


Abstract
Genes encoding toxin–antitoxin (TA) systems are near ubiquitous in bacterial genomes and they play key roles in important aspects of bacterial physiology, including genomic stability, formation of persister cells under antibiotic stress, and resistance to phage infection. The CptIN locus from Eubacterium rectale is a member of the recently-discovered Type III class of TA systems, defined by a protein toxin suppressed by direct interaction with a structured RNA antitoxin. Here, we present the crystal structure of the CptIN protein–RNA complex to 2.2 Å resolution. The structure reveals a new heterotetrameric quaternary organization for the Type III TA class, and the RNA antitoxin bears a novel structural feature of an extended A-twist motif within the pseudoknot fold. The retention of a conserved ribonuclease active site as well as traits normally associated with TA systems, such as plasmid maintenance, implicates a wider functional role for Type III TA systems. We present evidence for the co-variation of the Type III component pair, highlighting a distinctive evolutionary process in which an enzyme and its substrate co-evolve.
INTRODUCTION
Bacterial toxin–antitoxin (TA) systems were discovered nearly 30 years ago in studies of plasmid stabilization during cell division (1,2). Genes encoding TA systems are now known to be very widespread in prokaryotic genomes (3). TA systems have been classified into five types. All share common features in that they include a toxic protein component and are usually bicistronic in architecture, but are distinguished from each other by the manner in which this toxic protein is antagonized. In Type I systems, the antitoxin is an antisense RNA molecule that prevents translation of what is usually a small hydrophobic toxin. In Type II systems the antitoxin is a co-expressed protein that directly inhibits the toxin. Type IV and V TA systems also utilize protein antitoxins; Type IV TA components do not physically interact and in the prototype system both proteins act in opposing manners on a common substrate (4,5). The single described Type V TA system employs a ribonuclease antitoxin that targets the toxin transcript (6). The focus of the present study, Type III systems, comprise a ribonuclease (RNase) toxin that processes, and is then directly inhibited by, its structured and specific cognate RNA antitoxin.
Recent advances in sequencing technology and the associated exponential increase in available genomic data have enabled large-scale bioinformatic surveys of TA loci, aimed both at finding locations of known families and discovering new TA families within the many open reading frames (ORFs) of unknown function (7,8). As it became clear that TA system loci are near-ubiquitous in bacterial genomes (and often in very high numbers within a single species) the definition of their role(s) in bacterial physiology became a more pressing issue. TA systems have been associated with multiple important phenotypes, including phage resistance, maintenance of genomic islands and formation of bacterial persister cells (9). The widespread distribution and redundancy of the TA systems, especially in the Type II group, suggests that TA pairs are important for organism fitness (10–12).
The paradigmatic Type III TA locus, toxINPa, was discovered on a cryptic plasmid of Pectobacterium atrosepticum strain SCRI1039. A gene on this plasmid encoded a homologue of a known phage resistance protein, and this provided strong protection from some bacteriophages while also being extremely toxic to the cell (13). This toxic gene could only be cloned in concert with short tandem repeats located upstream; these repeats were shown to encode a small RNA that suppressed the activity of the toxin. The components were named ToxNPa and ToxIPa (for Toxin Inhibitor). Structural studies of toxINPa revealed that the ToxNPa ribonuclease cleaved the repetitive ToxIPa RNA into individual 36 base pseudoknot repeats, and then assembled with these catalytic products into a heterohexameric, triangular complex that inhibits the toxin (14). Interestingly, ToxNPa was shown to be a distant structural homologue of the Kid family of Type II TA system toxins.
The corresponding ToxINBt complex from Bacillus thuringiensis showed a largely similar architecture to that of P. atrosepticum, but with subtle structural changes that underpinned highly specific TA recognition between cognate partners (15). While the heterohexameric organization of three ToxNs and three ToxIs was retained in this homologue, the interface between the RNA and protein showed key changes in the variable loops outside of the structural core of the toxin that allow the selective recognition of its cognate antitoxic RNA.
Over 120 putative Type III TA loci have been identified bioinformatically, in very diverse bacterial species, and these systems fall into three separate families (named ToxIN, CptIN and TenpIN) based on sequence similarity of their toxin components (16). While it is presumed that each locus contains a structured RNA as its functional antitoxin, as in the toxINPa paradigm, the tandem repeats differ greatly in both primary sequence and length. Members of all three families have been shown to function as TA loci, and at least one other family (the TenpINs) can also mediate phage resistance (16).
It has also become increasingly clear that RNA species play a pivotal role in controlling gene expression in bacteria (17). Since the realization of the importance of Hfq as a regulator through its function as an RNA chaperone (18) as well as the discovery of riboswitches (19), RNA is no longer seen as a passive informational mediator. Similarly, the discovery of systems of acquired phage immunity (CRISPR) is another development that illuminates the ways in which cellular proteins are able to directly use the information contained within RNA species (20). While RNAs have been postulated to be natural mediators of protein activity, based on the precedent of artificial aptamers (21), most known examples show the RNA mimicking DNA (17). The distinctive case in which ToxN is specifically regulated by a pseudoknot RNA species which the toxin itself processes is a unique example in the growing repertoire of riboregulation (14).
Here, we present the first structural investigation of a member of the CptIN family of Type III TA systems. Many Type III TA systems are plasmid-encoded (16) but we decided to investigate the chromosomally-encoded CptIN system, from the human commensal Eubacterium rectale (CptINEr) as the first representative of the CptIN family. CptINEr forms a dramatically different complex from that of the previously described ToxIN systems, exhibiting a unique oligomeric structure. We demonstrate that, while CptIEr retains the pseudoknot core previously observed in other Type III antitoxins, the extended length of the RNA leads to the formation of unique tertiary features. However, the overall principles of toxin inhibition are shared between the CptIN and ToxIN families. The present study reveals the structural diversity that can exist in these loci and highlights the remarkable case in which an enzyme co-evolves with its cognate substrate.
