

Abstract
Background
In medical research, it is common to collect information of multiple continuous biomarkers to improve the accuracy of diagnostic tests. Combining the measurements of these biomarkers into one single score is a popular practice to integrate the collected information, where the accuracy of the resultant diagnostic test is usually improved. To measure the accuracy of a diagnostic test, the Youden index has been widely used in literature. Various parametric and nonparametric methods have been proposed to linearly combine biomarkers so that the corresponding Youden index can be optimized. Yet there seems to be little justification of enforcing such a linear combination.
Methods
This paper proposes a flexible approach that allows both linear and nonlinear combinations of biomarkers. The proposed approach formulates the problem in a large margin classification framework, where the combination function is embedded in a flexible reproducing kernel Hilbert space.
Results
Advantages of the proposed approach are demonstrated in a variety of simulated experiments as well as a real application to a liver disorder study.
Conclusion
Linear combination of multiple diagnostic biomarkers are widely used without proper justification. Additional research on flexible framework allowing both linear and nonlinear combinations is in need.
Electronic supplementary material
The online version of this article (doi:10.1186/s12874-015-0085-z) contains supplementary material, which is available to authorized users.
Keywords: Biomarker, Diagnostic accuracy, Margin, Receiver operating characteristic curve, Reproducing kernel Hilbert space, Youden index
Background
In medical research, continuous biomarkers have been commonly explored as diagnostic tools to distinguish subjects, such as diseased and non-diseased groups [1]. The accuracy of a diagnostic test is usually evaluated through sensitivity and specificity, or the probabilities of true positive and true negative for any given cut-point. Particularly, the receiver operating characteristic (ROC) curve is defined as sensitivity versus 1−specificity over all possible cut-points for a given biomarker [2, 3], which is a comprehensive plot that displays the influence of a biomarker as the cut-point varies. To summarize the overall information of an ROC curve, different summarizing indices have been proposed, including the Youden index [4] and the area under the ROC curve (AUC; [5]).
The Youden index, defined as the maximum vertical distance between the ROC curve and the 45° line, is an indicator of how far the ROC curve is from the uninformative test [3]. Normally, it ranges from 0 to 1 with 0 for an uninformative test and 1 for an ideal test. The Youden index has been successfully applied in many clinical studies and served as an appropriate summary for the diagnostic accuracy of a single quantitative measurement (e.g., [2, 6, 7]).
It has been widely accepted by medical researchers that diagnosis based on one single biomarker may not provide sufficient accuracy [8, 9]. Consequently, it is becoming more and more common that multiple biomarker tests are performed on each individual, and the corresponding measurements are combined into one single score to help clinicians make better diagnostic judgment. In literature, various statistical modeling strategies have been proposed to combine biomarkers in a linear fashion. For instance, Su and Liu [10] derived the analytical results of optimal linear combination based on AUC under multivariate normal assumption. Pepe and Thompson [11] proposed to relax the distributional assumption and perform a grid search for the optimal linear combination, while its computation becomes expensive when the number of biomarkers gets large. Recently, a number of alternatives were proposed to alleviate the computational burden. For instances, the min-max approach [12] combines only the minimum and maximum values of biomarker measurements linearly; the stepwise approach [13] combines all biomarker measurements in a stepwise manner. By targeting directly on the optimal diagnostic accuracy, Yin and Tian [14] extended these two methods to optimize the Youden index and demonstrated their improved performance in a number of numerical examples.
In recent years, nonlinear methods have been popularly employed to combine multiple biomarkers in various fields, including genotype classification [15], medical diagnosis [16], and treatment selection [17]. In this paper, a new model-free approach is proposed and formulated in a large margin classification framework, where the biomarkers are flexibly combined into one single diagnostic score so that the corresponding Youdex index [4] is maximized. Specifically, the combination function is modeled non-parametrically in a flexible reproducing kernel Hilbert space (RKHS; [18]), where both linear and nonlinear combinations could be accommodated via a pre-specified kernel function.
