

Abstract
Loss of ATRX leads to epigenetic alterations, including abnormal levels of DNA methylation at repetitive elements such as telomeres in murine cells. We conducted an extensive DNA methylation and mRNA expression profile study on a cohort of 82 patients with astrocytic tumors to study whether ATRX expression was associated with DNA methylation level in astrocytic tumors and in which cellular functions it participated. We observed that astrocytic tumors with lower ATRX expression harbored higher DNA methylation level at chromatin end and astrocytic tumors with ATRX-low had distinct gene expression profile and DNA methylation profile compared with ATRX-high tumors. Then, we uncovered that several ATRX associated biological functions in the DNA methylation and mRNA expression profile (GEP), including apoptotic process, DNA-dependent positive regulation of transcription, chromatin modification, and observed that ATRX expression was companied by MGMT methylation and expression. We also found that loss of ATRX caused by siRNA induced apoptotic cells increasing, reduced tumor cell proliferation and repressed the cell migration in glioma cells. Our results showed ATRX-related regulatory functions of the combined profiles from DNA methylation and mRNA expression in astrocytic tumors, and delineated that loss of ATRX impacted biological behaviors of astrocytic tumor cells, providing important resources for future dissection of ATRX role in glioma.
Keywords: ATRX, DNA methylation, chromosome end, MGMT, biological behaviors
INTRODUCTION
Astrocytic tumors are the most common primary malignant brain tumors in adults [1]. They are heterogeneous in cellular composition consisting of tumor stem cells, mesenchymal cells, and host stromal cells. Primary glioblastoma can arise de novo, whereas secondary glioblastoma is thought to arise from lower-grade gliomas [2]. Lots of studies have resulted in the identification of various chromosomes and genes that are frequently altered in astrocytic tumours [3].
Mutations in X-linked alpha thalassaemia mental retardation (ATRX), encoding a SWI/SNF-like protein, were first identified in patients bearing the X-linked alpha thalassemia/mental retardation syndrome [4, 5]. ATRX plays a variety of key role at tandem repeat sequences within the genome, including prevention of replication fork stalling, the deposition of a histone variant, and the suppression of a homologous recombination-based pathway of telomere maintenance [6–8]. Recent reports described that ATRX mutation or loss occurred at high percentages in multiple tumor types, including low grade astrocytoma and secondary glioblastoma, suggestive of a potential “driver” role in cancer [9–11]. And several teams demonstrated that ATRX alteration, combined with other classical biomarkers, refined the molecular classification of adult gliomas, providing a prognostic tool for clinicians [12–14]. In addition, loss of ATRX leads to epigenetic alterations, including abnormal levels of DNA methylation at repetitive elements such as telomeres in murine cells [15]. Epigenetic alterations are now accepted as having a role in tumorigenesis [16]. DNA methylation alterations have been widely reported in human glioblastoma multiform (GBM) and other glioma subtypes [17, 18]. In 2013, from The Cancer Genome Atlas (TCGA)'s report, GBMs harboring ATRX mutation were enriched in G-CIMP + subtype [3]. In current work, we aimed to study whether ATRX expression was associated with DNA methylation level in astrocytic tumors and in which cellular functions it participated. We conducted an extensive DNA methylation and mRNA expression profile study on a cohort of 82 patients with astrocytic tumors in China. We observed that astrocytic tumors with lower ATRX expression harbored higher DNA methylation level at chromatin end and astrocytic tumors with ATRX-low had distinct GEP and DNA methylation profile compared with ATRX-high tumors. Then, we uncovered that several ATRX associated biological functions in the DNA methylation and mRNA expression profile, for example, apoptotic process, metabolic process, DNA-dependent positive regulation of transcription chromatin and modification. Interestingly, we observed that astrocytic tumors with lower ATRX expression companied with MGMT (O6-methylguanine–DNA methyltransferase) hypermethylation and downregulation. Consistent with our report [14], ATRX expression characteristically decreased in grade II astrocytomas and secondary glioblastoma and low ATRX expression was correlated with favorable survival of patients in astrocytic tumors. And we also found that loss of ATRX caused by siRNA induced apoptotic cells increasing, reduced tumor cell proliferation and repressed the cell migration in glioma cells. Our results provided novel insights into ATRX-related regulatory functions at the DNA methylation and mRNA expression level in astrocytic tumors, and here could serve as important resources for future dissection of ATRX role in glioma.
RESULTS
ATRX mRNA expression characteristically decreased in grade II astrocytomas and secondary glioblastoma
In our mRNA array expression profile, there were 65 diffuse astrocytomas (As, Grade II), 15 anaplastic astrocytomas (AAs, Grade III), 10 secondary glioblastomas (sGBMs, Grade IV) and 118 primary glioblastomas (pGBMs, Grade IV). IDH1/2 mutations occurred in 77.4% (48/62) As, 40% (6/15) AAs, 60% (6/10) sGBMs and 12.7% (15/118) pGBMs. Consistent with our previous report, ATRX mRNA expression was significantly different in grade II-IV astrocytic tumors (Figure 1A; p < 0.0001) and reduced in grade II astrocytomas compared with in pGBMs and AAs (Figure 1A; p < 0.0001, p < 0.05, respectively). We also observed that secondary GBMs harbored lower ATRX expression than primary GBMs (Figure 1A; p < 0.01). These results suggested that ATRX expression was associated with malignancy in astrocytic tumors.
Figure 1. ATRX mRNA expression correlated with tumor grade and survival in astrocytic tumors.
