

Abstract
Background
Xenorhabdus bacteria engage in a beneficial symbiosis with Steinernema nematodes, in part by providing activities that help kill and degrade insect hosts for nutrition. Xenorhabdus strains (members of a single species) can display wide variation in host-interaction phenotypes and genetic potential indicating that strains may differ in their encoded symbiosis factors, including secreted metabolites.
Methods
To discern strain-level variation among symbiosis factors, and facilitate the identification of novel compounds, we performed a comparative analysis of the genomes of 10 Xenorhabdus bovienii bacterial strains.
Results
The analyzed X. bovienii draft genomes are broadly similar in structure (e.g. size, GC content, number of coding sequences). Genome content analysis revealed that general classes of putative host-microbe interaction functions, such as secretion systems and toxin classes, were identified in all bacterial strains. In contrast, we observed diversity of individual genes within families (e.g. non-ribosomal peptide synthetase clusters and insecticidal toxin components), indicating the specific molecules secreted by each strain can vary. Additionally, phenotypic analysis indicates that regulation of activities (e.g. enzymes and motility) differs among strains.
Conclusions
The analyses presented here demonstrate that while general mechanisms by which X. bovienii bacterial strains interact with their invertebrate hosts are similar, the specific molecules mediating these interactions differ. Our data support that adaptation of individual bacterial strains to distinct hosts or niches has occurred. For example, diverse metabolic profiles among bacterial symbionts may have been selected by dissimilarities in nutritional requirements of their different nematode hosts. Similarly, factors involved in parasitism (e.g. immune suppression and microbial competition factors), likely differ based on evolution in response to naturally encountered organisms, such as insect hosts, competitors, predators or pathogens. This study provides insight into effectors of a symbiotic lifestyle, and also highlights that when mining Xenorhabdus species for novel natural products, including antibiotics and insecticidal toxins, analysis of multiple bacterial strains likely will increase the potential for the discovery of novel molecules
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-2000-8) contains supplementary material, which is available to authorized users.
Keywords: Symbiosis, Competition, Xenorhabdus, Steinernema, Bacteria, Nematodes, Insects, Comparative genomics
Background
Xenorhabdus bacteria are beneficial symbionts of entomopathogenic (insect-parasitic) Steinernema nematodes. In addition to being effective biocontrol agents for a variety of insect pests [1, 2], Xenorhabdus – Steinernema complexes are tractable laboratory systems that facilitate investigation of ecological [3], evolutionary [4, 5] and symbiotic [6, 7] processes. The integrated life cycle of Xenorhabdus bacteria and Steinernema nematodes comprises alternating environments of the soil and insect hosts infected by the pair (Fig. 1) [6]. The infective juvenile (IJ) is the soil dwelling, environmental stage of the nematode that carries bacteria and infects insect hosts. Once within the insect, the nematodes and bacteria kill the insect and reproduce using the nutrients derived from the cadaver. During reproduction, the nematodes and bacteria are vulnerable to predation by scavenger insects [8, 9] and competition from other opportunistic organisms, such as nematodes, bacteria, or fungi [10–13]. After nutrients within the insect cadaver are consumed, and nematode density is high, the nematodes develop into progeny IJs that exit the cadaver to repeat the cycle [14]. In the association, the bacterial symbiont contributes to virulence against the insect host [5, 15, 16], support of nematode reproduction [4, 5, 17], and defense against encountered competitors, pathogens, and predators [8–10, 18]. In turn, the nematode partner serves as a vector to transmit bacteria between insect hosts [19, 20] and augments bacterial virulence against insects [5, 21, 22].
Fig. 1.
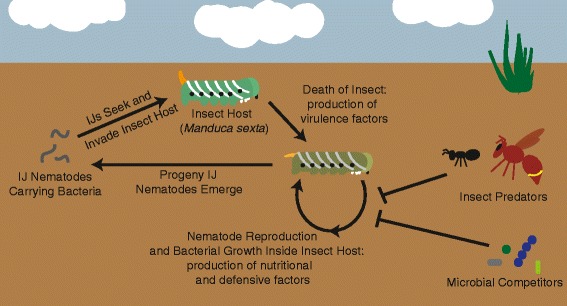
Xenorhabdus bacteria and Steinernema nematode life cycle. In the soil, Steinernema infective juvenile (IJ) nematodes containing their Xenorhabdus symbionts seek out and invade insect hosts. Once in the insect blood cavity, the nematodes and bacteria produce virulence factors and kill the insect host. The nematodes and bacteria then grow and reproduce using insect cadaver biomass, a process facilitated by the bacterial symbiont. During reproduction, the growing nematodes and bacteria are vulnerable to insect predators and microbial competitors, and therefore, defensive compounds are produced during reproduction. Once all nutrients within the cadaver are consumed, the nematodes form the next generation of IJs (progeny) that then exit the insect cadaver to seek new insect hosts
To accomplish symbiotic functions, Xenorhabdus bacteria encode a wide array of bioactive molecules that can serve as virulence factors [23–27], degradative enzymes for nutritional support [17], anti-predatory compounds [8, 9], and anti-microbial compounds [10, 11, 18]. Therefore, Xenorhabdus species have been utilized for the identification of novel metabolites with pharmaceutical properties [18, 28, 29], and it has been proposed that other Xenorhabdus compounds may be useful for development as insecticides, nematicides, and antimicrobials [18]. Our recent work has revealed intra-species variation (i.e. sub-species by standard metrics [30]) in the ability of Xenorhabdus bovienii bacterial strains to engage in symbiosis with S. feltiae nematode hosts [5], indicating that examination of multiple strains within a species has the potential to expose additional diversity of bioactive metabolites. Indeed, pan-genomic analysis of X. bovienii strains revealed the core genome to be approximately 55 % of total coding content, with 1–9 % of the coding content being unique to an individual strain [5]. The remaining 36–44 % is shared among some, but not all, bacterial strains studied. However, it has not been determined if the bioactive molecules that may contribute to symbiotic functions of X. bovienii are conserved or shared. To address this, we performed a comparative genomic analysis on ten previously published bacterial genomes [5, 31, 32] that were isolated from 6 different Steinernema spp. nematode hosts (Table 1), focusing on genes and predicted products that are likely to be involved in symbiotic interactions.
Table 1.
Xenorhabdus bovienii genomes used in this studya
| Genomea | Steinernema nematode hostb | Sourcec | Genome Accessiond Numberd | Studye | Genome Size (Mb)f | G + C content (%)g | Number of CDSh | Coding Density (%)i | Number of Contigsj | N50 Value (Kb)k |
|---|---|---|---|---|---|---|---|---|---|---|
| Xb-Sf-FL | S. feltiae | Florida, USA | [EMBL:PRJEB432] | 5 | 4.47 | 44.89 | 4508 | 83.43 | 436 | 45 |
| Xb-Sf-FR | S. feltiae | France | [EMBL:PRJEB4319] | 5 | 4.41 | 44.75 | 4441 | 84.06 | 449 | 45 |
| Xb-Sf-MD | S. feltiae | Moldova | [EMBL:PRJEB4321] | 5 | 4.66 | 44.36 | 4695 | 83.82 | 397 | 52 |
| Xb-Si | S. intermedium | North Carolina, USA | [EMBL:PRJEB4327] | 5 | 4.71 | 44.89 | 4719 | 83.39 | 467 | 51 |
| Xb-Sj | S. jollieti | Monsanto | [EMBL:PRJEB4326] | 5 | 3.93 | 44.48 | 3939 | 83.20 | 457 | 46 |
| Xb-Sj-2000 | S. jollieti | Monsanto | [EMBL:FN667741] | 31 | 4.23 | 44.97 | 4406 | 84.07 | 1 | NA |
| Xb-Sk-BU | S. kraussei | Becker Underwood | [EMBL:PRJEB4325] | 5 | 4.72 | 44.75 | 4860 | 83.10 | 752 | 33 |
| Xb-Sk-CA | S. kraussei | Quebec, CA | [EMBL:PRJEB4324] | 5 | 4.21 | 44.18 | 4097 | 83.96 | 422 | 34 |
| Xb-So | S. oregonense | Oregon, USA | [EMBL:PRJEB4323] | 5 | 4.13 | 44.90 | 4261 | 84.30 | 429 | 51 |
| Xb-Sp | S. puntauvense | Costa Rica | [EMBL:PRJEB4322] | 5 | 4.48 | 44.32 | 4584 | 84.34 | 443 | 51 |
aAbbreviation used for bacterial genomes
bThe Steinernema nematode host species from which the X. bovienii bacterial strain was isolated
cThe location or company from which the nematode was obtained
dThe accession number for the bacterial genome in EMBL
eStudy in which the genome sequence was originally reported. Number corresponds to the citation
fSize of the genome in megabases
gGC content percentage of the genome
hTotal number of coding sequences in the genome
iAmount of the genome that is coding sequences
jNumber of contigs in the genome sequence
kMedian size of the contigs in megabases. NA indicates not applicable for the finished genome
Results and discussion
General genomic features of Xenorhabdus bovienii
We recently reported a brief description of draft genomes of 9 X. bovienii strains, but did not provide an in depth comparison of the general genomic features relative to the finished X. bovienii genome [5, 31, 32]. Here we present a more thorough analysis, which indicated that all the examined X. bovienii genomes are similar to each other in genome in size, GC content, number of coding sequences and coding density (Table 1). On average the genomes are 4.4 Mbp, with 4451 coding sequences. No plasmids were detected in any of these strains through sequencing, DNA extraction, or plasmid extraction (data not shown).
To assess completeness of the draft genomes, we determined the number and size of contigs. All of the draft genomes have ~400 contigs, with an N50 value between 30 and 55 kilobasepairs (Table 1). Additionally, the draft genome sequence of the S. jollieti symbiont (Xb-Sj) shows genome-wide synteny to the finished genome sequence (Xb-Sj-2000), but the draft genome lacks portions found within the complete genome, approximately 0.3Mbp total (Fig. 2). Two regions of the draft Xb-Sj genome that were not associated with the ends of contigs lacked synteny with the complete Xb-Sj-2000 genome (6- and 14-Kb in size), indicating one or more rearrangements in the draft genome. The remaining regions that did not match the draft genome were associated with the ends of contigs, indicating that these breaks may be due to misassembly rather than rearrangements. Xb-Sj and Xb-Sj-2000 are bacterial strains that came from the same nematode host strain but were isolated 7 years apart [15], so the two genomes should be almost identical because these symbionts are transmitted with high fidelity and the nematodes were propagated in the laboratory without exposure to other Xenorhabdus species and strains [33]. It is more likely that differences in the size of Xb-Sj and Xb-Sj-2000 genomes is due to genome reduction in the symbiont that had been associated with the host through laboratory propagation for a longer period of time. Additionally, Xb-Sj contains a predicted open reading frame (XBJ2v2_1630007) that is absent from Xb-Sj-2000 based on BLASTn analysis, which indicates that it has gained at least some genetic material in the 7 years between these strains’ isolation. This gene has a GC content lower (17 %) than the remainder of the genome (44 %), suggesting potential horizontal transmission. However, the predicted product is a protein of unknown function and very small in size (69 amino acids) with no significant BLAST analysis hits to proteins or genes of known function.
Fig. 2.