The rest of the paper is organized as follows. In Section ‘Methods’, we provide some preliminary background of combining multiple biomarkers based on the Youden index. In Section ‘Results and discussion’, we discuss the motivation for flexible combinations and formulate the proposed flexible approach in a framework of large margin classification for combining multiple biomarkers. In Section ‘Results and discussion’, we conduct numerical experiments to demonstrate the advantages of the proposed approach, apply the proposed approach to a liver disorder study, and extend the proposed framework to incorporate the effect of covariates. Section ‘Conclusions’ contains some discussion.
Methods
Preliminaries
Suppose that every subject has m biomarker measurements X=(X(1),X(2),…,X(m))T with a probability density function f(X), where X(j) is a continuous measurement of the j-th biomarker. It also has a binary response variable Y∈{1,−1} indicating the subject is diseased or not. In literature, researchers from different fields [8, 9, 14] have discussed and explored the validity of combining m biomarker measurements into one single score function g(X) as a more powerful diagnostic tool. A subject is diagnosed as diseased if the combined score g(X) is higher than a given cut-point c, and non-diseased otherwise. To summarize its diagnostic accuracy, the Youden index is commonly used in practice. With sensitivity and specificity defined as sen(g,c)=Pr(g(X)≥c|Y=1) and spe(g,c)=Pr(g(X)<c|Y=−1) respectively, the Youden index is formulated as
The Youden index normally ranges from 0 to 1, where J=1 corresponds to a perfect separation, and J=0 corresponds to a random guess.
To estimate the Youden index, various modeling strategies have been proposed. Schisterman et al. [19] provided a closed form for the Youden index assuming the conditional distribution of X|Y=±1 follows a multivariate Gaussian distribution. Further relaxing the distributional assumption, kernel smoothing techniques were adopted by Yin and Tian [14] and Fluss et al. [20], where the sensitivity and specificity were estimated in a nonparametric fashion.
Note that the formulation of J can be rewritten as
| (1) |
where w(1)=1/π, w(−1)=1/(1−π), π=Pr(Y=1), and sign(u)=1 if u≥0 and −1 otherwise. Denote the ideal combination function g∗(x) and cut-point c∗ as the ones that maximize J over all possible functionals and cut-points. Following the proof of Proposition 1 in [21], the ideal g∗(x) and c∗ must satisfy
| (2) |
where p(x)=Pr(Y=1|x) is the conditional probability of disease given the biomarker measurements.
Linear or nonlinear combination
In (2), the ideal g∗(x) and c∗ are defined based on p(x) that is often unavailable in practice. Hence the expectation in (1) needs to be estimated based on the given sample . Specifically, a natural estimate can be obtained as
| (3) |
where , , , , and |·| denotes the set cardinality.
The optimization in (3) is generally intractable without a specified candidate space of g. In literature, linear functional space g(x)=βTx is often used [10–14], mainly due to its convenient implementation and natural interpretation. Yet there seems to be lack of scientific support for the use of linear combination of biomarkers.
Consider a toy example, where π=1/2, and , where I2 is a 2-dimensional identity matrix. Then for any given x,
where . Thus, the ideal combination of biomarkers g∗(x) can take the linear form g∗(x)=x1+x2, leading to sign(g∗(x)−c)=sign(p(x)−1/2) with c=1. However, if the biomarkers are heterocedastic in the positive and negative groups, the ideal combination would be no longer linear. For instance, when but ,
Clearly, the ideal combination of biomarkers is a quadratic function with . Furthermore, if the conditional distribution X|Y is unknown, then the ideal combination of biomarkers may take various forms, and thus a pre-specified assumption on linear combination can be too restrictive and lead to suboptimal combinations.