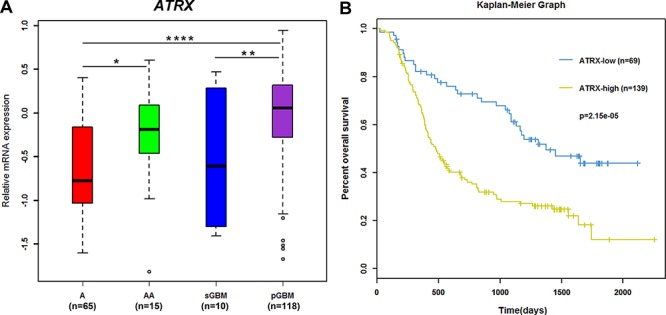
ATRX mRNA expression was significantly different in grade II-IV astrocytic tumors (A. p < 0.0001) and reduced in grade II astrocytomas than in pGBMs and AA (A. p < 0.0001, p < 0.05, respectively). Secondary GBMs harbored lower ATRX expression compared with primary GBMs (Figure 1A; p < 0.01). Patients in the ATRX-low group displayed significantly longer overall survival (OS) than patients in the ATRX-high group (B. log-rank test, p = 2.15e-05).
ATRX mRNA expression was correlated with survival in patients with astrocytic tumors
We defined the ATRX-high group and the ATRX-low group according to the cutoff point (−0.5368) (Supplementary Figure 1). There were 69 samples, harboring lower ATRX expression than the cutoff point, in the ATRX-low group, which included 41 As, 4 AAs, 5 sGBMs and 19 pGBMs. The ATRX-high group contained 24 As, 11 AAs, 5 sGBMs and 99 pGBMs (Supplementary Table 1). Decreased ATRX expression was associated with favorable survival of patients with astrocytic tumors (Figure 1B; p = 2.15e–05). To control the influence of age at diagnosis, gender, preoperative Karnofsky performance status (KPS) score, extent of surgical resection, chemotherapy, and radiotherapy on the stratification of astrocytic gliomas, we performed the Cox regression model as showed in Supplementary Table 2. The prognostic value of ATRX expression was still significant, independent of age at diagnosis, preoperative KPS score, extent of surgical resection, and radiotherapy.
ATRX-low astrocytic tumors had distinct GEP and DNA methylation profile compared with ATRX-high tumors
Matched genome-wide mRNA expression and DNA methylation profile was successfully obtained from these 82 astrocytic tumor samples (Table 1). To identify which genes methylation and expression level likely associated with ATRX expression in astrocytic tumors, pearson correlation analysis was performed, respectively. We screened top 500 positively correlated expression probes (r > 0.525682177, p < 3.95e–07) with ATRX and top 500 negatively correlated expression probes (r < −0.505967751, p < 1.24e–06). On the other hand, top 300 positively correlated methylation probes (r >0.340059, p < 0.001773) with ATRX and top 300 negatively correlated with methylation probes (r < −0.37802, p < 0.000463) were screen. Expression and methylation patterns of genes correlated with ATRX were showed using one-dimensional hierarchical clustering analysis in Figure 2A/2B. The four most representative biological processes for genes commonly altered by hypermethylation were apoptotic process (for example, TNFSF10 and TNFRSF19), metabolic process (for example, PDHA1 and UCK1), DNA-dependent positive regulation of transcription (for example, BRCA1 and RARA) and nervous system development (for example, METRN and DBN1) (Figure 2C). On the other hand, these genes regulated by hypomethylation in ATRX-low tumors were implicated with transport, including transmembrane transport and ion transport, and immune response, including inflammatory response, positive regulation of T cell proliferation, positive regulation of B cell proliferation and leukocyte cell-cell adhesion (Figure 2C). Among the genes involved in immune response, IL23R, ITGAL, CCL22 and IL22 were hypomethylated.
Table 1. Clinicopathological characteristics of 82 patients with astrocytic tumors.
| Characteristic | No. of Patients |
|---|---|
| Age at diagnosis | |
| < 45 | 58 |
| ≥ 45 | 24 |
| Gender | |
| Male | 50 |
| Female | 32 |
| Preoperative KPS score | |
| ≥ 80 | 49 |
| < 80 | 20 |
| Histology & Grade | |
| Astrocytoma (A, II) | 50 |
| Anaplastic astrocytoma (AA, III) | 8 |
| Primary glioblastoma (pGBM, IV) | 20 |
| Secondary glioblastoma (sGBM, IV) | 4 |
| TCGA Subtypes | |
| Proneural | 24 |
| Neural | 26 |
| Classical | 5 |
| Mesenchymal | 27 |
| Resection | |
| Total | 31 |
| Subtotal | 47 |
| Radiotherapy | |
| Yes | 61 |
| No | 15 |
| TMZ Chemotherapy | |
| Yes | 27 |
| No | 40 |
| IDH1/2 mutation | |
| Mutation | 46 |
| Wildtype | 34 |
Abbreviations: KPS, Karnofsky performance status.
Figure 2. Clustering analysis of both DNA methylation and mRNA expression profiles in astrocytic tumors.