MATERIALS AND METHODS
CptINEr purification, crystallization and structure determination
The ORF encoding CptNEr was cloned into pTYB1 (NEB) using primers TRB273 and TRB274 (Table 2), with a three-amino acid extension (LEG) to enable intein cleavage. The CptIEr gene was cloned into pACYC184 (22) along with a T7 promoter 3′ of the predicted start of the gene, identical to that present in pTYB1, using the primers TRB271 and TRB272 (Table 2). The resulting plasmids were introduced to Escherichia coli ER2566 (NEB) by electroporation. For native protein expression a 500 ml culture in 2×TY media was inoculated with an overnight culture of the expression strain at a 1:100 dilution and was grown at 37°C to OD600 0.8. The cells were then shifted to 18°C, expression of CptINEr was induced with a final concentration of 1 mM IPTG, and expression was continued for 16 h. Expression of the SeMet derivative was performed using a supplemented minimal medium, as described (23). The cells were resuspended in 20 mM Na2HPO4, 500 mM NaCl, 1% v/v Triton X-100, 1 mM ethylenediaminetetraacetic acid, pH 6.0 and lysed by pressure with four passes through an EmulsiFlex-C5 instrument at 10 000 psi. The CptINEr protein–RNA complex was purified initially by passing the clarified lysate through a chitin column followed by on-column cleavage of the intein fragment fused to the CptNEr protein under reducing alkaline conditions. The eluate from the cleavage step was then fractionated by anion exchange chromatography using a HiTrap Q 5 ml column (GE Healthcare), yielding one sharp peak identified as the CptINEr complex. The protein–RNA complex was then concentrated to ∼5 mg.ml−1 and used in crystallization trials.
Table 2. Strains, plasmids and primers used in this study.
| Strain | Genotype | Reference |
|---|---|---|
| Escherichia coli DH5α | F− λ−endA1 glnV44 thi-1 recA1 relA1 gyrA96 deoR nupG Φ80 lacZ ΔM15 Δ(lacZYA-argF) U169, hsdR17(rK− mK+) | Invitrogen |
| E. coli ER2566 | F− λ−fhuA2 [lon] ompT lacZ::T7 gene 1 gal sulA11 Δ(mcrC-mrr)114::IS10 R(mcr-73::miniTn10-TetS)2 R(zgb-210::Tn10)(TetS) endA1 [dcm] | NEB |
| E. coli W3110 | F− λ−rph-1 INV(rrnD, rrnE) | (39) |
| Plasmid | Description | Source |
| pALO3 | cptNEr in pTYB1 | This study |
| pALO5 | cptIEr with PT7 upstream in pACYC184 | This study |
| pFLS104 | cptIRt repeats and terminator in pTA100, SpR | (16) |
| pFLS105 | cptNRt in pBAD30, ApR | (16) |
| pFLS106 | cptICc repeats and terminator in pTA100, SpR | (16) |
| pFLS107 | cptNCc in pBAD30, ApR | (16) |
| pFLS108 | cptIEr repeats and terminator in pTA100, SpR | (16) |
| pFLS109 | cptNEr in pBAD30, ApR | (16) |
| pFLS110 | cptIEr single repeat in pTA100, SpR | This study |
| pFLS111 | cptIEr ΔU30 single repeat in pTA100, SpR | This study |
| pFLS112 | cptIEr A29U single repeat in pTA100, SpR | This study |
| pFLS122 | cptINEr locus in pRBJ200, ApR | This study |
| pRBJ200 | E. coli par-deficient single-copy vector, ApR | (40) |
| pTA100 | SpR derivative of pQE-80L | (13) |
| Primer | Sequence 5′-3prime;* (Restriction site) | |
| CptI-WT | tttaagcttTCAACCCGACCATTTATATACCACATATCGGTCAGTGGTAAACTTtcaattgaatctattataattgtta (HindIII) | |
| CptI-dU30 | tttaagcttTCAACCCGACCATTTTATACCACATATCGGTCAGTGGTAAACTTtcaattgaatctattataattgtta (HindIII) | |
| CptI-A29U | ttaagcttTCAACCCGACCATTTAAATACCACATATCGGTCAGTGGTAAACTTtcaattgaatctattataattgtta (HindIII) | |
| PF185 | AAACAAATAGGGGTTCCG | |
| TRB271 | TTGGATCCATAATCAGTATCACTGAGAAA (BamHI | |
| TRB272 | TTTCTAGAACTACGGCAAAAGACTTTTTC (XbaI) | |
| TRB273 | CCGGCATATGATAAGGAATGGTTTTTATATTATC (NdeI) | |
| TRB274 | GTGGTTGCTCTTCCGCACAAATGTGTTGCTGGTAAATC (SapI) |
*For CptI primers, the region encoding the repeat is shown in uppercase.
Initial crystallization trials were carried out in 96-well tray sitting drops of 200 nl of protein mixed with 200 nl of precipitant in a vapour diffusion system with 200 μl of the precipitant in a reservoir, using the Classics, JCSG+ and Protein Complex commercial screens (Qiagen). Hits were optimized using 24-well tray hanging drops of 1 μl of protein mixed with 1 μl of precipitant with 500 μl of precipitant in the reservoir. The best diffracting crystals for both the native and the SeMet derivative were from a condition containing 200 mM CaCl2, 100 mM CH3CO2Na, 20% v/v 2-propanol, pH 4.8. Data collection was performed on the I-02 beamline at the Diamond Light Source synchrotron facility (Harwell, Oxford, UK), with a single crystal of the native complex diffracting to 2.2 Å and two crystals of the SeMet derivative, both of which diffracted to 3.0 Å (summarized in Table 1). Data processing and reduction were performed on these datasets using iMOSFLM (24), SCALA (25) and TRUNCATE (26) from the CCP4 suite of programs (27). The combined SeMet data was input as a single-wavelength anomalous dispersion (SAD) set into the AutoSolve function in PHENIX (28), yielding a solution with 40 selenium sites (corresponding to eight protein monomers) with a Figure of Merit of 0.385 and a readily interpretable electron density map. It was noticed that where there was obvious density corresponding to RNA, protein residues had instead been placed by PHENIX. For this reason, the prebuilt chains were removed and chain re-traced using firstly BUCCANEER (29) for the protein and then NAUTILUS (30) for the RNA. Manual chain building was continued using COOT (31), as well as the manual addition of waters; very high positive peaks observed in the FO-FC map showed the presence of Ca2+ ions from the crystallization condition, also corroborated by the presence of clear octahedral coordination from adjacent waters and RNA phosphate groups. The model could then be directly refined using REFMAC5 (32) against the isomorphous native dataset, and the final 2.2 Å structure contained eight protein chains of CptNEr from residues 1 to 155, as well as eight full 45 nt chains of the CptIEr RNA (with Ramachandran statistics of 98.3% preferred, 1.6% allowed and 0.1% outliers).
