scholar]
- 29.Naranjo CA, Busto U, Sellers EM, Sandor P, Ruiz I, Roberts EA, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981 Aug;30(2):239–45. doi: 10.1038/clpt.1981.154. [DOI] [PubMed] [Google Scholar]
- 30.Hutchinson TA, Lane DA. Assessing methods for causality assessment of suspected adverse drug reactions. J Clin Epidemiol. 1989;42(1):5–16. doi: 10.1016/0895-4356(89)90020-6. [DOI] [PubMed] [Google Scholar]
- 31.Böttiger Y, Laine K, Andersson ML, Korhonen T, Molin B, Ovesjö ML, et al. SFINX-a drug-drug interaction database designed for clinical decision support systems. Eur J Clin Pharmacol. 2009 Jun;65(6):627–33. doi: 10.1007/s00228-008-0612-5. [DOI] [PubMed] [Google Scholar]
- 32.van Roon EN, Flikweert S, le Comte M, Langendijk PN, Kwee-Zuiderwijk WJ, Smits P, et al. Clinical relevance of drug-drug interactions : a structured assessment procedure. Drug Saf. 2005;28(12):1131–9. doi: 10.2165/00002018-200528120-00007. [DOI] [PubMed] [Google Scholar]
- 33.Boyce R, Collins C, Horn J, Kalet I. Computing with evidence Part II: An evidential approach to predicting metabolic drug-drug interactions. J Biomed Inform. 2009 Dec;42(6):990–1003. doi: 10.1016/j.jbi.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boyce RD, Handler SM, Karp JF, Hanlon JT. Age-related changes in antidepressant pharmacokinetics and potential drug-drug interactions: a comparison of evidence-based literature and package insert information. Am J Geriatr Pharmacother. 2012 Apr;10(2):139–50. doi: 10.1016/j.amjopharm.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boyce RD, Collins C, Clayton M, Kloke J, Horn JR. Inhibitory metabolic drug interactions with newer psychotropic drugs: inclusion in package inserts and influences of concurrence in drug interaction screening software. Ann Pharmacother. 2012 Oct;46(10):1287–98. doi: 10.1345/aph.1R150. [DOI] [PubMed] [Google Scholar]
- 36.McDonagh MS, Peterson K, Balshem H, Helfand M. US Food and Drug Administration documents can provide unpublished evidence relevant to systematic reviews. J Clin Epidemiol. 2013 Oct;66(10):1071–81. doi: 10.1016/j.jclinepi.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 37.Turner EH. How to access and process FDA drug approval packages for use in research. BMJ. 2013;347:f5992. doi: 10.1136/bmj.f5992. [DOI] [PubMed] [Google Scholar]
- 38.O’Connor AB. The need for improved access to FDA reviews. JAMA. 2009 Jul;302(2):191–3. doi: 10.1001/jama.2009.973. [DOI] [PubMed] [Google Scholar]
- 39.DiNicolantonio JJ, Serebruany VL. Challenging the FDA black box warning for high aspirin dose with ticagrelor in patients with diabetes. Diabetes. 2013 Mar;62(3):669–71. doi: 10.2337/db12-0746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anthony M, Romero K, Malone DC, Hines LE, Higgins L, Woosley RL. Warfarin interactions with substances listed in drug information compendia and in the FDA-approved label for warfarin sodium. Clin Pharmacol Ther. 2009 Oct;86(4):425–9. doi: 10.1038/clpt.2009.95. [DOI] [PubMed] [Google Scholar]
- 41.Chao SD, Maibach HI. Lack of drug interaction conformity in commonly used drug compendia for selected at-risk dermatologic drugs. Am J Clin Dermatol. 2005;6(2):105–11. doi: 10.2165/00128071-200506020-00005. [DOI] [PubMed] [Google Scholar]
- 42.Hines LE, Ceron-Cabrera D, Romero K, Anthony M, Woosley RL, Armstrong EP, et al. Evaluation of warfarin drug interaction listings in US product information for warfarin and interacting drugs. Clin Ther. 2011 Jan;33(1):36–45. doi: 10.1016/j.clinthera.2011.01.021. [DOI] [PubMed] [Google Scholar]
- 43.Guidance for industry: Drug interaction studies — study design, data analysis, and implications for dosing and labeling: Draft guidance. Rockville, MD: Food and Drug Administration, U.S. Department of Health and Human Services; 2006. [Google Scholar]
- 44.Administration USFaD. [Accessed October 29, 2010];Information for Healthcare Professionals on FDA’s New Prescribing Information for Drugs. 2009 Available from: http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/LawsActsandRules/ucm084189.htm.