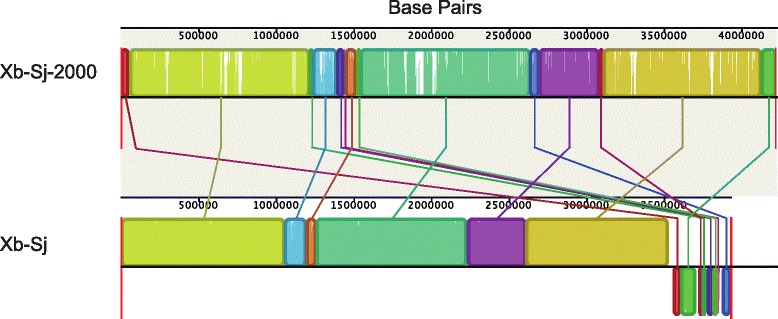
Genome synteny between finished and draft X. bovienii genomes. Genomes were aligned with progressive MAUVE to assess the large-scale synteny among the draft X. bovienii genomes and the finished X. bovienii genome (Xb-Sj-2000). The synteny of the whole length of the genomes is shown. Genome names are indicated on the left, and the scale is shown in base pairs. Colored boxes indicate regions of synteny, and colored lines connect like regions
Metabolic analysis of X. bovienii strains
Previous studies have shown that specific metabolic pathways (e.g. amino acid and vitamin biosynthesis) are integral to some interactions between Xenorhabdus nematophila bacterial symbionts and their Steinernema carpocapsae host nematodes [34]. To determine if X. bovienii bacterial strains differ in metabolic potential, we performed a global metabolic analysis on all 10 bacterial genomes. These analyses found very few differences among the strains (Additional file 1). There were many incomplete pathways, suggesting that annotations may not be entirely accurate. However, all genomes including the complete genome had approximately the same degree of pathway annotation, including biosynthetic pathways, degradative pathways, and energy metabolism. This supports that the absence of some genes within pathways is not due to incomplete draft genome assembly. Of note, the Xb-Sf-MD genome has an incomplete chitin degradation pathway, lacking intact genes to encode chitinase, chitotriose synthase, and diacetylchitobiose synthase, while all other strains have the complete set of genes within this pathway. However, the Xb-Sf-MD genome does contain several pseudogenes (due to early stop codons) with homology to portions of the missing enzymes. Although these bioinformatic data suggest that Xb-Sf-MD may have a deficiency in chitin utilization relative to other X. bovienii strains we were unable to verify this experimentally, since under laboratory conditions none of the strains grew on chitin as a sole carbon source (data not shown).
Secretion systems present in X. bovienii strains
For our comparative genomic analyses, we focused on factors likely to contribute to the interaction between X. bovienii strains and hosts. Bacterial products that directly interact with host cells must be exported out of the bacterial cell through secretion systems, and disruption of secretion systems in bacterial symbionts can cause defects in pathogenesis [35, 36] and mutualism [37, 38]. To determine which secretion system types are present among X. bovienii genomes we searched for gene clusters known to encode systems responsible for secretion of host-interacting factors (Additional file 2). Consistent with published observations [39], none of the X. bovienii strains encoded an obvious Type III secretion system, one of the systems most commonly associated with pathogenesis [40]. Furthermore, none of the X. bovienii strains encoded a complete Type IV secretion system [41], although various Type IV-system-related genes were present in all of the genomes, as previously noted for the complete genome of X. bovienii [39] (data not shown). All X. bovienii strains analyzed encode a flagellar export apparatus, which is evolutionarily related to Type III secretion systems. In other bacteria, including X. nematophila the flagellar system exports non-flagellar factors such as xenocin (an antibiotic) and virulence determinants [42–44]. All of the X. bovienii strains examined here also encode a type II secretion system, which in other Gram-negative bacteria transport folded proteins (e.g. toxins and degradative enzymes) from the periplasm to the extracellular environment [45]. All X. bovienii strains analyzed in this study also encode a type VI secretion system. These systems transfer effectors (e.g. hemolysins) directly from the bacterial cell into the target host cell (which can be bacterial or eukaryotic) through injection [46, 47]. Also, each genome contains additional type VI secretion system structural genes, such as multiple vgrG genes (data not shown). Finally, all of the examined X. bovienii strains encode alkaline protease secretion systems, which export the protease PrtA [48]. Overall, the types and numbers of intact secretion systems of X. bovienii appear to be conserved, with each genome possessing syntenous regions containing all required secretion system components.
The presence of the same secretion systems (and the absence of others) in all X. bovienii strains examined likely reflects their similar symbiotic lifestyles interacting with nematode and insect hosts. However, these bacterial strains associate with divergent nematode host species and likely encounter different insect host species within the environment. Therefore, the secreted effector proteins delivered by the secretion systems are expected to vary. To address this idea, we further investigated bacterial factors, or putative secreted effector molecules, that may be involved in symbiotic interactions.
Phenotypic testing of X. bovienii strains
Some Xenorhabdus factors that are likely secreted and involved in symbiosis have readily monitored activities. For example, lipase activity in X. nematophila contributes to nutritional support of its nematode host, Steinernema carpocapsae, and is secreted through the flagellar apparatus [44, 49, 50]. To assess the diversity of phenotypes associated with symbiosis among the sequenced X. bovienii strains, we performed phenotypic testing. We measured activities implicated in bacterial or nematode nutrition, including iron acquisition through siderophore activity [51, 52] and the enzymatic activities of lipase [17, 44], lecithinase [17], and protease [17, 48] that help degrade insect tissues. In addition, we monitored hemolytic activity [25] and motility [49], which are both associated with virulence towards the insect host or subsequent degradation [53–55]. Finally, we assessed antibiotic activity, which likely plays a defensive role in removing competitor or nematode-pathogenic microbes from the insect cadaver [11].
In Xenorhabdus species, including X. bovienii, assessment of the activities listed above is complicated by phenotypic variation. All Xenorhabdus spp. studied to date undergo phenotypic variation between primary and secondary form cells that have differing levels of some activities, such as lipase and production of antibiotics [15, 29]. In the laboratory, primary form cells give rise to secondary form cells after long-term cultivation in liquid media. However, it remains unclear if or when phenotypic variation occurs within the natural life cycle. To accurately determine the production potential of activities in X. bovienii strains, we generated secondary form bacterial isolates from primary form isolates and used both for phenotypic testing (Table 2).
Table 2.
Phenotypes of X. bovienii bacterial strainsa
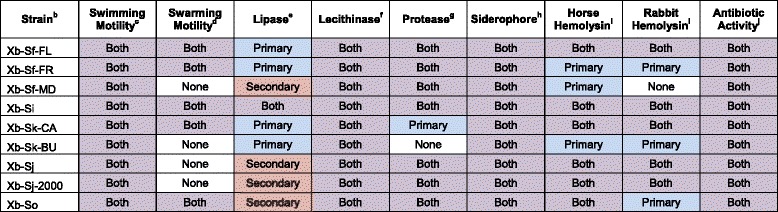
aPhenotypic testing of primary and secondary form X. bovienii bacterial strains. Each result is given as the form that produces that activity: Primary – activity in primary form only (blue), Secondary – activity in secondary form only (pink), Both – activity in primary and secondary forms (purple), or None – no activity in either form (no fill)
bThe bacterial strain tested
cSwimming motility detected as motility through soft agar (0.5 % LB agar)
dSwarming motility detected as motility on top of semi-solid agar (0.7 % LB agar)
eLipase activity as detected on Tween 20 agar
fLecithinase activity as detected on egg yolk agar
gProtease activity as detected on milk agar
hSiderophore activity as detected activity on CAS plates
iHemolysin activity as detected on horse blood agar or rabbit blood agar;
jAntibiotic activity as detected by antibiotic overlay assays
Almost all tested X. bovienii strains have swimming motility, lipase, lecithinase, protease, siderophore, hemolysin, and antibiotic activities in at least one form, except that Xb-Sk-Bu lacks protease activity, and Xb-Sp lacks hemolysin activity (Table 2). Several strains (i.e. Xb-Sf-MD, Xb-Sk-BU, Xb-Sj, Xb-Sj-2000, and Xb-Sp) lack swarming motility (Table 2). Some activities, such as lecithinase and siderophore activity are not affected by phenotypic variation in any of the X. bovienii strains. Other activities, such as lipase and hemolysin production, differ between the two forms, and which form produces the activity was inconsistent among the strains. These differences could be due to variations in coding potential among the strains or differences in regulatory control of the genes encoding the activities tested.
To assess if observed activity differences are due to variation in the coding potential, we examined the genomes of the tested X. bovienii strains for relevant genes (Additional file 3), such as those encoding hemolysin (xhlA, xhlA2, and xaxAB), lipase (xlpA) [17], lecithinase (estA) [17], protease (prtA) [48], and motility factors (e.g. operons encoding flagellar structural genes and motility regulators) [56]. All of the strains, including those that lack detectable levels of the associated activity, encode intact homologs of these genes (Additional file 3). This indicates that diversity in activity phenotypes is not due to variation in coding potential for structural genes, and we therefore turned our attention to possible differences in regulatory pathways.
Among Xenorhabdus species, gene regulation pathways of X. nematophila are the best characterized. In this species, the leucine-responsive regulatory protein (Lrp) [57], two-component signal transduction systems CpxRA [6] and OmpR-EnvZ [50], LysR like homolog A (LrhA) [44], flagellar transcriptional regulators (FlhDC) [49, 53, 54], and nematode intestinal localization gene repressor (NilR) [17] regulatory proteins have been implicated in controlling the expression of the phenotypic activities listed in Table 2. Each X. bovienii strain genomes contained homologs predicted to encode all of these regulators except NilR (Additional file 4). This is consistent with the fact that in X. nematophila NilR functions synergistically to negatively regulate the nilA, B, and C genes, nematode-host range specificity determinants that are not present in the X. bovienii genomes [17, 31, 58].
LysR type transcriptional regulators, of which LrhA is a member, are widely distributed among bacteria, and can respond to specific signals to regulate narrow and broad regulons, which can include genes involved in virulence, metabolism, and other behaviors [59]. We assessed putative LysR type regulators encoded by X. bovienii and identified 19 lysR-type genes, which are present within all the sequenced X. bovienii strains and seven with homologs in one or more strains (Additional file 5). While our inability to identify certain homologs may be due to their absence in draft assemblies, we did identify a lysR-type gene within several draft genomes (e.g. XBFFL1v2_2160021) that is absent in the complete Xb-Sj genome. In all of the X. bovienii strains examined in which this gene is present, it co-occurs with genes encoding a putative aspartate racemase and a putative glutamate symporter. This analysis indicates that distinctive LysR-type transcription factor regulation of specific metabolic pathways may exist in some strains but not others.
An additional class of regulators common in bacteria is the two-component regulatory systems (TCSs), such as CpxRA and EnvZ/OmpR as mentioned above. These systems transduce specific signals, such as those indicative of a host environment, to various output responses, frequently a change in transcription. In canonical TCSs, the histidine kinase (HK) protein recognizes a stimulus, such as an antimicrobial peptide or quorum sensing molecule, and transmits this signal to the response regulator (RR), which directly or indirectly influences transcription [60]. These transcriptional changes can affect bacterial phenotypic variation [61] and production of virulence factors [62, 63] and degradative enzymes, such as lipase [64]. We assessed the distribution of TCSs among the X. bovienii strains, and identified 23 TCSs and 2 orphan RRs that are present in all the strains, and 1 orphan HK and 4 orphan RRs that are within some but not all the sequenced X. bovienii strains (Additional file 6). This suggests that differences in TCS-dependent regulation occur among X. bovienii strains and could impact their bioactivity.
Our analysis indicates broad conservation of coding potential for transcriptional regulators, and the limited observed variation in the presence or absence of encoded regulators is unlikely to explain the breadth of observed phenotypic differences. Therefore, phenotypic differences among strains may be due to variation in the expression or modulation of regulatory factors. The transcription factor Lrp is of particular interest in this regard as there is an established link between Lrp-dependent regulation and phenotypic variation [57]. However, a detailed understanding of the basis of the observed phenotype differences among the tested X. bovienii strains awaits further experimentation examining their individual regulatory hierarchies. The identification of distinct signals to which regulators respond, as well as variations in the constituents of their regulons likely will provide further insights into niche diversification among these bacteria.