Table 1. Crystallographic and structural refinement data.
| Native | SeMet | |
|---|---|---|
| Diffraction Statistics | ||
| Space group | P21 | P21 |
| Cell dimensions | ||
| a, b, c (Å) | 63.1, 185.9, 138.8 | 63.2, 186.5, 139.2 |
| α, β, γ (°) | 90, 92.63, 90 | 90, 92.58, 90 |
| Resolution (Å) | 2.2 (2.24–2.20) | 3.0 (3.07–3.00) |
| Rmerge | 0.048 (0.338) | 0.164 (0.612) |
| I/σI | 11.6 (2.4) | 13.9. (4.8) |
| Completeness (%) | 97.9 (97.0) | 100 (100) |
| Redundancy | 2.3 (2.1) | 13.5 (12.6) |
| Number of unique reflections | 157 960 (7761) | 64 337 (4487) |
| Wilson B factor (Å2) | 28.8 | 44.1 |
| Refinement statistics | ||
| Rfactor | 0.20 | |
| Rfree | 0.23 | |
| Number of reflections used | 161 398 | |
| Number of atoms | ||
| Protein | 10409 | |
| RNA | 7696 | |
| Ion | 52 | |
| Water | 1330 | |
| Average B factors | ||
| Protein | 41.4 | |
| RNA | 42.6 | |
| Ion | 59.9 | |
| Water | 39.8 | |
| Rmsd (bonds) | 0.013 | |
| Rmsd (angles) | 1.72 | |
RNA structural searches
AMIGOS II (33) was used to calculate RNA torsional angles and to perform structural searches. Structural searches were performed using the ‘Worm search’ function on all available RNA structures in the PDB.
Construction of CptIEr mutants
Overexpression vectors encoding either a single CptIEr repeat, or a mutated repeat, were constructed using a strategy described previously (13), where a PCR was performed with pQE-80L as the template, using PF185 together with a reverse primer encoding the repeat sequence. An additional 5′-GA and 3′-AAG was included at the boundaries of the repeat to allow processing by CptNEr. Primer and plasmid details are in Table 2.
CptI antitoxicity and cross-inhibition assays
Antitoxicity assays were performed in E. coli DH5α that had been transformed with separately-inducible CptI and CptN plasmids (Table 2). Assays were performed as described previously (13,16).
Plasmid loss assays
Plasmid loss assays were performed in E. coli W3110 carrying either pFLS122 or pRBJ200 (Table 2). Experiments were performed essentially as described (34), with non-selective exponential growth maintained for 24 h.
RESULTS
CptINEr is a heterotetramer
The cptIN locus of E. rectale (Figure 1A) was previously shown to have TA function (16). To explore the structural basis for this activity, and the extent to which its mechanism is shared with ToxIN systems, the structure of the CptINEr complex was solved by X-ray crystallography. CptIEr co-purified with the CptNEr protein following overexpression in E. coli, and the stable protein–RNA complex was crystallized in both the native and SeMet forms. The best diffracting crystals (1 native and 2 SeMet) were isomorphous, and, following phasing through SAD, the final structure was solved to a resolution of 2.2 Å (Table 1).
Figure 1.

The crystal structure of CptINEr to 2.2Å resolution. (A) The architecture of the CptINEr locus, showing the tandem repeats upstream of the toxic CptNEr ORF that become processed into CptIEr monomers. (B) When analysed with symmetry cells in conjunction with data from size exclusion chromatography, the biological unit was found to be a heterotetramer—an oval structure with two protein monomers at the poles (CptNEr) held together by two pseudoknotted RNAs (CptIEr). The surface representation shows the electrostatic potential (red: electronegative, blue: electropositive. (PDB: 4RMO) (C) The structural core of the protein is a highly twisted β-sheet surrounded by α-helices. (D) The overlay with carbon-α traces of ToxNPa (magenta, PDB: 2XDD) and Kid, a Type II RNase (cyan, PDB: 1M1F), shows the lack of the kinked helix seen in ToxNPa in CptNEr, but still an overall similarity in fold.
The unit cell contained a total of eight protein (CptNEr) monomers each complexed with a single CptIEr RNA repeat of 45 nt. Examination of the symmetry units (Supplementary Figure S1) reveals that each protein–RNA heterodimer formed a self-closing, heterotetrameric ring-shaped complex with a heterodimer from the neighbouring asymmetric unit (Figure 1B). The idea that the biological CptIN complex was the heterotetramer (of two protein monomers and two RNA ligands) observed in the crystal structure was corroborated by analytical size exclusion chromatography (Supplementary Figure S2 and Table S1). Note that both crystallization and size exclusion chromatography use high concentrations of CptN and CptI; the concentration of these molecules in the cell is unknown, and complex assembly may be strongly influenced by the turnover of these components and their interactions with other molecules. Studies on a homologous system found that the results from gel filtration could be further corroborated by analytical ultracentrifugation. The heterotetrameric architecture is a major deviation from the heterohexameric complexes formed by the other structurally-defined Type III TA systems.
The structural core of the CptN protein is similar to the structures of the three previously solved Type III toxins, being made up of a highly twisted antiparallel β-sheet (Figure 1C). This is surrounded by four α-helices, which, along with the many loops in the structure, make extensive contacts with the cognate RNA (Figure 1B). Of note is the absence of the kink in the third helix that is seen in the structures of ToxNPa (14), ToxNBt (15) and AbiQ (35); the equivalent helix H4 in CptNEr is visibly shorter in comparison (Figure 1C and D) and, as a result, CptNEr more closely resembles the structure of the Type II Toxin Kid.
Each CptNEr protein binds two CptIEr RNAs, and the protein–RNA interfaces are considerable for both. Interface 1, which connects the protein to the 5′ end of the RNA, is the larger of the two and spans over 1250 Å2 (Figure 2A). Of note here is the strong electropositive tract (shown in blue) created by the RNA-facing side of helix H3, stabilizing the phosphate backbone and allowing the bases to stack and face into the protein. Also of note are the bases U4 and C6, which do not stack with adjacent bases, but are instead bound in hydrophobic pockets (residues Ile60, Ile74, Leu88 and Ile73, Val129, Phe139, Met132 respectively) that would otherwise be exposed to solvent. Interface 2, which spans an area of just over 1000 Å2, is made up of largely polar contacts between the protein and exposed bases at the 3′ end of CptIEr (Figure 2B). A predicted hydrophobic interaction with Ile106 presumably helps to stabilize the proximal end of the S2 duplex, and the Tyr104 residue appears to be important both in helping to stabilize the position of the scissile A45 nucleotide, as well as stacking onto G44.