Model-free estimation formulation
To allow more flexible g(x) than linear functions, it is natural to optimize (3) over a bigger functional space consisting of nonlinear functions. The objective function in (3) can be simplified as
where ui=yi(g(xi)−c). However, it involves a sign operator, which makes it discontinuous in g and thus difficult to optimize in general [22]. To circumvent the difficulty, surrogate loss functions are often used to facilitate the computation, so that the estimation formulation becomes
Popularly used surrogate loss functions include the hinge loss L(u)=(1−u)+ [23], the logistic loss [24], the ψ-loss [22, 25], and the extended ψδ-loss [26]. It is showed that all these surrogate loss functions are Fisher consistent in estimating the 0-1 loss 1−sign(u). The general proofs are given in [22, 27, 28], and thus omitted here. Figure 1 displays the 0-1 loss, the hinge loss, the logistic loss, the ψ-loss, and the ψ0.5-loss as functions of u. For illustration, we focus on the ψδ-loss in the sequel considering its extendability to a more flexible framework with covariate effects as discussed in Section 6.
Fig. 1.
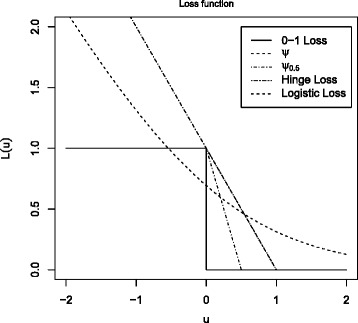
Various loss functions, including the 0–1 loss, the hinge loss, the logistic loss, the ψ loss and the ψ 0.5 loss
With the ψδ-loss, the proposed model-free estimation framework for (g(x),c) is formulated as
| (4) |
where λ is a tuning parameter, is set as a RKHS associated with a pre-specified kernel function K(·,·), and is the RKHS norm penalizing the complexity of g(x). The popular kernel functions include the linear kernel K(u,v) = uTv, the m-th order polynomial kernel , and the Gaussian kernel with a scale parameter τ2. When the linear kernel is used, the resultant contains all linear functions; when the Gaussian kernel is used, becomes much richer and admits more flexible nonlinear functions.
More interestingly, the representer theorem [18] implies that the solution to (4) must be of the form , and thus with a=(a1,⋯,an)T and . The representor theorem greatly simplifies the optimization task by turning the minimization over a functional space into the minimization over a finite-dimensional vector space. Specifically, the minimization task in (4) becomes
| (5) |
where is an (n+1)-dim vector.
The minimization task in (5) involves a non-convex function Lδ(·), and thus we employ the difference convex algorithm (DCA; [29]) to tackle the non-convex optimization task. The DCA decomposes the non-convex objective function in to the difference of two convex functions, and iteratively approximates it through a refined convex objective function. It has been widely used for non-convex optimization and delivers superior numerical performance [17, 21, 30]. Its computational complexity is of order o(log(1/ε)n3) [30], where ε denotes the computational precision. The details of solving (5) are similar to that in [21], and thus omitted here.
Results and discussion
Simulation examples
This section examines the proposed estimation method for combining biomarkers in a number of simulated examples. The numerical performance of the proposed kernel machine estimation (KME) method is compared against some existing popular alternatives, including the min-max method (MMM) [12], the parametric method under multivariate normality assumption (MVN) [31], the non-parametric kernel smoothing method (KSM) with Gaussian kernel [14], the stepwise method (SWM) [13], and the other two classification methods in [15], the logistic regression (LR) and the classification tree (TREE).
For illustration, the kernel function used in all methods is set as the linear kernel and the Gaussian kernel , where the scale parameter τ2 is set as the median of pairwise Euclidean distances between the positive and negative instances within the training set [32]. The tuning parameter λ for our proposed method is selected by 5-fold cross validation that maximizes the empirical Youden index
| (6) |
where I(·) is an indicator function and Vk is the validation set of k-th folder. The maximization is conducted via a grid search, where the grid for selecting λ is set as . The optimal solutions of MVN and KSM are searched by routine optim() in R as suggested in Ying and Tian [14]. SWM and MMM are based on the grid search with the same grid. TREE is tuned by default in R. Furthermore, for the proposed KME method, δ is set as 0.1 for all simulated examples as suggested in Hedayat et al. [26].