Toxins encoded by X. bovienii strains
Bacteria encode a wide range of toxins that are exported by various secretions systems [65]. In symbioses, these toxins can be involved in defensive mutualism (i.e. protection against predators, pathogens, or competitors) or in pathogenesis. For example, lysogenic-phage-encoded toxins expressed by the bacterial secondary symbionts of aphids protect the aphid host from parasitism by parasitoid wasps [66], while lysogenic-phage-encoded Cholera toxin [67] and Shiga toxin [68] are produced during human infection by Vibrio cholerae and Escherichia coli respectively. In the case of X. bovienii bacterial strains, bacterially derived toxins could play a defensive role in protecting the insect cadaver from predators and competitors or could aid in killing the insect host.
Genome analyses indicated that all of the sequenced X. bovienii strains contain genes with homology to makes caterpillars floppy toxin 1 (mcf1), metalloprotease MARTX toxin (MARTX), zinc alkaline metalloprotease similar to RTX toxins (prtA), and hemolysins Xenorhabdus alpha-xenorhabdolysin (xaxAB) and one or two homologs of Xenorhabus hemolysin (xhlA and xhlA2) (Additional file 7). Homologs of mcf have been identified in diverse organisms, including other species of Xenorhabdus [39], Photorhabdus bacterial symbionts of Heterorhabditis nematodes [69, 70] and Epichloë fungal symbionts of grasses [71] In the Photorhabdus – Heterorhabditis symbiosis, Mcf contributes to virulence towards the insect host and causes the insect to lose body turgor [69, 72]. A common feature of the Mcf toxin homologs is a central TcdA/B translocating domain, but other regions of each Mcf protein contain distinct domains depending on the homolog. The finished X. bovienii genome encodes a single homolog [15, 31, 39] that also is present in each X. bovienii strain genome analyzed here. Like other mcf homologs, these X. bovienii homologs encode the conserved TcdA/B like domain as well as a C-terminal RTX toxin-like domain predicted to be involved in export (Fig. 3a) [69]. Additionally, the X. bovienii Mcf proteins analyzed here have a putative C58 peptidase domain similar to those in RTX toxins that activate toxins via cleavage (Fig. 3a) [73, 74]. Because of differences between the X. bovienii Mcf homologs and the experimentally verified Photorhabdus Mcf protein, we refer to the X. bovienii homologs as McfXb (Fig. 3a).
Fig. 3.
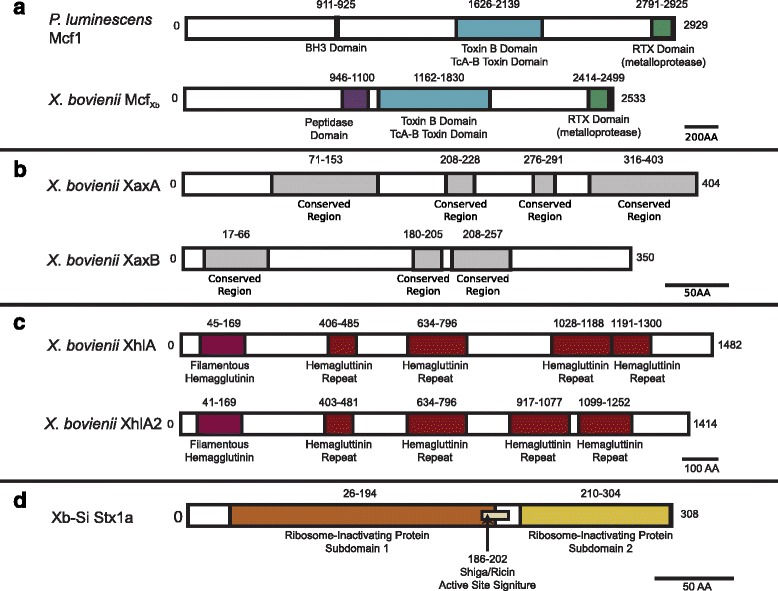
Annotated toxin genes in X. bovienii genomes. Schematics represent the domains identified in the proteins of toxin genes: McfXb(a), hemolysins XaxAB (b) and XhlA homologs (c), and Shiga toxin Stx1a (d). Name of the bacterium encoding each protein and the protein name are listed to the left of each schematic. Colored boxes represent different protein domains or conserved regions, and similar domains are colored the same. The predicted type of domain is listed below, and the amino acids spanning each domain are labeled above. Scale bars represent the amino acid number per distance for each panel
Two types of metalloprotease toxins were observed in all of the X. bovienii genomes we analyzed. One is homologous to and has the same domain structure as previously identified PrtA proteins from nematode-associated bacterial symbionts P. luminescens [48] and X. nematophila [75]. The PrtA proteins from P. luminescens and X. nematophila cleave insect hemolymph proteins and are most likely involved in immunosuppression [76]. The other metalloprotease is a MARTX toxin that has been previously analyzed from the finished X. bovienii genome [77, 78]. MARTX metalloprotease toxin proteins, are a family of large RTX toxins containing >40 repeats [79]. Many MARTX proteins are virulence factors, including the cytotoxic RtxA from Vibrio vulnificus [80], which is the closest homolog by BLASTp to X. bovienii MARTX proteins (named MARTXXb by convention) [48, 75, 76].
All X. bovienii strains analyzed here encode single copies each of Xenorhabdus alpha-xenorhabdolysin A and B (XaxA and XaxB), proteins homologous to the XaxAB binary toxin hemolysins of X. nematophila [24, 81] and P. luminescens [82]. These multiple variants of XaxA and XaxB proteins were used to assess regions of conservation and divergence. We found that each protein had regions (four and three for XaxA and XaxB respectively) with 100 % identity across all homologs (Fig. 3b), while the rest of the regions within the proteins had higher amino acid diversity (Additional file 8D, E). Similar regions of high and low amino acid diversity were present when the XaxAB proteins from X. nematophila and P. luminescens were included in the analysis (data not shown). These highly conserved regions may be important for XaxAB function, an idea that awaits experimental investigation.
In addition to the XaxAB hemolysin, all analyzed X. bovienii strains contain one or two homologs of the X. nematophila hemolysin XhlA [25] (Additional file 7). One homolog (XhlA) is present in all of the X. bovienii strains, while the other (XhlA2) is present only within a subset of strains (i.e. Xb-Sf-FL, Xb-Sf-FR, Xb-Sf-MD, Xb-Sp and Xb-Si). Both the XhlA and XhlA2 proteins have a similar size (1400 amino acids) (Additional file 8 F) and similar filamentous hemolysin and hemagglutinin repeat domain structures (Fig. 3c).
To assess if divergence occurs in the toxins discussed above, we assessed each toxin family for amino acid divergence (Additional file 8), recombination, and phylogenetic history (Fig. 4). No intragenic recombination was detected in the nucleotide sequences of any of the toxin families, as analyzed using the break-point analysis method implemented in TOPALi v2 [69]. Also, amino acid sequence divergence was spread evenly throughout each protein, suggesting that each protein region experiences the same level of selection (Additional file 8A-D).
Fig. 4.
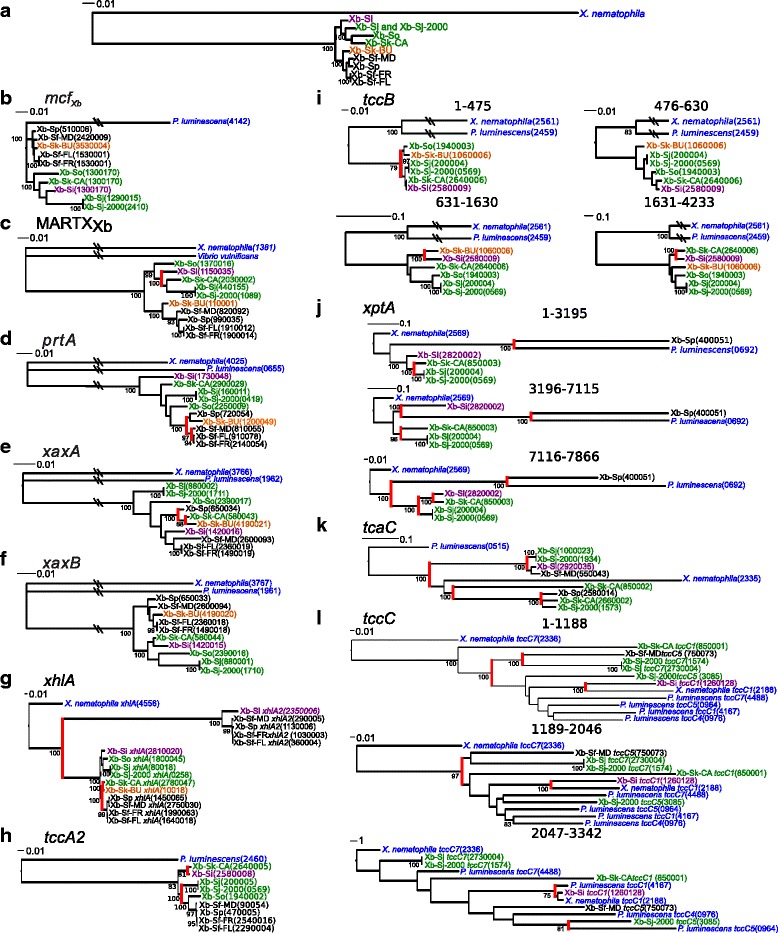
Phylogenies of partitioned nucleic acid sequence. The nucleic acid sequences of genes were analyzed for recombination, and for each piece, phylogenies were built. The gene phylogenies were compared to the whole genome phylogeny of the bacterial strains (a) [5]. Shown above are the trees for X. bovienii genes mcf Xb (b), MARTX Xb (c), prtA (d), xaxA (e), xaxB (f), xhlA (g), tccA2 (h), tccB2 (i), xptA2 (j), tcaC (k), and tccC (l). Values indicate bootstrap values. The leaves are color-coded by clade determination from the core X. bovienii phylogeny, and red highlighting indicates strongly supported branches in the gene tree that do not match the previously reported bacterial phylogeny [5]
Maximum likelihood nucleotide phylogenetic trees were constructed using full-length sequences from each toxin family, branching orders were compared to the previously published core X. bovienii genome phylogeny (Fig. 4a) [5]. The phylogenies for Mcf-family toxin A, xaxB, and xhlA2 matched the previously published core genome phylogeny for these strains at all strongly supported nodes (Fig. 4bf, g), indicating that these genes are orthologs unaffected by horizontal gene transfer (HGT) and likely have conserved functions. Further analyses of Mcf-family toxin A, xaxB, and xhlA2 using PAML v4 [83] revealed that all branches of each phylogeny had dN/dS < 0.30 consistent with negative selection and functional conservation within each class of toxin. Phylogenetic analysis of MARTXXb, prtA, xaxA, and xhlA showed that not all strongly supported splits were consistent with the core genome phylogeny, suggesting that HGT had occurred and members of these gene sets are xenologs (Fig. 4 c–e, g). This is consistent with previous findings that toxin genes, such as xaxAB are located in regions of genome plasticity (i.e. regions of recombination) in Xenorhabdus spp. and are likely transferring between bacteria frequently [39, 84]. Other events that might have caused divergence from the core phylogeny are duplication with subsequent loss of the gene and different rates of evolution, although these are less parsimonious explanations. All of these xenolog sets share the same genomic context, indicating xenologous replacement, and perhaps suggesting functional conservation. Phylogenetic analysis of xhlA and xhlA2 indicate that these genes likely arose from a gene duplication event and are paralogs of one another (Fig. 4g). This supports the XhlA and XhlA2 proteins potentially having divergent functions. Taken together, our divergence, recombination, and phylogenetic analyses have revealed that all of the toxin classes we analyzed likely have conserved function in the encoding X. bovienii strains, with the exception being in the hemolytic enzymes encoded by xhlA and xhlA2.