Figure 2.

Diversity in the two protein–RNA interfaces in CptINEr. (A) The surface representation shows the electrostatic potential (red: electronegative, blue: electropositive. Interface 1 is dominated by a long electropositive tract created by helix H3 interacting with the phosphate backbone, as well as the stabilization of U4 and C6 by a hydrophobic groove created through the labelled side chains. (B) Interface 2 lacks any dominant electropositive surface from the protein and is instead formed through a number of specific polar interactions with hydrophilic residues on the outside of the protein. Key residues are labelled and shown as sticks, with hydrogen bonds shown as black dashed lines. An important hydrophobic interaction is made to the proximal end of the RNA duplex by Ile106.
The angle that the protein component makes with the antitoxin strands is related to the quaternary state as it forces the complex into a particular geometry; the angle that CptIEr makes with CptNEr is significantly smaller than that between ToxI and ToxN (Figure 3), and the number of amino acid residues that are involved in contacting the RNA ligand is high.
Figure 3.

The heterotetrameric quaternary structure and the geometry of the bound RNA. (A) It can be seen that the incoming and outgoing RNA strands in CptINEr are completely parallel, driven by a much more extensive single stranded region at both the 5′ and 3′ ends of CptIEr looping into the protein. (B) In ToxINPa, the interfaces are much more direct in relation to the ToxIPa duplex, creating a 60° angle between adjacent ToxIPa strands, leading to the formation of a heterohexamer.
The CptIEr RNA is an H-type pseudoknot with a functionally important extended A-minor twist and no major groove loop
The CptIEr RNA observed bound to CptNEr is a classic H-type pseudoknot of 45 nt, which is derived from the two genetic cptIEr repeats. The CptIEr pseudoknot comprises two coaxial stem (base-paired) regions S1 and S2, with two loops L1 and L2, with L1 being extremely short (one base), and a much longer L2 loop (Figure 4A). The previously-solved Type III antitoxin structures—the ToxI RNAs from Pectobacterium and Bacillus (14,15)—were found to form very similar H-type pseudoknots, and the tertiary structures of both were maintained by intricate triplex interactions (which were largely conserved between the two antitoxins) between their loop sections and the associated stems. While the Pectobacterium and Bacillus ToxI structures are very similar to each other, the fold of the CptIEr differs markedly from the ToxI paradigm, and is also structurally extremely unusual among known RNA pseudoknots notably in the structure of the loops.
Figure 4.
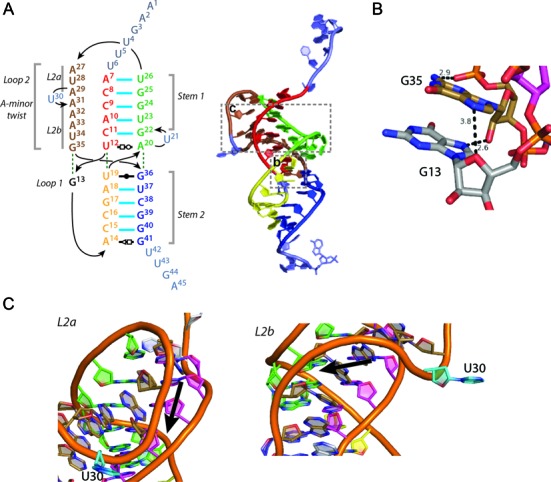
CptIEr is an H-Type pseudoknot with unique features in its L1 and L2 loop regions. (A) Left: a coloured schematic of CptIEr showing its two stems (S1 and S2), and two loop regions (L1 and L2); right: a representation of the schematic on a cartoon of a single CptIEr monomer. (B) A close-up of the L1 loop region, unique in its dissociation with the stems altogether. It instead stacks with the proximal end of the L2 loop. (C) Close-up cartoon representations of the L2 loop region (brown), which can be divided into two halves—L2a and L2b. Above: the upper half (L2a) is a unique counterclockwise A-minor twist; below: the lower half (L2b) is a clockwise A-minor twist, with the change in directionality mediated by the ‘kink’ nucleotide (U30, teal).
In most pseudoknots the L1 loop interacts with the major groove of the S2 helix, and L2 interacts with the minor groove of the S1 helix. This topology often leads to the triplex interactions seen in many pseudoknots. In CptIEr, the single base that forms the L1 loop, G13, does not interact with the major groove of S2 at all and is held in place by base stacking with the 3′ end of the L2 loop (G35), at the same time forming some unique intra-chain hydrogen bonds (Figure 4B). The L2 loop (A27-G35) in CptIEr is much longer than those of the ToxIs, and is very A-rich. Whilst L2 follows the classical association with the minor groove of S1, the details of the interaction are not standard (Figures 4C and 5). L2 forms a short, three-stranded helix which has been previously referred to as an A-minor twist motif (21). In other RNA species, the widely occurring A-minor motif involves interaction between an unpaired adenine base with the minor groove of an RNA duplex, usually involving interactions with both the exposed bases in the groove as well as the RNA backbone. A-minor twist motifs can be seen as extensions of the classical A-minor types and were first seen in the SAM-II riboswitch (36) and as part of the glmS ribozyme structure (37). Both structures reported a series of adenine bases associated with the minor groove of a duplex at an angle of ∼70° with respect to the duplex helical axis. The L2 loop in CptIEr is similar, but, being far longer than either of the previously described A-minor twists, shows further detail as to how this motif can be extended. The motifs in the SAM-II riboswitch and in glmS were shown to rotate around the helix clockwise. Similarly, the section of the loop marked L2b (A31-G35) also rotates around S1 in a clockwise manner (Figure 4C), with the polar interactions surrounding one particular nucleotide, A33, and the similarity of these interactions to those surrounding A37 in the riboswitch structure reflecting this (Figure 5C and D). As well as the nature of the interactions being made with multiple ‘tiers’ of the S1 duplex, a number of interesting features of L2b should be noted, including the intricate hydrogen bond network centred on the 2′ hydroxyl oxygen atoms of A33 and U23, as well as an unusual interaction between the 4′ oxygen of A33 and the 2′ hydroxyl of G24 (Figure 5A and C).