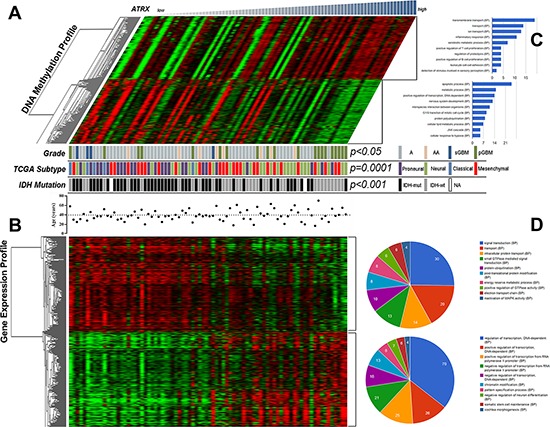
Patterns of mRNA expression and DNA methylation were associated with ATRX in astrocytic tumors using one-dimensional hierarchical clustering analysis A. and B. Functional enrichment analysis of associated genes, indicating the functional roles of gene sets in different subgroups. Enrichment results for biological processes were obtained from the GO database. The orders of biological processes listed in the circle or histogram are based on the number of targets annotated in the biological process C. and D.
A similar analysis approach was taken to investigate these correlated expression gene probes. One-dimensional hierarchical clustering resulted in a good separation between ATRX-low and ATRX-high (Figure 2B). These genes overexpressed in ATRX-low tumors also participated in transport and protein modification process, especially intracellular protein transport, protein ubiquitination and post translational protein modification, and signal transduction including small GTPase mediated signal transduction and positive regulation of GTPase activity. In addition, regulation of transcription and chromatin modification were the most frequently deregulated biological process (Figure 2D). In this respect, a total of 13 genes involved in the chromatin modification, including BANP, PHF2, FOXA2, HMG20B, CABIN1, DOT1L, CHD8, KDM5A, CCDC101, EP400, EPC1, KDM5B and USP3.
Moreover, we observed that proneural or IDH-mut tumors mainly enriched in the ATRX-low group (p = 0.0001 and p < 0.001, respectively).
Decreased ATRX expression was associated with DNA methylation level of chromatin end
According to Sturm et al's work [18], we screened the probes within 4Mb bases to chromosome end, and then we observed that tumors in ATRX-low cluster specifically showed widespread hypermethylation at chromosome end, when compared with ATRX-high subgroup, potentially linking subtelomeric methylation pattern to alternative lengthening of telomeres (ALT) (Figure 3A). 1929 probes from gene expression profile and 2407 probes from DNA methylation profile were analyzed, respectively at the chromosome end (Figure 3B). Significantly differential probes (p < 0.005) were screened via t test. 441 probes from gene expression profile and 53 probes from DNA methylation profile were identified. Multiple probes were corresponding to a single a gene, the expression/methylation values of these redundant probes were averaged. Integrative analysis involving methylation and expression profiling was used to characterize genomic changes between ATRX-low tumors and ATRX-high tumors. Comparison of the 52 putative target genes of differential methylation and the 374 genes of differential expression allowed the detection of gene loci that experienced both concurrent changes in ATRX-low tumors. In total, 18 genes were both differentially methylated and differentially expressed: 17 of these (94%) were hypermethylated, and 1 (6%) were hypomethylated. Among them, a high proportion of hypermethylated genes was downregulated. Thirteen of the genes that were hypermethylated in ATRX-low also featured downregulated gene expression (76%). Using functional enrichment, we observed that cell death (CLN8, PANK2, BNIP3) and DNA repair (TYMS, MGMT) were the most significant-represented terms (Table 2).
Figure 3. Strategy to identify significant genes of both DNA methylation and gene expression profiles.
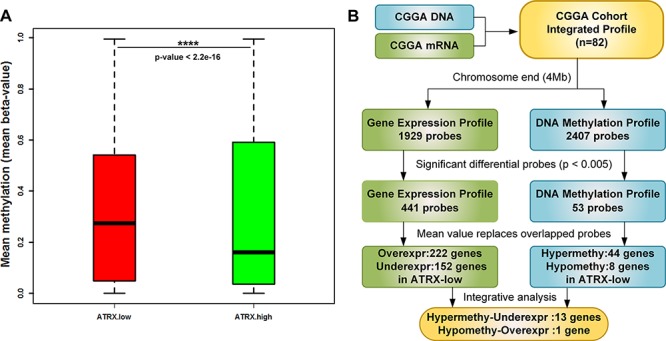
According to Sturm et al.'s work [18], we screened the probes within 4Mb bases to chromosome end. Mean methylation value within 4 Mb to the chromosome end normalized to the mean overall methylation value. Significant differences (p < 2.2e-16) between these two subgroups are indicated A. Tumors in ATRX-low cluster specifically showed widespread hypermethylation at chromosome end compared with ATRX-high subgroup.1929 probes from gene expression profile and 2407 probes from DNA methylation profile were analyzed, respectively at the chromosome end. We identified that 441 differential probes from gene expression profile and 53 differential probes from DNA methylation profile (p < 0.005). Comparison of the 52 putative target genes of differential methylation and the 374 genes of differential expression allowed the detection of gene loci that experienced both concurrent changes in ATRX-low tumors. In total, 18 genes were both differentially methylated and differentially expressed: 17 of these were hypermethylated, and 1 were hypomethylated. Thirteen of the genes that were hypermethylated in ATRX-low also featured downregulated gene expression B.