In addition to the shared toxins that are described above, the symbiont from S. intermedium, Xb-Si also encodes a putative Shiga toxin chain A (Additional file 7). This gene was previously identified through analyses of proteins unique to each X. bovienii strain [5]. The top matches for this protein from BLASTp indicate that it is most similar (e-value = 1.0E-37) to Shiga toxin 1 variant A from Escherichia coli (Additional file 9), and analysis of its domain structure revealed two ribosome-inactivating protein subdomains as well as a signature Shiga-type active site (Fig. 3d) [85]. Examination of the surrounding proteins and total genome content did not reveal an obvious Shiga toxin B chain homolog, which is responsible for targeting the toxin complex to the correct mammalian cell type by binding a cell-surface ganglioside [86]. As X. bovienii is not known to colonize a mammalian or other vertebrate host, we hypothesize that it instead encodes an invertebrate-specific targeting protein partner for the Shiga toxin variant A homolog. The genes surrounding the Xb-Si Shiga toxin are of unknown function or are phage associated (i.e. holin and transposase). This suggests that the toxin may have been transferred via a phage, similar to Shiga toxin producing E. coli or Vibrio cholerae [87]. Analysis of the predicted holin and transposase did not conclusively identify the type of phage from which they were derived.
Insecticidal toxin complexes are diverse among X. bovienii bacterial strains
In addition to the shared annotated toxins described in the previous section, Xenorhabdus and Photorhabdus bacteria encode insecticidal toxin complex (Tc) proteins [88, 89]. Tc toxins are large molecular weight toxins comprised of three protein subunits: A, B, and C. For each subunit, there are multiple genes that can encode a similar function (e.g. xptA2 and tccA2 both encode A subunit proteins) [88, 89]. Recent literature suggests that all three protein subunits function together for secretion from the cell, binding of the target membrane, and translocation into the cell to deliver the C-terminal end of the C subunit, which is the functional toxin [90–92]. The B subunit has also been proposed to function in linking the A and C subunits. The A subunit mediates host cell targeting and specificity of the toxin complex through membrane receptor binding of insect intestinal cells [89, 91, 93]. Additionally, some A subunit proteins (XptA1 and XptA2) have independent oral toxicity against insects [89, 93].
The finished genome of X. bovienii (Xb-Sj-2000) encodes three intact A subunits, two intact B subunits, and two intact C subunits [31]. To determine if genes encoding Tc toxins have diversified within different strains of X. bovienii, we identified homologs in the 9 draft genomes using a keyword search of the genome annotations and a BLASTp search of the Tc toxin proteins previously identified in the Xb-Sj-2000, X. nematophila, and P. luminescens genomes (Additional file 10). We identified 118 potential A, B, and C subunit genes within the 10 X. bovienii bacterial genomes with 4–20 genes in each genome, but further analysis revealed that many of the genes are not predicted to encode full length protein subunits (i.e. were truncated versions of the proteins identified in other X. bovienii strains) (Additional file 10). The non-full-length genes are located in the middle of contigs that are otherwise syntenous with Xb-Sj-2000, indicating that truncation likely is not due to incorrect assembly. Additionally, Xb-Sj lacked one of the seven Tc toxin genes that were found in Xb-Sj-2000, supporting that the majority of differences are not due to misassembly. All genomes had fragmented open reading frames corresponding to each subunit type, but it is unclear if these fragments would be able to function as a complete protein when combined. When considering only full length coding regions, all of the sequenced X. bovienii strains encoded at least one of three potential A subunits (XptA2, TccA2, TccB2), five strains encoded an intact B subunit (TcaC), and six strains encoded an intact C subunit (TccC) (Additional file 10). Only four (Xb-Sf-MD, Xb-Sk-CA, Xb-Sj, Xb-Si) of the nine draft genomes encode at least one intact protein of each subunit type.
To assess the variability among intact Tc protein sequences, we measured amino acid divergence between these proteins (Additional file 8) and constructed nucleotide and amino acid sequence phylogenies (Additional file 11). Separate analysis of the three A subunit types (i.e. XptA2, TccA2, TccB2) revealed that substitutions in amino acid sequences among the homologs are distributed evenly throughout the amino acid sequence (i.e. no regions of the protein showed greater number of substitutions than others) (Additional file 8G-I). There are two areas of XptA2 amino acid sequence that are somewhat less conserved than the rest of the protein, but the majority of this diversity is driven by Xb-Sp (Additional file 8G). For B subunit proteins, amino acid substitutions were distributed along the length of the protein (Additional file 8J). Phylogenetic analysis showed that these proteins clustered into two clades (Additional file 11D), indicating some combination of horizontal transfer and/or gene duplication events in the evolutionary history of these genes and potential functional divergence. Consistent with previous reports, tccC genes had greater sequence divergence near the C-terminal end of the protein (Additional file 8K) [31]. This portion of the protein is the functional domain. BLASTp analysis found that each TccC protein showed the highest similarity to one of four subtypes (TccC1, TccC4, TccC5, TccC7) (Additional file 10). However, different portions of each TccC protein matched different subtypes, including some multiple matches (although with different strength). These data indicate that overall amino acid similarity does not accurately annotate the correct subtype for an individual TccC protein.
To further characterize protein divergence, we performed phylogenetic and dN/dS analyses of Tc toxin amino acid and nucleotide sequences. Using TOPALi analysis, there are 2 recombination points leading to 3 distinct recombination blocks in xptA2. In bacterial pathogens, recombination leads to shuffling of virulence genes and can confer new ecological niches on recipient bacteria [94, 95]. Phylogenetic analysis of the xptA2 nucleotide sequences encoded by each recombination block from TOPALi analysis identified that the phylogenetic tree for each portion was distinct and each phylogeny had some branching patterns not congruent with the X. bovienii core genome phylogenetic tree, consistent with recombination [5] (Fig. 4j). The xptA2 genes in Xb-Sp and Xb-Sk-CA shared the same genomic context, as did those in Xb-Sj-2000, Xb-Sj, and Xb-Si. In contrast, the analyzed full-length X. bovienii tccA2 and tccB2 (A subunit-encoding) genes have not undergone recombination according to TOPALi, but have potentially undergone HGT, as not all strongly supported nodes were congruent with the X. bovienii core genome phylogeny (Fig. 4h, i). However, The tccA2 and tccB2 xenologs have the same genomic context and therefore may have conserved functions.
Phylogenetic analysis of the Tc toxin B subunit proteins showed that proteins clustered into two clades (Additional file 11D), indicating potential horizontal transfer and/or gene duplication. We did not identify recombination among B subunit genes using TOPALi [5] (Fig. 4k). However, all tcaC (B subunit-encoding) genes except that of Xb-Sp have the same genomic context, and therefore may have conserved functions.
Analysis of the Tc toxin C subunit tccC genes indicated that a recombination breakpoint occurred 1188 bp from the 3’-end of these genes, consistent with increased sequence diversity within this region, as well at 2047 from the 3’-end of these genes [31] (Fig. 4l). Phylogenetic analyses of nucleotide sequences from each of these recombination blocks had different topologies, consistent with recombination. Parallel amino acid phylogenies lacked sufficient phylogenetic to display such incongruities (Additional file 4E-G). Additionally, not all strongly supported splits were consistent with the core genome, suggesting potential horizontal transfer of whole genes. Xb-Sj-2000 has two copies of the tccC genes, suggesting potentially divergent functions between them. The tccC genes from Xb-Sj, Xb-Sj-2000, and Xb-Sk-CA have the same genomic context, unlike those from Xb-Sf-FR, Xb-Sf-MD, and Xb-Si, which each has a distinct genomic context. Together, these data indicate that X. bovienii tccC genes have complicated recent evolutionary histories and that their functions likely vary among X. bovienii strains.
There is considerable divergence among the previously characterized Tc toxin proteins and those identified in X. bovienii, indicating that X. bovienii toxin proteins may target different insects or have different effects on target cells and molecules. As insecticidal toxins, these proteins may function in the symbiosis to aid in killing the insect host or to protect the insect cadaver against insect scavengers. Indeed, one study has shown that Tc toxins are dispensable for virulence in Xenorhabdus strains, at least in particular insect hosts, suggesting they may play other roles in the life cycle in addition to or instead of insecticidal activity [39]. Therefore, variation in the Tc toxins among X. bovienii strains likely reflects differences in the insect hosts or scavengers encountered during their lifecycle.
Non-ribosomal peptide synthetase and polyketide synthetase cluster variation
A class of molecules likely involved in symbiotic interactions is produced by non-ribosomal peptide synthetase (NRPS) and polyketide synthetase (PKS) systems: multi-gene, modular enzymes that synthesize small molecules with a variety of biological functions. Prior studies have reported extensive diversity in NRPS and PKS clusters among entomopathogenic nematode symbiont species, such as X. nematophila and P. luminescens [18, 31]. Among X. bovienii bacterial strains, variation in NRPS and PKS coding potential also occurs (Table 3). A total of 29 distinct NRPS and PKS clusters were identified among all the analyzed strains combined. Of these clusters, six were identified in all of the strains (conserved) and seven were found within only one strain (unique). The remaining sixteen clusters were found in a subset of some, but not all, strains (shared). While some differences in NRPS and PKS clusters could be due to genome misassembly, as these are repetitive sequences, the two like genomes (Xb-Sj and Xb-Sj-2000) only differ by one cluster. This suggests that the majority of clusters did assemble correctly.
Table 3.
NRPS an PKS clustersa
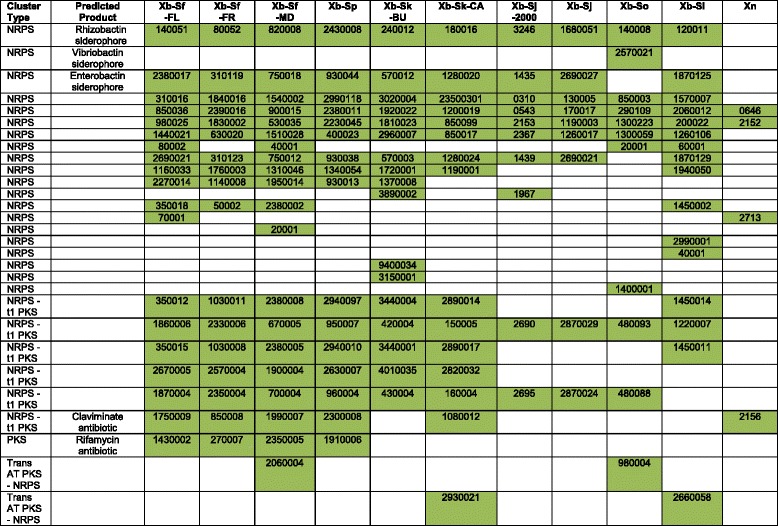
aDistribution of NRPS and PKS clusters among the X. bovienii genomes listed by the biosynthesis gene of the cluster that js numerically first in the genome annotation. The number designation for each gene is given without the prefixes: Xb-Sf-FL (XBFFL1), Xb-Sf-FR (XBFFR1), Xb-Sf-MD (XBFMD1), Xb-Sp (XBP1), Xb-Sk-BU (XBKB1), Xb-Sk-CA (XBKQ1), Xb-Sj-2000 (XBJ1), Xb-Sj (XBJ2), Xb-So (XBO1), Xb-Si (XBI1), and Xn (XNC1)
bTypes of clusters: NRPS (non-ribosomal peptide synthetase), PKS (polyketide synthetase), T1 PKS (type 1 PKS), or Trans AT PKS (trans-acyltransferase PKS)
cPredicted product from the NRPS and PKS clusters, if known
One of the conserved clusters, one of the unique clusters, and four shared clusters have known siderophore (i.e. metal scavenging compounds) or antibiotic products (Table 3). The remaining clusters (five conserved, six unique, and 12 shared clusters) do not have a known product. When compared to the published genomes of X. nematophila and P. luminescens [31, 32] we found that of the 29 identified clusters, six (two conserved, one unique, and three shared clusters) were also shared by X. nematophila and none were shared by P. luminescens (Table 3). This indicates that entomopathogenic Xenorhabdus and Photorhabdus symbiont species all encode many NRPS and PKS clusters and likely produce many different secondary metabolite products.