Figure 5.
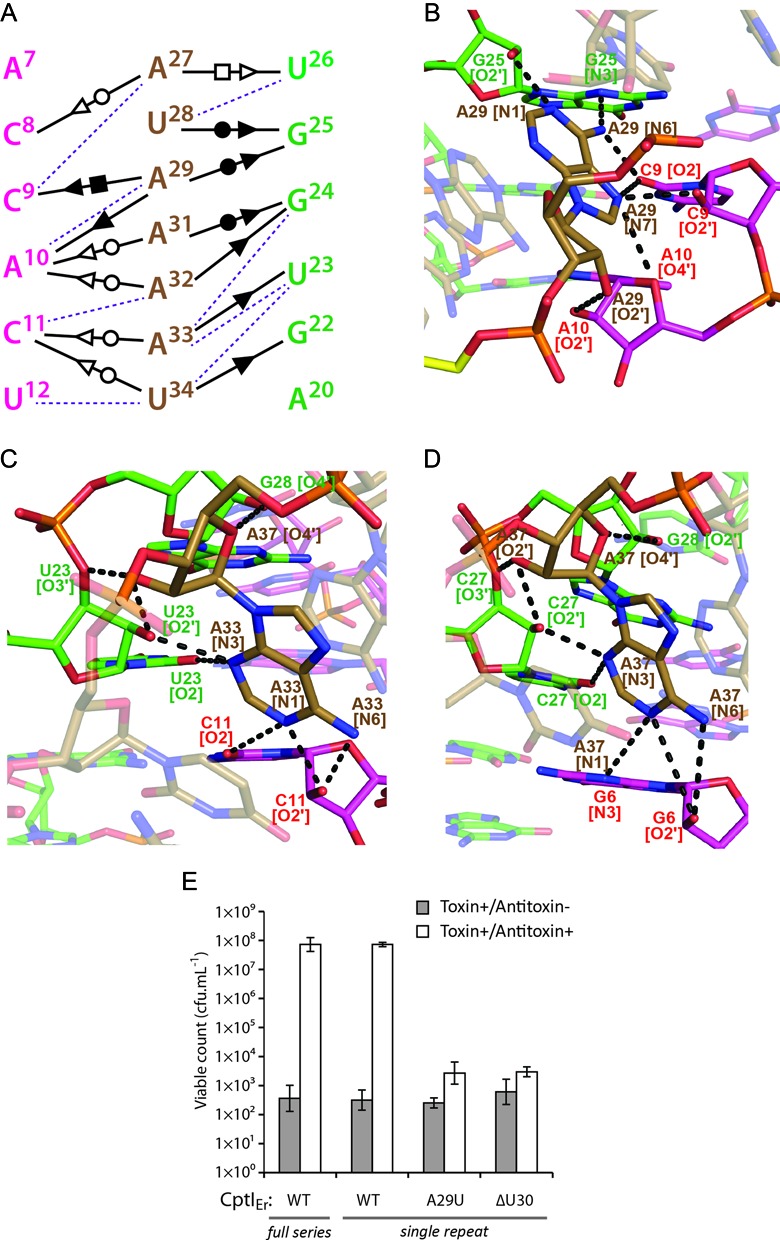
The A-minor twist motif is stabilized by extensive, multi-tiered, hydrogen bond networks and is required for antitoxicity. (A) A schematic of the complex and interactions observed in the A-minor twist motif seen in isolation from the rest of the pseudoknot. Purple dashes represent unusual polar interactions with no known notation, representing interactions involving the O3’ or O4’ of the ribose sugar. (B) Interactions in the counterclockwise A-minor twist are dominated by interactions with the backbone of the red half of the stem (A7–10). Black dashes represent hydrogen bonds. The U30 kink nucleotide is again highlighted here in yellow. (C) The opposite is seen in the clockwise A-minor twist, where the strongest networks are formed on the green half of the stem (G22–24). (D) An identical network can be seen in a previously identified A-minor motif in an unrelated structured RNA, the SAM-II riboswitch (PDB: 2QWY). (E) Growth of Escherichia coli DH5α strains overexpressing CptNEr and CptIEr. Overexpression of CptNEr prevents growth, which is rescued by co-overexpression of the full series of CptIEr repeats. A single CptIEr repeat, corresponding to the processed unit observed in the crystal structure, shows full antitoxicity. However, mutants CptIEr-A29U and CptIEr-ΔU30 are unable to counteract the toxicity of CptNEr. Results shown are mean ± standard deviation for three biological replicates.
In contrast to L2b, L2a (A27–A29) is a counter-clockwise twist of 3 nt with its own unique set of polar interactions (Figure 5A); this displays an equally extended network of hydrogen bonds as exemplified by the schematic shown for the nucleotide A29 (Figure 5B). Of note is the fact that this counter-clockwise twist was not seen adjacent to the A-minor twist in glmS or the SAM-II riboswitch. To determine whether this counter-clockwise twist had been seen in any previously published RNA structures, a search was performed using the calculated torsional angles (Supplementary Table S2) for nucleotides A27–A29. No examples of nucleotide stretches with similar torsional angles (total deviation <40) that also interacted with the minor groove of a double helix were found in any previously described RNA structure. This suggests that the counter-clockwise A-minor twist is a novel motif.
A schematic of all the interactions in the CptI A-minor twist summarizes the extensive network encompassing the entirety of L2 (Figure 5A). The only bases that do not participate in the network directly are U30, which faces outwards (and therefore makes no polar contacts with the pseudoknot itself) but is instrumental in changing the directionality of the twist, and G35, which appears to not make any interactions with the minor groove itself yet stacks with the rest of the bases in the twist, and acts to bridge the twist with the sole L1 nucleotide, G13 (Figure 4B). In both the reported examples of A-minor twist motifs (36,37), the segment was small. However, the potential for it to be lengthened further with the aid of a ‘kink’ nucleotide such as U30, as shown here, reveals the capacity for the A-minor twist to be extended into a dominant structural feature. To test the functional significance of the extended A-minor twist, site-directed mutants of CptIEr were tested for antitoxicity against CptNEr (Figure 5E). As shown, deletion of the ‘kink’ nucleotide U30, or substitution of the adjacent A29 for U, rendered CptIEr unable to rescue the growth inhibition caused by CptNEr. This result suggests that the extended A-minor twist seen in the structure is important for CptIEr function, and that the structural integrity of this feature depends on the presence of a ‘kink’ nucleotide to facilitate a change of direction of the twist, and on extensive hydrogen-bonding between A29 and the RNA duplex to stabilize the counter-clockwise portion of this motif.