Table 2. Integrative analysis of hypermethylated and underexpressed genes in ATRX-low patients.
| Gene symbol | Expression | Methylation | ||||||
|---|---|---|---|---|---|---|---|---|
| ATRX-low | ATRX-high | fold change | p value | ATRX-low | ATRX-high | fold change | p value | |
| GAS8 | 0.8874 | 1.3309 | 0.6668 | 0.0001 | 0.3448 | 0.2098 | 1.6432 | 0.0001 |
| MGMT | 1.8678 | 2.5767 | 0.7249 | 0.0000 | 0.1869 | 0.1122 | 1.6662 | 0.0007 |
| CLN8 | 1.8429 | 2.3516 | 0.7837 | 0.0016 | 0.2118 | 0.1093 | 1.9379 | 0.0010 |
| LMBR1 | 2.4871 | 2.8192 | 0.8822 | 0.0046 | 0.2236 | 0.1321 | 1.6932 | 0.0014 |
| BNIP3 | 1.4292 | 1.7909 | 0.7980 | 0.0020 | 0.2122 | 0.1149 | 1.8470 | 0.0016 |
| LY6E | 0.9572 | 1.4992 | 0.6385 | 0.0003 | 0.2377 | 0.1251 | 1.8994 | 0.0020 |
| ZFP28 | 1.4123 | 2.1194 | 0.6663 | 0.0018 | 0.2386 | 0.1653 | 1.4439 | 0.0022 |
| GLT1D1 | 2.7025 | 3.9834 | 0.6784 | 0.0001 | 0.3092 | 0.2020 | 1.5309 | 0.0030 |
| TYMS | 2.6947 | 3.6345 | 0.7414 | 0.0029 | 0.2429 | 0.1472 | 1.6504 | 0.0035 |
| ANKRD11 | 2.4346 | 2.7741 | 0.8776 | 0.0011 | 0.1684 | 0.0889 | 1.8943 | 0.0036 |
| PANK2 | 1.4702 | 1.7699 | 0.8307 | 0.0021 | 0.1469 | 0.0663 | 2.2139 | 0.0037 |
| TUBB2A | 1.9709 | 3.0462 | 0.6470 | 0.0000 | 0.3389 | 0.2299 | 1.4743 | 0.0045 |
| PRKAR1B | 0.9218 | 1.5021 | 0.6137 | 0.0002 | 0.2416 | 0.1342 | 1.8002 | 0.0048 |
To validate the association between ATRX and MGMT expression, we detected the MGMT protein expression via immunohistochemistry. We observed that ATRX-low astrocytic tumors harbored lower MGMT expression (Figure 4).
Figure 4. MGMT expression by immunohistochemical staining.
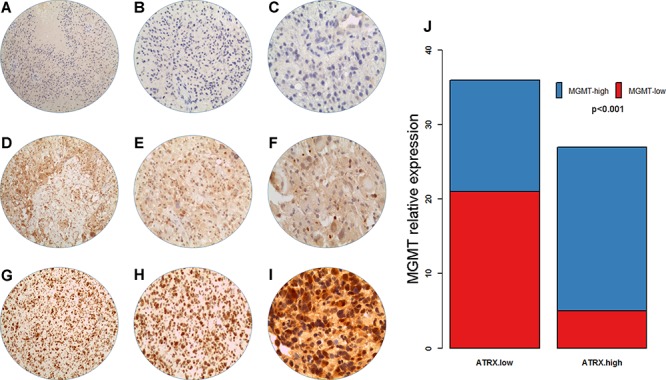
Negative (A. 100 ×; B. 200 ×; C. 400 ×); Weakly positive (D. 100 ×; E. 200 ×; F. 400 ×); Strongly positive (G. 100 ×; H. 200 ×; I. 400 ×). ATRX-low astrocytic tumors harbored lower MGMT expression (J. p < 0.001).
The knockdown of ATRX affected migration, apoptosis and proliferation of LN229 cells
In the present study, to examine effect of ATRX on glioma cells, the intrinsic expression of ATRX was repressed by siRNA 589i, 590i and 592i. Repression efficiency was confirmed by western blot analysis (Figure 5A) and real-time PCR (Supplementary Figure 2A), which indicated that ATRX expression inhibition was the most significant in 590i transfected cells. The effect on migration of ATRX was verified by transwell chamber assay. Cell apoptosis was monitored by evaluating the percentage of Annexin V-positive and PI-positive cells by flow cytometry. Cell counting kit-8 (CCK-8) assay was performed to identify cell viability. The results of the present study showed that ATRX siRNA 590i markedly reduced LN229 cells migration, but in contrast, the negative control and untransduced had no inhibitory effect on it (Figure 5B and Supplementary Figure 2B). Following 590i transfection, flow cytometry analysis revealed a significantly higher percentage of Annexin V- and PI-positive cells among the LN229 cells (Figure 5C and Supplementary Figure 2C) compared with the cells transfected with negative control. CCK-8 assay showed that the knockdown of ATRX inhibited cells proliferation. The inhibitory effect of 590i was significant at 24 h, and it was maximal at 48 h, 72 h and 96 h (Figure 5D). These results indicated that reduced expression of ATRX inhibited the migration, promoted apoptosis and reduced proliferation of LN229 cells. This suggested important role of ATRX for glioma cells.
Figure 5. The knockdown of ATRX affected migration, apoptosis and proliferation of LN229 cells.