The function of Xenorhabdus NRPS and PKS products are diverse, and include anti-microbial [11, 18, 96–98], anti-predation [8, 9, 99], immunosuppressive [100], hemolytic [98], and metal acquisition [101] activities. Several of these functions have the potential to provide symbiotic benefits. Products that provide anti-microbial or anti-predation activity could play a defensive role within the symbiosis by protecting the insect cadaver, and therefore the developing nematodes and bacteria, from invasion by pathogens, competitors, or predators. Additionally, products that provide immunosuppressive or hemolytic activity could aid in the killing of the insect host, thereby providing nutrition to the nematodes and bacteria. Although it is possible that the predicted NRPS and PKS cluster products provide these benefits, defining the activities and role of the molecules awaits further experimental testing and functional characterization.
Conclusions
Overall, the presented analyses highlight that fundamental processes underlying symbiotic interactions in X. bovienii (e.g. secretion systems and degradative activities) are largely conserved between strains. However, phenotypic testing indicates that despite conserved coding potential, strain-level differences in symbiosis factor expression occurs (Table 2). Further, we observed notable variation in certain classes of genes, such as those encoding NRPS and PKS clusters and Tc toxins (Table 3, Additional file 10). This variation may contribute to previously identified differences in symbiotic fitness [5], but confirmation of this possibility awaits further experimental assessment.
By standard molecular metrics (e.g. average nucleotide identity and 16S rRNA analyses) the X. bovienii strains analyzed here previously were identified as members of the same species [5, 30]. Our data demonstrate that within the X. bovienii species, there is diversity in the coding potential of bioactive molecules (Table 3, Additional file 10) and the regulation of some conserved products [5] (Table 2). These data in combination with the finding that these strains differ in symbiotic fitness [5] suggest that using ecotype definitions, rather than bacterial species or strains, might be more useful in categorizing diversity among bacterial symbionts and their potential impact on host fitness. Bacterial ecotypes are defined as evolutionarily and ecologically distinct groups [102] and within symbiosis, this could be defined as groups that produce particular molecules to interact with specific hosts. The use of bacterial ecotype definitions would allow for detection and discussion of the nuanced differences among symbiont groups.
Variation in the ability of the bacterial strains to engage in symbiosis with the nematode likely results from coevolution, and therefore co-adaptation, between nematode host species and bacterial symbiont strains [4, 5]. It is also possible that differences in the ability of symbionts to engage in symbiosis could be due to differential gene loss or access to gene pools, not necessarily dependent on coevolution. Activities predicted to diversify in this way are those that benefit the nematode host, such as those involved in nutrient acquisition. Our data indicate that while many of the nutritional factors do not differ among strains, their regulation does (e.g. lipase). This demonstrates that while overall nutritional requirements may not vastly differ between nematode hosts, the timing of expression of metabolic pathways varies and may be important for optimal symbiotic benefits. We did not detect any phenotypic pattern that was consistent with the published nematode or bacterial phylogenies [5] (data not shown), suggesting that expression of the measured activities is dependent on variables other than or in addition to the nematode host identity, such as the identity of the insect host. Likewise, bacterial factors that contribute to virulence towards the insect host or to defense against predators likely vary based on selective pressures of environmental differences encountered by the bacterial strains, such as the insect host species, endogenous bacterial competitors within these insects, or the predators, competitors, and pathogens naturally encountered. Our genomic analyses demonstrate diversity in many of these types of compounds (e.g. NRPS derived molecules, Tc toxins, and hemolysins), suggesting that the nematode – bacterial pairs encounter different insect hosts and competitors, as some of these have been demonstrated to function differently against divergent insect hosts [93]. However, many of the large molecular weight toxins (e.g. McfXb, MARTXXb toxin, and XaxAB) are conserved in all X. bovienii strains, indicating that these toxins may not be specific to particular insect host-ranges and instead functional against many insect hosts. Unfortunately, little is known about the identity of X. bovienii insect hosts, predators, competitors or pathogens that occur in natural conditions, precluding concrete conclusions correlating the diversity of gene products to naturally encountered organisms. However, our comparative genomics and phenotypic analyses do support that diversity in symbiotic functions of the X. bovienii bacterial strains differ due to the diversity of effectors and their regulation rather than the utilization of different mechanisms for interacting with hosts.
In addition to providing insight into the diversity of potential symbiotic functions, the data presented here highlight that strain variability is an important consideration when exploiting Xenorhabdus bacteria for discovery of compounds for application purposes. For the discovery of novel antibiotics or other NRPS- and PKS-derived compounds from Xenorhabdus spp., it will be useful to assess multiple bacterial strains, as we observed large strain-level diversity in coding potential for these systems. This is also the case when assessing Tc toxin clusters. However, for the application of many other large molecular weight toxins (e.g. XaxAB, McfXb, MARTXXb), the activities determined from a single bacterial strain likely will be similar among the members of that species but divergent from the toxins identified in different Xenorhabdus species.
In summary, the comparative genomic analysis presented here provides an assessment of X. bovienii bacterial strain variation in factors that could be involved in symbiotic interactions and may be utilized for applications. This analysis provides a foundation for understanding how bacterial strain variability affects symbiosis and for the discovery of novel compounds within Xenorhabdus spp.
Methods
Genome features
Genomes were submitted to MaGe [103, 104] for annotation and analysis. The genomes were analyzed for size, GC content, number of coding sequences, and the percentage of the genome covered by coding content. The genomes were assessed for synteny using MAUVE 2.3.1 [105] relative to the finished genome of X. bovienii (Table 1) [32]. This analysis was done excluding contigs <1000 bp.
Plasmid detection
No plasmids were detected in any of the X. bovienii strains. The experimental methods utilized were, plasmid extraction kit Zippy Plasmid Miniprep Kit (Zymo, Irvine, CA), plasmid boil prep [106], and Wizard Genomic DNA Purification Kit (Promega, Madison, WI). After extraction, DNA was run on a 1 % agarose gel and observed for bands. No bands corresponding to potential plasmid were observed. Additionally, within DNA sequences, no genes corresponding to know plasmid genes (e.g. origin of replication) were identified using BLASTp. This BLAST analyses, and all others performed in this study were done using default parameters, and a significant match was considered 75 % coverage with an e-value <1E-4.
Global metabolism analysis
The metabolic pathways of each strain were determined using two methods. The MaGe platform [103, 104] was utilized to interpret the percentage of each pathway that was intact, and this platform uses KEGG and EC annotations. Additionally, pathways were analyzed through mapping to MetaCyc [107], which is shown as the number of pathways present for each class. Genomes were compared for metabolic coding potential by identifying if pathways were divergent among the strains in both analyses. Incompleteness of a pathway was confirmed through BLASTp analysis using missing pathway enzymes.
Secretion systems
Secretion systems were identified through keyword searching the MaGe annotations for secretion system components (Additional file 1). For a secretion system to called intact, it must have had all known necessary secretion system genes [35, 36, 40, 42, 45–47, 108]. The presence or absence of other secretion systems were assessed through comparing known necessary secretion system components to the genomes using BLASTp [109].
NRPS and PKS clusters
NRPS and PKS clusters were identified by analyzing each genome using antiSMASH 3.0 [110]. Clusters were confirmed as intact by assessing that each cluster was in the middle of a contig and the genes encoding the cluster were surrounded by genes annotated as other functions. Exclusion of clusters on the edge of contigs resulted in the exclusion of 23 clusters (1–3 per genome). The NRPS and PKS clusters were compared to determine their distribution among X. bovienii genomes by assessing the cluster proteins for local synteny within the genomes, using MaGe and MAUVE 2.3.1 alignments. The presence or absence of the X. bovienii NRPS and PKS clusters in X. nematophila and P. luminescens was determined by searching for the gene clusters in the finished genomes using MaGE synteny mapping.
Toxins
Genes for putative toxin proteins were revealed by searching for genes annotated as toxins by the MaGe platform. Annotations were further supported by BLASTp, Interpro 51.0 [111], and Swiss Prot [103, 104] analyses. Toxin domains were analyzed using Interpro, and in the case of Mcf1, comparison to known protein domains in homologs. Assessment of toxin subtypes (i.e. Shiga toxin and Tc toxin) was performed based on BLASTp results [109]. For assessing similarities among X. bovienii, amino acid sequences were aligned using MUSCLE 3.7 [112, 113], and protein distances were calculated using Phylip 3.695 with the Jones-Taylor-Thornton model in Protdist [114]. Protein distances are given in point accepted mutation (PAM), representing the number of point mutation events in 100 amino acids. Protein trees were built using Maximum Likelihood and bootstrapped in Phylip [114], and trees were visualized in iTol [115]. For visualization of amino acid differences along the length of the proteins, the alignment was visualized in MegAlign Pro from DNASTAR 11.0 (www.dnastar.com). Regions of dissimilarity were considered when at least four amino acids in a row were not conserved among the homologs.
Testing of recombination, horizontal gene transfer, and selection
Nucleotide sequences were aligned in MEGA v6.0 [116]. Analyses of nucleotide sequences was done in TOPALi v2 [117]. Assessment of recombination was done in TOPALi using DSS and the sequence alignment was divided up into portions with a similar evolutionary history. Horizontal gene transfer was identified by comparing gene phylogenies to the bacterial whole genome phylogeny [5]. Phylogenetic trees were built in TOPALi from the full length or sub-sections of the nucleotide sequences as determined by DSS. Bootstrapping was done using the maximum likelihood GTR substitution model in Topali. Bootstrap values were used to determine strongly supported splits (>75 %). Selection was determined using PAML in TOPALi to calculate dN/dS ratios [118].
Phenotypic testing
Stable secondary form (variant 2) bacterial strains were isolated from stable primary form (variant 1) bacteria through repeated passage. Briefly, bacterial strains were grown at 30 °C in lysogeny broth with aeration in the dark. Bacterial strains were grown approximately 24 h and sub-cultured into fresh media. Sub-culturing occurred for a period of 2–4 weeks, until bacteria spread on NBTA agar [119] no longer bound bromothymol bule dye and was red in color with repeated restreaking. Phenotypic tests were performed using previously published assays for swimming [120] and swarming [54] motility on LB agar, lipase on tween 20 agar [121], lecithinase on egg yolk agar [122], protease on milk agar [122], siderophore on CAS agar [52], and hemolysin on horse and rabbit blood agar plates [123]. For all assays, 5 μL of overnight bacterial culture was spotted onto the agar plate and dried, and the plates were incubated at 30 °C in the dark for 48 h prior to reading. Antibiotic activity was determined through overlay assays using Escherichia coli, Micrococcus luteus, and Bacillus subtilis as the overlayed test strains [124]. For antibiotic activity, 5 μL of overnight X. bovienii bacterial culture was spotted onto LB agar plates supplemented with pyruvate and dried, and the plates were incubated for at 30 °C in the dark for 24 h prior to overlaying with the test bacterial strain. After overlay, the plates were incubated for at 37 °C for 24 h prior to reading. For these experiments, all test strains were inhibited. For all activities, the relative activity was measured through measuring the diameter of the output (e.g. zone of clearing). However, no significant difference was observed between strains with measurable activity. Therefore the activity is reported as yes or no rather than numerically.
Assessment of X. bovienii genes contributing to phenotypic activity
Homologs of genes known to contribute to the identified activity were assessed through BLASTp analysis of the homologs in each X. bovienii bacterial strain. NRPS and PKS clusters were determined as described above.
Availability of data and materials
Genomes have been deposited in GenBank (http://www.ncbi.nlm.nih.gov/genbank/) (Table 1).
