

Abstract
The aggregation of specific proteins is hypothesized to underlie several degenerative diseases, collectively called amyloid disorders. However, the mechanistic connection between the process of protein aggregation and tissue degeneration is not yet fully understood. Here, we review current and emerging strategies to ameliorate aggregation-associated degenerative disorders, with a focus on disease-modifying strategies that prevent the formation of and/or eliminate protein aggregates. Persuasive pharmacologic and genetic evidence now support protein aggregation as the cause of post-mitotic tissue dysfunction or loss. However, a more detailed understanding of the factors that trigger and sustain aggregate formation, as well as the structure-activity relationships underlying proteotoxicity are needed to develop future disease-modifying therapies.
Transthyretin (TTR)1, immunoglobulin light chain (LC)2, serum amyloid A (SAA)3, and amyloid-β (Aβ)4 are examples of more than thirty human proteins whose misfolding and/or misassembly into a variety of aggregate structures appear to cause a spectrum of degenerative disorders5. These so-called amyloid diseases are named after the cross-β-sheet aggregates, or amyloid fibrils, that are the pathological hallmarks of these maladies6, 7. Amyloid fibrils in a specific disease are generally composed predominantly of one protein5. Amyloid fibrils from different diseases and composed of different proteins exhibit similar structural features6.
In affected patient tissues, protein aggregation and deposition mainly occurs at the normal extracellular or intracellular location of the aggregation-prone protein. However, there is increasing evidence for the presence of both intra- and extracellular aggregates in nearly all of the aggregation-associated degenerative diseases8-10. Moreover, evidence indicates that aggregates can travel between intracellular and extracellular locations, suggesting that intracellular toxicity might also contribute to the pathology once thought to result exclusively from extracellular aggregation, e.g., Aβ aggregates in Alzheimer's disease (AD)11-16. Furthermore, cellular uptake and release of protein aggregates appears to contribute to their spreading within a multicellular organism and the associated pathology and tissue damage17-20. However, the mechanism(s) by which the process of intra- and/or extracellular aggregation cause pathology remains unclear.
Strong genetic, pharmacologic, biochemical and pathologic evidence support the hypothesis that human amyloid diseases result from the process of protein aggregation or amyloidogenesis (Fig. 1)21-28. By the “process of protein aggregation” we are referring to aggregation in a multicellular organism wherein physical chemical forces and biological modifiers together influence the aggregate structural ensembles afforded. It is important to recognize that there is an incomplete understanding of aggregation, both in vitro and in a multicellular organism, because probes to monitor the different types of aggregates formed or the structures afforded during this dynamic process are not available. In the absence of more detailed information about the ensemble of aggregate structures present in a patient, it is probably useful to think about aggregates as a spectrum of structures ranging from small relatively unstructured oligomers to structurally well-defined cross-β-sheet amyloid fibrils, recognizing that some structures may only be significantly populated in an organism or in certain cellular compartments. It is also unclear, which of the aggregate types are toxic and what the mechanism of cytotoxicity is. A current hypothesis suggests that smaller diffusible oligomers, exhibiting a spectrum of structures, rather than the insoluble cross-β-sheet amyloid fibrils are driving the degenerative pathology29. However, these diffusible oligomers could result from fragmentation of fibrils into small pieces no longer capable of supporting a cross-β-sheet amyloid structure or from unsuccessful degradation of amyloid by the lysosome or proteasome. Thus, we hypothesize that it is important to consider protein aggregation in vivo as a dynamic process with many players. Even with this incomplete knowledge of the aggregated structures present in patients, preventing active protein aggregation and/or removing diffusible proteotoxic aggregates, as well as ameliorating the toxic effects of aggregates while maximizing the physiological function of these proteins, are the focus of therapeutic strategies currently being developed22-27, 30-32.
Figure 1. Amyloidogenesis—a process of aggregation influenced by the physical chemistry of the protein as well as cellular and extracellular components.
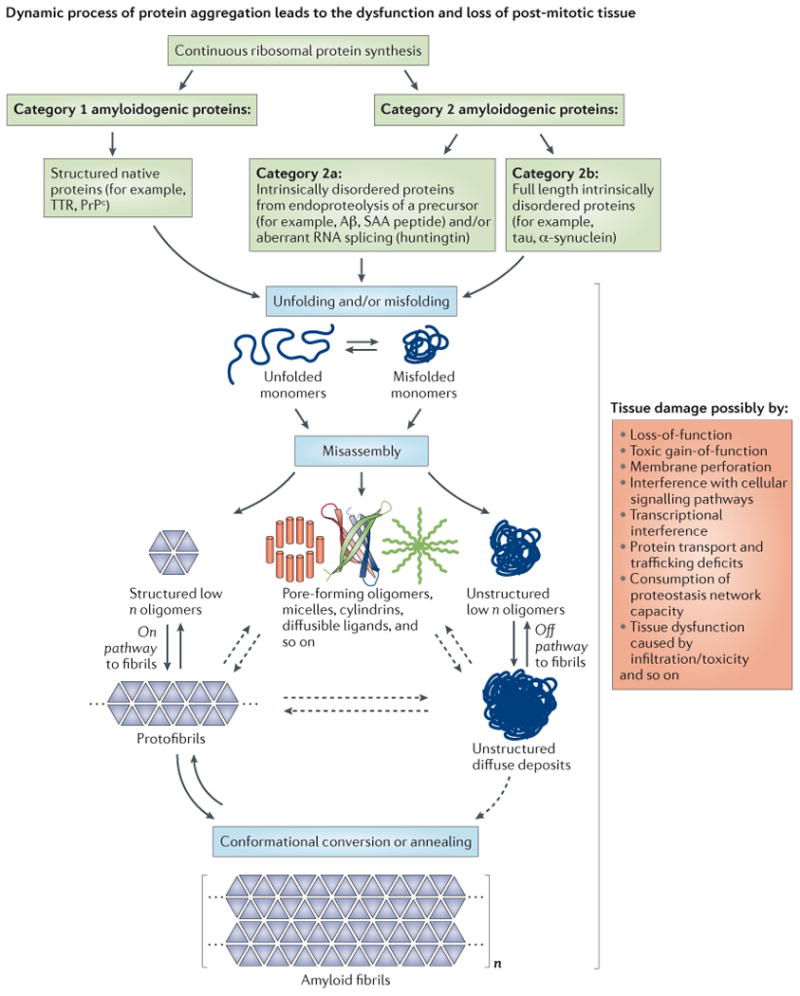
Amyloidogenic proteins associated with degenerative disorders can be subdivided into two categories based on their native structure. Category 1 proteins, such as transthyretin (TTR) and the prion protein (PrPc), exhibit a well-defined native state three-dimensional structure, whereas category 2 proteins are intrinsically disordered. Both, intrinsically disordered polypeptides generated by endoproteolytic processing of a precursor protein (category 2a), such as Aβ generated by cleavage of the amyloid precursor protein (APP), as well as full-length intrinsically disordered proteins (category 2b), such as tau and α-synuclein, can be amyloidogenic. The critical step in amyloidogenesis is misfolding and aggregation of category 1 proteins or misassembly of category 2 proteins into a spectrum of aggregate structures, including β-sheet-rich structures and amyloid fibrils. The structures associated with the amyloid cascade are depicted along with their hypothesized mechanisms of proteotoxicity (shown on the far right). The ensemble of structures is likely influenced and some may be generated by the biology of the organism, e.g., incomplete degradation of amyloid could afford novel structures, or aggregation on cell membranes could afford aggregate structures that can only form in the presence of certain lipids and/or carbohydrates.
Amyloidogenesis: The Process of Protein Aggregation
There are two categories of amyloidogenic proteins (Fig. 1). In the case of proteins that initially adopt a well-defined, folded, three-dimensional structure (category 1), substantial evidence supports the idea that a partial loss of this well-defined structure is required for their aggregation33-36. Early studies on TTR and LC demonstrate that conformational changes alone are sufficient to enable these proteins to misassemble into a spectrum of aggregate structures, including amyloid fibrils33, 34. These observations provide substantial evidence supporting the “conformation change hypothesis” as the basis for their aggregation. The same forces that drive intramolecular protein folding, including hydrogen bonding and the hydrophobic effect, also mediate intermolecular aggregation or concentration-dependent amyloidogenesis when the misfolded protein is present at sufficient concentrations33, 37.
Intrinsically disordered polypeptides are the second category of amyloidogenic proteins (category 2)38, 39. Generally speaking, category 2 polypeptides undergo thermodynamically linked aggregation and conformational changes, affording more β-sheet-rich structures, often requiring a nucleation step (Fig. 2). Category 2 polypeptides can be further separated into two subcategories. The first of these, category 2a, are intrinsically disordered polypeptides that result from the endoproteolysis of a large precursor protein. The best-studied example is the amyloid-β protein (Aβ), which is actually a family of peptides arising from β- and γ-secretase endoproteolysis of the amyloid precursor protein (APP)40, 41. Under pathological conditions, Aβ forms extracellular aggregates in the brain parenchyma and/or the walls of blood vessels in the brain, and, to a lesser extent, inside of brain neurons—these processes have been linked to AD and cerebral β-amyloid angiopathy. The SAA peptide, derived from endoproteolysis of the SAA protein, that is produced in high concentration as a consequence of chronic inflammation, is also a well-studied intrinsically disordered category 2a protein whose aggregation causes AA amyloidosis3. The aggregation propensity of intrinsically disordered proteins, such as SAA and Aβ, is strongly influenced by their concentration, post-translational modifications, and the exact sequence afforded by the endoproteolytic processing event(s).
Figure 2. Mechanisms of Protein Aggregation.
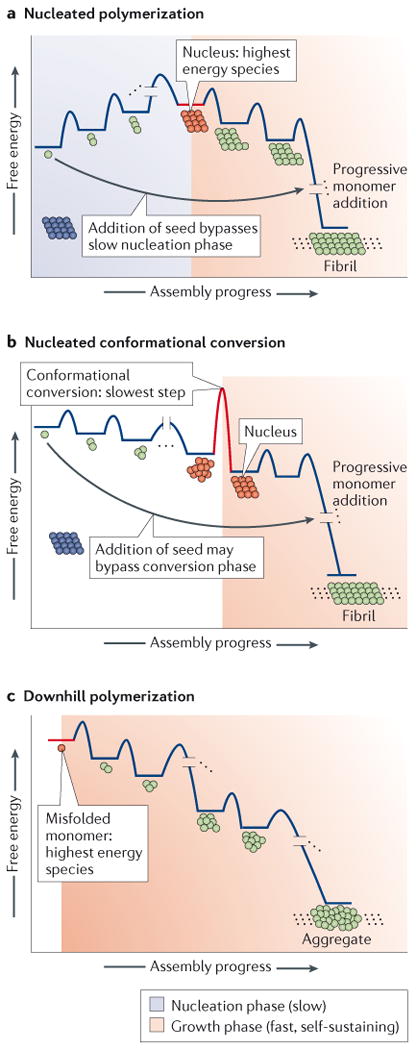
(A) In a nucleation-dependent polymerization, the initiating step of aggregation involves the formation of a nucleus, a sparsely populated, high energy species that has a conformation that differs from that of the soluble protein. The nucleus is typically rich in β-sheet structure and presumably is oligomeric, although monomeric species have been implicated. Once the nucleus is formed, monomers are added rapidly to the growing non-covalent polymer to generate thermodynamically more stable aggregates that can also act as seeds. Monomer aggregation can proceed very rapidly by the addition of preformed aggregates, or seeds, because nucleation is no longer required in a “seeded” aggregation reaction. (B) In a nucleated conformational conversion, an equilibrium exists between monomers and structurally heterogeneous oligomers, which are typically (but not always) more stable than monomers. Over time, the oligomers are converted into a nucleus. This nucleus then initiates conformational conversion of neighboring monomers comprising the oligomer into amyloid fibers. Seeding can bypass the requirement for a slow nucleated conformational conversion step. (C) In a downhill polymerization, formation of the aggregation-prone misfolded monomer from a natively folded protein is the rate limiting step (not shown). Subsequent addition of monomers to the growing polymer is energetically favorable and leads to amorphous aggregate formation, as well as the generation of cross-β-sheet amyloid fibrils. Seeding does not accelerate the rate of aggregation in a downhill polymerization, at least in transthyretin amyloidogenesis studied largely in vitro.
The huntingtin (htt) protein42 - whose aggregation underlies Huntington's disease - is also thought to be a category 2a protein. The majority of publications suggest that an N-terminal fragment of htt, including the polyglutamine (polyQ) stretch, is released following proteolysis and/or aberrant RNA splicing, triggering the formation of aggregates, and is more toxic than the full-length htt43, 44. Other mechanisms underlying htt cytoxicity have been suggested, including altered protein structure of the polyQ stretch within the full-length htt and possibly altered protein-protein interactions of full-length htt. Other polyQ disorders may result from the misfolding and misassembly of category 1 proteins, e.g., spinal and bulbar muscular atrophy, a neuromuscular disorder apparently caused by the aggregation of an expanded polyQ tract in the androgen receptor45. It is clear that more thorough observations are needed with regard to whether full-length and/or fragment polyQ proteins are driving pathology. Nonetheless, the number of CAG repeats in several polyQ expanded proteins, including htt, correlates with the age of onset and disease severity, suggesting that the increased probability for misassembly correlates with proteotoxicity46.
Category 2b polypeptides are full-length intrinsically disordered proteins that aggregate without the requirement for endoproteolysis. This classification includes proteins such as tau47 whose aggregation is associated with forms of frontotemporal dementia and AD, and α-synuclein48 whose amyloidogenesis is thought to cause Parkinson's disease. These category 2b proteins aggregate intracellularly. The propensity of tau and α-synuclein to aggregate is influenced principally by concentration, and by post-translational modifications like phosphorylation and nitrosylation at certain residues in sporadic forms of these diseases or by point mutations in familial categories of these maladies49, 50. It should be noted that recent findings suggest that the α-synuclein protein may also adopt a quaternary structure within neurons under specific conditions, thus challenging the previous notion that α-synuclein is solely an intrinsically disordered protein51-53.
Typically, the misfolding and/or aggregation of category 1 or 2 proteins are thought to cause a “gain-of-proteotoxicity” phenotype, disturbing cellular function(s) and initiating toxicity cascades (Fig. 1). However, it is worth noting that loss of function may also contribute to aggregation disease pathogenesis. This seems especially relevant for nuclear proteins like TDP-43 (TAR DNA binding protein of 43kDa) and FUS (Fused in Sarcoma, an RNA binding protein) involved in amyotrophic lateral sclerosis and frontotemporal lobar degeneration type dementia. Both TDP-43 and FUS relocate to and aggregate in the cytoplasm, where they can also be recruited to stress granules, and cause translational alterations54, 55. In the case of C9orf72 hexanucleotide repeat expansion-associated amyotrophic lateral sclerosis and frontotemporal dementia, it appears that sense and antisense RNA aggregates as well as aggregation of proteins resulting from unconventional translation into polydipeptides are jointly responsible for the neurodegenerative phenotypes56. Importantly, protein aggregates can sequester folded proteins and RNA, altering additional cellular functions57. In addition, the buildup of protein aggregates can impair proteostasis network capacity and thereby compromise the quality of other cellular protein components.
We define the process of protein aggregation or amyloidogenesis as encompassing all misassembled structures formed, irrespective of their conformations. Aggregates include low n oligomers of heterogeneous structure, rather disordered micelle-type structures, amorphous aggregates of various sizes, β-sheet-rich structures, and cross-β-sheet amyloid structures (Fig. 1)29, 58, 59. Misfolding of the monomer could also contribute to proteotoxicity and should not be overlooked. At least three distinct mechanisms of protein aggregation are supported by the literature (Fig. 2). These mechanisms are largely based on in vitro data and their consistency with mathematical models of protein aggregation.
The first mechanism posits that protein aggregation occurs by a nucleated polymerization, wherein a high-energy, sparsely populated species (typically oligomeric, but monomers have also been implicated) is formed that can subsequently efficiently add monomers to form a thermodynamically more stable aggregate structure60-62 (Fig. 2A). A nucleus can have a cross-β-sheet structure, or it can have an alternative structure that may or may not later conformationally convert to a cross-β-sheet structure63. As mentioned above, nucleation is often required for the aggregation of both category 1 and 2 proteins64, 65. The requirement for nucleus formation can be bypassed by adding aggregate seeds (often cross-β-sheet aggregates) of a particular protein, such as Aβ, to a solution of monomers of the same protein (Fig. 2A). This enables efficient aggregation by exogenous seeding–thereby accelerating aggregation62. Indeed, there is now ample evidence that this type of seeding mechanism might be an important step for the progression and spreading of aggregation in neurodegenerative disorders (discussed in more detail below). Thus, aggregation could start in a single rogue cell or tissue and spread throughout the brain by cellular uptake of seeds followed by cellular secretion of the amplified aggregates either by canonical or alternative pathways8, 17, 66, 67. Based on in vitro and ex vivo data, it is commonly assumed that seeded polymerizations afford only structures identical to the seeds and not other structures that currently cannot easily be monitored. However, this may not be true in a multicellular organism where biological modifiers influence the process. In fact, it is hard to explain why oligomers can be isolated from tissue containing amyloid fibrils if seeded polymerizations only afford cross-β-sheet amyloid fibrils.
In the second accepted mechanism of aggregation - a nucleated conformational conversion - an equilibrium exists between monomers and structurally heterogeneous oligomers that are generally more stable than the monomers, but this does not always have to be the case (Fig. 2B)63. Over time, the oligomers are converted into a nucleus and then into amyloid fibrils. The fibrils could conformationally convert the proximal monomers into amyloid fibers, but this does not appear to occur with all proteins.
A third mechanism of protein aggregation posits that category 1 proteins sometimes aggregate by a downhill mechanism (Fig. 2C), a process that is not seedable68. In the downhill polymerization scenario, formation of the aggregation-prone misfolded monomer from a natively folded protein is the rate-limiting step (a step not shown in Fig. 2C). Partially folded monomeric proteins aggregate to afford thermodynamically more stable higher order aggregates including amyloid fibrils, thus high energy nucleus formation is not required for efficient aggregation68. Subsequent addition of misfolded monomers to the growing polymer is always energetically favorable in this mechanism of aggregation. Transthyretin appears to form amyloid fibrils and other aggregate structures in parallel by a pathway analogous to that shown in Fig. 2C, wherein tetramer dissociation is rate limiting.
While these models of protein aggregation provide a framework for our understanding, it seems likely that conditions in living organisms are more complex and it is possible that several different aggregate structures may coexist, e.g., due in part to unsuccessful lysosomal or proteasomal degradation. Different cellular environments (e.g., lysosomes vs. endoplasmic reticulum vs. cytoplasm vs. the extracellular space) provide distinct conditions for the formation and sustainment of different aggregate structures, likely influencing the ensemble of aggregate structures present. Efforts are underway to improve detection and quantification of aggregate structures in addition to cross-β-sheet amyloid. The availability of these probes is expected to greatly advance our understanding of structure-proteotoxicity relationships.