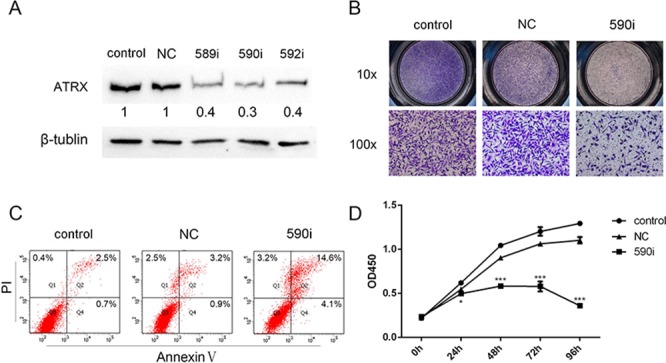
The expression levels of ATRX were detected by western blotting in LN229 cells that were transiently transfected with negative control (NC), 589i, 590i and 592i. β-tublin was used as a control for equal protein loading. The bands were scanned using Image Lab 5.1 software. The densitometric values normalized to β-tublin expression are indicated below the corresponding lanes, and are shown as the fold change relative to that in the LN229 cells transfected with negative control A. Contrasted with control and NC, notably less LN229 cells transfected with 590i penetrated the transwell at 24 h after cells seeded. (up) 10 × ; (down) 100 × B. Flow cytometry analysis of Annexin V and PI levels in the LN229 cells untreated or transfected with NC or 590i. Results are a representative experiment of the three conducted. The percentages of Annexin V and PI-positive cells are shown C. Transfection of LN229 cells with 590i inhibited cell proliferation D. *p < 0.05, **p < 0.01 and ***p < 0.001.
DISCUSSION
Aberrant gene function and altered patterns of gene expression are key features of glioma [18]. Increasing evidences showed that acquired epigenetic abnormalities participated with genetic alterations to cause this dysregulation [16]. Localized hypermethylation of gene-associated CpG islands and a more extensive genome-wide reduction in 5-methylcytosine are epigenetic events that typify many tumors [19]. Methylation is the known covalent modification of genomic DNA in humans and occurs at cytosines followed by guanines (CpG) [20]. Aberrant methylation of some CpG islands in cancer has been associated with silencing of tumor suppressor genes [21]. According to the TCGA report, patients with G-CIMP tumors, a distinct subset of samples displays concerted hypermethylation at a large number of loci, experience significantly improved outcome [17]. An important link between DNA methylation and these covalent histone modifications was illustrated by the pioneering experiments of Nan et al. [22]. Mutations in ATRX, encoding a SWI/SNF-like protein, lead to changes in the pattern of methylation of several highly repeated sequences including the rDNA arrays, a Y-specific satellite and subtelomeric repeats [15]. In the present study, we integratively analyzed genome-wide DNA methylation and mRNA expression profiling of 82 astrocytic tumor samples. We screened top 500 positively/negatively correlated expression probes with ATRX and top 300 positively/negatively correlated CpG locis with ATRX. We observed that ATRX-low astrocytic tumors had distinct GEP and DNA methylation profile compared with ATRX-high tumors. To our knowledge, this is the first time the same cohort of patients with both profiles has been used to analyze ATRX associated functions in astrocytic tumors. ATRX played a key role in the incorporation of the histone variant H3.3 into pericentromeric DNA, ribosomal and telomeric [8]. In addition, ATRX functioned in maintaining telomere integrity and facilitating normal telomere replication during DNA synthesis [6, 23]. Previous study showed that loss of SWI/SNF-mediated transcriptional activation was implicated in direct interactions with promoter sequences of affected genes and increased DNA methylation in cancer cells, providing insight into the mechanisms underlying aberrant gene induction and repression during tumor progression [24]. Our functional analysis results also confirmed that ATRX was associated with apoptotic process, DNA-dependent positive regulation of transcription, protein transport and chromatin modification in astrocytic tumors through Gene Ontology analysis, suggesting a functional role of ATRX alteration in astrocytic tumors development. Then, we compared the mean methylation level of chromosome end between ATRX-low tumors and ATRX-high tumors and investigated the combined profiles of DNA methylation and mRNA expression at chromosome end. The integrative study identified DNA methylation markers that could lead to the downregulation of some genes involved in important cellular functions in ATRX-low tumors: MGMT, TYMS, CLN8, PANK2 and BNIP3. We also identified that, in glioma cell line, the knockdown of ATRX expression induced apoptotic cells increasing, reduced tumor cell proliferation and repressed the cell migration. Similarly, a diagram depicting how loss of ATRX could affect neuroprogenitor cell apoptosis was provided by a report [25].
In conclusion, our results showed ATRX-related regulatory functions of the combined profiles from DNA methylation and mRNA expression in astrocytic tumors, and delineated that loss of ATRX impacted biological behaviors of astrocytic tumor cells, providing important resources for future dissection of ATRX role in glioma.
MATERIALS AND METHODS
Datasets collection
Whole genome mRNA expression profile (GEP) of 208 astrocytic tumor samples of all grades and DNA methylation profile of 105 astrocytic glioma samples, by using Agilent Whole Human Genome Array and Illumina Infinium Human Methylation 27K Bead Chip, respectively, were obtained from Chinese Glioma Genome Atlas (CGGA) database (http://www.cgga.org.cn) [26, 27]. For molecular subtype annotation of the CGGA dataset, we applied prediction analysis of microarrays (PAM) as previously reported [28]. All these samples were histologically graded according to current WHO classification of tumours of the nervous systems [29]. Written informed consent was obtained from all donors. Clinical investigations were performed after approval by the local research ethics committee and in accordance with the ethical principles.
Molecular analysis
IDH mutation
Genomic DNA was isolated from frozen tissues with a QIAamp DNA Mini Kit (Qiagen) as the manufacturer's protocol. DNA concentration and quality were evaluated with a Nano-Drop ND-1000 spectrophotometer (NanoDrop Technologies, Houston, TX). Pyrosequencing of IDH1/2 mutations was supported by Gene-tech (Shanghai, China) and performed on a Pyro-Mark Q96 ID System (Qiagen, Valencia, Calif). The primers 5′-GCTTGTGAGTGGATGGGTAAAAC-3′, 5′-BiotinTTGCCAACATGAC TTACTTGATC-3′ for IDH1 and 5′-ATCCTGGGGGGGACTGTCTT-3′, 5′-Biotin-CTCTCCACCCTGGCCT ACCT-3′ for IDH2 were used for PCR amplification, and the primers 5′-TGGATGGGTAAAACCT-3′ for IDH1 and 5′-AGCCCATCACCATTG-3′ for IDH2 were used for pyrosequencing [14].