Acknowledgements
This study was supported by grants from the National Science Foundation to HGB (IOS-0920631 and IOS-1353674). KEM also was supported by the National Institutes of Health (NIH) National Research Service Award T32 AI55397, a Louis and Elsa Thomsen Distinguished Predoctoral Fellowship, and a Betley-Allen Predoctoral Fellowship. The authors wish to thank Karla Satchell (Northwestern University) for her help with classification of RTX and Mcf toxins, Thomas Sorenson (UW-Madison) for his contributions to the analysis presented in Additional file 4 (LysR regulators) and S. Patricia Stock (University of Arizona) and Steven Forst (UW-Milwaukee) for their contributions to the acquisition of the genomes analyzed in this study.
Additional files
Global metabolic analysis of Xenorhabdus bovienii strains. Description: Table of all metabolic pathways in X. bovienii and the relative completeness of each pathway. (XLSX 76 kb)
Secretion system genes. Description: Table of all genes identified encoding the complete secretion systems in X. bovienii bacterial strains. (DOC 78 kb)
Bacterial genes predicted to encode select symbiotic activities. Description: Table of all genes identified within the bacterial strains that could confer the activities observed in Table 2. (DOC 32 kb)
Regulatory proteins found within X. bovienii strains. Description: Table of all genes predicted to encode the well-characterized regulatory proteins in Xenorhabdus species. (DOC 35 kb)
Xenorhabdus bovienii genes predicted to encode LysR family transcription factors. Description: Table of all genes within all X. bovienii strains predicted to encode LysR family regulators. (DOC 62 kb)
Xenorhabdus bovienii genes predicted to encode two-component regulatory systems. Description: Table of all genes within all X. bovienii strains predicted to encode two-component regulators. (DOC 74 kb)
Annotated toxin genes. Description: Table of all genes within all X. bovienii strains predicted to encode toxin proteins excluding Tc toxin subunits. (DOC 34 kb)
Distribution of amino acid sequence divergence in proteins. Description: Image of the amino acid sequence divergence among X. bovienii homologs of McfXb, MARTXXb, PrtA, XaxA, XaxB, XhlA/XhlA2, XptA2, TccA2, TccB2, TcaC, and TccC. (PDF 181 kb)
Best BLASTp hits of Xb-Si putative Shiga toxin. Description: Table of the top 5 BLASTp hits for the putative Shiga toxin from Xb-Si (XbI1v2_2730004). (DOC 30 kb)
Tc subunit genes from X. bovienii genomes. Description: Table of all predicted Tc toxin subunit proteins from all X. bovienii genomes, including the intact and fragmented open reading frames. (DOC 128 kb)
Phylogenies of TC toxin amino acid sequences. Description: Image of the phylogenies built from amino acid sequences of X. bovienii homologs XptA2, TccA2, TccB2, TcaC, and TccC. (PDF 52 kb)
Footnotes
Competing interest
The authors declare that they have no competing interests.
Authors’ contributions
HGB and KEM conceived of the study and its design. KEM did all comparative genomics analyses. ACW and KEM completed phenotypic experiments. KEM, JLK, and HGB analyzed data and wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Kristen E. Murfin, Email: kmurfin@wisc.edu
Amy C. Whooley, Email: whooleyac@gmail.com
Jonathan L. Klassen, Email: jonathan.klassen@uconn.edu
Heidi Goodrich-Blair, Email: hgblair@bact.wisc.edu.
References
- 1.Grewal PS, Ehlers RU, Shapiro-Ilan DI, editors. Nematodes as biocontrol agents. Wallingford, UK: CABI Publishing; 2005. [Google Scholar]
- 2.Griffin CT. Perspectives on the behavior of entomopathogenic nematodes from dispersal to reproduction: traits contributing to nematode fitness and biocontrol efficacy. J Nematol. 2012;44(2):177–84. [PMC free article] [PubMed] [Google Scholar]
- 3.Campos-Herrera R, Barbercheck M, Hoy CW, Stock SP. Entomopathogenic nematodes as a model system for advancing the frontiers of ecology. J Nematol. 2012;44(2):162–76. [PMC free article] [PubMed] [Google Scholar]
- 4.Chapuis E, Emelianoff V, Paulmier V, Le Brun N, Pages S, Sicard M, et al. Manifold aspects of specificity in a nematode-bacterium mutualism. J Evol Biol. 2009;22(10):2104–17. doi: 10.1111/j.1420-9101.2009.01829.x. [DOI] [PubMed] [Google Scholar]
- 5.Murfin KE, Lee MM, Klassen JL, McDonald BR, Larget B, Forst S, et al. Xenorhabdus bovienii strain diversity impacts coevolution and symbiotic maintenance with Steinernema spp. nematode hosts. MBio. 2015;6(3):e00076. doi: 10.1128/mBio.00076-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herbert EE, Cowles KN, Goodrich-Blair H. CpxRA regulates mutualism and pathogenesis in Xenorhabdus nematophila. Appl Environ Microbiol. 2007;73(24):7826–36. doi: 10.1128/AEM.01586-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Richards GR, Goodrich-Blair H. Masters of conquest and pillage: Xenorhabdus nematophila global regulators control transitions from virulence to nutrient acquisition. Cell Microbiol. 2009;11(7):1025–33. doi: 10.1111/j.1462-5822.2009.01322.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gulcu B, Hazir S, Kaya HK. Scavenger deterrent factor (SDF) from symbiotic bacteria of entomopathogenic nematodes. J Invertebr Pathol. 2012;110(3):326–33. doi: 10.1016/j.jip.2012.03.014. [DOI] [PubMed] [Google Scholar]
- 9.Zhou X, Kaya HK, Heungens K, Goodrich-Blair H. Response of ants to a deterrent factor(s) produced by the symbiotic bacteria of entomopathogenic nematodes. Appl Environ Microbiol. 2002;68:6202–9. doi: 10.1128/AEM.68.12.6202-6209.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morales-Soto N, Forst SA. The xnp1 P2-like tail synthesis gene cluster encodes xenorhabdicin and is required for interspecies competition. J Bacteriol. 2011;193(14):3624–32. doi: 10.1128/JB.00092-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singh S, Orr D, Divinagracia E, McGraw J, Dorff K, Forst S. Role of secondary metabolites in establishment of the mutualistic partnership between Xenorhabdus nematophila and the entomopathogenic nematode Steinernema carpocapsae. Appl Environ Microbiol. 2015;81(2):754–64. doi: 10.1128/AEM.02650-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singh S, Reese JM, Casanova-Torres AM, Goodrich-Blair H, Forst S. Microbial population dynamics in the hemolymph of Manduca sexta infected with Xenorhabdus nematophila and the entomopathogenic nematode Steinernema carpocapsae. Appl Environ Microbiol. 2014;80(14):4277–85. doi: 10.1128/AEM.00768-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koppenhofer AM, Kaya HK. Coexistence of two steinernematid nematode species (Rhabditida:Steinernematidae) in the presence of two host species. Appl Soil Ecol. 1996;4:221–30. doi: 10.1016/S0929-1393(96)00121-7. [DOI] [Google Scholar]
- 14.Popiel I, Grove DL, Friedman MJ. Infective juvenile formation in the insect parasitic nematode Steinernema feltiae. Parasitology. 1989;99(01):77–81. doi: 10.1017/S0031182000061047. [DOI] [Google Scholar]
- 15.Sugar DR, Murfin KE, Chaston JM, Andersen AW, Richards GR, DeLeon L, et al. Phenotypic variation and host interactions of Xenorhabdus bovienii SS-2004, the entomopathogenic symbiont of Steinernema jollieti nematodes. Environ Microbiol. 2012;14(4):924–39. doi: 10.1111/j.1462-2920.2011.02663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Orchard SS, Goodrich-Blair H. Identification and functional characterization of a Xenorhabdus nematophila oligopeptide permease. Appl Environ Microbiol. 2004;70(9):5621–7. doi: 10.1128/AEM.70.9.5621-5627.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richards GR, Goodrich-Blair H. Examination of Xenorhabdus nematophila lipases in pathogenic and mutualistic host interactions reveals a role for xlpA in nematode progeny production. Appl Environ Microbiol. 2010;76(1):221–9. doi: 10.1128/AEM.01715-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bode HB. Entomopathogenic bacteria as a source of secondary metabolites. Curr Opin Chem Biol. 2009;13(2):224–30. doi: 10.1016/j.cbpa.2009.02.037. [DOI] [PubMed] [Google Scholar]
- 19.Martens EC, Heungens K, Goodrich-Blair H. Early colonization events in the mutualistic association between Steinernema carpocapsae nematodes and Xenorhabdus nematophila bacteria. J Bacteriol. 2003;185(10):3147–54. doi: 10.1128/JB.185.10.3147-3154.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sicard M, Brugirard-Ricaud K, Pages S, Lanois A, Boemare NE, Brehelin M, et al. Stages of infection during the tripartite interaction between Xenorhabdus nematophila, its nematode vector, and insect hosts. Appl Environ Microbiol. 2004;70(11):6473–80. doi: 10.1128/AEM.70.11.6473-6480.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ehlers RU, Wulff A, Peters A. Pathogenicity of axenic Steinernema feltiae, Xenorhabdus bovienii, and the bacto-helminthic complex to larvae of Tipula oleracea (Diptera) and Galleria mellonella (Lepidoptera) J Invertebr Pathol. 1997;69(3):212–7. doi: 10.1006/jipa.1996.4647. [DOI] [PubMed] [Google Scholar]
- 22.Brivio MF, Moro M, Mastore M. Down-regulation of antibacterial peptide synthesis in an insect model induced by the body-surface of an entomoparasite (Steinernema feltiae) Dev Comp Immunol. 2006;30(7):627–38. doi: 10.1016/j.dci.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 23.Sheets JJ, Hey TD, Fencil KJ, Burton SL, Ni W, Lang AE, et al. Insecticidal toxin complex proteins from Xenorhabdus nematophilus: structure and pore formation. J Biol Chem. 2011;286(26):22742–9. doi: 10.1074/jbc.M111.227009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vigneux F, Zumbihl R, Jubelin G, Ribeiro C, Poncet J, Baghdiguian S, et al. The xaxAB genes encoding a new apoptotic toxin from the insect pathogen Xenorhabdus nematophila are present in plant and human pathogens. J Biol Chem. 2007;282(13):9571–80. doi: 10.1074/jbc.M604301200. [DOI] [PubMed] [Google Scholar]
- 25.Cowles KN, Goodrich-Blair H. Expression and activity of a Xenorhabdus nematophila haemolysin required for full virulence towards Manduca sexta insects. Cell Microbiol. 2005;7(2):209–19. doi: 10.1111/j.1462-5822.2004.00448.x. [DOI] [PubMed] [Google Scholar]
- 26.Eom S, Park Y, Kim Y. Sequential immunosuppressive activities of bacterial secondary metabolites from the entomopahogenic bacterium Xenorhabdus nematophila. J Microbiol. 2014;52(2):161–8. doi: 10.1007/s12275-014-3251-9. [DOI] [PubMed] [Google Scholar]
- 27.Reimer D, Cowles KN, Proschak A, Nollmann FI, Dowling AJ, Kaiser M, et al. Rhabdopeptides as insect-specific virulence factors from entomopathogenic bacteria. Chembiochem. 2013;14(15):1991–7. doi: 10.1002/cbic.201300205. [DOI] [PubMed] [Google Scholar]
- 28.Pidot SJ, Coyne S, Kloss F, Hertweck C. Antibiotics from neglected bacterial sources. Int J Med Microbiol. 2014;304(1):14–22. doi: 10.1016/j.ijmm.2013.08.011. [DOI] [PubMed] [Google Scholar]