CptI RNAs show selective antitoxicity
The nature of the interactions observed at the protein–RNA interfaces of the CptIN complex suggested that antitoxin recognition within this family would be highly specific, as precise CptI–CptN contacts would be needed to maintain the inactive complex. To explore this hypothesis, cross-inhibition experiments between CptINEr components and the toxin and antitoxin components of the two other validated Type III TA systems of the CptIN family were performed. The three CptIN systems (CptINEr; CptINRt from Ruminococcus torques L2–14; and CptINCc from Coprococcus catus GD-7) share a common locus organization and 35–44% sequence identity between their toxin components. Expression of any of the three CptN toxins was sufficient to cause a dramatic drop in viable count, and this was restored by co-overexpression of the cognate CptI antitoxin (Figure 6). Antitoxicity was not observed in any of the six strains expressing non-cognate CptI and CptN combinations. CptI RNA antitoxins are therefore highly selective toxin inhibitors. This exquisite selectivity was also observed in the ToxIN family (15) and supports the hypothesis that the enzyme and its substrate have co-evolved as a mutually compensating pair.
Figure 6.

CptN toxins are specific for their cognate CptI antitoxins. Kill/rescue assay with different combinations of CptI and CptN. All three toxins could be neutralized by their cognate antitoxin, but not by the non-cognate CptIs. Results shown are mean ± standard deviation for three biological replicates.
The active site of CptNEr is conserved with ToxN toxins
The RNA chains seen in the CptIN complex structure each correspond to single repeats from the repetitive CptI sequence and a cyclic phosphate is seen at the 3′ end of each RNA chain on the A45 nucleotide (Figure 7); these observations indicate that the CptN protein is, like ToxN, a ribonuclease that processes its own antitoxin. Although there is very little primary sequence identity between CptNEr and any of the ToxNs, the hydrogen bonding networks in their RNase active sites are well conserved. However, some deviations can be seen. One is the use of two serine residues in the conserved triad of [Ser]-[Ser/Thr]-[Arg] instead of threonine as in the ToxNs. Another is in the group of residues specifying the adenine base at the site of cleavage: the coordinating glutamine is instead replaced by two residues (Asp81 and Ser85), and the coordinating tyrosine (Tyr104) approaches from below the plane of the base as depicted, instead of above, as in the ToxN structures. This positioning also stabilizes the entry of the RNA by stacking with G44, as described above. The combined evidence from the structures of, now, three active sites in complex with their RNA products suggest a catalytic mechanism that is similar to that seen in Ribonuclease A (38), with the residues mentioned conserved specifically to stabilize the generation of the cyclic phosphate. A similar mechanism was postulated in the resolution of the AbiQ structure (35).
Figure 7.
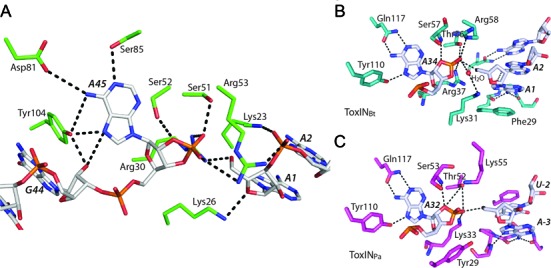
A conserved active site and mechanism for the RNase activity of CptNEr. (A) A stick representation of the active site residues surrounding the scissile adenine base (A45). Of note is Tyr104 which appears instrumental in both specifying for the adenine, as well as stacking onto the adjacent bases. (B and C) Show similar representations of the active sites from ToxNPa and ToxNBt, showing a conserved mechanism and hydrogen bond cage, despite significant primary sequence differences elsewhere.
The TA locus CptIN stabilizes plasmids, but does not abort phage infection in E. coli
Type III TA systems of the ToxIN family have roles in bacteriophage resistance and plasmid stabilization (13,15,35). The cptINEr locus was therefore tested for capacity to confer these phenotypes on a bacterial host. In the absence of known Eubacterium phages, we assessed impacts on phage of the CptINEr system reconstituted in E. coli. To examine bacteriophage resistance, an E. coli strain expressing CptINEr was screened with over 200 environmentally isolated ‘virgin’ coliphages. However, no difference was seen in the plaque forming ability of these phages in comparison to the control strain without plasmid. To explore plasmid stabilization by cptINEr, plasmid loss assays were performed in E. coli W3110 over the course of 24 h. As shown (Figure 8), the test vector was lost from ∼50% of the cells over this period, while the presence of cptINEr increased plasmid retention in the E. coli population to 100%. Although cptINEr is located on the chromosome in the original host, we suggest that its plasmid stabilization property is likely to also promote its retention in E. rectale and in other hosts that may receive this locus through horizontal transfer.
Figure 8.

CptINEr stabilizes plasmids in vivo. Proportion of plasmid-containing Escherichia coli W3110 cells before (white) or after (grey) 24 h of non-selective exponential growth. Results shown are mean ± standard deviation for three biological replicates.
DISCUSSION
The present work is the first structural study of a new family of Type III loci, the CptINs, originally identified in a bioinformatics search for the distribution of Type III TA systems. With the precedent set by the ToxIN system structures (the two ToxINs from Pectobacterium (14) and Bacillus (15), and the AbiQ structure from Lactococcus (35)) of the formation of a heterohexameric triangular oligomer, it was expected that the CptIN and TenpIN families would also exhibit this complex common architecture; indeed the other Type III families were found using the structure of ToxNPa as the initial seed in the bioinformatic search (16).
Unexpectedly, the CptINEr structure solved here forms a heterotetrameric complex (Figure 1B), which was validated by analytical size exclusion chromatography (Supplementary Figure S1). While the biological reason behind the differing organization of the tetrameric and hexameric Type III TA systems remains unknown, it is not unreasonable to suggest that their respective quaternary structures may play a role in their relative stabilities within the cell. A higher order complex necessitates a greater entropic penalty in its formation and thus may be more likely to be disrupted and dissociate in the cell.