MGMT protein expression
Immunohistochemical analysis was performed as described in our previous study [30]. Briefly, tumor tissues were formalin-fixed and embedded in paraffin. For detecting MGMT expression, 5 μm sections were incubated with monoclonal antibodies against MGMT (Santa Cruz Biotechnology, Santa Cruz, CA, USA). All samples were evaluated by two pathologists experienced in analysis of tumors of the central nervous system. Scoring on the expression level of MGMT protein was performed according to the percentage of positive MGMT cells. A score of less than 5% was classified as negative MGMT expression and a score of 5–100% was classified as positive MGMT expression. Scores of 5–25% were denoted as weakly positive MGMT expression, and scores of 25–100% was denoted as strongly positive MGMT expression.
Gene ontology (GO) and KEGG pathway analysis
After Pearson correlation analysis, gene ontology analysis or KEGG pathway of the correlated genes were analyzed by GeneCodis (http://genecodis.cnb.csic.es/) [31–33].
Cell lines and cell culture
The human glioma cell line LN229 was obtained from the Institute of Biochemistry and Cell Biology, Chinese Academy of Science. LN229 cell line was maintained in DMEM F12 medium (Gibco), 10% FBS (Gibco), 50 units/ml penicillin and 50 μg/ml streptomycin (Sigma) at 37°C in a 5% CO2 atmosphere.
Oligonucleotides and cell transfection
Stealth siRNA targeting ATRX 589i (GCAGATTGAT ATGAGAGGAAT), 590i (CGACAGAA ACTAACCCT GTAA), 592i (CCGGTGGTGAACATAAG AAAT) and negative control (UUCUCCGAACGUGUCACGUTT) was obtained from GenePharma. For the siRNA experiments, LN229 cells were transfected with either a control non-targeting human RNA or siRNA against ATRX at a final concentration of 200 nM. All cell transfections were introduced by the RNAi MAX reagent (Invitrogen) according to the manufacturer's instructions. The cell transfections were performed in six-well plates. For each transfection, 7 μl of transfection reagent was used and three replication experiments were performed.
Western blotting and antibodies
Whole-cell lysates were prepared using RIPA buffer. Equal amounts of total protein (30 μg) from cell lysates were loaded on a 6% SDS/PAGE gel, transferred to a PVDF membrane (Millipore), and detected using an ECL Western Blotting Detection System (Biorad). Primary antibodies were primary antibodies against ATRX (ab97508; Abcam; 1:1000), β-tublin (CW0098A; CWBIO; 1:5000). Secondary antibodies used were goat anti-rabbit IgG-HRP and goat anti-mouse IgG-HRP (Zhongshan Gold Bridge Biotechnology). Immunoblots were quantified using Image Lab 5.1 software.
RNA isolation and quantitative real-time RT-PCR
Total RNA was extracted from the harvested cells using RNAprep pure Cell Kit (Tian Gen) according to the manufacturer's instructions. cDNA was synthesized by M-MLV (Moloney murine leukaemia virus) reverse transcriptase (Invitrogen) from 2 μg of total RNA. Oligo (dT) 18 was used as the primer for reverse transcription of mRNA. Quantitative real-time RT-PCR was carried out in a 7500 real-time PCR System (Applied Biosystems) using the SYBR Select Master Mix (Applied Biosystems) according to the manufacturer's instructions. For each mRNA assay, the data were normalized using the endogenous actin cDNA control. The comparative CT method was used to quantify the target genes relative to the endogenous control. For each individual analysis one of the samples was designated as the calibrator and given a relative value of 1.0. All quantities were expressed as then-fold relative to the calibrator. The real-time PCR primers were as follows: ATRX forward-5′ GCTGAGCCCATGAGTGAAAG 3′, reverse-5′ CGTGA CGATCCTGAAGACTTG 3′; β-actin forward-5′ CATGTA CGTTGCTATCCAGGC 3′, reverse-5′ CTCCTTAATGT CACGCACGAT 3′.
Cell proliferation assay
LN229 cells plated at a final concentration of 104 cells /well in 96-well plates were transfected with NC or si-ATRX in exponential growth. The viability of cells was evaluated by cell counting kit-8 (CCK-8) assay after 0, 24, 48, 72 and 96 h of transfection. The optical density at 450 nm (OD450) of each well was measured with a microplate reader (Infinite® M1000 PRO, TECAN).
Cell migration assay
The lower Transwell chamber (Costar) contained cell appropriate medium (10% fetal calf serum as chemoattractants). Cells in appropriate medium (1% fetal calf serum) were seeded onto membranes of the upper Transwell chamber (6.5 mm diameter, 8 μm pores). After 48 h of transfection with NC or si-ATRX 105 LN229 cells were incubated for 24 h, cells were ethanol-fixed and stained (crystal violet). The non-invading cells were removed from the upper surfaces of the invasion membranes and the cells on four fields of the lower face were counted using an inverted microscope.
Cell apoptosis assay
Cell apoptosis detection was performed by fluorescence-activated cell sorting (FACS) analysis using a flow cytometer. LN229 cells were placed in six-well plates, and after transfection with NC or si-ATRX for 72 h, cells were harvested. After centrifugation, cell were washed twice with cold phosphate-buffered saline which then incubated with 5 μL of annexin V-FITC and 10 μL of PI at room temperature for 15 min in the dark. After incubation, 400 μL of 1× binding buffer was added to each tube. The cells were immediately analyzed by FACS Calibur flow cytometry (Becton Dickinson).