Amyloid versus the Aggregation Process as the Toxicity Driver
The extracellular and/or intracellular cross-β-sheet amyloid fibril deposits in tissues of patients afflicted with amyloid diseases are recognized as the histopathological hallmarks, and still serve as the basis for a definitive diagnosis of the systemic amyloidoses and diagnostic confirmation of brain disorders such as AD postmortem (Box 1)7. While the protein deposits in human amyloid diseases often consist of aggregates of diverse structure, the focus has been on the cross β-sheet amyloid fibrils because of their well-defined structure, and thus ease of detection6. Amyloid fibrils bind to Thioflavin T, Congo red, and other aromatic chromophores that exhibit fluorescence and/or birefringence. These attributes help pathologists identify amyloid in tissue. Amyloid fibrils are typically covered by or co-deposit with glycosaminoglycans, the amyloid P glycoprotein, and other cellular and extracellular macromolecules in humans.
Box 1: The amyloid hypothesis.
The amyloid cascade hypothesis was first postulated in the context of systemic amyloidosis in the 1960s265 and as a framework for Alzheimer's disease (AD) in the early 1990s266-268. AD is characterized by the aggregation of two proteins, namely Aβ, depositing in the form of extracellular amyloid plaques, and tau, becoming hyperphosphorylated and aggregating intracellularly to form neurofibrillary tangles269. The amyloid cascade hypothesis for AD, which incorporates genetic, biochemical and histological evidence, posits that the deposition of Aβ in the brain due to an imbalance between its production and clearance initiates a sequence of events that ultimately lead to AD dementia. It was proposed that the sequence of events is similar for the rare familial forms as well as for the common sporadic forms of AD. While it was originally thought that amyloid plaques containing fibrillar forms of Aβ were the pathogenic entities268, some findings appear to contradict this. For example, reports that the amount of amyloid does not correlate with disease status270. Moreover, clinically normal individuals can have a substantial amyloid load, similar in amount to severely symptomatic patients271. More recent evidence points to a stronger neurotoxic effect from smaller, diffusible protein aggregates, often referred to as oligomers, and suggests that they may represent the disease-driving force272, 273. It could be that oligomers and amyloid fibrils are part of a dynamic structure-proteotoxicity relationship and that numerous structures contribute to AD pathogenesis. The amyloid cascade hypothesis for AD proposes that Aβ aggregation is the primary event and that aggregation of tau, inflammation and other changes observed in AD brains are downstream272, 274. If correct, prohibiting the formation of Aβ aggregates, as well as reducing or removing them should be therapeutically useful. However, so far, clinical trials targeting Aβ have not exhibited the ability to slow AD progression. This may be due to several reasons, including that advanced patients too far along in the progression of AD have been enrolled in clinical trials and/or that drugs targeting the wrong Aβ species are being pursued. These results have led investigators to question whether Aβ is the right primary target, in addition to questioning the validity of the amyloid cascade hypothesis for AD. While the amyloid cascade hypothesis was postulated to explain the temporal relationship of Aβ and tau aggregation for AD, the amyloid hypothesis in a broader sense posits that the conversion of natively folded proteins into non-native protein aggregates is the primary event in amyloid pathogenesis. Moreover, this hypothesis states that aggregation results in alterations in the cellular microenvironment occur that lead to pathological changes and eventually cellular malfunction and degeneration. In this broader sense, the amyloid hypothesis provides a framework for all amyloid disorders. Recent advances in the treatment of familial amyloid polyneuropathy, a transthyretin amyloid disease (Box 2), supports the notion that indeed protein misfolding and aggregation is the primary culprit and that dramatically slowing the process of active aggregation is disease modifying.
It remains unclear to what extent amyloid fibrils are by themselves the toxic entities, or if other forms of misassembled proteins, e.g., non-uniformly structured oligomers, are contributors to cytotoxicity or are the primary drivers of proteotoxicity. Whether this cytotoxicity derives from intracellular and/or extracellular aggregates is unknown. It also remains unclear whether the sequestration of the smaller non cross-β aggregates into cross-β-sheet amyloid fibrils would possibly mediate some protection (Box 1). Amyloid load often does not correlate with disease status, calling into question whether amyloid fibrils are the main driver of neurodegeneration, a topic that has been discussed intensively in the AD field69. It has been suggested that amyloid formation may even be protective, the hypothesis being that the more toxic, structurally diverse aggregates are converted into less toxic cross-β-sheet amyloid fibrils, minimizing exposure of hydrophobic surface area and open hydrogen bond valencies that mediate aberrant interactions with proteins, nucleic acids and carbohydrates70. Experimental evidence suggests that neuronal survival is increased in cultured cells when mutant huntingtin is sequestered into inclusion bodies, concomitant with a decrease in the more diffuse aggregated forms of cellular mutant huntingtin71. Moreover, amyloid clearance occurs in some, but not in the majority of patients benefiting from disease-modifying therapies on the timescale of the clinical response. Amyloid clearance needs to be studied more systematically, especially in the peripheral amyloidoses where disease-modifying therapies exist so that clear conclusions can be drawn30, 72, 73. Numerous laboratories have hypothesized that smaller diffusible aggregates of poorly characterized structure, often called oligomers or protofibrils, that often escape histological detection, are the toxic species driving post-mitotic tissue degeneration in the amyloidoses29, 58. Acute cellular toxicity assessments in vitro and in vivo also indicate that smaller soluble and diffusible aggregates are notably more toxic than amyloid fibrils74-76, while conversion of toxic oligomers to amyloid fibrils can reduce toxicity75, 77. Our understanding of structure-proteotoxicity relationships is currently incomplete, largely due to technical limitations; thus we urgently need better technologies to specifically detect and quantify amyloid and non-amyloid aggregate types, which could also be useful as much needed biomarkers for diagnosing and monitoring response to therapy in the amyloidoses.
There is mounting evidence that the process of protein aggregation or amyloidogenesis is an important driver of neurodegeneration. Amyloidogenesis is dynamic, i.e., there is a constant flux of newly synthesized proteins aggregating into a spectrum of aggregates that are transient structures78, 79. These fleeting and structurally heterogeneous aggregates evolve into progressively larger aggregates80, and if the conditions are right, into amyloid fibrils. Presumably, most of the non-fibrillar aggregate structures (Fig. 1) are intrinsically unstable owing to exposed hydrophobic surfaces and/or the presence of unsatisfied hydrogen bonds, and can engage in aberrant molecular interactions. Amyloid fibrils are less able to engage in inappropriate molecular interactions because of the more extensive burial of hydrophobic residues and the formation of intermolecular hydrogen bonds6. It is now clear that nearly all proteins capable of forming amyloid are also capable of forming a spectrum of aggregates of diverse structure that appear to be more toxic than amyloid fibrils to cultured cells, but whether this is the case in humans remains unclear.
Autosomal dominantly inherited familial amyloid diseases are caused by mutations that can destabilize the native structure of category 1 proteins, enabling their partial unfolding and rendering them aggregation prone33, 34, 36. Similarly, mutations in intrinsically disordered category 2 proteins can facilitate aggregation and cause familial forms of amyloidosis81, 82. An increased concentration of category 2 proteins, e.g., of α-synuclein through gene duplication in Parkinson's disease and of APP and Aβ in Down's Syndrome-linked Alzheimer's disease, or variations within promoter or regulatory regions can lead to aggregation-associated degenerative pathologies83-85.
Strategies to Target The Process of Protein Aggregation
Protein reduction strategy
The first successful treatment of a human amyloid disease not caused by an underlying pathology (such as a cancer or inflammation) occurred in the context of the transthyretin (TTR) amyloid disease, familial amyloid polyneuropathy (Box 2). This strategy involved surgically-mediated gene therapy86, 87(Table 1). Familial TTR amyloid diseases are caused by the aggregation of predominantly mutant TTR and, to a lesser extent, wild type (WT) TTR88. Since the liver secretes 95% of WT and mutant TTR into the bloodstream, replacing the liver of a heterozygous polyneuropathy patient secreting V30M and WT TTR with a WT / WT TTR secreting liver from a normal subject by liver transplantation abrogates the secretion of mutant TTR into the blood. This resulted in increased serum concentrations of the more kinetically stable, less aggregation-prone WT TTR tetramers (Fig. 3A), which slowed the progression of peripheral and autonomic neuropathy and significantly extended the lifespan of familial amyloid polyneuropathy patients.89, 90. Amyloid clearance appears to occur slowly in transplanted patients. The newly available Positron Emission Tomography amyloid imaging agents would enable the rate of TTR amyloid clearance to be studied more rigorously at multiple centers and this should be done91.
Box 2: Transthyretin Amyloidogenesis.
Transthyretin (TTR) is a natively folded, β-sheet-rich, tetrameric category 1 protein secreted into the blood by the liver, into the vitreous by the retinal pigment epithelial cells, and into the cerebrospinal fluid (CSF) by the choroid plexus275-277. In most people, TTR tetramers are made up of four identical subunits; however, the TTR tetramers in heterozygotic amyloidosis patients comprise wild type (WT) and mutant subunits278. The established function of TTR in the bloodstream is to transport ≈ 0.5 equivalents of retinol binding protein bound to vitamin A279. Transthyretin carries a substantial quantity of thyroxine (T4) in the CSF, but in the blood, the two T4-binding sites of TTR are 99% unoccupied owing to the presence of additional T4 carriers280. Substantial biophysical data suggest that in the case of the 125+ distinct mutations linked to the familial TTR amyloidoses281 (http://amyloidosismutations.com/mut-attr.php), incorporation of mutant TTR subunits into a TTR tetramer otherwise composed of WT subunits destabilizes the heterotetramer—leading to faster TTR tetramer dissociation kinetics (kinetic destabilization), an increased population of misfolded aggregation-prone TTR monomer (thermodynamic destabilization), or both282, 283. Kinetic destabilization is especially relevant for aggregation and likely for disease progression, since TTR tetramer dissociation is the rate-limiting step in the amyloidogenesis cascade (see figure)25, 26. The subsequent monomer misfolding required for TTR aggregation is much faster than tetramer dissociation33, 68, 283-286. Misfolded TTR monomers efficiently aggregate by a downhill polymerization mechanism (Figure 2C). Thus, TTR aggregation is not susceptible to seeding68.
Depending on the destabilized mutant inherited in the familial TTR amyloidoses, TTR aggregation can compromise the function of the autonomic and/or peripheral nervous systems (familial amyloid polyneuropathy)1, 287, and/or the heart (familial amyloid cardiomyopathy)288-291, and/or the eyes97, and/or the meningocerebrovascular system in the brain292. The recently renamed WT TTR Amyloidosis—formally called senile systemic amyloidosis (SSA)—results in a cardiomyopathy that affects as much as 15% of the population exceeding 80 years of age288, 289, 293, 294. It was recently reported that WT TTR aggregation in and around the vasculature may be a more prominent cause of vascular diseases than previously thought295.
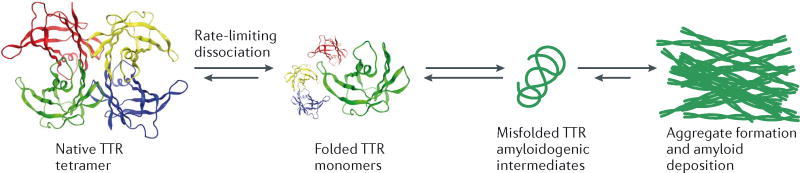
Table 1.
Emerging, potentially disease-modifying, therapeutic strategies.
| Therapeutic strategy / agent | Disease | State of development | References |
|---|---|---|---|
| Protein Reduction | |||
| Anti-inflammatory drugs | AA amyloidosis | Regulatory-agency approved | 107-112, 296, 297 |
| mRNA reduction | AA amyloidosis | Preclinical | 128, 129 |
| β-Secretase inhibitors/modulators; Especially Ligand Pharmaceuticals / Merck-8931 | Alzheimer's Disease | Clinical trials | 118, 119 |
| γ-Secretase modulators | Alzheimer's disease | 123, 298 | |
| Chemotherapy | Light chain amyloidosis | Regulatory-agency approved | 27, 103, 299, 300 |
| Proteasome inhibitors | Light chain amyloidosis | Regulatory-agency approved | 27, 103, 299, 300 |
| mRNA reduction | Light chain amyloidosis | Preclinical | 128 |
| Liver transplantation mediated gene therapy | TTR amyloidoses Hereditary fibrinogen amyloidosis | Regulatory-agency approved | 86, 87, 301, 95 |
| RNAi or antisense/RNAse | TTR amyloidoses | Clinical trials | 126, 127, 302 |
| Protein Stabilization | |||
| Kinetic stabilizers: tafamidis, diflunisal | TTR amyloidoses | Regulatory-agency approved | 22-24 |
| Protein Quality Control | |||
| Small molecule activators of the heat shock response | Huntington's disease / other degenerative disorders | Preclinical | 160, 161 |
| Small molecule activators of the unfolded protein response | TTR amyloidoses Light chain amyloidoses |
Preclinical |
147 148, 178 |
| Hsp 90 inhibition | Alzheimer's Disease Huntington's disease Parkinson's disease |
Preclinical |
303 163, 304 164 |
| Protein reduction and quality control | |||
| Small molecule Hsc70 inhibitors | Alzheimer's Disease | Preclinical | 175-177 |
| Deubiquitination enzyme inhibitors | Alzheimer's disease Parkinson's disease |
Preclinical |
179, 180 179, 180 |
| Amyloid Remodeling | |||
| EGCG | Different amyloidogenic proteins Transthyretin cardiomyopathy Parkinson's disease |
Preclinical Clinical report Preclinical |
191-197, 305 190 199 |
| Amyloid removal | |||
| Variants of Hsp104 disaggregase | Parkinson's disease Amyotropic lateral sclerosis Frontotemporal dementia |
Preclinical |
218, 219 218, 219 |
| Catalytic antibodies | TTR amyloidoses Alzheimer's disease |
Preclinical |
185 186 |
| Protein Reduction and Amyloid Removal | |||
| Passive immunization, Aβ and tau | Alzheimer's Disease | Clinical trials | 205, 209, 211-214, 306, 307 |
| Active Immunization | Alzheimer's Disease | Clinical trial halted | 203-207 |
| Passive immunization | Parkinson's disease | Preclinical | 211-216 |
Figure 3. Therapeutic Strategies to Ameliorate Amyloidosis.
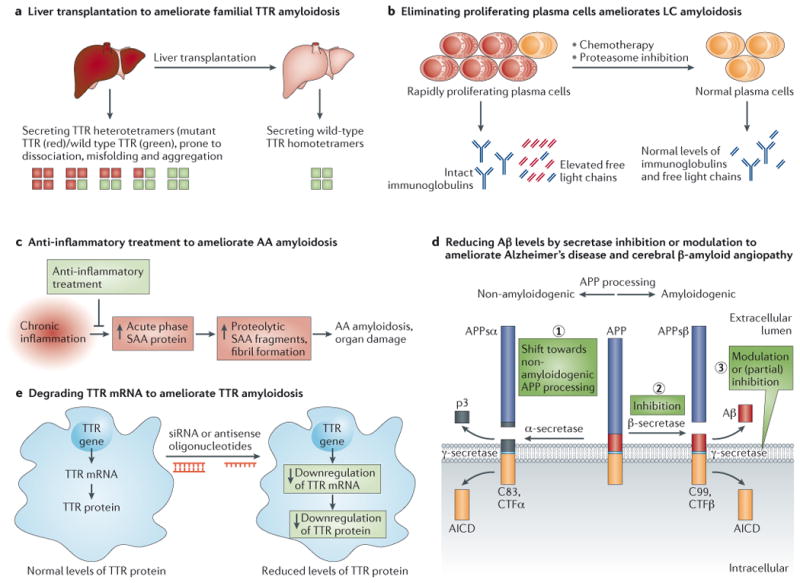
A. Gene therapy by liver transplantation to treat Familial Amyloid Polyneuropathy (FAP). Since TTR is largely produced by the liver, liver transplantation was introduced in the 1990s as a therapy for early stage patients with FAP. Wild type (WT) TTR tetramers (depicted with green squares) produced by the donor liver are much less amyloidogenic than the mutant TTR/WT TTR heterodimers (depicted with red and green squares, respectively) produced by the patient's liver before transplant. This approach successfully slows the progression of FAP and extends life span, but requires organ availability, life-long immunosuppression, and is associated with 10% mortality due to the transplant procedure.
B. Protein reduction by chemotherapy to treat Light Chain Amyloidosis (AL). The amyloidogenic protein in AL amyloidosis is an immunoglobulin light chain or a fragment thereof overproduced by a plasma cell dyscrasia. Removal of the proliferating cancerous plasma cells producing the amyloidogenic light chain by chemotherapy regimens, with or without autologous stem cell transplantation, is used for patients without major cardiac involvement that are not too sick to tolerate these aggressive regimens, which leads to apparently durable disease remission.
C. Anti-inflammatory treatment dramatically lowers acute phase serum amyloid A (SAA) protein production for the treatment of Amyloid A Amyloidosis (AA amyloidosis). Systemic amyloid A amyloidosis is a long-term complication in some patients suffering from chronic infection or chronic inflammation (e.g., chronic inflammatory arthritis). The amyloidogenic protein is derived by proteolysis of SAA, an acute-phase reactant protein transcriptionally upregulated during inflammation. Persistent high concentrations of SAA fragments in plasma above a critical threshold for aggregation can trigger AA deposition. Treatment of the underlying infectious or inflammatory trigger can reverse this type of amyloidosis, if diagnosed early.
D. Reduction of pathogenic Aβ levels by secretase inhibition or modulation has therapeutic potential for Alzheimer's disease (AD) and Cerebral β-Amyloid Angiopathy. The amyloid precursor protein (APP) is a transmembrane protein that is constitutively cleaved by enzymes called secretases. Endoproteolytic processing via the non-amyloidogenic pathway, i.e., α-secretase cleavage followed by γ-secretase cleavage (shown on left) generates non-amyloidogenic APP fragments. In contrast, processing by β-secretase and subsequent γ-secretase cleavage produces amyloidogenic Aβ peptides (shown in red on right). Aggregation of Aβ peptides into extracellular amyloid fibrils or plaques is a histopathological hallmark and a diagnostic criterion for Alzheimer's disease. Reduction of Aβ peptide concentration can be achieved by (1) shifting APP processing towards non-amyloidogenic endoproteolysis, or (2) inhibition of β-secretase , or (3) γ-secretase inhibition or modulation. Inhibition of γ-secretase was shown to be feasible experimentally, however, side effects are a concern due to its many substrates. Interestingly, γ-secretase cleavage generates Aβ peptides of various lengths—as a rule of thumb, the longer the peptide, the more amyloidogenic. Modulation of γ-secretase processing of APP to generate shorter, less amyloidogenic Aβ peptides is currently being investigated.
E. Gene silencing by siRNA or antisense oligonucleotides for the treatment of Familial Amyloid Polyneuropathy (FAP). Recently published early-stage, small, placebo-controlled clinical trial data has identified small interfering RNAs (siRNA, also RNAi) that lower mutant and WT transthyretin (TTR) protein production in patients with FAP. Analogous clinical data on antisense oligonucleotides that degrade TTR mRNA and thus lower TTR production and secretion into the blood from the liver are promising.