- 45.Administration USFaD. products Isplfhpdab. 2010. Indexing structured product labeling for human prescription drug and biologic products; Request for comments. comments Rf, editors. [Google Scholar]
- 46.Horn JR, Hansten PD. Predicting the Magnitude of Drug Interactions: The Final Frontier. Pharmacy Times. 2006 Oct72 [Google Scholar]
- 47.de Leon J, Spina E, Diaz FJ. Pharmacokinetic drug interaction studies must consider pharmacological heterogeneity, use of repeated dosing, and translation into a message understandable to practicing clinicians. J Clin Psychopharmacol. 2009 Jun;29(3):201–5. doi: 10.1097/JCP.0b013e3181a497f1. [DOI] [PubMed] [Google Scholar]
- 48.Anglin R, Yuan Y, Moayyedi P, Tse F, Armstrong D, Leontiadis GI. Risk of upper gastrointestinal bleeding with selective serotonin reuptake inhibitors with or without concurrent nonsteroidal anti-inflammatory use: a systematic review and meta-analysis. Am J Gastroenterol. 2014 Jun;109(6):811–9. doi: 10.1038/ajg.2014.82. [DOI] [PubMed] [Google Scholar]
- 49.Floor-Schreudering A, Geerts AF, Aronson JK, Bouvy ML, Ferner RE, De Smet PA. Checklist for standardized reporting of drug-drug interaction management guidelines. Eur J Clin Pharmacol. 2014 Mar;70(3):313–8. doi: 10.1007/s00228-013-1612-7. [DOI] [PubMed] [Google Scholar]
- 50.Verbeurgt P, Mamiya T, Oesterheld J. How common are drug and gene interactions? Prevalence in a sample of 1143 patients with CYP2C9, CYP2C19 and CYP2D6 genotyping. Pharmacogenomics. 2014 Apr;15(5):655–65. doi: 10.2217/pgs.14.6. [DOI] [PubMed] [Google Scholar]
- 51.Tamblyn R, Eguale T, Buckeridge DL, Huang A, Hanley J, Reidel K, et al. The effectiveness of a new generation of computerized drug alerts in reducing the risk of injury from drug side effects: a cluster randomized trial. J Am Med Inform Assoc. 2012 Jul-Aug;19(4):635–43. doi: 10.1136/amiajnl-2011-000609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Seidling HM, Klein U, Schaier M, Czock D, Theile D, Pruszydlo MG, et al. What, if all alerts were specific - Estimating the potential impact on drug interaction alert burden. Int J Med Inform. 2014 Apr;83(4):285–91. doi: 10.1016/j.ijmedinf.2013.12.006. [DOI] [PubMed] [Google Scholar]
- 53.Miura M, Tada H, Yasui-Furukori N, Uno T, Sugawara K, Tateishi T, et al. Effect of clarithromycin on the enantioselective disposition of lansoprazole in relation to CYP2C19 genotypes. Chirality. 2005 Jun;17(6):338–44. doi: 10.1002/chir.20159. [DOI] [PubMed] [Google Scholar]
- 54.Guyatt GH, Oxman AD, Kunz R, Vist GE, Falck-Ytter Y, Schunemann HJ. What is “quality of evidence” and why is it important to clinicians? BMJ. 2008 May 3;336(7651):995–8. doi: 10.1136/bmj.39490.551019.BE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guyatt GH, Oxman AD, Vist GE, Kunz R, Falck-Ytter Y, Alonso-Coello P, et al. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ. 2008 Apr 26;336(7650):924–6. doi: 10.1136/bmj.39489.470347.AD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Atkins D, Best D, Briss PA, Eccles M, Falck-Ytter Y, Flottorp S, et al. Grading quality of evidence and strength of recommendations. BMJ. 2004;328(7454):1490. doi: 10.1136/bmj.328.7454.1490. 2004–06–17 21:56:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Grade Working Group. [cited; Available from: http://www.gradeworkinggroup.org/index.htm.
- 58.Horn JR, Hansten Philip D. “Classy” Drug Interactions “Classy” Drug Interactions “Classy” Drug Interactions “Classy” Drug Interactions. Pharmacy Times; http://www.pharmacytimes.com/publications/issue/2005/2005-06/2005-06-9585:PharmacyTimes;2005. [Google Scholar]
- 59.Varhe A, Olkkola KT, Neuvonen PJ. Oral triazolam is potentially hazardous to patients receiving systemic antimycotics ketoconazole or itraconazole. Clin Pharmacol Ther. 1994 Dec;56(6 Pt 1):601–7. doi: 10.1038/clpt.1994.184. [DOI] [PubMed] [Google Scholar]
- 60.Varhe A, Olkkola KT, Neuvonen PJ. Effect of fluconazole dose on the extent of fluconazole-triazolam interaction. Br J Clin Pharmacol. 1996 Oct;42(4):465–70. doi: 10.1046/j.1365-2125.1996.45111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hughes BM, Small RE, Brink D, McKenzie ND. The effect of flurbiprofen on steady-state plasma lithium levels. Pharmacotherapy. 1997 Jan-Feb;17(1):113–20. [PubMed] [Google Scholar]
- 62.Phansalkar S, Desai AA, Bell D, Yoshida E, Doole J, Czochanski M, et al. High-priority drug-drug interactions for use in electronic health records. J Am Med Inform Assoc. 2012 Sep-Oct;19(5):735–43. doi: 10.1136/amiajnl-2011-000612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Abarca J, Malone DC, Armstrong EP, Grizzle AJ, Hansten PD, Van Bergen RC, et al. Concordance of severity ratings provided in four drug interaction compendia. J Am Pharm Assoc 2003. 2004 Mar-Apr;44(2):136–41. doi: 10.1331/154434504773062582. [DOI] [PubMed] [Google Scholar]
- 64.HIV-Drug Interactions. 2014 Mar 27; [cited; Available from: http://www.hiv-druginteractions.org.
- 65.HEP-Drug Interactions. 2014 Mar 27; [cited; Available from: http://www.hep-druginteractions.org/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.