Four simulated examples are examined. Example 1 is similar to Example 5.1.1 in [14]. Example 2 modifies Example 1 by using multivariate Gamma distribution, which appears to be a popular model assumption in literature [19]. Examples 3 and 4 are similar to Setting 2 in [17] and Example II(b) in [33], which simulate data from logistic models with nonlinear effect terms
Example1.
A random sample {(Xi,Yi);i=1,⋯,n} is generated as follows. First, Yi is generated from Bernoulli(0.5). Second, if Yi=1, then Xi is generated from MVN(μ1,Σ1), where μ1=(0.4,1.0,1.5,1.2)T and Σ1=0.3I4 + 0.7J4 with I4 a 4-dimensional identity matrix and J4 a 4×4 matrix of all 1’s; if Yi=−1, then Xi is generated from MVN(μ2,Σ1) with μ2=(0,0,0,0)T.
Example2.
A random sample {(Xi,Yi);i=1,⋯,n} is generated as follows. First, Yi is generated from Bernoulli(0.5). Second, if Yi=1, then Xi is generated from a multivariate gamma distribution with mean μ1=(0.55,0.7,0.85,1)T and covariance matrix Σ1=0.25J4+diag(0.025,0.1,0.175,0.25); if Yi=−1, then Xi is generated from multivariate gamma distribution with mean μ2=(0.55,0.55,0.55,0.55)T and covariance matrix Σ2=0.025I4+0.25J4. The multivariate gamma distributed samples are generated with normal copula.
Example3.
A random sample {(Xi,Yi);i=1,⋯,n} is generated as follows. First, Xi is generated from MVN(μ,Σ), where μ=(0,0,0,0)T and Σ=0.3I4+0.7J4. Second, Yi is generated from a logistic model with .
Example4.
A random sample {(Xi,Yi);i=1,⋯,n} is generated as follows. First, Xi is generated from t4(μ,Σ), where μ=(0,0,0,0)T and Σ=I4. Second, Yi is generated from a logistic model with +
In all examples, the sample sizes for training ntr and testing nte are set as ntr=100,250,500 and nte=2000, respectively. Each scenario is replicated 100 times. The averaged empirical Youden index estimated on the testing sets, as well as the corresponding standard deviations, are summarized in Table 1.
Table 1.
Simulation examples: estimated means and standard deviations (in parentheses) of the empirical Youden index J over 100 replications
| n=100 | n=250 | n=500 | |
|---|---|---|---|
| Example 1 | |||
| LKME | 0.604 (0.0042) | 0.628 (0.0019) | 0.641 (0.0018) |
| GKME | 0.572 (0.0063) | 0.604 (0.0029) | 0.623 (0.0023) |
| MMM | 0.455 (0.0032) | 0.470 (0.0021) | 0.483 (0.0020) |
| MVN | 0.633 (0.0018) | 0.638 (0.0014) | 0.647 (0.0012) |
| KSM | 0.388 (0.0180) | 0.458 (0.0104) | 0.490 (0.0106) |
| SWM | 0.555 (0.0065) | 0.594 (0.0044) | 0.611 (0.0035) |
| LR | 0.628 (0.0022) | 0.639 (0.0017) | 0.646 (0.0017) |
| TREE | 0.490 (0.0068) | 0.525 (0.0047) | 0.559 (0.0029) |
| Example 2 | |||
| LKME | 0.636 (0.0075) | 0.690 (0.0025) | 0.710 (0.0015) |
| GKME | 0.612 (0.0054) | 0.654 (0.0045) | 0.696 (0.0016) |
| MMM | 0.609 (0.0033) | 0.622 (0.0025) | 0.622 (0.0022) |
| MVN | 0.573 (0.0065) | 0.571 (0.0047) | 0.563 (0.0040) |