Since amyloid clearance does not appear to correlate with clinical benefit, amyloid fibrils per se are unlikely to directly cause the neuronal dysfunction and death associated with this malady92-94. However, the fragmentation of amyloid fibrils or incomplete catabolism could contribute to the spectrum of oligomers formed during the process of aggregation (Fig.1), which appear to be more proteotoxic76. Liver transplantation might also be useful to treat other familial amyloid diseases wherein the amyloidogenic protein is predominantly secreted by the liver, e.g., in hereditary fibrinogen amyloidosis95.
While the pioneering liver transplant strategy has improved the health, and prolonged the lives of thousands of familial amyloid polyneuropathy patients and demonstrated that intervention was possible after the start of neuropathy, it is not without its challenges. These include the 10% mortality due to surgical complications, limited organ availability, the risks of infection associated with life-long immunosuppression, and the emergence of WT TTR aggregation-associated cardiomyopathy upon aging96. In addition, because liver transplant-mediated gene therapy does not alter mutant TTR secretion by the choroid plexus into the CSF or by the retinal pigment epithelium cells into the vitreous of the eye, mutant TTR aggregation in the eye97 and in meningeal vessels98 appears to lead to ophthalmological and neurological symptoms in the patients treated by liver transplantation. These challenges warrant the development of alternative treatments discussed below.
The “protein reduction” strategy has also been used to successfully treat light chain (LC) amyloidosis (AL)27, 99. AL results when a proliferating plasma cell (a cancer cell similar to that causing multiple myeloma) secretes an immunoglobulin LC protein that is aggregation prone owing to its destabilization and misfolding and misassembly proclivity27, 34, 100. The misfolding and/or aggregation of LC within the circulatory system leads to multiple organ system dysfunction, including renal dysfunction and cardiomyopathy72, 101, 102. The concentration of LC is reduced by eliminating the clonal plasma cells secreting LC into the blood using a variety of chemotherapy agents, including proteasome inhibitors (Table 1)27, 103. This therapeutic strategy (Fig. 3B) leads to a dramatic and rapid improvement in patient health, especially in cardiac function, even though there is overwhelming evidence that the amyloid fibrils are not cleared from the hearts of these patients on this timescale72. Although the presence of amyloid in the heart is deleterious , it appears that it is the process of active LC aggregation - including the apparent uptake of proteotoxic misfolded LC monomers or oligomers by cardiomyocytes and other relevant cells - that causes the severe multiple organ system toxicity and pathology71, 95, 96, 98. Unfortunately, approximately 30% of the LC amyloidosis patients presenting with severe cardiomyopathy are too sick to tolerate the chemotherapy regimen99. Thus, other strategies to ameliorate aggregation-associated LC proteotoxicity are still needed.
An analogous “protein reduction” strategy has been employed to successfully treat systemic amyloid A, or AA amyloidosis, which arises in a subset of individuals facing chronic inflammatory disease, (e.g., rheumatoid arthritis, Crohn's disease, and recurring infections)104. The SAA protein is an acute phase reactant protein; therefore sustained high concentrations occur as a consequence of chronic inflammation. While high serum amyloid A (SAA) concentrations appear to be the prominent risk factor for the onset and progression of AA amyloidosis105, they are insufficient to cause AA. Apparently, other factors are needed to contribute to the development of AA, including endoproteolysis of the SAA protein, which generates an intrinsically disordered amyloidogenic SAA fragment106. Kidney damage appears to result from the aggregation of SAA protein3. Treating the underlying inflammatory disease with anti-inflammatory drugs (Table 1) lowers the concentration of SAA and its amyloidogenic fragments (Fig. 3C), ameliorating renal dysfunction, providing strong evidence that SAA fragment aggregation causes AA amyloidosis107-112.
Seeding of SAA fragment aggregation by injecting amyloid fibrils comprising an SAA fragment hastens the onset of AA amyloidosis in a rodent model, providing further compelling evidence that the process of aggregation is causing the characteristic renal dysfunction66. Seeded protein aggregation by a prion-like mechanism is now considered a major driver for propagation and intra-organismal spreading of protein aggregation for many amyloid diseases (discussed in more detail below)8, 17. Not surprisingly, disease severity is also influenced by the exact sequence of the SAA protein that is transcriptionally upregulated by inflammation and generated through aberrant endoproteolytic processing113.
The “protein reduction” strategy is also being pursued very aggressively in AD .One approach is to inhibit or modulate the activity of the β- and/or γ-secretases - endoproteases that cut Aβ out of the large amyloid precursor protein - in an effort to lower the Aβ concentration (Fig. 3D)40, 114, 115. After long development phases due to several challenges, including achieving effective blood-brain barrier penetration, a new generation of small molecule β-secretase (BACE) inhibitors are being evaluated in clinical trials (Table 1)116-119. BACE1 (β-site APP cleaving enzyme 1) is the major β-secretase enzyme in the brain initiating Aβ generation (Fig. 3D). Promising BACE inhibitors are emerging in phase I clinical trials, raising hope that a good safety profile, good pharmacokinetics, and significant Aβ lowering by BACE1 targeting is achievable, which will hopefully translate into an AD modifying strategy. Other compounds exhibiting BACE1 selectivity over BACE2, a homolog with low neuronal expression, are being developed by several companies and are starting to emerge in the patent literature. Complete inhibition of BACE1 will likely not be necessary to ameliorate AD pathogenesis, owing to the striking concentration dependence of Aβ aggregation, although BACE knockouts exhibit mild phenotypes in rodents120.
γ-Secretase is a multi-protein complex with four essential subunits conducting intramembrane cleavage (Fig. 3D) of numerous substrates121. The nicastrin and PEN-2 (presenilin enhancer 2) subunits are identical in every γ-secretase complex, whereas the APH-1 (anterior-pharynx defective) subunit comes in two varieties APH-1A or APH-1B, analogous to the catalytic subunit, which is either presenilin 1 (PSEN1) or presenilin 2 (PSEN2). Challenges arose when it was discovered that γ-secretase cleaves not only APP to afford the Aβ peptides, but also >100 protein substrates, one of which is Notch, an important cellular signaling protein122. Studies and clinical trials undertaken so far suggest that direct γ-secretase inhibition may be problematic as a long-term pharmacologic strategy for AD, due to the strong mechanism-based toxicity observed (Table 1). Current efforts focus on modulating γ-secretase substrate specificity using small molecules that function by an allosteric mechanism (Table 1), shifting Aβ generation towards shorter, non-amyloidogenic fragments without compromising the cleavage of other important γ-secretase substrates122, 123. Emerging knowledge about the assembly, regulation, and specificity of the γ-secretase complexes is expected to guide these endeavors and has been reviewed elsewhere121.
Another “protein reduction” approach is to inject antibodies (passive immunization) into AD patients that bind to Aβ peptides, promoting their clearance and thus inhibiting Aβ aggregation 124 (Table 1). Based on clinical trial data so far, this appears to be a promising approach for reducing amyloid load and continues to be pursued intensively. An active immunization strategy (vaccination) using the appropriate antigen could ultimately produce these “protein reduction” antibodies125. For a more detailed discussion of the immunization strategies for amyloid disorders, please see below.
Another “protein reduction” strategy being actively pursued involves gene silencing by RNA interference (RNAi) or antisense oligonucleotide technology (Fig. 3E). The reduction of both WT and mutant TTR protein levels in the blood stream has been successfully achieved by applying either of these technologies in the liver126, 127 (Table 1). TTR familial amyloid polyneuropathy clinical trials are currently being enrolled to test TTR RNAi (Alnylam) and to evaluate the efficacy of an antisense TTR oligonucleotide approach (Isis) (Fig. 3E). Analogous mRNA reduction approaches are also being explored in tissue culture and animal models for other amyloidoses, including AL128, 129.
Protein Stabilization Strategy
A different approach to preventing the detrimental effects of misfolding and aggregation of category 1 proteins is the “protein stabilization” strategy or “kinetic stabilization” strategy. Portuguese family members carrying the familial amyloid polyneuropathy V30M TTR mutation, but who exhibited a very mild polyneuropathy phenotype or no obvious pathology130, were found to be compound heterozygotes, expressing V30M TTR from one allele and T119M TTR from the other allele, which results in the secretion of TTR heterotetramers comprising both V30M and T119M subunits in the expected stoichiometries25. Biophysical studies revealed that T119M TTR subunit incorporation into tetramers otherwise composed of the disease-associated TTR subunits proportionately reduces the rate of tetramer dissociation and the rate of aggregation25, 26, by destabilizing the dissociative TTR transition state and thus increasing the activation energy for tetramer dissociation131, 132. This kinetic stabilization mechanism provides strong evidence that the process of aggregation or amyloidogenesis is the cause of these maladies, i.e., the V30M subunits within the context of natively folded tetramers are not in themselves proteotoxic33, 132. We hypothesize that V30M TTR aggregates expose hydrophobic surface area and unsatisfied hydrogen bonds that enable aberrant interactions with proteins, lipids, nucleic acids, and carbohydrates, rationalizing how the process of TTR aggregation becomes proteotoxic. While the TTR concentration is not reduced in this strategy, the concentration of TTR that can dissociate, misfold and aggregate is dramatically reduced by this interallelic trans-suppression kinetic stabilization mechanism133.
The data outlined directly above demonstrate that TTR aggregation is suppressed and a variety of pathologies are ameliorated via the “kinetic stabilization approach”. This human genetic evidence motivated efforts to discover small molecules that could prevent TTR aggregation through an analogous kinetic stabilization of the TTR tetramer26, 131, 132, 134, 135. Initial in vitro studies revealed that when thyroxine (T4) bound to the T4-binding sites within TTR, the tetramer was stabilized and TTR amyloidogenesis was inhibited134. This finding led to a screening136 and structure-based drug design effort137, 138 to discover small molecule ligands that selectively and avidly bind to the TTR tetramer over the dissociative transition state, kinetically stabilizing the native, non-amyloidogenic tetramer, slowing the rate-limiting step of amyloidogenesis26, 131, 132.
Over 1000 small molecules exhibiting structural complementarity to the T4-binding sites within TTR have been synthesized over the last 20 years and over 100 ligand2·TTR crystal structures have been solved, enabling the synthesis of very potent kinetic stabilizers131, 135, 137, 138. One of these, tafamidis,31, 133 was evaluated in a placebo-controlled clinical trial and in a follow-on open-label study22, 23. Both primary endpoints were met in the “efficacy-evaluable” population (n=87) and were just missed in the “intent-to-treat” population (n=125), apparently due to more patients than expected in the intent-to-treat population being selected for liver transplantation during the course of the trial, not as a consequence of treatment failure. However, these patients were classified as treatment failures in the conservative analysis used22, 23. Tafamidis (Vyndaqel® 20 mg, once daily) is now approved for the treatment of early stage transthyretin-related hereditary amyloidosis (polyneuropathy; all mutations) by the regulatory agencies of Europe, Japan, Mexico, and Argentina. Tafamidis is currently being considered for approval by the neurological division of the United States Food and Drug Administration (FDA) (Table 1).
In the process of searching for TTR kinetic stabilizers, we demonstrated that the non-steroidal anti-inflammatory drug (NSAID) diflunisal binds to and kinetically stabilizes the TTR tetramer139, 140. An international, randomized, double-blind, placebo-controlled clinical trial showed the ability of diflunisal to reduce the rate of progressive neurological impairment and preserve quality of life in patients suffering from familial amyloid polyneuropathy, thus enabling diflunisal to be repurposed as a TTR kinetic stabilizer (Table 1)24. Even though diflunisal is not very potent as a TTR kinetic stabilizer, it efficiently kinetically stabilizes TTR at a dose of 250 mg twice a day because of its very high oral bioavailability and its correspondingly very high plasma concentration (≈ 300 μM–1 mM). Since diflunisal is an NSAID, it slows renal blood flow, which is a contraindication for those patients exhibiting cardiomyopathy or renal compromise.
Collectively, the tafamidis and diflunisal clinical trial results provide compelling pharmacologic evidence that the process of TTR amyloidogenesis causes neurodegeneration, and that slowing tetramer dissociation and amyloidogenesis after symptom presentation by small molecule kinetic stabilization dramatically slows and in up to 60% of the patients stops the progression of neurological impairment. These data very strongly support the validity of the amyloid hypothesis, the notion that the process of aggregation causes post-mitotic tissue loss132. These data motivated the tafamidis cardiomyopathy clinical trial that is currently enrolling WT and familial patients (placebo vs. 20 mg vs. 80 mg tafamidis once daily).
Protein Quality Control Strategy
Aging is the most significant risk factor for the development of either sporadic or inherited amyloidoses, for reasons that are still not fully understood141. One explanation for this is that the capacity of the cellular and/or extracellular protein homeostasis networks declines with aging, a hypothesis gaining experimental support142. Another non-mutually exclusive possibility is that the capacity to activate the stress-responsive signaling pathways regulating these proteostasis network functions declines with aging142-145. The classic view of amyloid disease etiology is that the aggregation of a specific protein causes the aggregation-associated degenerative phenotype through a gain-of-toxic-function type mechanism (Fig. 1). However, there is mounting evidence that the intracytosolic aggregation of proteins linked to specific amyloid diseases compromises the folding of the endogenous proteome by sequestering chaperones and chaperonins—resulting in the aggregation of proteins required for critical cellular functions such as controlling transcription142, 146. While there is no doubt that the conformational integrity of one protein is compromised in specific amyloid diseases, it now appears that the native conformations, and thus function, of many additional proteins could be compromised due to consumption of proteostasis network capacity, suggesting that a breakdown in the ability to maintain the proteome is part of degenerative disease etiology, if not the cause57, 142, 146.
Thus, a “protein quality control strategy” could be beneficial for ameliorating the amyloidoses. This approach focuses on adapting the intracellular and/or extracellular proteostasis pathways involved in protein folding, trafficking and/or degradation to reduce the aggregation of disease-associated proteins and possibly additional cellular proteins147-153. Adaptation of these cellular proteostasis pathways alters the intracellular interactions between destabilized, aggregation-prone proteins and the proteostasis network components composing these pathways, providing the opportunity to directly or indirectly attenuate proteotoxic aggregation. This “protein quality control strategy” has been primarily pursued through the activation of organelle-selective stress-responsive signaling pathways such as the heat shock response (HSR) or the unfolded protein response (UPR) that transcriptionally regulate proteostasis capacity in specific intracellular environments, such as the cytosol or the secretory pathway, respectively154-158. Thus, genetic and small molecule stress-independent activators of individual stress-responsive signaling pathways are now being developed (Table 1)147, 159, 160.
Celastrol activation of the HSR, which increases the cytosolic proteostasis network capacity, has been used in the context of pre-clinical Huntington's disease models to validate the “protein quality control” therapeutic strategy161. This molecule primarily functions through the activation of heat shock factor 1 (HSF1; the predominant transcription factor responsible for stress-responsive upregulation of cytosolic proteostasis network components via the HSR)155, 156. These small molecules (Table 1) exhibit considerable promise for ameliorating pathologic intracellular protein aggregation, although not all reports agree on whether HSR activation is beneficial162.
Another strategy to induce the HSR is to inhibit the cytosolic ATP-dependent Hsp90 chaperone. Hsp90 binds to and negatively regulates HSF1. Inhibition of Hsp90 may prevent its binding to HSF1 and also leads to the misfolding and misassembly of many cytosolic proteins that in turn activate HSF1, leading to transcriptional reprogramming of the cytosolic proteostasis network via the HSR. Hsp90 inhibition ameliorates aggregation and proteotoxicity in several experimental models of aggregation-associated degenerative diseases, including Huntington's, Parkinson's and Alzheimer's diseases163, 164. HSF1 can also be activated indirectly by reducing insulin / insulin growth factor 1-like signaling (IIS), which provides significant benefits in Alzheimer's and Huntington's disease models, with likely contribution from the FOXO transcriptional program(s)75, 165. Chemical-genetic and pharmacologic approaches (Table 1) have recently become available to activate HSF1 in the absence of stress or IIS inhibition159, 160.
Increasing cytosolic proteostasis network capacity through the overexpression of the molecular chaperone Hsp70 was also found to significantly reduce proteotoxicity and neurodegeneration in models of Huntington's166 and Parkinson's diseases167. However, other studies did not observe the same effect168, possibly due to different relative stoichiometry of the proteostasis network components owing to differences in Hsp70 overexpression. Binding of Hsp70 to α-synuclein aggregates was shown to reduce toxicity169 and may even act extracellularly on oligomer formation and protect from trans-synaptic spreading of aggregates170.
While the Hsp70-Hsp40-nucleotide exchange factor pathway is often considered to be a pro-folding pathway, there is abundant evidence that this pathway makes critical folding versus degradation or quality control decisions153, 171-174. Thus, small molecule modulators of this pathway could be used to enhance the degradation of certain aggregation-prone client proteins175-177. This approach also falls into the “protein reduction” therapeutic strategy category. There is evidence that Hsc70 (the constitutive chaperone) stabilizes tau against degradation in the brain152, whereas Hsc72 recruits the co-chaperone ubiquitin ligase CHIP, which is known to ubiquitinate tau and facilitate its degradation177. Thus, inhibitors of Hsc70 should partition tau in the brain into the Hsc72-CHIP pathway, facilitating its degradation. The allosteric small molecule Hsc70 inhibitors, YM-01 and YM-08 (Table 1), both decreased tau levels in a variety of preclinical brain-relevant model systems, rendering this therapeutic strategy very promising176.
Extracellular protein aggregation can also be attenuated by activating the unfolded protein response (UPR), which functions to regulate proteostasis (quality control) in the endoplasmic reticulum (ER) and in downstream compartments of the secretory pathway157, 158, including in the extracellular space178. The UPR signaling pathway comprises three arms activated downstream of the ER transmembrane stress-sensor proteins, IRE1, ATF6, and PERK. Activation of these arms adapts ER proteostasis through both transient attenuation of new protein synthesis (downstream of PERK) and the activation of stress-responsive transcription factors including XBP1s (downstream of IRE1), ATF6 (a cleaved product of full-length ATF6), and ATF4 (downstream of PERK). These transcription factors upregulate ER protein folding, trafficking and degradation pathways comprising the ER proteostasis network.
Arm-selective activation of the UPR appears to be a very promising strategy for increasing the scrutiny of protein quality control in the secretory pathway of human cells by selectively reducing secretion of destabilized, amyloidogenic proteins147, 148. The hypothesis underlying this strategy is that UPR transcriptional remodeling of the ER proteostasis network could lead to increased partitioning of amyloidogenic mutant proteins (e.g., V30M TTR) to ER degradation pathways, including ER-associated degradation (ERAD) or autophagy, while still allowing efficient secretion of the more stable WT protein (e.g., WT TTR) at normal levels147. To date, preclinical results from this approach are very encouraging. For example, selective activation of the ATF6 arm of the UPR leads to an ≈ 40% reduction in the secretion of a destabilized A25T TTR variant, while permitting efficient secretion of WT TTR147. Importantly, the preferential reduction in destabilized TTR secretion afforded by ATF6 activation corresponds with increased partitioning of the destabilized TTR variants to ER degradation pathways, indicating that the reduced secretion of destabilized TTR does not lead to the intracellular accumulation of destabilized, aggregation-prone TTR that could aggregate in the ER. The ATF6-dependent reduction in destabilized TTR secretion lowers the extracellular concentration of destabilized TTR available for concentration-dependent aggregation 147. Both the selective degradation of an energetically compromised TTR variant and the global reduction in extracellular concentrations of destabilized TTR will reduce the TTR aggregation propensity in the extracellular space. Moreover, ATF6 activation results in the secretion of an Hsp40 chaperone, ERdj3, which further reduces aggregation of amyloidogenic proteins in the extracellular space178. Stress-independent activation of UPR-associated transcription factors has also recently been shown to reduce the secretion of a destabilized amyloidogenic LC without affecting the secretion of an energetically normal LC148.