The CptIEr structure adds evidence to the now increasing perception that RNA molecules, like proteins, can form diverse structures to meet requirements for molecular recognition (17). The A-minor triple helix in the form of two adjacent A-minor twist motifs (Figures 4 and 5) is truly remarkable in the multiple levels of interactions that are formed in order to stabilize the duplex, and along with it, the pseudoknot itself. The occurrence of the A-minor twist in the SAM-II riboswitch lends strength to the idea that there are a limited number of RNA folds used in nature. With all Type III antitoxins to date found to have a pseudoknot structure, it could be that the features of pseudoknots are both sufficient and necessary for its functionality; the complex intramolecular interactions allow for the generation of a large interacting surface, while the single stranded nature of the 5′ and 3′ ends allow for processing by a co-factor. Nonetheless, taking into account the number of unusual interactions seen (hydrogen bonding to the 3′ and 4′ ribose oxygens), this CptIEr structure illustrates the variety of intra-strand RNA interactions that can occur.
This particular structure also raises some interesting questions regarding the evolution of the Type III TA systems. The conservation of the cage of residues in the active site shows the importance of this core to the toxic function of the system, but the low primary sequence identity (<15% between CptNEr to either of the ToxNs) overall implies a high degree of malleability when nucleating the secondary structure around it. The three Type III families seen appear to therefore represent three energetically favourable ‘solutions’ to forming this protein–RNA complex. The lack of cross-reactivity between even extremely similar antitoxin RNAs (Figure 6) show that anything that would disrupt the TA association while maintaining toxicity should ultimately lead to a lower fitness for the cell. It therefore seems that any deleterious mutation in one component must be compensated by mutation in the same or partner component. As one component is an enzyme and the other the target substrate, this system represents a distinctive case in molecular evolution in which substrates co-vary with the enzyme.
Functionally, there are still many mysteries surrounding TA systems. The plasmid stabilization shows the selfish nature of these loci, independent of host context. While no phage resistance was seen in the experiments conducted in E. coli, the fact that such structural diversity is seen, even with the integrity of the TA functionality being so critical, hints at the importance of these loci in bacterial physiology.
Supplementary Material
Acknowledgments
We thank Dr Keith Turner for providing the genomic DNA of E. rectale DSM 17629. We thank the beamline scientists at station I-02 at Diamond Light Source, Harwell, UK for use of facilities and assistance with diffraction data collection.
Footnotes
Present addresses:
Francesca L. Short, College of Life Sciences, University of Dundee, Dow Street, Dundee DD1 5EH, U.K.
Jarrod E. Voss, Department of Biochemistry and Molecular Biology, School of Biomedical Sciences, Monash University, Clayton, Victoria 3800, Australia.
Tim R. Blower, School of Biological and Biomedical Sciences and Department of Chemistry, Durham University, Durham DH1 3LE, U.K.
Anastasia L. Orme, John Innes Centre, Norwich Research Park, Norwich NR4 7UH, U.K.
Tom E. Whittaker, School of Mines Building, Imperial College London, London SW7 2BP, U.K.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Biotechnology and Biological Sciences Research Council [BB/H002677/1 to G.P.C.S and B.F.L.]; Wellcome Trust [RG61065 to B.F.L.]; Biotechnology and Biological Sciences Research Council studentship (to F.R.); Commonwealth Scholarships Commission (UK) Scholarship (to F.L.S.); Herschel Smith Scholarship, University of Cambridge (to J.E.V.). Funding for open access charge: Biotechnology and Biological Sciences Research Council.
Conflict of interest statement. None declared.
REFERENCES
- 1.Gerdes K., Larsen J.E., Molin S. Stable inheritance of plasmid R1 requires two different loci. J. Bacteriol. 1985;161:292–298. doi: 10.1128/jb.161.1.292-298.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ogura T., Hiraga S. Mini-F plasmid genes that couple host cell division to plasmid proliferation. Proc. Natl. Acad. Sci. U.S.A. 1983;80:4784–4788. doi: 10.1073/pnas.80.15.4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hayes F., Melderen L. Toxins-antitoxins: diversity, evolution and function. Crit. Rev. Biochem. Mol. Biol. 2011;46:386–408. doi: 10.3109/10409238.2011.600437. [DOI] [PubMed] [Google Scholar]
- 4.Dy R.L., Przybilski R., Semeijn K., Salmond G.P.C., Fineran P.C. A widespread bacteriophage abortive infection system functions through a Type IV toxin–antitoxin mechanism. Nucleic Acids Res. 2014;42:4590–4605. doi: 10.1093/nar/gkt1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Masuda H., Tan Q., Awano N., Wu K.-P., Inouye M. YeeU enhances the bundling of cytoskeletal polymers of MreB and FtsZ, antagonizing the CbtA (YeeV) toxicity in Escherichia coli. Mol. Microbiol. 2012;84:979–989. doi: 10.1111/j.1365-2958.2012.08068.x. [DOI] [PubMed] [Google Scholar]
- 6.Wang X., Lord D.M., Cheng H.-Y., Osbourne D.O., Hong S.H., Sanchez-Torres V., Quiroga C., Zheng K., Herrmann T., Peti W., et al. A new type V toxin-antitoxin system where mRNA for toxin GhoT is cleaved by antitoxin GhoS. Nat. Chem. Biol. 2012;8:855–861. doi: 10.1038/nchembio.1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fozo E.M., Makarova K.S., Shabalina S.A., Yutin N., Koonin E.V., Storz G. Abundance of type I toxin-antitoxin systems in bacteria: searches for new candidates and discovery of novel families. Nucleic Acids Res. 2010;38:3743–3759. doi: 10.1093/nar/gkq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leplae R., Geeraerts D., Hallez R., Guglielmini J., Dreze P., Van Melderen L. Diversity of bacterial type II toxin-antitoxin systems: a comprehensive search and functional analysis of novel families. Nucleic Acids Res. 2011;39:5513–5525. doi: 10.1093/nar/gkr131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamaguchi Y., Park J.-H., Inouye M. Toxin-antitoxin systems in bacteria and archaea. Annu. Rev. Genet. 2011;45:61–79. doi: 10.1146/annurev-genet-110410-132412. [DOI] [PubMed] [Google Scholar]