| KSM | 0.214 (0.0281) | 0.046 (0.0164) | 0.047 (0.0171) |
| SWM | 0.447 (0.0094) | 0.426 (0.0078) | 0.429 (0.0065) |
| LR | 0.648 (0.0054) | 0.675 (0.0028) | 0.678 (0.0025) |
| TREE | 0.433 (0.0052) | 0.512 (0.0039) | 0.555 (0.0036) |
| Example 3 | |||
| LKME | 0.296 (0.0091) | 0.367 (0.0053) | 0.389 (0.0049) |
| GKME | 0.511 (0.0052) | 0.568 (0.0028) | 0.592 (0.0022) |
| MMM | 0.423 (0.0035) | 0.434 (0.0021) | 0.443 (0.0018) |
| MVN | 0.344 (0.0050) | 0.371 (0.0045) | 0.377 (0.0041) |
| KSM | 0.192 (0.0085) | 0.193 (0.0084) | 0.202 (0.0086) |
| SWM | 0.370 (0.0057) | 0.406 (0.0028) | 0.417 (0.0025) |
| LR | 0.307 (0.0043) | 0.316 (0.0030) | 0.320 (0.0026) |
| TREE | 0.424 (0.0059) | 0.477 (0.0042) | 0.528 (0.0031) |
| Example 4 | |||
| LKME | 0.103 (0.0102) | 0.150 (0.0098) | 0.209 (0.0089) |
| GKME | 0.529 (0.0078) | 0.626 (0.0050) | 0.682 (0.0028) |
| MMM | 0.184 (0.0084) | 0.227 (0.0034) | 0.236 (0.0026) |
| MVN | 0.109 (0.0071) | 0.152 (0.0056) | 0.189 (0.0054) |
| KSM | 0.188 (0.0050) | 0.213 (0.0035) | 0.220 (0.0028) |
| SWM | 0.255 (0.0078) | 0.293 (0.0050) | 0.307 (0.0039) |
| LR | 0.002 (0.0023) | 0.004 (0.0008) | 0.011 (0.0007) |
| TREE | 0.257 (0.0143) | 0.364 (0.0111) | 0.368 (0.0101) |
It is evident that our proposed methods, linear kernel machine estimation method (LKME) and Gaussian kernel machine estimation method (GKME), yield competitive performance in all examples. The performance of MVN, SWM, and LR is competitive in Example 1 as the data within each class indeed follows a Gaussian distribution sharing a common covariance structure, and thus the linear combination is optimal. Their performance becomes less competitive in other examples when linear combination is no longer optimal. It is evident that in Examples 3 and 4, with nonlinear patterns specified, the GKME outperforms all other methods. Especially, in Example 4, the performance of GKME is outstanding due to a strong nonlinear pattern specified. In general, the performance of KSM is less competitive. It could be due to the over-fitting issue when applying the Gaussian kernel to estimate sensitivity and specificity. With similar exhaustive grid search, the performance of SWM is better than MMM in Examples 1 and 4 but worse in Examples 2 and 3. As for the two classification methods, LR yields competitive performance in Examples 1 and 2 and becomes less competitive when logistic models with nonlinear patterns are applied in Examples 3 and 4. The performance of TREE is modest considering the nature of recursive partition.
Furthermore, it is of interest to conduct a numerical comparison on the performance of various surrogate loss functions in estimating the Youden index J. Figure 2 displays their estimated empirical Youden index in Example 3 with training size 500 over 100 replications. It is evident that the performances of all loss functions are similar.
Fig. 2.