Such a dual “quality control and protein reduction strategy” is also a very attractive approach for enhancing the degradation capacity of the cytosolic proteostasis network, which is currently being investigated in patients with degenerative diseases179, 180. For example, the concentration of α-synuclein or tau can be reduced by diminishing the negative regulation of the proteasome172,180. The deubiquitination enzymes, or DUBs, are proteasome-associated enzymes that inhibit ubiquitin-mediated proteasomal degradation by removing the ubiquitin from the client protein. Thus, DUB inhibitors are being developed by several companies to enhance the degradation of client proteins, including α-synuclein and tau to treat neurodegenerative diseases 172,180. Small molecules that inhibit USP14 (ubiquitin-specific peptidase 14, a DUB) (Table 1) are being actively pursued by more than one company to enhance clearance of aggregation-prone proteins.
Proteolysis Strategy
Proteases such as the Aβ-degrading enzyme, neprilysin, were shown to degrade monomeric Aβ, and possibly also low n oligomeric Aβ181— an observation which may potentially be exploited therapeutically 182, 183. A concern is that Aβ amyloid fibrils and possibly also late stage oligomers are no longer enzymatically degradable owing to their kinetic stability. However, it is not inconceivable that depletion of the monomer pool could not only prevent aggregate formation, but also promote fibril dissociation owing to the change in equilibrium concentrations (see ‘Targeting Amyloid Fibrils’ below). Consistent with this hypothesis, upregulation of neprilysin levels can delay Aβ deposition in APP transgenic mice182. Such an approach may be most useful as a preventive treatment or in the early stages of AD184.
Catalytic antibodies that bind and proteolytically degrade amyloidogenic proteins are being explored as possible biologic drugs. If subsets of such “catabodies” could recognize a toxic/amyloidogenic epitope of a protein not exposed in the natively folded form, i.e., differentiating the native form from the toxic form, then such catabodies could hold promise for specific degradation of toxic protein structures. Importantly, degradation by catabodies should not elicit an inflammatory response. A recent study reported physiologic IgM class catabodies that degrade misfolded soluble and particulate forms of transthyretin (Table 1), but not the physiological form, suggesting a protective function of such catabodies against TTR amyloidogenesis185. The same lab recently engineered catabodies capable of degrading aggregated Aβ186 (Table 1).
Strategies that Target Amyloid Fibrils
In addition to therapeutic strategies that aim to prevent protein misfolding, and thereby the initiation and propagation of aggregation, approaches to remove or inactivate amyloid fibrils and other higher order aggregates are emerging. Due to the long presymptomatic phase of amyloid diseases, initiation of treatment may only be feasible after substantial protein deposition has occurred, especially in sporadic cases. Removal of existing aggregates is advisable, since these aggregates can themselves be toxic. Moreover, amyloid fibrils are known to dissociate187-189, and thus it is possible that amyloid fibril dissociation to diffusible more toxic structures could continue to drive pathology in some patients treated by a “protein reduction strategy”, “kinetic stabilizer”, or “protein quality control” strategy. It might therefore be necessary to combine one of the aforementioned approaches with strategies that target the amyloid fibrils themselves, such as amyloid crosslinking/remodeling, removal or disaggregation coupled to degradation. Elimination or inactivation of the existing amyloid load should also stop the self-sustaining cascade of seeded or prion-like misfolding hypothesized to play a significant role in driving the propagation of many neurodegenerative disorders (see below).
Amyloid Remodeling
A recent observational report suggests that epigallocatechin-3-gallate (EGCG), the principal polyphenol present in green tea and an amyloid fibril binder and crosslinker, is effective at slowing the progression of cardiac transthyretin amyloidosis190. EGCG (Table 1) is able to remodel protein aggregates, including amyloid fibrils and oligomers, comprising different amyloidogenic proteins, although the mechanistic underpinnings of this process are not entirely clear191-198. Ample evidence shows that the EGCG amyloid remodeling activity in vitro is dependent on auto-oxidation of the EGCG molecule. The amyloid remodeling observed by EGCG treatment appears to be driven by hydrophobic binding of oxidized EGCG molecules to amyloid fibrils, followed by the oxidized EGCG reacting with free amines in the amyloid fibril, which cross-links the fibrils194, 197. Amyloid remodeling appears to stabilize the aggregates and prevent the formation of toxic oligomers, possibly explaining the reduction in proteotoxicity in the patients taking EGCG who have experienced clinical benefit190. A recent study suggests that EGCG can also reduce toxicity of α-synuclein oligomers, presumably by binding to the oligomers and thereby moderately reducing their toxic interaction with membranes199. It was also shown that EGCG is able to direct α-synuclein aggregation towards the formation of off-pathway, nontoxic oligomers, to prevent seeding of α-synuclein aggregation, and to remodel α-synuclein fibrils to smaller, nontoxic aggregates 191, 192.
Amyloid Removal by Immunization
Another approach to rid the tissue of amyloid deposits is passive or active immunization against the amyloidogenic protein. Antibody binding to the monomeric protein, to oligomers, and/or to fibrils can direct the degradation of these structures by phagocytic cells, resulting in their clearance. This strategy is being widely explored in the context of Alzheimer's disease. Indeed, in APP transgenic mice, immunization against Aβ peptides elicits an anti-Aβ immune response and is effective at removing β-amyloid deposits200. Importantly, active immunization was also effective in preventing Aβ deposition when initiated during the predepositing stage (before amyloid deposition had occurred). Additional studies confirmed these findings, found improved behavioral/cognitive performance201, 202, and suggested that this approach was suitable for removal of Aβ deposits in such mice32. Unfortunately, an Aβ active immunization clinical trial in late-stage AD patients revealed serious adverse events in approximately 6% of the patients, specifically aseptic meningoencephalitis, forcing Elan Pharmaceuticals to halt the trial in 2002203. Postmortem analysis of the brains of patients who participated in this trial confirmed lower Aβ load and evidence that amyloid was removed by immunotherapy, showing that the biological outcome was met204, 205. Closer examination suggests that extracellular plaques are cleared first, while vascular Aβ deposits remain longer or may even increase due to clearance of solubilized Aβ along perivascular drainage pathways206, 207.
Passive immunization trials with monoclonal antibodies against Aβ also demonstrate that the Aβ plaque burden in mice is cleared124, 208. Hopes remain high that the passive immunization approach will be a safe alternative, as it does not elicit an uncontrolled autoimmune response (reviewed in 209). Even though the AD passive immunization trials completed so far have demonstrated removal of Aβ deposits, neither significant improvement of cognitive performance, nor slower progression of the disorder, nor improved survival was observed in these later stage AD patients205, 209. However, recent data from a Phase Ib study has shown that Aducanumab (Biogen) treatment resulted in statistically significant reductions in amyloid load and a reduction in cognitive decline in patients with a prodromal or mild AD, providing hope for the use of this strategy. Thus, new passive immunization trials are focusing on early treatment of patients carrying AD risk genes210.
Similar immunization strategies are also being developed for intracellular amyloid disorders such as tauopathies and synucleinopathies. Promising results were obtained in transgenic rodent models and these therapies are progressing toward clinical trials 211-216. A Phase 1 study targeting tau by immunotherapy is ongoing (AADvac1, Axon Neuroscience SE) with results expected in 2015. With regard to α-synuclein immunization, AFFiRis (Austria) conducted a small Phase I trial and reported in a recent press release that the immunization regimen (vaccine PD01A) appears to be well tolerated, safe, and that most participants developed anti-α-synuclein antibodies. As the development of immunization strategies against tau and α-synuclein are in early stages, it is currently unknown if there is a disease-modifying effect in humans.
Macromolecular Disaggregation of Amyloid Fibrils
Enzymatic aggregate disaggregation linked to degradation may be a possible strategy to clear amyloid deposits from tissue. The proteostasis networks of yeast and bacteria have established disaggregases, Hsp104 and ClpB, respectively217. Despite intensive efforts, the mammalian equivalent of these disaggregases has not yet been identified and may not exist. However, it may still be possible to engineer yeast Hsp104 or bacterial ClpB to disaggregate certain amyloids and use these variants as injectable protein drugs. For example, Jackrel et al. showed that “potentiated” variants of Hsp104 (Table 1) discovered by screening were able to reduce TDP-43, FUS, and α-synuclein proteotoxicity in model organisms218, 219.
Similarly, it was recently demonstrated that α-synuclein aggregates could be disassembled by the Hsp70-40-nucleotide exchange factor pathway; however, the more kinetically stable Aβ amyloid fibrils could not be disassembled220. It may be the case that multicellular organisms abandoned a disaggregation strategy as the primary detoxifier, instead utilizing autophagy or other pathways ending in lysosomal degradation to purge aggregates because of the risk of dissociating amyloid fibrils to more toxic structures. Strategies to use autophagy enhancement therapeutically are currently being explored, but are presently limited by our incomplete understanding of this process221, 222.
Inhibition of Amyloid Seeding and Aggregate Cell-to-Cell Spreading
For several amyloidogenic proteins, the transition from their native structures to amyloid appears to follow a nucleation-dependent polymerization process61, 65, 223. In this mechanistic paradigm, the formation of the nucleus (a putative β-sheet-rich species) is energetically unfavorable and its formation is the rate-limiting step for efficient aggregation (Fig. 2A). The formation of such a nucleus may not occur during the lifetime of a healthy individual and if nucleus formation does occur, the nucleus should typically be identified by and neutralized via one of the multiple mechanisms used by the intracellular or extracellular proteostasis networks149. However, if the amyloid nucleus escapes neutralization or degradation in one rogue cell or in one extracellular location within the body, it can act as a template for the further aggregation of soluble homotypic proteins (Fig. 2A)224. The resulting higher order aggregates are referred to as “seeds” since their addition to a monomeric solution immediately initiates aggregation of the homotypic protein without a requirement for nucleus formation. This templated or seeded aggregation is relatively efficient, relentlessly progressive and initiates a fatal cascade of progressive aggregation that also appears to drive proteotoxicity and tissue degeneration (Fig. 4).
Figure 4. Seeded or Prion-like Protein Aggregate Spreading: Disease Initiation, Progression and Therapeutic Strategies.
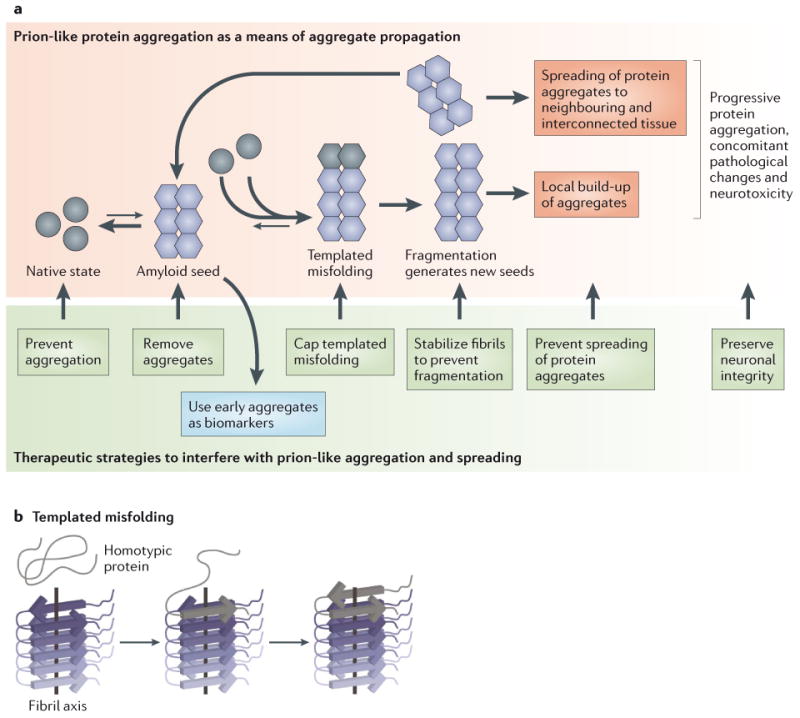
(A) Recent advances in our understanding of protein aggregation disorders have revealed that several amyloidogenic proteins once aggregated into amyloid fibrils can seed or catalyze the misfolding and/or misassembly of homotypic monomers, leading to accelerated aggregate formation and aggregate spreading from tissue to tissue. This type of mechanism was first proposed for prion disorders. Aggregate spreading by this mechanism is currently termed “templated misfolding” or “seeded aggregation” (see Figure 2). Once an amyloid seed is formed, it can catalyze the misfolding and/or misassembly of a monomeric homotypic protein into growing aggregates. Amyloid fibril fragmentation generates new seeds, and the vicious cycle of template misfolding/aggregation leads to the spreading of amyloid to neighboring and interconnected tissue, assisted by cellular aggregate uptake, which can result in subsequent template misfolding followed by aggregate secretion. This mechanistic paradigm has important therapeutic implications. Most importantly, it argues for an early intervention or preventive treatment before the amyloid spreading cascade progresses out of control. Several therapeutic strategies are possible (shown in the green box), ranging from stopping active aggregation, to removing aggregates, to inhibiting templated misfolding, to stabilizing seeds against fragmentation and/or altering their structure, to preventing the cellular uptake or secretion of aggregates. Early detection of amyloid diseases, which is still problematic, is important for these therapeutic strategies to have maximal impact (B) Amyloid fibrils often adopt an in-register β-sheet arrangement, wherein the strands are oriented perpendicular to the fibril axis. The amyloid fibril serves as a seed or template to incorporate the homotypic monomer into the growing fibril. The same protein can adopt distinct amyloid structures or strains, which can be reliably propagated by template misfolding, and which appear to be associated with different pathogenic phenotypes. Figure adapted from ref 224.
This concept of seeded aggregation was first hypothesized to explain the pathogenesis of prion diseases62, 225. It is now widely accepted that prion disorders arise from misfolded and misassembled prion protein (PrPSc) that is sufficient to act as a seed, thus promoting sustained aggregation of the normal, cellular prion protein (PrPC)225. In sporadic prion disorders, e.g., Creutzfeld Jacob Disease (CJD) in humans, PrPSc nucleus and subsequent seed formation occurs spontaneously, followed by aggregate spreading from cell to cell by a seeding mechanism. Prion seeds (PrPSc) are also known to be transmissible between individuals, as seen in very rare instances of surgical instrument contamination that occurred in the past before adequate testing and precautions were implemented, or by funerary cannibalism as in Kuru, or more commonly by uptake of prions from the environment in bovine spongiform encephalopathy (BSE, mad cow disease), and in scrapie in sheep225.
Several human amyloid diseases are now hypothesized to spread within the affected tissue by a seeded aggregation or prion-like mechanism, however the seeds do not appear to be transmissible between humans in contrast to the very rare transmissible prion disorders226. The evidence for seeded spreading of Aβ, tau, and α-synuclein aggregates comes mainly from studies in vitro and from animal models. However, the pathology observed during the course of the disease in human brain provides additional support for this hypothesis, as pathology seem to spread via neurons known to be connected to one another.
Multiple research groups have now demonstrated that injecting a tissue homogenate from a human or mouse harboring an amyloid burden into an amyloid-free mouse can dramatically accelerate amyloid deposition of the homotypic protein in the recipient mouse. This seeding phenomenon for non-prion diseases was first shown in vivo in animal models of AA amyloidosis227. More recently, this approach of injecting homotypic aggregates has been shown to accelerate Aβ228, 229, tau230, and α-synuclein20, 231 aggregation. The idea that aggregation can begin in one rogue cell or extracellular area and then spread within an organism is also under discussion for several other amyloid diseases232, 233.
The experimental seeded induction of amyloidogenesis by injection of seeds is first evident within the injected brain area; however, the induced pathology subsequently spreads to neighboring and interconnected regions along neuronal pathways with increasing incubation time234-238. This is especially interesting and important because spreading of amyloid seeds along neuroanatomical routes would also explain why and how neurodegenerative diseases target neuronal networks239. Although the mechanisms of cellular release and uptake of seeds, as well as of intra- and extracellular spreading are poorly understood, it appears that seeded active aggregation on cell membranes can compromise membrane integrity, rendering the cells quite permeable, even for fibrillar aggregates240-242. Recent studies also show that injection of amyloid in the periphery, i.e., intraperitoneal injections of brain extracts containing Aβ or tau aggregates, can induce progressive aggregation in the brains of transgenic mice17, 243, 244.
The aggregation-inducing seed is almost certainly the aggregated protein (e.g., Aβ) itself, in a conformation generated most effectively in the living brain229, 245. However, amyloid seeds can also be generated in vitro from synthetic or recombinant proteins237, 238, 246-249. Convincing evidence for seeded spreading comes from recent studies showing strain-like phenomena for several amyloidogenic proteins (Fig. 4B). Strains or specific misassembled conformations of a protein were first defined in prion disorders and were found to correlate with distinct biochemical, biophysical and pathological characteristics250-254. Recent studies showed similar strain-like phenomena for seeded Aβ255-257, tau258, 259 and α-synuclein260, 261.
The availability of the soluble, homotypic protein for incorporation into the growing aggregate is important, as is the seed concentration, in determining the efficiency of propagation of protein aggregation229, 236, 243. Small soluble protein aggregates are the most potent inducers of cerebral Aβ-amyloidosis; however, seeds range in size and also comprise large, insoluble, amyloid fibrils262,263. Importantly, seeded protein aggregation in these animal models induces the typical pathological signature that also arises from spontaneous aggregation of the respective protein in animals and humans. This suggests that the process of seeded spreading initiates the typical ensemble of aggregate structures, ranging from relatively disordered to cross-β-sheet amyloid fibrils. This process of aggregation generates proteotoxicity241 and induces the typical tissue response, including inflammation and eventually tissue dysfunction and degeneration.
These exciting findings raise many questions, perhaps the most important being, in addition to amyloid fibrils, which other types of aggregates are formed? That the field cannot answer this question reflects how much remains to be learned. We need tools to discern whether other aggregate structures are present in these mice and in human patients in addition to cross-β-sheet amyloid structures that appear to be acting as seeds. A related question that also needs to be answered by the field is, if there are non-cross-β-sheet aggregates present, how are they formed? Is seeded growth also possible for non-cross-β-sheet aggregates or are they formed in parallel with cross-β-sheets? It is possible that incomplete degradation of amyloid fibrils or fibril fragmentation could yield the diffusible aggregate structures. A complete inventory of aggregates formed as a function of aging in non-seeded and seeded animal models of amyloid disease, as well as in human amyloidosis patients, should provide the much needed structure-proteotoxicity relationships required to understand how the process of aggregation causes post-mitotic tissue loss.