Statistical analysis
According to the cutoff value (−0.5368), patients in our dataset were stratified into the ATRX-high group and the ATRX-low group. The threshold was determined by receiver operating characteristics (ROC) analysis with an area under the curve of 0.6396 (Supplementary Figure 1).
We screened the probes within 4Mb bases to chromosome end as corresponding genomic coordinates of the probes according to Sturm et al's work [18]. Overall survival was estimated from the date of diagnosis to the date of either death or last follow-up. Patients were censored at the time they were last known to be alive. Kaplan-Meier analysis was performed to estimate the survival time of different subgroups and a log-rank test was used to test for differences of more than one survival curve. Student's t-test and analysis of variance (ANOVA) were used to determine significant differences. Comparisons of binary and categorical patient characteristics between subgroups were performed by the use of the Chi-Square test. P value < 0.05 was considered statistically significant. All statistical computations were performed with the statistical software environment R, version 3.1.0 (http://www.r-project.org/) or GraphPad Prism Version 6.01.
SUPPLEMENTARY FIGURES AND TABLES
Acknowledgments
We thank Yuling Yang for tissue sample collection and clinical data retrieval.
Footnotes
GRANT SUPPORT
- National High Technology Research and Development Program (863) (No. 2012AA02A508);
- The Research Special Fund For Public Welfare Industry of Heath (No. 201402008);
- International Science and Technology Cooperation Program (No.2012DFA30470);
- National Natural Science Foundation of China (No. 91229121, No. 81201993);
- Beijing Science and Technology Plan (No. Z1311000 06113018);
- National Key Technology Research and Development Program of the Ministry of Science and Technology of China (No. 2013BAI09B03).
- Beijing Science and Technology Plan (No. Z131100006113018)
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
REFERENCES
- 1.Reifenberger G, Collins VP. Pathology and molecular genetics of astrocytic gliomas. Journal of molecular medicine. 2004;82:656–670. doi: 10.1007/s00109-004-0564-x. [DOI] [PubMed] [Google Scholar]
- 2.Seyfried TN, Flores R, Poff AM, D'Agostino DP, Mukherjee P. Metabolic Therapy: A New Paradigm for Managing Malignant Brain Cancer. Cancer Lett. 2014 doi: 10.1016/j.canlet.2014.07.015. [DOI] [PubMed] [Google Scholar]
- 3.Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, Beroukhim R, Bernard B, Wu CJ, Genovese G, Shmulevich I, Barnholtz-Sloan J, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ratnakumar K, Bernstein E. ATRX: the case of a peculiar chromatin remodeler. Epigenetics: official journal of the DNA Methylation Society. 2013;8:3–9. doi: 10.4161/epi.23271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clynes D, Higgs DR, Gibbons RJ. The chromatin remodeller ATRX: a repeat offender in human disease. Trends in Biochemical Sciences. 2013;38:461–466. doi: 10.1016/j.tibs.2013.06.011. [DOI] [PubMed] [Google Scholar]
- 6.Wong LH, McGhie JD, Sim M, Anderson MA, Ahn S, Hannan RD, George AJ, Morgan KA, Mann JR, Choo KHA. ATRX interacts with H3.3 in maintaining telomere structural integrity in pluripotent embryonic stem cells. Genome research. 2010;20:351–360. doi: 10.1101/gr.101477.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lewis PW, Elsaesser SJ, Noh KM, Stadler SC, Allis CD. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc Natl Acad Sci U S A. 2010;107:14075–14080. doi: 10.1073/pnas.1008850107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clynes D, Gibbons RJ. ATRX and the replication of structured DNA. Current opinion in genetics & development. 2013;23:289–294. doi: 10.1016/j.gde.2013.01.005. [DOI] [PubMed] [Google Scholar]
- 9.Heaphy CM, de Wilde RF, Jiao Y, Klein AP, Edil BH, Shi C, Bettegowda C, Rodriguez FJ, Eberhart CG, Hebbar S, Offerhaus GJ, McLendon R, Rasheed BA, He Y, Yan H, Bigner DD, et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science. 2011;333:425. doi: 10.1126/science.1207313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiao Y, Shi C, Edil BH, de Wilde RF, Klimstra DS, Maitra A, Schulick RD, Tang LH, Wolfgang CL, Choti MA, Velculescu VE, Diaz LA, Jr., Vogelstein B, Kinzler KW, Hruban RH, Papadopoulos N. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011;331:1199–1203. doi: 10.1126/science.1200609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tonjes M, Hovestadt V, Albrecht S, Kool M, Nantel A, Konermann C, Lindroth A, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–231. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- 12.Jiao Y, Killela PJ, Reitman ZJ, Rasheed AB, Heaphy CM, de Wilde RF, Rodriguez FJ, Rosemberg S, Oba-Shinjo SM, Nagahashi Marie SK, Bettegowda C, Agrawal N, Lipp E, Pirozzi C, Lopez G, He Y, et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget. 2012;3:709–722. doi: 10.18632/oncotarget.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wiestler B, Capper D, Holland-Letz T, Korshunov A, von Deimling A, Pfister SM, Platten M, Weller M, Wick W. ATRX loss refines the classification of anaplastic gliomas and identifies a subgroup of IDH mutant astrocytic tumors with better prognosis. Acta Neuropathol. 2013;126:443–451. doi: 10.1007/s00401-013-1156-z. [DOI] [PubMed] [Google Scholar]