- 10.Gerdes K., Christensen S.K., Løbner-Olesen A. Prokaryotic toxin–antitoxin stress response loci. Nat. Rev. Microbiol. 2005;3:371–382. doi: 10.1038/nrmicro1147. [DOI] [PubMed] [Google Scholar]
- 11.Van Melderen L. Toxin-antitoxin systems: why so many, what for. Curr. Opin. Microbiol. 2010;13:781–785. doi: 10.1016/j.mib.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 12.Unterholzner S.J., Poppenberger B., Rozhon W. Toxin-antitoxin systems. Mob. Genet. Elem. 2013;3:e26219. doi: 10.4161/mge.26219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fineran P.C., Blower T.R., Foulds I.J., Humphreys D.P., Lilley K.S., Salmond G.P.C. The phage abortive infection system, ToxIN, functions as a protein–RNA toxin–antitoxin pair. Proc. Natl. Acad. Sci. U.S.A. 2009;106:894–899. doi: 10.1073/pnas.0808832106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blower T.R., Pei X.Y., Short F.L., Fineran P.C., Humphreys D.P., Luisi B.F., Salmond G.P.C. A processed noncoding RNA regulates an altruistic bacterial antiviral system. Nat. Struct. Mol. Biol. 2011;18:185–190. doi: 10.1038/nsmb.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Short F.L., Pei X.Y., Blower T.R., Ong S.-L., Fineran P.C., Luisi B.F., Salmond G.P.C. Selectivity and self-assembly in the control of a bacterial toxin by an antitoxic noncoding RNA pseudoknot. Proc. Natl. Acad. Sci. U.S.A. 2013;110:E241–E249. doi: 10.1073/pnas.1216039110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blower T.R., Short F.L., Rao F., Mizuguchi K., Pei X.Y., Fineran P.C., Luisi B.F., Salmond G.P.C. Identification and classification of bacterial Type III toxin–antitoxin systems encoded in chromosomal and plasmid genomes. Nucleic Acids Res. 2012;40:6158–6173. doi: 10.1093/nar/gks231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Storz G., Vogel J., Wassarman K.M. Regulation by small RNAs in bacteria: expanding frontiers. Mol. Cell. 2011;43:880–891. doi: 10.1016/j.molcel.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vogel J., Luisi B.F. Hfq and its constellation of RNA. Nat. Rev. Microbiol. 2011;9:578–589. doi: 10.1038/nrmicro2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Serganov A., Nudler E. A decade of riboswitches. Cell. 2013;152:17–24. doi: 10.1016/j.cell.2012.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Westra E.R., Swarts D.C., Staals R.H.J., Jore M.M., Brouns S.J.J., van der Oost J. The CRISPRs, they are A-Changin’: how prokaryotes generate adaptive immunity. Annu. Rev. Genet. 2012;46:311–339. doi: 10.1146/annurev-genet-110711-155447. [DOI] [PubMed] [Google Scholar]
- 21.Ellington A.D., Szostak J.W. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 22.Chang A.C., Cohen S.N. Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J. Bacteriol. 1978;134:1141–1156. doi: 10.1128/jb.134.3.1141-1156.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paterson N.G., Riboldi-Tunnicliffe A., Mitchell T.J., Isaacs N.W. Purification, crystallization and preliminary X-ray diffraction analysis of RafE, a sugar-binding lipoprotein from Streptococcus pneumoniae. Acta Crystallograph. Sect. F Struct. Biol. Cryst. Commun. 2006;62:676–679. doi: 10.1107/S1744309106021695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Battye T.G.G., Kontogiannis L., Johnson O., Powell H.R., Leslie A.G.W. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 2011;67:271–281. doi: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Evans P.R. An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr. D Biol. Crystallogr. 2011;67:282–292. doi: 10.1107/S090744491003982X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.French S., Wilson K. On the treatment of negative intensity observations. Acta Crystallogr. Sect. A. 1978;34:517–525. [Google Scholar]
- 27.Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G.W., McCoy A., et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.-W., Kapral G.J., Grosse-Kunstleve R.W., et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cowtan K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr. 2006;62:1002–1011. doi: 10.1107/S0907444906022116. [DOI] [PubMed] [Google Scholar]
- 30.Cowtan K. Fitting molecular fragments into electron density. Acta Crystallogr. D Biol. Crystallogr. 2008;64:83–89. doi: 10.1107/S0907444907033938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murshudov G.N., Vagin A.A., Dodson E.J. Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 33.Wadley L.M., Keating K.S., Duarte C.M., Pyle A.M. Evaluating and learning from RNA pseudotorsional space: quantitative validation of a reduced representation for RNA structure. J. Mol. Biol. 2007;372:942–957. doi: 10.1016/j.jmb.2007.06.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Christensen-Dalsgaard M., Gerdes K. Two higBA loci in the Vibrio cholerae superintegron encode mRNA cleaving enzymes and can stabilize plasmids. Mol. Microbiol. 2006;62:397–411. doi: 10.1111/j.1365-2958.2006.05385.x. [DOI] [PubMed] [Google Scholar]
- 35.Samson J.E., Spinelli S., Cambillau C., Moineau S. Structure and activity of AbiQ, a lactococcal endoribonuclease belonging to the type III toxin-antitoxin system. Mol. Microbiol. 2013;87:756–768. doi: 10.1111/mmi.12129. [DOI] [PubMed] [Google Scholar]
- 36.Gilbert S.D., Rambo R.P., Van Tyne D., Batey R.T. Structure of the SAM-II riboswitch bound to S-adenosylmethionine. Nat. Struct. Mol. Biol. 2008;15:177–182. doi: 10.1038/nsmb.1371. [DOI] [PubMed] [Google Scholar]
- 37.Klein D.J., Ferré-D'Amaré A.R. Structural basis of glmS Ribozyme activation by glucosamine-6-phosphate. Science. 2006;313:1752–1756. doi: 10.1126/science.1129666. [DOI] [PubMed] [Google Scholar]
- 38.Raines R.T. Active site of ribonuclease A. In: Zenkova DMA, editor. Artificial Nucleases: Nucleic Acids and Molecular Biology. Berlin Heidelberg: Springer; 2004. pp. 19–32. [Google Scholar]
- 39.Bachmann B.J. Pedigrees of some mutant strains of Escherichia coli K-12. Bacteriol. Rev. 1972;36:525–557. doi: 10.1128/br.36.4.525-557.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jensen R.B., Grohmann E., Schwab H., Díaz-Orejas R., Gerdes K. Comparison of ccd of F, parDE of RP4, and parD of R1 using a novel conditional replication control system of plasmid R1. Mol. Microbiol. 1995;17:211–220. doi: 10.1111/j.1365-2958.1995.mmi_17020211.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.