Taken together, the findings of seeded propagation of aggregates and pathology suggest therapeutic opportunities for the treatment and the prevention of these degenerative diseases (Fig. 4A). Since the formation of proteopathic seeds is a self-sustaining process, neutralizing or removing existing seeds should represent promising therapeutic strategies. Since seeded protein aggregation is concentration dependent, the formation of seeds could be prevented by reducing the concentration of the native amyloidogenic protein by the various “protein reduction” strategies discussed above. For well-folded category 1 proteins, the “protein stabilizer” strategy should prevent seed formation as well as the progression of aggregation by reducing the concentration of aggregation-competent protein. The “amyloid removal” strategies should reduce the efficiency of seeded spreading, and potentially the neurotoxic effects of protein aggregates if applied early. Immunotherapy seems to be especially promising for removing extracellular protein seeds and was shown to be effective in reducing seeded spreading of Aβ and tau in animal models10, 229, 243. In addition, stabilizing or remodeling fibrils to prevent fragmentation and/or seeding may slow the progression of prion-like spreading. Progression of the disease should be further prevented or restrained by inhibiting aggregate spreading within and between cells and tissues. An effective approach towards this end will likely require a better understanding of how aggregates enter cells, how they move along axons and how they are secreted from one neuron and taken up by others. All of these strategies will likely be most effective if applied early, before widespread protein aggregation and tissue damage has occurred, hence the importance of early disease detection, which is currently difficult.
Perspective
While the exact mechanism(s) by which protein aggregation causes loss of post-mitotic tissues remains unclear, the genetic and pharmacologic evidence summarized above makes a compelling case for the hypothesis that the process of protein aggregation drives degenerative disease pathology.
Which of the therapeutic strategies discussed above will be most appropriate, most easily applicable and most effective will likely depend on the nature of the target protein, i.e., intrinsically disordered versus natively folded, extracellular versus intracellular amyloid, nucleation-dependent polymerization versus downhill polymerization. The disease stage is also an important factor in discerning which strategy will be most effective. We envision that drugs functioning to reduce aggregation through distinct mechanisms of action will be used together in the future as combination therapies to more effectively treat amyloid diseases (Fig. 5). For example, we envision small molecule TTR kinetic stabilizers131, 132 will be used in combination with drugs that enhance the capacity of the proteostasis network to achieve proteome maintenance149-151 or possibly with drugs that lower the concentration of the amyloidogenic protein of interest126, 127, 153, 160, 180, 264 (Fig. 5).
Figure 5. Combining Therapeutic Strategies to Ameliorate Protein-misfolding / Aggregation Diseases.
We envision that amyloid diseases featuring degenerative phenotypes will be treated in the future using combinations of drugs exhibiting distinct mechanisms of action. For example, we envision treating the TTR amyloidoses with kinetic stabilizers (tafamidis; already in clinical use) and TTR messenger RNA degradation drugs (currently in clinical trials), or using a kinetic stabilizer in combination with drugs that enhance the capacity of the proteostasis network (currently being developed) to achieve proteome maintenance. Given the myriad proteins that lead to aggregation-associated degenerative diseases, strategies such as proteostasis network adaptation that could be useful for multiple maladies are particularly appealing. The goal is to achieve a fully functional proteome without pathogenic amyloidogenicity.
A major challenge in assessing successful therapeutic strategies for the amelioration of human amyloid disease is the lack of biomarkers for early diagnosis and for monitoring response to candidate therapies in human clinical trials. We envision that these biomarkers will be identified through basic research efforts focused on detecting and quantifying all the aggregate structures present before and after clinical symptom presentation. We also expect such biomarkers to emerge from much needed natural history studies on individual amyloid diseases. Natural history or disease progression studies facilitate the design of better clinical trials, ideally utilizing patients with early stage pathology.
Currently, we also lack an understanding of how protein aggregates induce cell toxicity and tissue degeneration, the prevention of which remains the ultimate goal of any therapeutic strategy or combination strategy. Understanding the structures of the aggregates mediating proteotoxicity and elucidating their mechanism(s) of tissue degeneration will undoubtedly provide new insights into therapeutic strategies to ameliorate these maladies, which as a group represent a significant economic burden to modern society and are therefore arguably the most urgent unmet medical need.
Acknowledgments
The authors would like to thank the reviewers for their critical and insightful comments that shaped our thinking and helped improve the manuscript. YSE expresses particular thanks to Mathias Jucker, Tübingen, Germany, for helpful discussions and for his mentorship. We appreciate NIH support from DK46335 (JWK), AG46495 (JWK) and GM101644 (ETP). SEE is supported by an Ellison Medical Foundation New Scholar in Aging Award. YSE is supported by a postdoctoral fellowship from the German Academic Exchange Service (DAAD). JWK and ETP discovered tafamidis and JWK founded FoldRx to commercialize it. FoldRx is now owned by Pfizer, which both JWK and ETP have a financial interest in. We apologize to our colleagues whose work we were unable to include due to space limitations.
Glossary
- Amyloid fibrils
Lateral assemblies of protein aggregates adopting a cross-β-sheet structure. These aggregates bind Congo red and thioflavin T and analogous fluorophores
- Amyloidogenesis
The process of protein aggregation in a multicellular organism wherein physical chemical forces and biological modifiers together influence the aggregate structural ensembles afforded
- Nucleus
An energetically unfavorable, sparsely populated, typically oligomeric species that is thought to be rich in β-sheet structure. Nucleus formation is typically the rate-limiting step for efficient aggregation: a step that is followed by rapid monomer addition, affording a seed
- Proteostasis Network
The macromolecular machinery that generates, folds, moves, and degrades the proteome. Proteostasis network components include chaperones, the proteasome, trafficking machinery, and a variety of enzymes that act on the proteome, such as disulfide isomerases
- Seed
A stable aggregate resulting from the addition of monomers to a nucleus. Seeds can also arise from the fragmentation of fibrils. Seeds enable homotypic protein aggregation without a requirement for nucleus formation, as seeded aggregation bypasses the requirement for a nucleation step
References
- 1.Andrade C. A peculiar form of peripheral neuropathy; familiar atypical generalized amyloidosis with special involvement of the peripheral nerves. Brain. 1952;75:408–27. doi: 10.1093/brain/75.3.408. [DOI] [PubMed] [Google Scholar]
- 2.Glenner GG, Terry W, Harada M, Isersky C, Page D. Amyloid fibril proteins - proof of homology with immunoglobulin light chains by sequence analyses. Science. 1971;172:1150. doi: 10.1126/science.172.3988.1150. &. [DOI] [PubMed] [Google Scholar]
- 3.Linke RP, et al. Characteristics of a serum substance (SAA) antigenically related to amyloif fibril protein AA. Zeitschrift Fur Immunitats-Forschung Experimentelle Und Klinische Immunologie. 1975;150:219–219. [Google Scholar]
- 4.Glenner GG, Wong CW. Alzheimer's disease and Down's syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun. 1984;122:1131–5. doi: 10.1016/0006-291x(84)91209-9. [DOI] [PubMed] [Google Scholar]
- 5.Sipe JD, et al. Nomenclature 2014: Amyloid fibril proteins and clinical classification of the amyloidosis. Amyloid. 2014;21:221–4. doi: 10.3109/13506129.2014.964858. [DOI] [PubMed] [Google Scholar]
- 6.Eisenberg D, Jucker M. The amyloid state of proteins in human diseases. Cell. 2012;148:1188–203. doi: 10.1016/j.cell.2012.02.022. In this review, the authors connect observations on amyloid polymorphism, amyloid strains, and co-aggregation of pathogenic proteins in tissues with possible mechanisms of toxicity and intra-organismal transmissibility of amyloid disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bonar L, Cohen AS, Skinner MM. Characterization of amyloid fibril as a cross-beta protein. Proc Soc Exp Biol Med. 1969;131:1373. doi: 10.3181/00379727-131-34110. &. [DOI] [PubMed] [Google Scholar]
- 8.Holmes BB, et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci U S A. 2013;110:E3138–E3147. doi: 10.1073/pnas.1301440110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Udan-Johns M, et al. Prion-like nuclear aggregation of TDP-43 during heat shock is regulated by HSP40/70 chaperones. Hum Mol Genet. 2014;23:157–170. doi: 10.1093/hmg/ddt408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yanamandra K, et al. Anti-Tau Antibodies that Block Tau Aggregate Seeding In Vitro Markedly Decrease Pathology and Improve Cognition In Vivo. Neuron. 2013;80:402–414. doi: 10.1016/j.neuron.2013.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oddo S, et al. Triple-transgenic model of Alzheimer's disease with plaques and tangles: Intracellular A beta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 12.Knauer MF, Soreghan B, Burdick D, Kosmoski J, Glabe CG. Intracellular accumulation and resistance to degradation of the Alzheimer amyloid A4/beta protein. Proc Natl Acad Sci U S A. 1992;89:7437–7441. doi: 10.1073/pnas.89.16.7437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferretti MT, Bruno MA, Ducatenzeiler A, Klein WL, Cuello AC. Intracellular A beta-oligomers and early inflammation in a model of Alzheimer's disease. Neurobiol Aging. 2012;33:1329–1342. doi: 10.1016/j.neurobiolaging.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 14.Hong MG, Alexeyenko A, Lambert JC, Amouyel P, Prince JA. Genome-wide pathway analysis implicates intracellular transmembrane protein transport in Alzheimer disease. J Hum Gen. 2010;55:707–709. doi: 10.1038/jhg.2010.92. [DOI] [PubMed] [Google Scholar]
- 15.Sahlin C, et al. The Arctic Alzheimer mutation favors intracellular amyloid-beta production by making amyloid precursor protein less available to alpha-secretase. J Neurochem. 2007;101:854–862. doi: 10.1111/j.1471-4159.2006.04443.x. [DOI] [PubMed] [Google Scholar]
- 16.Page LJ, et al. Secretion of amyloidogenic gelsolin progressively compromises protein homeostasis leading to the intracellular aggregation of proteins. Proc Natl Acad Sci U S A. 2009;106:11125–11130. doi: 10.1073/pnas.0811753106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eisele YS, et al. Peripherally Applied A beta-Containing Inoculates Induce Cerebral beta-Amyloidosis. Science. 2010;330:980–982. doi: 10.1126/science.1194516. The paper shows that intraperitoneal inoculation with Aβ-amyloid-rich extracts induced amyloidogenesis within the brains of transgenic mice after prolonged incubation times. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kfoury N, Holmes BB, Jiang H, Holtzman DM, Diamond MI. Trans-cellular propagation of Tau aggregation by fibrillar species. J Biol Chem. 2012;287:19440–51. doi: 10.1074/jbc.M112.346072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem. 2009;284:12845–52. doi: 10.1074/jbc.M808759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Luk KC, et al. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338:949–53. doi: 10.1126/science.1227157. This manuscript demonstrates that a single intrastriatal inoculation of synthetic α-synuclein fibrils led to the cell-to-cell transmission of pathologic α-synuclein Parkinson's-like Lewy pathology in anatomically interconnected regions. The pathology recapitulated the neurodegenerative cascade of Parkinson's disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tanzi RE, Bertram L. Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell. 2005;120:545–55. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 22.Coelho T, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. 2012;79:785–792. doi: 10.1212/WNL.0b013e3182661eb1. The data from a placebo-controlled double-blind clinical trial is summarized, demonstrating the efficacy of tafamidis in slowing the progression of a transthyretin familial amyloid polyneuropathy in the efficacy-evaluable population. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coelho T, et al. Long-term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. J Neurol. 2013;260:2802–2814. doi: 10.1007/s00415-013-7051-7. This follow-on study demonstrates that the efficacy of tafamidis demonstrated in (22) is durable for an additional 18 months. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berk JL, et al. Repurposing Diflunisal for Familial Amyloid Polyneuropathy A Randomized Clinical Trial. JAMA-J Am Med Assoc. 2013;310:2658–2667. doi: 10.1001/jama.2013.283815. The positive diflunisal clinical trial data provides complementary evidence that transthyretin kinetic stabilization is an effective therapeutic strategy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hammarstrom P, Schneider F, Kelly JW. Trans-suppression of misfolding in an amyloid disease. Science. 2001;293:2459–62. doi: 10.1126/science.1062245. This study demonstrates interallelic trans-suppression in an amyloid disease, the idea that a transthyretin mutation that is disease associated can be suppressed by a mutation within transthyretin on the second allele. This happens because TTR heterotetramer dissociation, the rate-limiting step of amyloidogenesis, is slowed dramatically, thus the concentration of misfolded monomer and aggregates is lower, and hence pathology is ameliorated. [DOI] [PubMed] [Google Scholar]
- 26.Hammarstrom P, Wiseman RL, Powers ET, Kelly JW. Prevention of transthyretin amyloid disease by changing protein misfolding energetics. Science. 2003;299:713–6. doi: 10.1126/science.1079589. This paper demonstrates how interallelic trans-suppression (25), which relies on increasing the activation barrier for dissociation, can be mimicked by a small molecule that selectively binds to and stabilizes the native state over the dissociative transition state, thus slowing the rate-limiting step of amyloidogenesis. These small molecules are a special class of pharmacologic chaperones called kinetic stabilizers. [DOI] [PubMed] [Google Scholar]
- 27.Kastritis E, et al. Bortezomib with or without dexamethasone in primary systemic (light chain) amyloidosis. J Clin Oncol. 2010;28:1031–1037. doi: 10.1200/JCO.2009.23.8220. This manuscript demonstrates that reducing the light chain concentration and consequently its aggregation by killing clonal plasma cells is efficacious for ameliorating light chain amyloidosis. [DOI] [PubMed] [Google Scholar]
- 28.Merlini G, Comenzo RL, Seldin DC, Wechalekar A, Gertz MA. Immunoglobulin light chain amyloidosis. Expert Rev Hematol. 2014;7:143–156. doi: 10.1586/17474086.2014.858594. [DOI] [PubMed] [Google Scholar]
- 29.Caughey B, Lansbury PT. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci. 2003;26:267–98. doi: 10.1146/annurev.neuro.26.010302.081142. This review introduces the spectrum of non-amyloid aggregates and the basis for suspecting them as agents of proteotoxicity and post-mitotic tissue loss. [DOI] [PubMed] [Google Scholar]
- 30.Lachmann HJ, et al. Outcome in systemic AL amyloidosis in relation to changes in concentration of circulating free immunoglobulin light chains following chemotherapy. Br J Haematol. 2003;122:78–84. doi: 10.1046/j.1365-2141.2003.04433.x. [DOI] [PubMed] [Google Scholar]
- 31.Bulawa CE, et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci U S A. 2012;109:9629–9634. S9629/1–S9629/9. doi: 10.1073/pnas.1121005109. This paper introduces the preclinical data and rationale for using tafamidis as a kinetic stabilizer to ameliorate the TTR amyloidoses. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bacskai BJ, et al. Imaging of amyloid-beta deposits in brains of living mice permits direct observation of clearance of plaques with immunotherapy. Nat Med. 2001;7:369–72. doi: 10.1038/85525. [DOI] [PubMed] [Google Scholar]
- 33.Colon W, Kelly JW. Partial denaturation of transthyretin is sufficient for amyloid fibril formation in vitro. Biochemistry. 1992;31:8654–60. doi: 10.1021/bi00151a036. This paper demonstrates that conformational changes are sufficient to enable the process of amyloidogenesis. [DOI] [PubMed] [Google Scholar]
- 34.Hurle MR, Helms LR, Li L, Chan W, Wetzel R. A role for destabilizing amino acid replacements in light-chain amyloidosis. Proc Natl Acad Sci U S A. 1994;91:5446–50. doi: 10.1073/pnas.91.12.5446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pan KM, et al. Conversion of alpha-helices into beta-sheets features in the formation of the Scrapie prion proteins. Proc Natl Acad Sci U S A. 1993;90:10962–10966. doi: 10.1073/pnas.90.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Booth DR, et al. Instability, unfolding and aggregation of human lysozyme variants underlying amyloid fibrillogenesis. Nature. 1997;385:787–93. doi: 10.1038/385787a0. [DOI] [PubMed] [Google Scholar]
- 37.Dobson CM. Protein folding and misfolding. Nature. 2003;426:884–90. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 38.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 39.Lee JP, et al. H-1-NMR of A-beta amyloid peptide congeners in water solution - conformational changes correlate with plaque competence. Biochemistry. 1995;34:5191–5200. doi: 10.1021/bi00015a033. [DOI] [PubMed] [Google Scholar]
- 40.De Strooper B, et al. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature. 1999;398:518–522. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- 41.Haass C, et al. The Swedish mutation causes early-onset Alzheime'rs disease by beta-secretase cleavage within the secretory pathway. Nature Medicine. 1995;1:1291–1296. doi: 10.1038/nm1295-1291. [DOI] [PubMed] [Google Scholar]
- 42.Ross CA. Polyglutamine pathogenesis: Emergence of unifying mechanisms for Huntington's disease and related disorders. Neuron. 2002;35:819–822. doi: 10.1016/s0896-6273(02)00872-3. [DOI] [PubMed] [Google Scholar]
- 43.Sathasivam K, et al. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc Natl Acad Sci U S A. 2013;110:2366–70. doi: 10.1073/pnas.1221891110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Graham RK, et al. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell. 2006;125:1179–91. doi: 10.1016/j.cell.2006.04.026. [DOI] [PubMed] [Google Scholar]
- 45.La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991;352:77–9. doi: 10.1038/352077a0. [DOI] [PubMed] [Google Scholar]