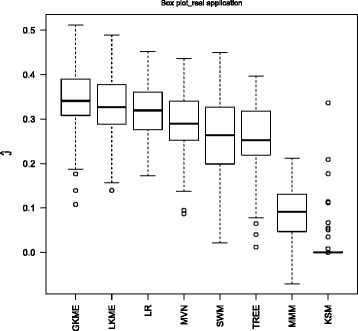
The boxplot of the empirical Youden index J for the hinge loss, the logistic loss, ψ-loss, and ψ 0.1-loss in Example 3 with n tr=500 over 100 replications
Real application
In this section, our proposed method is applied to a study of liver disorder. The dataset consists of 345 male subjects with 200 subjects in the control group and 145 subjects in the case group. For each subject, there are five blood tests (mean corpuscular volume, alkaline phosphotase, alamine aminotransferase, aspartate aminotransferase, and gamma-glutamyl transpeptidase) which are thought to be sensitive to liver disorders that may be related to excessive alcohol consumption, and another covariate with the average daily alcoholic beverages consumption information. The corresponding empirical estimates of the Youden index of all six markers are 0.141, 0.178, 0.174, 0.144, 0.240, and 0.121, respectively. The dataset was created by BUPA Medical Research Ltd., and is publicly available at University of California at Irvine Machine Learning Repository (https://archive.ics.uci.edu/ml/datasets/Liver+Disorders).
The total 345 samples are randomly split into a training set of 200 samples and a testing set of 145 samples. We also set δ=0.1 and select the tuning parameter λ by 5-fold cross validation targeting on maximizing (6). The experiment is replicated 100 times, and Fig. 3 summarizes the averaged performance measures of our proposed method, MMM, MVN, KSM, SWM, LR, and TREE.
Fig. 3.
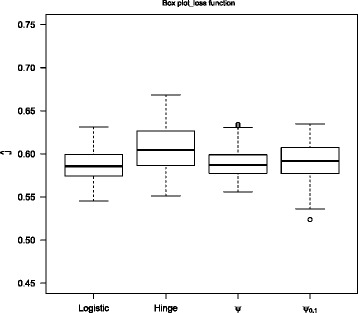
Real application: boxplot of the empirical Youden index J over 100 replications
It is evident that our proposed method delivers competitive performance in comparison with other methods. It is also interesting to notice the significant improvement on diagnostic accuracy by combining biomakers nonlinearly. It is encouraging to note that our proposed methods with Gaussian kernel outperforms all other methods.
Combining biomarkers with covariate-adjusted formulation
In many situations, the accuracy of diagnostic tests could be largely influenced by various factors, which population-based cut-point c does not take into account. To incorporate the effect of covariates, a natural idea is to consider personalized cut-point function c(z) as proposed in [21]. The covariate-adjusted formulation of J is then expressed as
| (7) |
where w(1,z)=1/πz, w(−1,z)=1/(1−πz), and πz=Pr(Y=1|Z=z).
Under this extended framework, the hinge loss, the logistic loss, and the ψ-loss are not longer Fisher consistent in estimating sign(g(x)−c(z)), as the candidate function is restricted to the form of g(x)−c(z) [26]. Proposition 1 shows that the surrogate ψδ-loss can still achieve the Fisher consistency when δ approaches 0.
Proposition1.
Denote , where , and . Given any ε>0, let , then as ,
where △ denotes the symmetric difference of two sets.
With the surrogate ψδ-loss, the covariate-adjusted estimation formulation becomes
| (8) |
where K1(·,·) and K2(·,·) are two per-specified kernel functions, and are their corresponding RKHS’s, and and are the corresponding RKHS norms. The optimization in (8) can be solved by DCA as for the population-based framework, and the details are omitted here.
Conclusions
This paper proposes a flexible model-free framework for combining multiple biomarkers. As opposed to most existing methods focusing on the optimal linear combinations, the framework admits both linear and nonlinear combinations. The superior numerical performance of the proposed approach is demonstrated in a number of simulated examples and a real application to the liver disorder study, especially when the sample size is relatively large. Furthermore, the proposed method is especially efficient with a relatively large number of biomarkers present, where most existing methods relying on grid search are often inefficient. An extension of the proposed framework to the covariate-adjusted formulation is also included. Further development on estimating confidence interval using perturbation resampling procedure [34] and variable selection for biomarkers are still under investigation.
Acknowledgments
JW’s research is partly supported by HK GRF Grant 11302615, CityU SRG Grant 7004244 and CityU Startup Grant 7200380. The authors thank the Associate Editor and two referees for their constructive comments and suggestions.