- 29.Vizcaino MI, Guo X, Crawford JM. Merging chemical ecology with bacterial genome mining for secondary metabolite discovery. J Ind Microbiol Biotechnol. 2014;41(2):285–99. doi: 10.1007/s10295-013-1356-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Konstantinidis KT, Tiedje JM. Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci U S A. 2005;102(7):2567–72. doi: 10.1073/pnas.0409727102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chaston JM, Suen G, Tucker SL, Andersen AW, Bhasin A, Bode E, et al. The entomopathogenic bacterial endosymbionts Xenorhabdus and Photorhabdus: convergent lifestyles from divergent genomes. PLoS One. 2011;6(11):e27909. doi: 10.1371/journal.pone.0027909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Latreille P, Norton S, Goldman BS, Henkhaus J, Miller N, Barbazuk B, et al. Optical mapping as a routine tool in bacterial genome sequencing. BMC Genomics. 2007;8(1):321. doi: 10.1186/1471-2164-8-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chaston JM, Murfin KE, Heath-Heckman EA, Goodrich-Blair H. Previously unrecognized stages of species-specific colonization in the mutualism between Xenorhabdus bacteria and Steinernema nematodes. Cell Microbiol. 2013;15(9):1545–59. doi: 10.1111/cmi.12134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martens EC, Russell FM, Goodrich-Blair H. Analysis of Xenorhabdus nematophila metabolic mutants yields insight into stages of Steinernema carpocapsae nematode intestinal colonization. Mol Microbiol. 2005;58(1):28–45. doi: 10.1111/j.1365-2958.2005.04742.x. [DOI] [PubMed] [Google Scholar]
- 35.Coulthurst SJ. The Type VI secretion system - a widespread and versatile cell targeting system. Res Microbiol. 2013;164(6):640–54. doi: 10.1016/j.resmic.2013.03.017. [DOI] [PubMed] [Google Scholar]
- 36.Cianciotto NP. Type II, secretion and Legionella virulence. Curr Top Microbiol Immunol. 2013;376:81–102. doi: 10.1007/82_2013_339. [DOI] [PubMed] [Google Scholar]
- 37.Jani AJ, Cotter PA. Type VI secretion: not just for pathogenesis anymore. Cell Host Microbe. 2010;8(1):2–6. doi: 10.1016/j.chom.2010.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Okazaki S, Kaneko T, Sato S, Saeki K. Hijacking of leguminous nodulation signaling by the rhizobial type III secretion system. Proc Natl Acad Sci U S A. 2013;110(42):17131–6. doi: 10.1073/pnas.1302360110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ogier JC, Pages S, Bisch G, Chiapello H, Medigue C, Rouy Z, et al. Attenuated virulence and genomic reductive evolution in the entomopathogenic bacterial symbiont species. Xenorhabdus poinarii Genome Biol Evol. 2014;6(6):1495–513. doi: 10.1093/gbe/evu119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abrusci P, McDowell MA, Lea SM, Johnson S. Building a secreting nanomachine: a structural overview of the T3SS. Curr Opin Struct Biol. 2014;25:111–7. doi: 10.1016/j.sbi.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Christie PJ, Whitaker N, Gonzalez-Rivera C. Mechanism and structure of the bacterial type IV secretion systems. Biochim Biophys Acta. 2014;1843(8):1578–91. doi: 10.1016/j.bbamcr.2013.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Singh P, Park D, Forst S, Banerjee N. Xenocin export by the flagellar type III pathway in Xenorhabdus nematophila. J Bacteriol. 2013;195(7):1400–10. doi: 10.1128/JB.01532-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barrero-Tobon AM, Hendrixson DR. Flagellar biosynthesis exerts temporal regulation of secretion of specific Campylobacter jejuni colonization and virulence determinants. Mol Microbiol. 2014;93(5):957–74. doi: 10.1111/mmi.12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Richards GR, Herbert EE, Park Y, Goodrich-Blair H. Xenorhabdus nematophila lrhA is necessary for motility, lipase activity, toxin expression, and virulence in Manduca sexta insects. J Bacteriol. 2008;190(14):4870–9. doi: 10.1128/JB.00358-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nivaskumar M, Francetic O. Type II secretion system: a magic beanstalk or a protein escalator. Biochim Biophys Acta. 2014;1843(8):1568–77. doi: 10.1016/j.bbamcr.2013.12.020. [DOI] [PubMed] [Google Scholar]
- 46.Cascales E. The type VI secretion toolkit. EMBO Rep. 2008;9(8):735–41. doi: 10.1038/embor.2008.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Silverman JM, Brunet YR, Cascales E, Mougous JD. Structure and regulation of the type VI secretion system. Annu Rev Microbiol. 2012;66:453–72. doi: 10.1146/annurev-micro-121809-151619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bowen DJ, Rocheleau TA, Grutzmacher CK, Meslet L, Valens M, Marble D, et al. Genetic and biochemical characterization of PrtA, an RTX-like metalloprotease from Photorhabdus. Microbiology. 2003;149(Pt 6):1581–91. doi: 10.1099/mic.0.26171-0. [DOI] [PubMed] [Google Scholar]
- 49.Givaudan A, Lanois A. flhDC, the flagellar master operon of Xenorhabdus nematophilus: requirement for motility, lipolysis, extracellular hemolysis, and full virulence in insects. J Bacteriol. 2000;182(1):107–15. doi: 10.1128/JB.182.1.107-115.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park D, Forst S. Co-regulation of motility, exoenzyme and antibiotic production by the EnvZ-OmpR-FlhDC-FliA pathway in Xenorhabdus nematophila. Mol Microbiol. 2006;61(6):1397–412. doi: 10.1111/j.1365-2958.2006.05320.x. [DOI] [PubMed] [Google Scholar]
- 51.An R, Sreevatsan S, Grewal PS. Comparative in vivo gene expression of the closely related bacteria Photorhabdus temperata and Xenorhabdus koppenhoeferi upon infection of the same insect host. Rhizotrogus majalis BMC Genomics. 2009;10:433. doi: 10.1186/1471-2164-10-433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schwyn B, Neilands JB. Universal chemical assay for the detection and determination of siderophores. Anal Biochem. 1987;160(1):47–56. doi: 10.1016/0003-2697(87)90612-9. [DOI] [PubMed] [Google Scholar]
- 53.Jubelin G, Pages S, Lanois A, Boyer MH, Gaudriault S, Ferdy JB, et al. Studies of the dynamic expression of the Xenorhabdus FliAZ regulon reveal atypical iron-dependent regulation of the flagellin and haemolysin genes during insect infection. Environ Microbiol. 2011;13(5):1271–84. doi: 10.1111/j.1462-2920.2011.02427.x. [DOI] [PubMed] [Google Scholar]
- 54.Kim DJ, Boylan B, George N, Forst S. Inactivation of ompR promotes precocious swarming and flhDC expression in Xenorhabdus nematophila. J Bacteriol. 2003;185(17):5290–4. doi: 10.1128/JB.185.17.5290-5294.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chapuis E, Arnal A, Ferdy JB. Trade-offs shape the evolution of the vector-borne insect pathogen Xenorhabdus nematophila. Proc Biol Sci. 2012;279(1738):2672–80. doi: 10.1098/rspb.2012.0228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Macnab RM. Genetics and biogenesis of bacterial flagella. Annu Rev Genet. 1992;26:131–58. doi: 10.1146/annurev.ge.26.120192.001023. [DOI] [PubMed] [Google Scholar]
- 57.Cowles KN, Cowles CE, Richards GR, Martens EC, Goodrich-Blair H. The global regulator Lrp contributes to mutualism, pathogenesis and phenotypic variation in the bacterium Xenorhabdus nematophila. Cell Microbiol. 2007;9(5):1311–23. doi: 10.1111/j.1462-5822.2006.00873.x. [DOI] [PubMed] [Google Scholar]
- 58.Bhasin A, Chaston JM, Goodrich-Blair H. Mutational analyses reveal overall topology and functional regions of NilB, a bacterial outer membrane protein required for host association in a model of animal-microbe mutualism. J Bacteriol. 2012;194(7):1763–76. doi: 10.1128/JB.06711-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maddocks SE, Oyston PC. Structure and function of the LysR-type transcriptional regulator (LTTR) family proteins. Microbiology. 2008;154(Pt 12):3609–23. doi: 10.1099/mic.0.2008/022772-0. [DOI] [PubMed] [Google Scholar]
- 60.Jung K, Fried L, Behr S, Heermann R. Histidine kinases and response regulators in networks. Curr Opin Microbiol. 2012;15(2):118–24. doi: 10.1016/j.mib.2011.11.009. [DOI] [PubMed] [Google Scholar]
- 61.Derzelle S, Ngo S, Turlin E, Duchaud E, Namane A, Kunst F, et al. AstR-AstS, a new two-component signal transduction system, mediates swarming, adaptation to stationary phase and phenotypic variation in Photorhabdus luminescens. Microbiology. 2004;150(Pt 4):897–910. doi: 10.1099/mic.0.26563-0. [DOI] [PubMed] [Google Scholar]
- 62.Derzelle S, Turlin E, Duchaud E, Pages S, Kunst F, Givaudan A, et al. The PhoP-PhoQ two-component regulatory system of Photorhabdus luminescens is essential for virulence in insects. J Bacteriol. 2004;186(5):1270–9. doi: 10.1128/JB.186.5.1270-1279.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Reboul A, Lemaitre N, Titecat M, Merchez M, Deloison G, Ricard I, et al. Yersinia pestis requires the 2-component regulatory system OmpR-EnvZ to resist innate immunity during the early and late stages of plague. J Infect Dis. 2014;210(9):1367–75. doi: 10.1093/infdis/jiu274. [DOI] [PubMed] [Google Scholar]
- 64.Krzeslak J, Gerritse G, van Merkerk R, Cool RH, Quax WJ. Lipase expression in Pseudomonas alcaligenes is under the control of a two-component regulatory system. Appl Environ Microbiol. 2008;74(5):1402–11. doi: 10.1128/AEM.01632-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang D, de Souza RF, Anantharaman V, Iyer LM, Aravind L. Polymorphic toxin systems: Comprehensive characterization of trafficking modes, processing, mechanisms of action, immunity and ecology using comparative genomics. Biol Direct. 2012;7:18. doi: 10.1186/1745-6150-7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Oliver KM, Russell JA, Moran NA, Hunter MS. Facultative bacterial symbionts in aphids confer resistance to parasitic wasps. Proc Natl Acad Sci U S A. 2003;100(4):1803–7. doi: 10.1073/pnas.0335320100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jobling MG, Holmes RK. Identification of motifs in cholera toxin A1 polypeptide that are required for its interaction with human ADP-ribosylation factor 6 in a bacterial two-hybrid system. Proc Natl Acad Sci U S A. 2000;97(26):14662–7. doi: 10.1073/pnas.011442598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Obrig TG, Karpman D. Shiga toxin pathogenesis: kidney complications and renal failure. Curr Top Microbiol Immunol. 2012;357:105–36. doi: 10.1007/82_2011_172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Daborn PJ, Waterfield N, Silva CP, Au CP, Sharma S, Ffrench-Constant RH. A single Photorhabdus gene, makes caterpillars floppy (mcf), allows Escherichia coli to persist within and kill insects. Proc Natl Acad Sci U S A. 2002;99(16):10742–7. doi: 10.1073/pnas.102068099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Waterfield NR, Wren BW, Ffrench-Constant RH. Invertebrates as a source of emerging human pathogens. Nat Rev Microbiol. 2004;2(10):833–41. doi: 10.1038/nrmicro1008. [DOI] [PubMed] [Google Scholar]
- 71.Ambrose KV, Koppenhofer AM, Belanger FC. Horizontal gene transfer of a bacterial insect toxin gene into the Epichloe fungal symbionts of grasses. Sci Rep. 2014;4:5562. doi: 10.1038/srep05562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ullah I, Jang EK, Kim MS, Shin JH, Park GS, Khan AR, et al. Identification and characterization of the insecticidal toxin “makes caterpillars floppy” in Photorhabdus temperata M1021 using a cosmid library. Toxins (Basel) 2014;6(7):2024–40. doi: 10.3390/toxins6072024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sheahan KL, Cordero CL, Satchell KJ. Autoprocessing of the Vibrio cholerae RTX toxin by the cysteine protease domain. EMBO J. 2007;26(10):2552–61. doi: 10.1038/sj.emboj.7601700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Agarwal S, Agarwal S, Biancucci M, Satchell KJ. Induced autoprocessing of the cytopathic makes caterpillars floppy-like effector domain of the Vibrio vulnificus MARTX toxin. Cell Microbiol. 2015. doi:10.1111/cmi.12451 [DOI] [PMC free article] [PubMed]