- 46.Chen S, Ferrone FA, Wetzel R. Huntington's disease age-of-onset linked to polyglutamine aggregation nucleation. Proc Natl Acad Sci U S A. 2002;99:11884–9. doi: 10.1073/pnas.182276099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kosik KS, Joachim CL, Selkoe DJ. MicrotubuIe-associated protein tau (tau) is a major antigneic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci U S A. 1986;83:4044–4048. doi: 10.1073/pnas.83.11.4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Polymeropoulos MH, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 49.Wolfe MS. Tau mutations in neurodegenerative diseases. J Biol Chem. 2009;284:6021–5. doi: 10.1074/jbc.R800013200. [DOI] [PubMed] [Google Scholar]
- 50.Drewes G, et al. Mitogen activated protein (MAP) kinase transforms tau protein into an Alzheimer-like state. EMBO J. 1992;11:2131–2138. doi: 10.1002/j.1460-2075.1992.tb05272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Burre J, Sharma M, Sudhof TC. alpha-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc Natl Acad Sci U S A. 2014;111:E4274–83. doi: 10.1073/pnas.1416598111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bartels T, Choi JG, Selkoe DJ. alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011;477:107–U123. doi: 10.1038/nature10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang L, et al. alpha-Synuclein Multimers Cluster Synaptic Vesicles and Attenuate Recycling. Curr Biol. 2014;24:2319–26. doi: 10.1016/j.cub.2014.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Neumann M, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–3. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 55.Shelkovnikova TA, Robinson HK, Southcombe JA, Ninkina N, Buchman VL. Multistep process of FUS aggregation in the cell cytoplasm involves RNA-dependent and RNA-independent mechanisms. Hum Mol Genet. 2014;23:5211–26. doi: 10.1093/hmg/ddu243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.DeJesus-Hernandez M, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–56. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Olzscha H, et al. Amyloid-like Aggregates Sequester Numerous Metastable Proteins with Essential Cellular Functions. Cell. 2011;144:67–78. doi: 10.1016/j.cell.2010.11.050. [DOI] [PubMed] [Google Scholar]
- 58.Laganowsky A, et al. Atomic View of a Toxic Amyloid Small Oligomer. Science. 2012;335:1228–1231. doi: 10.1126/science.1213151. This paper reports an oligomeric cylindrical barrel structure, formed from six antiparallel strands of a segment of the amyloid-forming aB crystalline protein, that the authors call a cylindrin structure–a structure that allows hypotheses about proteotoxicity of the type discussed in ref. 59 to be generated and tested. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lashuel HA, Hartley D, Petre BM, Walz T, Lansbury PT., Jr Neurodegenerative disease: amyloid pores from pathogenic mutations. Nature. 2002;418:291. doi: 10.1038/418291a. [DOI] [PubMed] [Google Scholar]
- 60.Ferrone F. Analysis of protein aggregation kinetics. Methods Enzymol. 1999;309:256–74. doi: 10.1016/s0076-6879(99)09019-9. [DOI] [PubMed] [Google Scholar]
- 61.Powers ET, Powers DL. The kinetics of nucleated polymerizations at high concentrations: amyloid fibril formation near and above the “supercritical concentration”. Biophys J. 2006;91:122–32. doi: 10.1529/biophysj.105.073767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jarrett JT, Lansbury PT., Jr Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie? Cell. 1993;73:1055–8. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- 63.Serio TR, et al. Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science. 2000;289:1317–21. doi: 10.1126/science.289.5483.1317. [DOI] [PubMed] [Google Scholar]
- 64.Hofrichter J, Ross PD, Eaton WA. Kinetics and mechanism of deoxyhemogloblin-S gelation - new approach to understanding sickle cell disease. Proc Natl Acad Sci U S A. 1974;71:4864–4868. doi: 10.1073/pnas.71.12.4864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ferrone FA, Hofrichter J, Eaton WA. Kinetics of sickle hemoglobin ploymerization. 2. A double nucleation mechansim. J Mol Biol. 1985;183:611–631. doi: 10.1016/0022-2836(85)90175-5. [DOI] [PubMed] [Google Scholar]
- 66.Lundmark K, et al. Transmissibility of systemic amyloidosis by a prion-like mechanism. Proc Natl Acad Sci U S A. 2002;99:6979–6984. doi: 10.1073/pnas.092205999. This pioneering paper demonstrated the acceleration of AA amyloidosis by the injection or oral administration of AA amyloid fibrils. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Aguzzi A, Rajendran L. The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron. 2009;64:783–90. doi: 10.1016/j.neuron.2009.12.016. [DOI] [PubMed] [Google Scholar]
- 68.Hurshman AR, White JT, Powers ET, Kelly JW. Transthyretin aggregation under partially denaturing conditions is a downhill polymerization. Biochemistry. 2004;43:7365–81. doi: 10.1021/bi049621l. [DOI] [PubMed] [Google Scholar]
- 69.Terry RD, et al. Physical basis of cognitive alterations in Alzheimers disease - synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 70.Cheng IH, et al. Accelerating amyloid-beta fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J Biol Chem. 2007;282:23818–28. doi: 10.1074/jbc.M701078200. [DOI] [PubMed] [Google Scholar]
- 71.Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–10. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- 72.Palladini G, et al. Circulating amyloidogenic free light chains and serum N-terminal natriuretic peptide type B decrease simultaneously in association with improvement of survival in AL. Blood. 2006;107:3854–8. doi: 10.1182/blood-2005-11-4385. This manuscript demonstrates that a drop in circulating soluble light chain levels correlates with an improvement in health without a correlation with a decrease in light chain amyloid fibrils. [DOI] [PubMed] [Google Scholar]
- 73.Simsek I, et al. No regression of renal amyloid mass despite remission of nephrotic syndrome in a patient with TRAPS following etanercept therapy. J Nephrol. 2010;23:119–123. [PubMed] [Google Scholar]
- 74.Winner B, et al. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc Natl Acad Sci U S A. 2011;108:4194–9. doi: 10.1073/pnas.1100976108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cohen E, Bieschke J, Perciavalle RM, Kelly JW, Dillin A. Opposing Activities Protect Against Age-Onset Proteotoxicity. Science. 2006;313:1604–1610. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- 76.Sousa MM, Cardoso I, Fernandes R, Guimaraes A, Saraiva MJ. Deposition of transthyretin in early stages of familial amyloidotic polyneuropathy: evidence for toxicity of nonfibrillar aggregates. Am J Pathol. 2001;159:1993–2000. doi: 10.1016/s0002-9440(10)63050-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bieschke J, et al. Small-molecule conversion of toxic oligomers to nontoxic beta-sheet-rich amyloid fibrils. Nat Chem Biol. 2012;8:93–101. doi: 10.1038/nchembio.719. This paper demonstrates that the acceleration of Aβ fibrillogenesis through the action of the orcein-related small molecule O4 decreases the concentration of small, toxic Aβ oligomers in aggregation reactions, as O4 treatment suppresses inhibition of long-term potentiation by Aβ oligomers in hippocampal brain slices. [DOI] [PubMed] [Google Scholar]
- 78.Lee J, Culyba EK, Powers ET, Kelly JW. Amyloid-beta forms fibrils by nucleated conformational conversion of oligomers. Nat Chem Biol. 2011;7:602–609. doi: 10.1038/nchembio.624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Garai K, Frieden C. Quantitative analysis of the time course of Aβ oligomerization and subsequent growth steps using tetramethylrhodamine-labeled Aβ. Proc Natl Acad Sci U S A. 2013;110:3321–3326. doi: 10.1073/pnas.1222478110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lashuel HA, Wurth C, Woo L, Kelly JW. The most pathogenic transthyretin variant, L55P, forms amyloid fibrils under acidic conditions and protofilaments under physiological conditions. Biochemistry. 1999;38:13560–73. doi: 10.1021/bi991021c. [DOI] [PubMed] [Google Scholar]
- 81.Goate A, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991;349:704–6. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 82.Polymeropoulos MH, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–7. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 83.Theuns J, et al. Promoter mutations that increase amyloid precursor-protein expression are associated with Alzheimer disease. Am J Hum Genet. 2006;78:936–46. doi: 10.1086/504044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zigman WB, Lott IT. Alzheimer's disease in Down syndrome: neurobiology and risk. Ment Retard Dev Disabil Res Rev. 2007;13:237–46. doi: 10.1002/mrdd.20163. [DOI] [PubMed] [Google Scholar]
- 85.Chartier-Harlin MC, et al. Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet. 2004;364:1167–9. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- 86.Herlenius G, Wilczek HE, Larsson M, Ericzon BG. Ten years of international experience with liver transplantation for familial amyloidotic polyneuropathy: results from the Familial Amyloidotic Polyneuropathy World Transplant Registry. Transplantation. 2004;77:64–71. doi: 10.1097/01.TP.0000092307.98347.CB. [DOI] [PubMed] [Google Scholar]
- 87.Holmgren G, et al. Clinical improvement and amyloid regression after liver transplantation in hereditary transthyretin amyloidosis. Lancet. 1993;341:1113–6. doi: 10.1016/0140-6736(93)93127-m. This pioneering approach of surgically-mediated gene therapy for successfully treating transthyretin amyloid disease demonstrated the concept of protein reduction therapy. [DOI] [PubMed] [Google Scholar]
- 88.Ihse E, et al. Amyloid fibril composition is related to the phenotype of hereditary transthyretin V30M amyloidosis. J Pathol. 2008;216:253–261. doi: 10.1002/path.2411. [DOI] [PubMed] [Google Scholar]
- 89.Wilczek HE, Larsson M, Ericzon BG, Fapwtr Long-term data from the Familial Amyloidotic Polyneuropathy World Transplant Registry (FAPWTR) Amyloid. 2011;18:193–195. doi: 10.3109/13506129.2011.574354072. [DOI] [PubMed] [Google Scholar]
- 90.Yamashita T, et al. Long-term survival after liver transplantation in patients with familial amyloid polyneuropathy. Neurology. 2012;78:637–643. doi: 10.1212/WNL.0b013e318248df18. [DOI] [PubMed] [Google Scholar]
- 91.Antoni G, et al. In Vivo Visualization of Amyloid Deposits in the Heart with C-11-PIB and PET. J Nuclear Med. 2013;54:213–220. doi: 10.2967/jnumed.111.102053. [DOI] [PubMed] [Google Scholar]
- 92.Delahaye N, et al. Impact of liver transplantation on cardiac autonomic denervation in familial amyloid polyneuropathy. Medicine. 2006;85:229–238. doi: 10.1097/01.md.0000232559.22098.c3. [DOI] [PubMed] [Google Scholar]
- 93.Suhr OB. Impact of liver transplantation on familial amyloidotic polyneuropathy (FAP) patients' symptoms and complications. Amyloid. 2003;10:77–83. [PubMed] [Google Scholar]
- 94.Rydh A, et al. Serum amyloid P component scintigraphy in familial amyloid polyneuropathy: regression of visceral amyloid following liver transplantation. Eur J Nucl Med. 1998;25:709–713. doi: 10.1007/s002590050273. [DOI] [PubMed] [Google Scholar]
- 95.Stangou AJ, et al. Hereditary fibrinogen A alpha-chain amyloidosis: phenotypic characterization of a systemic disease and the role of liver transplantation. Blood. 2010;115:2998–3007. doi: 10.1182/blood-2009-06-223792. [DOI] [PubMed] [Google Scholar]
- 96.Olofsson BO, Backman C, Karp K, Suhr OB. Progression of cardiomyopathy after liver transplantation in patients with familial amyloidotic polyneuropathy, Portuguese type. Transplantation. 2002;73:745–51. doi: 10.1097/00007890-200203150-00015. [DOI] [PubMed] [Google Scholar]
- 97.Munar-Ques M, et al. Vitreous amyloidosis after liver transplantation in patients with familial amyloid polyneuropathy: Ocular synthesis of mutant transthyretin. Amyloid. 2000;7:266–269. doi: 10.3109/13506120009146440. [DOI] [PubMed] [Google Scholar]
- 98.Maia LF, et al. CNS involvement in V30M transthyretin amyloidosis: clinical, neuropathological and biochemical findings. J Neurol Neurosurg Psychiatry. 2014 doi: 10.1136/jnnp-2014-308107. [DOI] [PubMed] [Google Scholar]
- 99.Gertz MA. Immunoglobulin light chain amyloidosis: 2011 update on diagnosis, risk-stratification, and management. Am J Hematol. 2011;86:181–186. doi: 10.1002/ajh.21934. [DOI] [PubMed] [Google Scholar]
- 100.Arendt BK, et al. Biologic and genetic characterization of the novel amyloidogenic lambda light chain-secreting human cell lines, ALMC-1 and ALMC-2. Blood. 2008;112:1931–1941. doi: 10.1182/blood-2008-03-143040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Monis GF, et al. Role of endocytic inhibitory drugs on internalization of amyloidogenic light chains by cardiac fibroblasts. Am J Pathol. 2006;169:1939–52. doi: 10.2353/ajpath.2006.060183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Shi J, et al. Amyloidogenic light chains induce cardiomyocyte contractile dysfunction and apoptosis via a non-canonical p38alpha MAPK pathway. Proc Natl Acad Sci U S A. 2010;107:4188–93. doi: 10.1073/pnas.0912263107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Comenzo RL. Current and emerging views and treatments of systemic immunoglobulin light-chain (AL) amyloidosis. Contrib Nephrol. 2007;153:195–210. doi: 10.1159/000096768. [DOI] [PubMed] [Google Scholar]
- 104.Obici L, Merlini G. Amyloidosis in autoinflammatory syndromes. Autoimmunity Reviews. 2012;12:14–17. doi: 10.1016/j.autrev.2012.07.016. [DOI] [PubMed] [Google Scholar]
- 105.Gillmore JD, Lovat LB, Persey MR, Pepys MB, Hawkins PN. Amyloid load and clinical outcome in AA amyloidosis in relation to circulating concentration of serum amyloid A protein. Lancet. 2001;358:24–29. doi: 10.1016/S0140-6736(00)05252-1. [DOI] [PubMed] [Google Scholar]
- 106.Kisilevsky R, Narindrasorasak S, Tape C, Tan R, Boudreau L. During AA-amyloidogenesis is proteolytic attack on serum amyloid-A a pre-fibrillogenic or post-fibrillogenic event. Amyloid. 1994;1:174–183. [Google Scholar]
- 107.Denis MA, et al. Control of AA amyloidosis complicating Crohn's disease: a clinico-pathological study. Eur J Clin Invest. 2013;43:292–301. doi: 10.1111/eci.12045. [DOI] [PubMed] [Google Scholar]
- 108.Ishii W, et al. A case with rheumatoid arthritis and systemic reactive AA amyloidosis showing rapid regression of amyloid deposition on gastroduodenal mucosa after a combined therapy of corticosteroid and etanercept. Rheumatol Int. 2011;31:247–250. doi: 10.1007/s00296-009-1205-z. [DOI] [PubMed] [Google Scholar]
- 109.Kuroda T, et al. A case of AA amyloidosis associated with rheumatoid arthritis effectively treated with Infliximab. Rheumatol Int. 2008;28:1155–1159. doi: 10.1007/s00296-008-0590-z. [DOI] [PubMed] [Google Scholar]
- 110.Lesnyak Q, et al. Beneficial effect of eprodisate (NC-503) on the preservation of kidney function in AA amyloidosis patients: 3-year follow-up results. Ann Rheum Dis. 2007;66:248–248. [Google Scholar]
- 111.Matsuda M, Morita H, Ikeda S. Long-term follow-up of systemic reactive AA amyloidosis secondary to rheumatoid arthritis: Successful treatment with intermediate-dose corticosteroid. Internal Medicine. 2002;41:403–407. doi: 10.2169/internalmedicine.41.403. This work shows that a reduction in inflammation, which lowers the AA amyloidogenic protein concentration, is effective at ameliorating AA amyloidosis. [DOI] [PubMed] [Google Scholar]
- 112.Nakamura T, et al. Efficacy of cyclophosphamide combined with prednisolone in patients with AA amyloidosis secondary to rheumatoid arthritis. Clinical Rheumatology. 2003;22:371–375. doi: 10.1007/s10067-003-0763-9. [DOI] [PubMed] [Google Scholar]
- 113.Srinivasan S, et al. Pathogenic Serum Amyloid A 1.1 Shows a Long Oligomer-rich Fibrillation Lag Phase Contrary to the Highly Amyloidogenic Non-pathogenic SAA2.2. J Biol Chem. 2013;288:2744–2755. doi: 10.1074/jbc.M112.394155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Borchelt DR, et al. Familial Alzheimer's disease-linked presenilin 1 variants elevate A beta 1-42/1-40 ratio in vitro and in vivo. Neuron. 1996;17:1005–1013. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- 115.Wolfe MS. Structure, mechanism and inhibition of gamma-secretase and presenilin-like proteases. Biol Chem. 2010;391:839–847. doi: 10.1515/BC.2010.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sambamurti K, et al. Targets for AD treatment: conflicting messages from Œ≥-secretase inhibitors. J Neurochem. 2011;117:359–374. doi: 10.1111/j.1471-4159.2011.07213.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.De Strooper B, Vassar R, Golde T. The secretases: enzymes with therapeutic potential in Alzheimer disease. Nat Rev Neurol. 2010;6:99–107. doi: 10.1038/nrneurol.2009.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yan R, Vassar R. Targeting the beta secretase BACE1 for Alzheimer's disease therapy. Lancet Neurol. 2014;13:319–29. doi: 10.1016/S1474-4422(13)70276-X. A few companies, including Merck, have BACE1-selective small molecule inhibitors that hold great promise for ameliorating Alzheimer's disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ghosh AK, Osswald HL. BACE1 (beta-secretase) inhibitors for the treatment of Alzheimer's disease. Chem Soc Rev. 2014;43:6765–813. doi: 10.1039/c3cs60460h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ohno M, et al. BACE1 deficiency rescues memory deficits and cholinergic dysfunction in a mouse model of Alzheimer's disease. Neuron. 2004;41:27–33. doi: 10.1016/s0896-6273(03)00810-9. [DOI] [PubMed] [Google Scholar]
- 121.De Strooper B, Iwatsubo T, Wolfe MS. Presenilins and gamma-secretase: structure, function, and role in Alzheimer Disease. Cold Spring Harb Perspect Med. 2012;2:a006304. doi: 10.1101/cshperspect.a006304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.De Strooper B, Gutierrez LC. Learning by Failing: Ideas and Concepts to Tackle gamma-Secretases in Alzheimer Disease and Beyond. Annu Rev Pharmacol Toxicol. 2014 doi: 10.1146/annurev-pharmtox-010814-124309. [DOI] [PubMed] [Google Scholar]
- 123.Golde TE, Koo EH, Felsenstein KM, Osborne BA, Miele L. gamma-Secretase inhibitors and modulators. Biochim Biophys Acta. 2013;1828:2898–907. doi: 10.1016/j.bbamem.2013.06.005. Allosteric regulators of γ-secretase which impede Aβ generation without inhibiting its processing of critical substrates is an appealing strategy to ameliorate Alzheimer's disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bard F, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nature Medicine. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- 125.Masliah E, et al. A beta vaccination effects on plaque pathology in the absence of encephalitis in Alzheimer disease. Neurology. 2005;64:129–131. doi: 10.1212/01.WNL.0000148590.39911.DF. Amyloid disease vaccination in the absence of an immune or inflammatory reaction holds great promise for preventing the human amyloid diseases. [DOI] [PubMed] [Google Scholar]
- 126.Coelho T, et al. Safety and Efficacy of RNAi Therapy for Transthyretin Amyloidosis. N Engl J Med. 2013;369:819–829. doi: 10.1056/NEJMoa1208760. RNAi treatment is effective at reducing protein levels in some tissues and is a promising strategy for ameliorating amyloidosis in the systemic amyloidoses, see also refs. 127 and 128. [DOI] [PubMed] [Google Scholar]
- 127.Benson MD, et al. Targeted suppression of an amyloidogenic transthyretin with antisense oligonucleotides. Muscle Nerve. 2006;33:609–618. doi: 10.1002/mus.20503. [DOI] [PubMed] [Google Scholar]
- 128.Zhou P, Ma X, Iyer L, Chaulagain C, Comenzo RL. One siRNA pool targeting the lambda constant region stops lambda light-chain production and causes terminal endoplasmic reticulum stress. Blood. 2014;123:3440–51. doi: 10.1182/blood-2013-10-535187. [DOI] [PubMed] [Google Scholar]