- 14.Cai J, Yang P, Zhang C, Zhang W, Liu Y, Bao Z, Liu X, Du W, Wang H, Jiang T, Jiang C. ATRX mRNA expression combined with IDH1/2 mutational status and Ki-67 expression refines the molecular classification of astrocytic tumors: evidence from the whole transcriptome sequencing of 169 samples samples. Oncotarget. 2014 doi: 10.18632/oncotarget.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gibbons RJ, McDowell TL, Raman S, O'Rourke DM, Garrick D, Ayyub H, Higgs DR. Mutations in ATRX, encoding a SWI/SNF-like protein, cause diverse changes in the pattern of DNA methylation. Nat Genet. 2000;24:368–371. doi: 10.1038/74191. [DOI] [PubMed] [Google Scholar]
- 16.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, Verhaak RG, Hoadley KA, Hayes DN, Perou CM, Schmidt HK, Ding L, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, Pfaff E, Tonjes M, Sill M, Bender S, Kool M, Zapatka M, Becker N, Zucknick M, Hielscher T, Liu XY, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22:425–437. doi: 10.1016/j.ccr.2012.08.024. [DOI] [PubMed] [Google Scholar]
- 19.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nature reviews Genetics. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 20.Cadieux B, Ching TT, VandenBerg SR, Costello JF. Genome-wide hypomethylation in human glioblastomas associated with specific copy number alteration, methylenetetrahydrofolate reductase allele status, and increased proliferation. Cancer Res. 2006;66:8469–8476. doi: 10.1158/0008-5472.CAN-06-1547. [DOI] [PubMed] [Google Scholar]
- 21.McGarvey KM, Fahrner JA, Greene E, Martens J, Jenuwein T, Baylin SB. Silenced tumor suppressor genes reactivated by DNA demethylation do not return to a fully euchromatic chromatin state. Cancer Res. 2006;66:3541–3549. doi: 10.1158/0008-5472.CAN-05-2481. [DOI] [PubMed] [Google Scholar]
- 22.Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- 23.Huh MS, Price O'Dea T, Ouazia D, McKay BC, Parise G, Parks RJ, Rudnicki MA, Picketts DJ. Compromised genomic integrity impedes muscle growth after Atrx inactivation. The Journal of clinical investigation. 2012;122:4412–4423. doi: 10.1172/JCI63765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Banine F, Bartlett C, Gunawardena R, Muchardt C, Yaniv M, Knudsen ES, Weissman BE, Sherman LS. SWI/SNF chromatin-remodeling factors induce changes in DNA methylation to promote transcriptional activation. Cancer Research. 2005;65:3542–3547. doi: 10.1158/0008-5472.CAN-04-3554. [DOI] [PubMed] [Google Scholar]
- 25.Iwase S, Xiang B, Ghosh S, Ren T, Lewis PW, Cochrane JC, Allis CD, Picketts DJ, Patel DJ, Li H, Shi Y. ATRX ADD domain links an atypical histone methylation recognition mechanism to human mental-retardation syndrome. Nature structural & molecular biology. 2011;18:769–776. doi: 10.1038/nsmb.2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang W. Genome-wide DNA methylation profiling identifies ALDH1A3 promoter methylation as a prognostic predictor in G-CIMPprimary glioblastoma. Cancer Letters. 2013 doi: 10.1016/j.canlet.2012.08.033. [DOI] [PubMed] [Google Scholar]
- 27.Bao ZS, Chen HM, Yang MY, Zhang CB, Yu K, Ye WL, Hu BQ, Yan W, Zhang W, Akers J, Ramakrishnan V, Li J, Carter B, Liu YW, Hu HM, Wang Z, et al. RNA-seq of 272 gliomas revealed a novel, recurrent PTPRZ1-MET fusion transcript in secondary glioblastomas. Genome research. 2014 doi: 10.1101/gr.165126.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yan W, Zhang W, You G, Zhang J, Han L, Bao Z, Wang Y, Liu Y, Jiang C, Kang C, You Y, Jiang T. Molecular classification of gliomas based on whole genome gene expression: a systematic report of 225 samples from the Chinese Glioma Cooperative Group. Neuro Oncol. 2012;14:1432–1440. doi: 10.1093/neuonc/nos263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathologica. 2007;114:97–109. doi: 10.1007/s00401-007-0243-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Y, Fan X, Zhang C, Zhang T, Peng X, Li S, Wang L, Ma J, Jiang T. Anatomical specificity of O6-methylguanine DNA methyltransferase protein expression in glioblastomas. J Neurooncol. 2014 doi: 10.1007/s11060-014-1555-6. [DOI] [PubMed] [Google Scholar]
- 31.Tabas-Madrid D, Nogales-Cadenas R, Pascual-Montano A. GeneCodis3: a non-redundant and modular enrichment analysis tool for functional genomics. Nucleic acids research. 2012;40:W478–483. doi: 10.1093/nar/gks402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nogales-Cadenas R, Carmona-Saez P, Vazquez M, Vicente C, Yang X, Tirado F, Carazo JM, Pascual-Montano A. GeneCodis: interpreting gene lists through enrichment analysis and integration of diverse biological information. Nucleic acids research. 2009;37:W317–322. doi: 10.1093/nar/gkp416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carmona-Saez P, Chagoyen M, Tirado F, Carazo JM, Pascual-Montano A. GENECODIS: a web-based tool for finding significant concurrent annotations in gene lists. Genome Biol. 2007;8:R3. doi: 10.1186/gb-2007-8-1-r3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.