Additional file
The Appendix includes the proof of Proposition 1. (PDF 28.6 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All authors have made contribution to the development of the proposed methodology. TX carried out the simulations and real data analysis and drafted the first version of the paper. YF, AR and JW are actively involved in revisions and have read and approved the final manuscript.
Contributor Information
Tu Xu, Email: Tu.Xu@gilead.com.
Yixin Fang, Email: Yixin.Fang@nyumc.org.
Alan Rong, Email: Alan.Rong@astellas.com.
Junhui Wang, Email: j.h.wang@cityu.edu.hk.
References
- 1.Shapiro D. The interpretation of diagnostic tests. Stat Methods Med Res. 1999;8:113–34. doi: 10.1191/096228099666928387. [DOI] [PubMed] [Google Scholar]
- 2.Zhou X, McClish D, Obuchowski N. Statistical methods in diagnostic medicine. New York: Wiley; 2002. [Google Scholar]
- 3.Pepe M. The statistical evaluation of medical tests for classification and prediction. Oxford, UK: Oxford University Press; 2003. [Google Scholar]
- 4.Youden W. An index for rating diagnostic tests. Cancer. 1950;3:32–5. doi: 10.1002/1097-0142(1950)3:1<32::AID-CNCR2820030106>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 5.Bamber D. The area above the ordinal dominance graph and the area below the receive operating characteristic graph. J Math Psychol. 1975;12:387–415. doi: 10.1016/0022-2496(75)90001-2. [DOI] [Google Scholar]
- 6.Aoki K, Misumi J, Kimura T, Zhao W, Xie T. Evaluation of cutoff levels for screening of gastric cancer using serum pepsinogens and distributions of levels of serum pepsinogens I, II and of PG I/PG II ratios in a gastric cancer case-control study. J Epidemiol. 1997;7:143–51. doi: 10.2188/jea.7.143. [DOI] [PubMed] [Google Scholar]
- 7.Perkins N, Schisterman E. The inconsistency of optimal cutpoints obtained using two criteria based on the receiver operating characteristic curve. J Epidemiol. 2006;163:670–5. doi: 10.1093/aje/kwj063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sidransky D. Emerging molecular markers of cancer. Nat Rev Cancer. 2002;2:210–9. doi: 10.1038/nrc755. [DOI] [PubMed] [Google Scholar]
- 9.Kumar S, Mohan A, Guleria R. Biomarkers in cancer screening, research and detection: present and future: a review. Biomarkers. 2006;11:385–405. doi: 10.1080/13547500600775011. [DOI] [PubMed] [Google Scholar]
- 10.Su J, Liu J. Linear combinations of multiple diagnostic markers. J Am Stat Assoc. 1993;88:1350–5. doi: 10.1080/01621459.1993.10476417. [DOI] [Google Scholar]
- 11.Pepe M, Thompson M. Combining diagnostic test results to increase accuracy. Biostatistics. 2000;1:123–40. doi: 10.1093/biostatistics/1.2.123. [DOI] [PubMed] [Google Scholar]
- 12.Liu C, Liu A, Halabi S. A min-max combination of biomarkers to improve diagnostic accuracy. Stat Med. 2011;30:2005–14. doi: 10.1002/sim.4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kang L, Liu A, Tian L. Linear combination methods to improve diagnostic/prognostic accuracy on future observations. Stat Methods Med Res. 2013; 22. In press. [DOI] [PMC free article] [PubMed]
- 14.Yin J, Tian L. Optimal linear combinations of multiple diagnostic biomarkers based on Youden index. Stat Med. 2014;33:1426–40. doi: 10.1002/sim.6046. [DOI] [PubMed] [Google Scholar]
- 15.Kouskoumvekaki I, Yang Z, Jónsdóttir S, Olsson L, Panagiotu G. Identification of biomarkers for genotyping Aspergilli using non-linear methods for clustering and classification. BMC Bioinformatics. 2008;9:59. doi: 10.1186/1471-2105-9-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Turck C. Biomarkers for psychiatric disorders. USA: Springer-Verlag; 2009. [Google Scholar]
- 17.Huang Y, Fong Y. Identifying optimal biomarker combinations for treatment selection via a robust kernel method. Biometrics. 2014;70:891–901. doi: 10.1111/biom.12204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wahba G. Spline models for observational data: CBMS-NSF Regional Conference Series in Applied Mathematics; 1990.