- 129.Hovey BM, et al. Preclinical development of siRNA therapeutics for AL amyloidosis. Gene Ther. 2011;18:1150–6. doi: 10.1038/gt.2011.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Coelho T, et al. A strikingly benign evolution of FAP in an individual found to be a compound heterozygote for two TTR mutations: TTR MET 30 and TTR MET 119. J Rheumatol. 1993;20:179. [Google Scholar]
- 131.Johnson SM, et al. Native state kinetic stabilization as a strategy to ameliorate protein misfolding diseases: a focus on the transthyretin amyloidoses. Acc Chem Res. 2005;38:911–21. doi: 10.1021/ar020073i. [DOI] [PubMed] [Google Scholar]
- 132.Johnson SM, Connelly S, Fearns C, Powers ET, Kelly JW. The Transthyretin Amyloidoses: From Delineating the Molecular Mechanism of Aggregation Linked to Pathology to a Regulatory-Agency-Approved Drug. J Mol Biol. 2012;421:185–203. doi: 10.1016/j.jmb.2011.12.060. This review outlines the genetic and pharmacologic evidence supporting the hypothesis that the process of transthyretin aggregation causes the TTR amyloidoses. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rappley I, et al. Quantification of Transthyretin Kinetic Stability in Human Plasma Using Subunit Exchange. Biochemistry. 2014;53:1993–2006. doi: 10.1021/bi500171j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Miroy GJ, et al. Inhibiting transthyretin amyloid fibril formation via protein stabilization. Proc Natl Acad Sci U S A. 1996;93:15051–6. doi: 10.1073/pnas.93.26.15051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Razavi H, et al. Benzoxazoles as transthyretin amyloid fibril inhibitors: synthesis, evaluation, and mechanism of action. Angewandte Chemie. 2003;42:2758–61. doi: 10.1002/anie.200351179. [DOI] [PubMed] [Google Scholar]
- 136.Baures PW, Peterson SA, Kelly JW. Discovering transthyretin amyloid fibril inhibitors by limited screening. Bioorg Med Chem. 1998;6:1389–401. doi: 10.1016/s0968-0896(98)00130-8. [DOI] [PubMed] [Google Scholar]
- 137.Klabunde T, et al. Rational design of potent human transthyretin amyloid disease inhibitors. Nat Struct Biol. 2000;7:312–21. doi: 10.1038/74082. [DOI] [PubMed] [Google Scholar]
- 138.Connelly S, Choi S, Johnson SM, Kelly JW, Wilson IA. Structure-based design of kinetic stabilizers that ameliorate the transthyretin amyloidoses. Curr Opin Struct Biol. 2010;20:54–62. doi: 10.1016/j.sbi.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sekijima Y, Dendle MA, Kelly JW. Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid. 2006;13:236–49. doi: 10.1080/13506120600960882. [DOI] [PubMed] [Google Scholar]
- 140.Tojo K, Sekijima Y, Kelly JW, Ikeda S. Diflunisal stabilizes familial amyloid polyneuropathy-associated transthyretin variant tetramers in serum against dissociation required for amyloidogenesis. Neurosci Res. 2006;56:441–9. doi: 10.1016/j.neures.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 141.Amaducci L, Tesco G. Aging as a major risk for degenerative diseases of the central nervous system. Curr Opin Neurol. 1994;7:283–6. doi: 10.1097/00019052-199408000-00001. [DOI] [PubMed] [Google Scholar]
- 142.Ben-Zvi A, Miller EA, Morimoto RI. Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging. Proc Natl Acad Sci U S A. 2009;106:14914–14919. doi: 10.1073/pnas.0902882106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gavilan MP, et al. Dysfunction of the unfolded protein response increases neurodegeneration in aged rat hippocampus following proteasome inhibition. Aging Cell. 2009;8:654–665. doi: 10.1111/j.1474-9726.2009.00519.x. [DOI] [PubMed] [Google Scholar]
- 144.Takeda N, et al. Altered Unfolded Protein Response Is Implicated in the Age-Related Exacerbation of Proteinuria-Induced Proximal Tubular Cell Damage. Am J Pathol. 2013;183:774–785. doi: 10.1016/j.ajpath.2013.05.026. [DOI] [PubMed] [Google Scholar]
- 145.Singh R, et al. Reduced heat shock response in human mononuclear cells during aging and its association with polymorphisms in HSP70 genes. Cell Stress & Chaperones. 2006;11:208–215. doi: 10.1379/CSC-184R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Gupta R, et al. Firefly luciferase mutants as sensors of proteome stress. Nature Methods. 2011;8:879–U155. doi: 10.1038/nmeth.1697. [DOI] [PubMed] [Google Scholar]
- 147.Shoulders MD, et al. Stress-Independent Activation of XBP1s and/or ATF6 Reveals Three Functionally Diverse ER Proteostasis Environments. Cell Reports. 2013;3:1279–1292. doi: 10.1016/j.celrep.2013.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Cooley CB, et al. Unfolded protein response activation reduces secretion and extracellular aggregation of amyloidogenic immunoglobulin light chain. Proc Natl Acad Sci U S A. 2014;111:13046–13051. doi: 10.1073/pnas.1406050111. Activation of the unfolded protein response (UPR) stress-responsive signaling pathway transcriptionally reprograms the proteostasis network in the endoplasmic reticulum, enabling the detection and destruction of amyloidogenic light chain, but not energetically normal light chains. Ref 147 demonstrates that UPR activation can also lead to decreased secretion of amyloidogenic but not wild type transthyretin. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916–9. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- 150.Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 2009;78:959–91. doi: 10.1146/annurev.biochem.052308.114844. [DOI] [PubMed] [Google Scholar]
- 151.Mu TW, et al. Chemical and biological approaches synergize to ameliorate protein-folding diseases. Cell. 2008;134:769–781. doi: 10.1016/j.cell.2008.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Jinwal UK, et al. Imbalance of Hsp70 family variants fosters tau accumulation. FASEB J. 2013;27:1450–1459. doi: 10.1096/fj.12-220889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wang AM, et al. Activation of Hsp70 reduces neurotoxicity by promoting polyglutamine protein degradation. Nat Chem Biol. 2013;9:112–118. doi: 10.1038/nchembio.1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Craig EA. The Heat-shock Response. CRC Crit Rev Biochem. 1985;18:239–280. doi: 10.3109/10409238509085135. [DOI] [PubMed] [Google Scholar]
- 155.Lindquist S. The Heat-shock Response. Annu Rev Biochem. 1986;55:1151–1191. doi: 10.1146/annurev.bi.55.070186.005443. [DOI] [PubMed] [Google Scholar]
- 156.Morimoto RI. Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 1998;12:3788–3796. doi: 10.1101/gad.12.24.3788. [DOI] [PubMed] [Google Scholar]
- 157.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 158.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 159.Shoulders MD, Ryno LM, Cooley CB, Kelly JW, Wiseman RL. Broadly Applicable Methodology for the Rapid and Dosable Small Molecule-Mediated Regulation of Transcription Factors in Human Cells. J Am Chem Soc. 2013;135:8129–8132. doi: 10.1021/ja402756p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Calamini B, et al. Small-molecule proteostasis regulators for protein conformational diseases. Nat Chem Biol. 2012;8:185–196. doi: 10.1038/nchembio.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zhang YQ, Sarge KD. Celastrol inhibits polyglutamine aggregation and toxicity though induction of the heat shock response. J Mol Med. 2007;85:1421–1428. doi: 10.1007/s00109-007-0251-9. The authors demonstrate that heat shock response activation, which transcriptionally reprograms the cytosolic proteostasis network, significantly decreases killing of cells expressing mutant polyglutamine protein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Bersuker K, Hipp MS, Calamini B, Morimoto RI, Kopito RR. Heat Shock Response Activation Exacerbates Inclusion Body Formation in a Cellular Model of Huntington Disease. J Biol Chem. 2013;288:23633–23638. doi: 10.1074/jbc.C113.481945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Sittler A, et al. Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of Huntington's disease. Hum Mol Genet. 2001;10:1307–1315. doi: 10.1093/hmg/10.12.1307. [DOI] [PubMed] [Google Scholar]
- 164.Aridon P, et al. Protective Role of Heat Shock Proteins in Parkinson's Disease. Neurodegenerative Diseases. 2011;8:155–168. doi: 10.1159/000321548. [DOI] [PubMed] [Google Scholar]
- 165.Morley JF, Morimoto RI. Regulation of longevity in Caenorhabditis elegans by heat shock factor and molecular chaperones. Mol Biol Cell. 2004;15:657–664. doi: 10.1091/mbc.E03-07-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Warrick JM, et al. Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat Genet. 1999;23:425–8. doi: 10.1038/70532. [DOI] [PubMed] [Google Scholar]
- 167.Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM. Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson's disease. Science. 2002;295:865–8. doi: 10.1126/science.1067389. [DOI] [PubMed] [Google Scholar]
- 168.Shimshek DR, Mueller M, Wiessner C, Schweizer T, van der Putten PH. The HSP70 molecular chaperone is not beneficial in a mouse model of alpha-synucleinopathy. PLoS One. 2010;5:e10014. doi: 10.1371/journal.pone.0010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Pemberton S, et al. Hsc70 protein interaction with soluble and fibrillar alpha-synuclein. J Biol Chem. 2011;286:34690–9. doi: 10.1074/jbc.M111.261321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Danzer KM, et al. Heat-shock protein 70 modulates toxic extracellular alpha-synuclein oligomers and rescues trans-synaptic toxicity. FASEB J. 2011;25:326–36. doi: 10.1096/fj.10-164624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475:324–332. doi: 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- 172.Muchowski PJ, et al. Hsp70 and Hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc Natl Acad Sci U S A. 2000;97:7841–7846. doi: 10.1073/pnas.140202897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Mayer MP, et al. Multistep mechanism of substrate binding determines chaperone activity of Hsp70. Nat Struct Biol. 2000;7:586–593. doi: 10.1038/76819. [DOI] [PubMed] [Google Scholar]
- 174.Chafekar SM, et al. Pharmacological Tuning of Heat Shock Protein 70 Modulates Polyglutamine Toxicity and Aggregation. ACS Chem Biol. 2012;7:1556–1564. doi: 10.1021/cb300166p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Abisambra J, et al. Allosteric Heat Shock Protein 70 Inhibitors Rapidly Rescue Synaptic Plasticity Deficits by Reducing Aberrant Tau. Biological Psychiatry. 2013;74:367–374. doi: 10.1016/j.biopsych.2013.02.027. The authors showed that nM concentrations of the Hsc70-modulating small molecule YM-01 administered to brain slices of tau transgenic mice were able to rapidly and potently reduce tau levels. These data along with that presented in refs 176 demonstrates that small molecules altering the Hsp70-40-nucleotide exchange factor pathway can affect amyloidogenic intrinsically disordered protein degradation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Miyata Y, et al. Synthesis and Initial Evaluation of YM-08, a Blood-Brain Barrier Permeable Derivative of the Heat Shock Protein 70 (Hsp70) Inhibitor MKT-077, Which Reduces Tau Levels. ACS Chem Neurosci. 2013;4:930–939. doi: 10.1021/cn300210g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Smith MC, et al. The E3 Ubiquitin Ligase CHIP and the Molecular Chaperone Hsc70 Form a Dynamic, Tethered Complex. Biochemistry. 2013;52:5354–5364. doi: 10.1021/bi4009209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Genereux JC, et al. Unfolded protein response-induced ERdj3 secretion links ER stress to extracellular proteostasis. EMBO J. 2014 doi: 10.15252/embj.201488896. pii: e201488896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Villella AT, et al. Inhibition of Usp14 Stimulates the Proteolytic Degradation and Clearance of Misfolded Proteins Associated with Neurodegenerative Diseases. FASEB J. 2013;27:lb131. [Google Scholar]
- 180.Lee BH, et al. Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature. 2010;467:179–184. doi: 10.1038/nature09299. This paper reveals that inhibiting the deubiquitinase enzymes has the potential to lower the concentration of amyloidogenic proteins via enhanced proteasome degradation of misfolded, ubiquitinated proteins. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kanemitsu H, Tomiyama T, Mori H. Human neprilysin is capable of degrading amyloid beta peptide not only in the monomeric form but also the pathological oligomeric form. Neurosci Lett. 2003;350:113–6. doi: 10.1016/s0304-3940(03)00898-x. [DOI] [PubMed] [Google Scholar]
- 182.Leissring MA, et al. Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron. 2003;40:1087–93. doi: 10.1016/s0896-6273(03)00787-6. [DOI] [PubMed] [Google Scholar]
- 183.Iwata N, et al. Metabolic regulation of brain Abeta by neprilysin. Science. 2001;292:1550–2. doi: 10.1126/science.1059946. [DOI] [PubMed] [Google Scholar]
- 184.Meilandt WJ, et al. Neprilysin overexpression inhibits plaque formation but fails to reduce pathogenic Abeta oligomers and associated cognitive deficits in human amyloid precursor protein transgenic mice. J Neurosci. 2009;29:1977–86. doi: 10.1523/JNEUROSCI.2984-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Planque SA, et al. Physiological IgM class catalytic antibodies selective for transthyretin amyloid. J Biol Chem. 2014;289:13243–58. doi: 10.1074/jbc.M114.557231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Nishiyama Y, et al. Metal-dependent amyloid beta-degrading catalytic antibody construct. J Biotechnol. 2014;180:17–22. doi: 10.1016/j.jbiotec.2014.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Miller HI, Rotman Y, Benshaul Y, Ashkenaz Y. The dissociation of amyloid filament to subunits. Israel J Med Sci. 1968;4:982–986. [PubMed] [Google Scholar]
- 188.Safar J, Roller PP, Gajdusek DC, Gibbs CJ. Conformational transitions, dissociation, and unfolding of scrapie amyloid (prion) protein. J Biol Chem. 1993;268:20276–20284. [PubMed] [Google Scholar]
- 189.Hasegawa K, Ono K, Yamada M, Naiki H. Kinetic modeling and determination of reaction constants of Alzheimer's beta-amyloid fibril extension and dissociation using surface plasmon resonance. Biochemistry. 2002;41:13489–13498. doi: 10.1021/bi020369w. [DOI] [PubMed] [Google Scholar]
- 190.Kristen AV, et al. Green tea halts progression of cardiac transthyretin amyloidosis: an observational report. Clin Res Cardiol. 2012;101:805–13. doi: 10.1007/s00392-012-0463-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Bieschke J, et al. EGCG remodels mature alpha-synuclein and amyloid-beta fibrils and reduces cellular toxicity. Proc Natl Acad Sci U S A. 2010;107:7710–5. doi: 10.1073/pnas.0910723107. This manuscript and refs 192-199 demonstrate that aggregate remodeling small molecules could be useful for ameliorating amyloid diseases, as indicated by a clinical report outlined in ref 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Ehrnhoefer DE, et al. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat Struct Mol Biol. 2008;15:558–66. doi: 10.1038/nsmb.1437. [DOI] [PubMed] [Google Scholar]
- 193.Meng F, Abedini A, Plesner A, Verchere CB, Raleigh DP. The flavanol (-)-epigallocatechin 3-gallate inhibits amyloid formation by islet amyloid polypeptide, disaggregates amyloid fibrils, and protects cultured cells against IAPP-induced toxicity. Biochemistry. 2010;49:8127–33. doi: 10.1021/bi100939a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Cao P, Raleigh DP. Analysis of the inhibition and remodeling of islet amyloid polypeptide amyloid fibers by flavanols. Biochemistry. 2012;51:2670–83. doi: 10.1021/bi2015162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Young LM, Cao P, Raleigh DP, Ashcroft AE, Radford SE. Ion mobility spectrometry-mass spectrometry defines the oligomeric intermediates in amylin amyloid formation and the mode of action of inhibitors. J Am Chem Soc. 2014;136:660–70. doi: 10.1021/ja406831n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Hyung SJ, et al. Insights into antiamyloidogenic properties of the green tea extract (-)-epigallocatechin-3-gallate toward metal-associated amyloid-beta species. Proc Natl Acad Sci U S A. 2013;110:3743–8. doi: 10.1073/pnas.1220326110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Palhano FL, Lee J, Grimster NP, Kelly JW. Toward the Molecular Mechanism(s) by Which EGCG Treatment Remodels Mature Amyloid Fibrils. J Am Chem Soc. 2013;135:7503–7510. doi: 10.1021/ja3115696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Attar A, Rahimi F, Bitan G. Modulators of Amyloid Protein Aggregation and Toxicity: EGCG and Clr01. Translational Neuroscience. 2013;4:385–409. [Google Scholar]
- 199.Lorenzen N, et al. How epigallocatechin gallate can inhibit alpha-synuclein oligomer toxicity in vitro. J Biol Chem. 2014 doi: 10.1074/jbc.M114.554667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Schenk D, et al. Immunization with amyloid-beta attenuates Alzheimer disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 201.Janus C, et al. A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer's disease. Nature. 2000;408:979–82. doi: 10.1038/35050110. [DOI] [PubMed] [Google Scholar]
- 202.Morgan D, et al. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature. 2000;408:982–5. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- 203.Check E. Nerve inflammation halts trial for Alzheimer's drug. Nature. 2002;415:462. doi: 10.1038/415462a. [DOI] [PubMed] [Google Scholar]
- 204.Nicoll JA, et al. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med. 2003;9:448–52. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- 205.Holmes C, et al. Long-term effects of Abeta42 immunisation in Alzheimer's disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372:216–23. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- 206.Boche D, et al. Consequence of Abeta immunization on the vasculature of human Alzheimer's disease brain. Brain. 2008;131:3299–310. doi: 10.1093/brain/awn261. [DOI] [PubMed] [Google Scholar]
- 207.Nicoll JA, et al. Abeta species removal after abeta42 immunization. J Neuropathol Exp Neurol. 2006;65:1040–8. doi: 10.1097/01.jnen.0000240466.10758.ce. [DOI] [PubMed] [Google Scholar]
- 208.DeMattos RB, et al. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2001;98:8850–5. doi: 10.1073/pnas.151261398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Wisniewski T, Goni F. Immunotherapy for Alzheimer's disease. Biochem Pharmacol. 2014;88:499–507. doi: 10.1016/j.bcp.2013.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Bateman RJ, et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med. 2012;367:795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Masliah E, et al. Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of Lewy body disease. PLoS One. 2011;6:e19338. doi: 10.1371/journal.pone.0019338. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 212.Castillo-Carranza DL, et al. Passive immunization with Tau oligomer monoclonal antibody reverses tauopathy phenotypes without affecting hyperphosphorylated neurofibrillary tangles. J Neurosci. 2014;34:4260–72. doi: 10.1523/JNEUROSCI.3192-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Lindstrom V, et al. Immunotherapy targeting alpha-synuclein protofibrils reduced pathology in (Thy-1)-h[A30P] alpha-synuclein mice. Neurobiol Dis. 2014;69C:134–143. doi: 10.1016/j.nbd.2014.05.009. [DOI] [PubMed] [Google Scholar]
- 214.Chai X, et al. Passive immunization with anti-Tau antibodies in two transgenic models: reduction of Tau pathology and delay of disease progression. J Biol Chem. 2011;286:34457–67. doi: 10.1074/jbc.M111.229633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Masliah E, et al. Effects of alpha-synuclein immunization in a mouse model of Parkinson's disease. Neuron. 2005;46:857–68. doi: 10.1016/j.neuron.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 216.Bae EJ, et al. Antibody-aided clearance of extracellular alpha-synuclein prevents cell-to-cell aggregate transmission. J Neurosci. 2012;32:13454–69. doi: 10.1523/JNEUROSCI.1292-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Doyle SM, Wickner S. Hsp104 and ClpB: protein disaggregating machines. Trends Biochem Sci. 2009;34:40–8. doi: 10.1016/j.tibs.2008.09.010. [DOI] [PubMed] [Google Scholar]
- 218.Jackrel ME, et al. Potentiated Hsp104 variants antagonize diverse proteotoxic misfolding events. Cell. 2014;156:170–82. doi: 10.1016/j.cell.2013.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Jackrel ME, Shorter J. Reversing deleterious protein aggregation with re-engineered protein disaggregases. Cell Cycle. 2014;13:1379–83. doi: 10.4161/cc.28709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Winkler J, Tyedmers J, Bukau B, Mogk A. Chaperone networks in protein disaggregation and prion propagation. J Struct Biol. 2012;179:152–160. doi: 10.1016/j.jsb.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 221.Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19:983–97. doi: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- 222.Hidvegi T, et al. An autophagy-enhancing drug promotes degradation of mutant alpha 1-antitrypsin Z and reduces hepatic fibrosis. Science. 2010;329:229–232. doi: 10.1126/science.1190354. [DOI] [PubMed] [Google Scholar]
- 223.Harper JD, Lansbury PT., Jr Models of amyloid seeding in Alzheimer's disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu Rev Biochem. 1997;66:385–407. doi: 10.1146/annurev.biochem.66.1.385. [DOI] [PubMed] [Google Scholar]
- 224.Eisele YS. From soluble abeta to progressive abeta aggregation: could prion-like templated misfolding play a role? Brain Pathol. 2013;23:333–41. doi: 10.1111/bpa.12049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Colby DW, Prusiner SB. Prions. Cold Spring Harb Perspect Biol. 2011;3:a006833. doi: 10.1101/cshperspect.a006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Irwin DJ, et al. Evaluation of Potential Infectivity of Alzheimer and Parkinson Disease Proteins in Recipients of Cadaver-Derived Human Growth Hormone. JAMA Neurol. 2013:1–7. doi: 10.1001/jamaneurol.2013.1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Johan K, et al. Acceleration of amyloid protein A amyloidosis by amyloid-like synthetic fibrils. Proc Natl Acad Sci U S A. 1998;95:2558–63. doi: 10.1073/pnas.95.5.2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Kane MD, et al. Evidence for seeding of beta -amyloid by intracerebral infusion of Alzheimer brain extracts in beta -amyloid precursor protein-transgenic mice. J Neurosci. 2000;20:3606–11. doi: 10.1523/JNEUROSCI.20-10-03606.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Meyer-Luehmann M, et al. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science. 2006;313:1781–4. doi: 10.1126/science.1131864. This paper demonstrates that the intracerebral injection of Aβ-amyloid-containing brain extracts can induce cerebral Aβ amyloidogenesis. The phenotype of the amyloidosis depended on both the host and the source of the agent, suggesting the existence of polymorphic Aβ strains, indicative of prion strains. This paper and others make the case that reducing anatomical spreading would likely be an efficacious therapeutic strategy. [DOI] [PubMed] [Google Scholar]
- 230.Clavaguera F, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11:909–13. doi: 10.1038/ncb1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Mougenot AL, et al. Prion-like acceleration of a synucleinopathy in a transgenic mouse model. Neurobiol Aging. 2012;33:2225–8. doi: 10.1016/j.neurobiolaging.2011.06.022. [DOI] [PubMed] [Google Scholar]
- 232.Polymenidou M, Cleveland DW. Prion-like spread of protein aggregates in neurodegeneration. J Exp Med. 2012;209:889–93. doi: 10.1084/jem.20120741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Prusiner SB. Cell biology. A unifying role for prions in neurodegenerative diseases. Science. 2012;336:1511–3. doi: 10.1126/science.1222951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Ahmed Z, et al. A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: the pattern of spread is determined by connectivity, not proximity. Acta Neuropathol. 2014;127:667–83. doi: 10.1007/s00401-014-1254-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Eisele YS, et al. Induction of cerebral beta-amyloidosis: intracerebral versus systemic Abeta inoculation. Proc Natl Acad Sci U S A. 2009;106:12926–31. doi: 10.1073/pnas.0903200106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Hamaguchi T, et al. The presence of Abeta seeds, and not age per se, is critical to the initiation of Abeta deposition in the brain. Acta Neuropathol. 2012;123:31–7. doi: 10.1007/s00401-011-0912-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Iba M, et al. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer's-like tauopathy. J Neurosci. 2013;33:1024–37. doi: 10.1523/JNEUROSCI.2642-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Luk KC, et al. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med. 2012;209:975–86. doi: 10.1084/jem.20112457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Zhou J, Gennatas ED, Kramer JH, Miller BL, Seeley WW. Predicting regional neurodegeneration from the healthy brain functional connectome. Neuron. 2012;73:1216–27. doi: 10.1016/j.neuron.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Ren PH, et al. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat Cell Biol. 2009;11:219–25. doi: 10.1038/ncb1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Eleuteri S, et al. Novel therapeutic strategy for neurodegeneration by blocking Abeta seeding mediated aggregation in models of Alzheimer's disease. Neurobiol Dis. 2015;74:144–57. doi: 10.1016/j.nbd.2014.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Yang W, Dunlap JR, Andrews RB, Wetzel R. Aggregated polyglutamine peptides delivered to nuclei are toxic to mammalian cells. Hum Mol Genet. 2002;11:2905–17. doi: 10.1093/hmg/11.23.2905. [DOI] [PubMed] [Google Scholar]
- 243.Eisele YS, et al. Multiple factors contribute to the peripheral induction of cerebral beta-amyloidosis. J Neurosci. 2014;34:10264–10273. doi: 10.1523/JNEUROSCI.1608-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Clavaguera F, et al. Peripheral administration of tau aggregates triggers intracerebral tauopathy in transgenic mice. Acta Neuropathol. 2014;127:299–301. doi: 10.1007/s00401-013-1231-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Duran-Aniotz C, et al. Aggregate-depleted brain fails to induce Abeta deposition in a mouse model of Alzheimer's disease. PLoS One. 2014;9:e89014. doi: 10.1371/journal.pone.0089014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Stohr J, et al. Purified and synthetic Alzheimer's amyloid beta (Abeta) prions. Proc Natl Acad Sci U S A. 2012;109:11025–30. doi: 10.1073/pnas.1206555109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Zhang Z, et al. De novo generation of infectious prions with bacterially expressed recombinant prion protein. FASEB J. 2013;27:4768–75. doi: 10.1096/fj.13-233965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Wang F, Wang X, Yuan CG, Ma J. Generating a prion with bacterially expressed recombinant prion protein. Science. 2010;327:1132–5. doi: 10.1126/science.1183748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Guo JL, Lee VM. Neurofibrillary tangle-like tau pathology induced by synthetic tau fibrils in primary neurons over-expressing mutant tau. FEBS Lett. 2013;587:717–23. doi: 10.1016/j.febslet.2013.01.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science. 2007;318:930–6. doi: 10.1126/science.1138718. [DOI] [PubMed] [Google Scholar]
- 251.Aguzzi A, Heikenwalder M, Polymenidou M. Insights into prion strains and neurotoxicity. Nat Rev Mol Cell Biol. 2007;8:552–61. doi: 10.1038/nrm2204. [DOI] [PubMed] [Google Scholar]
- 252.Bessen RA, Marsh RF. Biochemical and physical properties of the prion protein from 2 strains of the transmissible mink encephalopathy agent. J Virol. 1992;66:2096–2101. doi: 10.1128/jvi.66.4.2096-2101.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Casacciabonnefil P, Kascsak RJ, Fersko R, Callahan S, Carp RI. Brain regional distribution of prion protein PrP27-30 in mice stereotaxically microinjected with different strains of scrapie. J Infect Dis. 1993;167:7–12. doi: 10.1093/infdis/167.1.7. [DOI] [PubMed] [Google Scholar]
- 254.Dearmond SJ, Yang SL, Prusiner SB. The sites of PrP(Sc) deposition in the brain are prion strain-specific. J Neuropathol Exp Neurol. 1993;52:293–293. [Google Scholar]
- 255.Heilbronner G, et al. Seeded strain-like transmission of beta-amyloid morphotypes in APP transgenic mice. EMBO Rep. 2013;14:1017–22. doi: 10.1038/embor.2013.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Stohr J, et al. Distinct synthetic Abeta prion strains producing different amyloid deposits in bigenic mice. Proc Natl Acad Sci U S A. 2014;111:10329–10334. doi: 10.1073/pnas.1408968111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Watts JC, et al. Serial propagation of distinct strains of Abeta prions from Alzheimer's disease patients. Proc Natl Acad Sci U S A. 2014;111:10323–10328. doi: 10.1073/pnas.1408900111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Sanders DW, et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron. 2014;82:1271–88. doi: 10.1016/j.neuron.2014.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Clavaguera F, et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci U S A. 2013;110:9535–40. doi: 10.1073/pnas.1301175110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Guo JL, et al. Distinct alpha-synuclein strains differentially promote tau inclusions in neurons. Cell. 2013;154:103–17. doi: 10.1016/j.cell.2013.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Bousset L, et al. Structural and functional characterization of two alpha-synuclein strains. Nat Commun. 2013;4:2575. doi: 10.1038/ncomms3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Langer F, et al. Soluble Abeta seeds are potent inducers of cerebral beta-amyloid deposition. J Neurosci. 2011;31:14488–95. doi: 10.1523/JNEUROSCI.3088-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Jucker M, Walker LC. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature. 2013;501:45–51. doi: 10.1038/nature12481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Tsaytler P, Harding HP, Ron D, Bertolotti A. Selective Inhibition of a Regulatory Subunit of Protein Phosphatase 1 Restores Proteostasis. Science. 2011;332:91–94. doi: 10.1126/science.1201396. [DOI] [PubMed] [Google Scholar]
- 265.Cohen AS. Amyloidosis. N Engl J Med. 1967;277:522–30. doi: 10.1056/NEJM196709072771006. contd. [DOI] [PubMed] [Google Scholar]
- 266.Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol Sci. 1991;12:383–8. doi: 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- 267.Selkoe DJ. The molecular pathology of Alzheimer's disease. Neuron. 1991;6:487–98. doi: 10.1016/0896-6273(91)90052-2. [DOI] [PubMed] [Google Scholar]
- 268.Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science. 1992;256:184–5. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 269.Holtzman DM, Morris JC, Goate AM. Alzheimer's disease: the challenge of the second century. Sci Transl Med. 2011;3:77sr1. doi: 10.1126/scitranslmed.3002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Giannakopoulos P, et al. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer's disease. Neurology. 2003;60:1495–500. doi: 10.1212/01.wnl.0000063311.58879.01. [DOI] [PubMed] [Google Scholar]
- 271.Rowe CC, et al. Imaging beta-amyloid burden in aging and dementia. Neurology. 2007;68:1718–25. doi: 10.1212/01.wnl.0000261919.22630.ea. [DOI] [PubMed] [Google Scholar]
- 272.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 273.Walsh DM, Selkoe DJ. A beta oligomers - a decade of discovery. J Neurochem. 2007;101:1172–84. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- 274.Jack CR, Jr, et al. Lancet Neurol. 2010;9:119–28. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Blake CC, Geisow MJ, Oatley SJ, Rerat B, Rerat C. Structure of prealbumin: secondary, tertiary and quaternary interactions determined by Fourier refinement at 1.8 A. J Mol Biol. 1978;121:339–56. doi: 10.1016/0022-2836(78)90368-6. [DOI] [PubMed] [Google Scholar]
- 276.Hornberg A, Eneqvist T, Olofsson A, Lundgren E, Sauer-Eriksson AE. A comparative analysis of 23 structures of the amyloidogenic protein transthyretin. J Mol Biol. 2000;302:649–69. doi: 10.1006/jmbi.2000.4078. [DOI] [PubMed] [Google Scholar]
- 277.Hamilton JA, Benson MD. Transthyretin: a review from a structural perspective. Cell Mol Life Sci. 2001;58:1491–521. doi: 10.1007/PL00000791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Schneider F, Hammarstrom P, Kelly JW. Transthyretin slowly exchanges subunits under physiological conditions: A convenient chromatographic method to study subunit exchange in oligomeric proteins. Protein Sci. 2001;10:1606–13. doi: 10.1110/ps.8901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Monaco HL, Rizzi M, Coda A. Structure of a complex of two plasma proteins: transthyretin and retinol-binding protein. Science. 1995;268:1039–41. doi: 10.1126/science.7754382. [DOI] [PubMed] [Google Scholar]
- 280.Purkey HE, Dorrell MI, Kelly JW. Evaluating the binding selectivity of transthyretin amyloid fibril inhibitors in blood plasma. Proc Natl Acad Sci U S A. 2001;98:5566–71. doi: 10.1073/pnas.091431798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Saraiva MJM. Transthyretin Mutations in Health and Disease. Human Mutation. 1995;5:191–196. doi: 10.1002/humu.1380050302. [DOI] [PubMed] [Google Scholar]
- 282.Sekijima Y, et al. The biological and chemical basis for tissue-selective amyloid disease. Cell. 2005;121:73–85. doi: 10.1016/j.cell.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 283.Hammarstrom P, Jiang X, Hurshman AR, Powers ET, Kelly JW. Sequence-dependent denaturation energetics: A major determinant in amyloid disease diversity. Proc Natl Acad Sci U S A. 2002;99(Suppl 4):16427–32. doi: 10.1073/pnas.202495199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Lai Z, Colon W, Kelly JW. The acid-mediated denaturation pathway of transthyretin yields a conformational intermediate that can self-assemble into amyloid. Biochemistry. 1996;35:6470–82. doi: 10.1021/bi952501g. [DOI] [PubMed] [Google Scholar]
- 285.Jiang X, et al. An engineered transthyretin monomer that is nonamyloidogenic, unless it is partially denatured. Biochemistry. 2001;40:11442–52. doi: 10.1021/bi011194d. [DOI] [PubMed] [Google Scholar]
- 286.Hurshman Babbes AR, Powers ET, Kelly JW. Quantification of the thermodynamically linked quaternary and tertiary structural stabilities of transthyretin and its disease-associated variants: the relationship between stability and amyloidosis. Biochemistry. 2008;47:6969–84. doi: 10.1021/bi800636q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Benson MD. Familial Amyloidotic polyneuropathy. TIBS. 1989;12:88–92. doi: 10.1016/0166-2236(89)90162-8. [DOI] [PubMed] [Google Scholar]
- 288.Rapezzi C, et al. Transthyretin-related amyloidoses and the heart: a clinical overview. Nat Rev Cardiol. 2010;7:398–408. doi: 10.1038/nrcardio.2010.67. [DOI] [PubMed] [Google Scholar]
- 289.Dharmarajan K, Maurer MS. Transthyretin Cardiac Amyloidoses in Older North Americans. J Am Geriatrics Soc. 2012;60:765–774. doi: 10.1111/j.1532-5415.2011.03868.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Miller AL, Falk RH, Levy BD, Loscalzo J. A heavy heart. N Engl J Med. 2010;363:1464–1470. doi: 10.1056/NEJMcps0804916. [DOI] [PubMed] [Google Scholar]
- 291.Ng B, Connors LH, Davidoff R, Skinner M, Falk RH. Senile systemic amyloidosis presenting with heart failure: a comparison with light chain-associated amyloidosis. Arch Intern Med. 2005;165:1425–9. doi: 10.1001/archinte.165.12.1425. [DOI] [PubMed] [Google Scholar]
- 292.Vidal R, et al. Meningocerebrovascular amyloidosis associated with a novel transthyretin mis-sense mutation at codon 18 (TTRD18G) Am J Pathol. 1996;148:361–366. [PMC free article] [PubMed] [Google Scholar]
- 293.Westermark P, Sletten K, Johansson B, Cornwell GG., 3rd Fibril in senile systemic amyloidosis is derived from normal transthyretin. Proc Natl Acad Sci U S A. 1990;87:2843–5. doi: 10.1073/pnas.87.7.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Falk RH. Cardiac amyloidosis: a treatable disease, often overlooked. Circulation. 2011;124:1079–85. doi: 10.1161/CIRCULATIONAHA.110.010447. [DOI] [PubMed] [Google Scholar]
- 295.Hornstrup LS, Frikke-Schmidt R, Nordestgaard BG, Tybjaerg-Hansen A. Genetic Stabilization of Transthyretin, Cerebrovascular Disease, and Life Expectancy. Arterioscler Thromb Vas Biol. 2013;33:1441–1447. doi: 10.1161/ATVBAHA.113.301273. [DOI] [PubMed] [Google Scholar]
- 296.Kuroda T, et al. Treatment with Biologic Agents Improves the Prognosis of Patients with Rheumatoid Arthritis and Amyloidosis. J Rheumatol. 2012;39:1348–1354. doi: 10.3899/jrheum.111453. [DOI] [PubMed] [Google Scholar]
- 297.Nakamura T, Higashi S, Tomoda K, Tsukano M, Baba S. Efficacy of etanercept in patients with AA amyloidosis secondary to rheumatoid arthritis. Clin Exp Rheumatol. 2007;25:518–522. [PubMed] [Google Scholar]
- 298.Hall A, Patel TR. In: Progress in Medicinal Chemistry. Lawton G, Witty DR, editors. Elsevier; 2014. pp. 101–145. [Google Scholar]
- 299.Kastritis E, et al. Treatment of light chain (AL) amyloidosis with the combination of bortezomib and dexamethasone. Haematologica. 2007;92:1351–1358. doi: 10.3324/haematol.11325. [DOI] [PubMed] [Google Scholar]
- 300.Sitia R, Palladini G, Merlini G. Bortezomib in the treatment of AL amyloidosis: targeted therapy? Haematologica. 2007;92:1302–1307. doi: 10.3324/haematol.12136. [DOI] [PubMed] [Google Scholar]
- 301.Holmgren G, et al. Biochemical effect of liver transplantation in two Swedish patients with familial amyloidotic polyneuropathy (FAP-met30) Clin Genet. 1991;40:242–6. doi: 10.1111/j.1399-0004.1991.tb03085.x. [DOI] [PubMed] [Google Scholar]
- 302.Benson MD, et al. Suppression of choroid plexus transthyretin levels by antisense oligonucleotide treatment. Amyloid. 2010;17:43–49. doi: 10.3109/13506129.2010.483121. [DOI] [PubMed] [Google Scholar]
- 303.DeTure M, Hicks C, Petrucelli L. Targeting Heat Shock Proteins in Tauopathies. Current Alzheimer Research. 2010;7:677–684. doi: 10.2174/156720510793611565. [DOI] [PubMed] [Google Scholar]
- 304.Herbst M, Wanker EE. Small molecule inducers of heat-shock response reduce polyQ-Mediated huntingtin aggregation - A possible therapeutic strategy. Neurodegenerative Diseases. 2007;4:254–260. doi: 10.1159/000101849. [DOI] [PubMed] [Google Scholar]
- 305.Wang Q, Guo J, Jiao P, Liu H, Yao X. Exploring the influence of EGCG on the beta-sheet-rich oligomers of human islet amyloid polypeptide (hIAPP1-37) and identifying its possible binding sites from molecular dynamics simulation. PLoS One. 2014;9:e94796. doi: 10.1371/journal.pone.0094796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Schenk D. Opinion: Amyloid-beta immunotherapy for Alzheimer's disease: the end of the beginning. Nat Rev Neurosci. 2002;3:824–828. doi: 10.1038/nrn938. [DOI] [PubMed] [Google Scholar]
- 307.Schenk D. Hopes remain for an Alzheimer's vaccine. Nature. 2004;431:398. doi: 10.1038/431398b. [DOI] [PubMed] [Google Scholar]