- 19.Schisterman E, Perkins N, Liu A, Bondell H. Optimal cut-point and its corresponding Youden Index to discriminate individuals using pooled blood samples. Epidemiology. 2005;16:73–81. doi: 10.1097/01.ede.0000147512.81966.ba. [DOI] [PubMed] [Google Scholar]
- 20.Fluss R, Faraggi D, Reiser B. Estimation of the Youden index and its associated cutoff point. Biom J. 2005;47:458–72. doi: 10.1002/bimj.200410135. [DOI] [PubMed] [Google Scholar]
- 21.Xu T, Wang J, Fang Y. A model-free estimation for the covariate-adjusted Youden index and its associated cut-point. Stat Med. 2014. in press. [DOI] [PubMed]
- 22.Shen X, Tseng G, Zhang X, Wong W. On ψ-learning. J Am Stat Assoc. 2003;98:724–34. doi: 10.1198/016214503000000639. [DOI] [Google Scholar]
- 23.Vapnik V. Statistical learning theory. Chichester, UK: Wiley; 1998. [Google Scholar]
- 24.Zhu J, Hastie T. Kernel logistic regression and the import vector machine. J Comput Graph Stat. 2005;14:185–205. doi: 10.1198/106186005X25619. [DOI] [Google Scholar]
- 25.Liu Y, Shen X. Multicategory psi-learning. J Am Stat Assoc. 2006;101:500–9. doi: 10.1198/016214505000000781. [DOI] [Google Scholar]
- 26.Hedayat AS, Wang J, Xu T. Minimum clinically important difference in medical studies. Biometrics. 2014;71:33–41. doi: 10.1111/biom.12251. [DOI] [PubMed] [Google Scholar]
- 27.Wang J, Shen X. Probability estimation for large-margin classifiers. Biometrika. 2008;95:149–67. doi: 10.1093/biomet/asm077. [DOI] [Google Scholar]
- 28.Lin Y. Support vector machines and the Bayes rule in classification. Data Mining Knowl Discov. 2002;6:259–75. doi: 10.1023/A:1015469627679. [DOI] [Google Scholar]
- 29.An L, Tao P. Solving a class of linearly constrained indefinite quadratic problems by D.C. algorithms. J Glob Optim. 1997;11:253–85. doi: 10.1023/A:1008288411710. [DOI] [Google Scholar]
- 30.Liu S, Shen X, Wong W. Computational development of ψ-learning. In: Proceedings of the SIAM International Conference on Data Mining. Newport, CA: 2005. p. 1–12.
- 31.Schisterman EF, Perkins N. Confidence intervals for the Youden index and corresponding optimal cut-point. Communications in Statistics-Simulation and Computation. 2007;36:549–63. doi: 10.1080/03610910701212181. [DOI] [Google Scholar]
- 32.Jaakkola T, Diekhans M, Haussler D. Using the Fisher kernel method to detect remote protein homologies. In: Proceedings of the Seventh International Conference on Intelligent Systems for Molecular Biology: 1999. p. 149–158. [PubMed]
- 33.Fong Y, Yin S, Huang Y. Combining biomarkers linearly and nonlinearly for classification using the area under the ROC curve; 2014. http://works.bepress.com/yfong/3. [DOI] [PMC free article] [PubMed]
- 34.Jiang B, Zhang X, Cai T. Estimating the confidence interval for prediction errors of support vector machine classifiers. J Mach Learn Res. 2008;9:521–40. [Google Scholar]