- 75.Caldas C, Cherqui A, Pereira A, Simoes N. Purification and characterization of an extracellular protease from Xenorhabdus nematophila involved in insect immunosuppression. Appl Environ Microbiol. 2002;68(3):1297–304. doi: 10.1128/AEM.68.3.1297-1304.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Massaoud MK, Marokhazi J, Fodor A, Venekei I. Proteolytic enzyme production by strains of the insect pathogen Xenorhabdus and characterization of an early-log-phase-secreted protease as a potential virulence factor. Appl Environ Microbiol. 2010;76(20):6901–9. doi: 10.1128/AEM.01567-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Satchell KJ. Structure and function of MARTX toxins and other large repetitive RTX proteins. Annu Rev Microbiol. 2011;65:71–90. doi: 10.1146/annurev-micro-090110-102943. [DOI] [PubMed] [Google Scholar]
- 78.Satchell KJ. Multifunctional-autoprocessing repeats-in-toxin (MARTX) Toxins of Vibrios. Microbiol Spectr. 2015;3(3). doi:10.1128/microbiolspec.VE-0002-2014. [DOI] [PMC free article] [PubMed]
- 79.Satchell KJ. MARTX, multifunctional autoprocessing repeats-in-toxin toxins. Infect Immun. 2007;75(11):5079–84. doi: 10.1128/IAI.00525-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kwak JS, Jeong HG, Satchell KJ. Vibrio vulnificus rtxA1 gene recombination generates toxin variants with altered potency during intestinal infection. Proc Natl Acad Sci U S A. 2011;108(4):1645–50. doi: 10.1073/pnas.1014339108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Brillard J, Ribeiro C, Boemare N, Brehelin M, Givaudan A. Two distinct hemolytic activities in Xenorhabdus nematophila are active against immunocompetent insect cells. Appl Environ Microbiol. 2001;67(6):2515–25. doi: 10.1128/AEM.67.6.2515-2525.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang X, Hu X, Li Y, Ding X, Yang Q, Sun Y, et al. XaxAB-like binary toxin from Photorhabdus luminescens exhibits both insecticidal activity and cytotoxicity. FEMS Microbiol Lett. 2014;350(1):48–56. doi: 10.1111/1574-6968.12321. [DOI] [PubMed] [Google Scholar]
- 83.Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 1997;13(5):555–6. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- 84.Ogier JC, Calteau A, Forst S, Goodrich-Blair H, Roche D, Rouy Z, et al. Units of plasticity in bacterial genomes: new insight from the comparative genomics of two bacteria interacting with invertebrates. Photorhabdus Xenorhabdus BMC genomics. 2010;11:568. doi:10.1186/1471-2164-11-568. [DOI] [PMC free article] [PubMed]
- 85.Hovde CJ, Calderwood SB, Mekalanos JJ, Collier RJ. Evidence that glutamic acid 167 is an active-site residue of Shiga-like toxin I. Proc Natl Acad Sci U S A. 1988;85(8):2568–72. doi: 10.1073/pnas.85.8.2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Stein PE, Boodhoo A, Tyrrell GJ, Brunton JL, Read RJ. Crystal structure of the cell-binding B oligomer of verotoxin-1 from E. coli. Nature. 1992;355(6362):748–50. doi: 10.1038/355748a0. [DOI] [PubMed] [Google Scholar]
- 87.Brussow H, Canchaya C, Hardt WD. Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiol Mol Biol Rev. 2004;68(3):560–602. doi: 10.1128/MMBR.68.3.560-602.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ffrench-Constant R, Waterfield N. An ABC guide to the bacterial toxin complexes. Adv Appl Microbiol. 2006;58:169–83. doi: 10.1016/S0065-2164(05)58005-5. [DOI] [PubMed] [Google Scholar]
- 89.Waterfield N, Hares M, Yang G, Dowling A. ffrench-Constant R. Potentiation and cellular phenotypes of the insecticidal toxin complexes of Photorhabdus bacteria. Cell Microbiol. 2005;7(3):373–82. doi: 10.1111/j.1462-5822.2004.00467.x. [DOI] [PubMed] [Google Scholar]
- 90.Lang AE, Ernst K, Lee H, Papatheodorou P, Schwan C, Barth H, et al. The chaperone Hsp90 and PPIases of the cyclophilin and FKBP families facilitate membrane translocation of Photorhabdus luminescens ADP-ribosyltransferases. Cell Microbiol. 2014;16(4):490–503. doi: 10.1111/cmi.12228. [DOI] [PubMed] [Google Scholar]
- 91.Lang AE, Schmidt G, Schlosser A, Hey TD, Larrinua IM, Sheets JJ, et al. Photorhabdus luminescens toxins ADP-ribosylate actin and RhoA to force actin clustering. Science. 2010;327(5969):1139–42. doi: 10.1126/science.1184557. [DOI] [PubMed] [Google Scholar]
- 92.Yang G. Waterfield NR. The role of TcdB and TccC subunits in secretion of the Photorhabdus Tcd toxin complex. PLoS Pathog. 2013;9(10):e1003644. doi: 10.1371/journal.ppat.1003644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sergeant M, Jarrett P, Ousley M, Morgan JA. Interactions of insecticidal toxin gene products from Xenorhabdus nematophilus PMFI296. Appl Environ Microbiol. 2003;69(6):3344–9. doi: 10.1128/AEM.69.6.3344-3349.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Didelot X, Achtman M, Parkhill J, Thomson NR, Falush D. A bimodal pattern of relatedness between the Salmonella Paratyphi A and Typhi genomes: convergence or divergence by homologous recombination? Genome Res. 2007;17(1):61–8. doi: 10.1101/gr.5512906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Didelot X, Meric G, Falush D, Darling AE. Impact of homologous and non-homologous recombination in the genomic evolution of Escherichia coli. BMC Genomics. 2012;13:256. doi: 10.1186/1471-2164-13-256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Garcia-Gonzalez E, Muller S, Ensle P, Sussmuth RD, Genersch E. Elucidation of sevadicin, a novel non-ribosomal peptide secondary metabolite produced by the honey bee pathogenic bacterium Paenibacillus larvae. Environ Microbiol. 2014;16(5):1297–309. doi: 10.1111/1462-2920.12417. [DOI] [PubMed] [Google Scholar]
- 97.Cochrane SA, Vederas JC. Lipopeptides from Bacillus and Paenibacillus spp.: A gold mine of antibiotic candidates. Med Res Rev. 2014. doi:10.1002/med.21321 [DOI] [PubMed]
- 98.Luo C, Liu X, Zhou H, Wang X, Chen Z. Identification of four NRPS gene clusters in Bacillus subtilis 916 for four families of lipopeptides biosynthesis and evaluation of their intricate functions to the typical phenotypic features. Appl Environ Microbiol. 2014. doi:10.1128/AEM.02921-14.
- 99.Tanaka A, Tapper BA, Popay A, Parker EJ, Scott B. A symbiosis expressed non-ribosomal peptide synthetase from a mutualistic fungal endophyte of perennial ryegrass confers protection to the symbiotum from insect herbivory. Mol Microbiol. 2005;57(4):1036–50. doi: 10.1111/j.1365-2958.2005.04747.x. [DOI] [PubMed] [Google Scholar]
- 100.Schwecke T, Aparicio JF, Molnar I, Konig A, Khaw LE, Haydock SF, et al. The biosynthetic gene cluster for the polyketide immunosuppressant rapamycin. Proc Natl Acad Sci U S A. 1995;92(17):7839–43. doi: 10.1073/pnas.92.17.7839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Raymond KN, Dertz EA, Kim SS. Enterobactin: an archetype for microbial iron transport. Proc Natl Acad Sci U S A. 2003;100(7):3584–8. doi: 10.1073/pnas.0630018100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cohan FM, Perry EB. A systematics for discovering the fundamental units of bacterial diversity. Curr Biol. 2007;17(10):R373–86. doi: 10.1016/j.cub.2007.03.032. [DOI] [PubMed] [Google Scholar]
- 103.Vallenet D, Belda E, Calteau A, Cruveiller S, Engelen S, Lajus A, et al. MicroScope--an integrated microbial resource for the curation and comparative analysis of genomic and metabolic data. Nucleic Acids Res. 2013;41:D636–47. doi: 10.1093/nar/gks1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Vallenet D, Labarre L, Rouy Z, Barbe V, Bocs S, Cruveiller S, et al. MaGe: a microbial genome annotation system supported by synteny results. Nucleic Acids Res. 2006;34(1):53–65. doi: 10.1093/nar/gkj406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Darling AC, Mau B, Blattner FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14(7):1394–403. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Holmes DS, Quigley M. A rapid boiling method for the preparation of bacterial plasmids. Anal Biochem. 1981;114(1):193–7. doi: 10.1016/0003-2697(81)90473-5. [DOI] [PubMed] [Google Scholar]
- 107.Caspi R, Altman T, Billington R, Dreher K, Foerster H, Fulcher CA, et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 2014;42:D459–71. doi: 10.1093/nar/gkt1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Brugirard-Ricaud K, Givaudan A, Parkhill J, Boemare N, Kunst F, Zumbihl R, et al. Variation in the effectors of the type III secretion system among Photorhabdus species as revealed by genomic analysis. J Bacteriol. 2004;186(13):4376–81. doi: 10.1128/JB.186.13.4376-4381.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 110.Blin K, Medema MH, Kazempour D, Fischbach MA, Breitling R, Takano E, et al. antiSMASH 2.0--a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res. 2013;41:W204–12. doi: 10.1093/nar/gkt449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mitchell A, Chang HY, Daugherty L, Fraser M, Hunter S, Lopez R, et al. The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res. 2015;43:D213–21. doi: 10.1093/nar/gku1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinf. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–7. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Retief JD. Phylogenetic analysis using PHYLIP. Methods Mol Biol. 2000;132:243–58. doi: 10.1385/1-59259-192-2:243. [DOI] [PubMed] [Google Scholar]
- 115.Letunic I, Bork P. Interactive Tree Of Life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res. 2011;39:W475–8. doi: 10.1093/nar/gkr201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–9. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Milne I, Lindner D, Bayer M, Husmeier D, McGuire G, Marshall DF, et al. TOPALi v2: a rich graphical interface for evolutionary analyses of multiple alignments on HPC clusters and multi-core desktops. Bioinformatics. 2009;25(1):126–7. doi: 10.1093/bioinformatics/btn575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hurst LD. The Ka/Ks ratio: diagnosing the form of sequence evolution. Trends Genet. 2002;18(9):486. doi: 10.1016/S0168-9525(02)02722-1. [DOI] [PubMed] [Google Scholar]
- 119.Boemare NE, Akhurst RJ. Biochemical and physiological characterization of colony form variants in Xenorhabdus spp. (Enterobacteriaceae) J Gen Microbiol. 1988;134:751–61. doi: 10.1099/00221287-134-7-1835. [DOI] [PubMed] [Google Scholar]
- 120.Vivas EI, Goodrich-Blair H. Xenorhabdus nematophilus as a model for host-bacterium interactions: rpoS is necessary for mutualism with nematodes. J Bacteriol. 2001;183(16):4687–93. doi: 10.1128/JB.183.16.4687-4693.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sierra G. A simple method for the detection of lipolytic activity of micro-organisms and some observations on the influence of the contact between cells and fatty substrates. Antonie Van Leeuwenhoek. 1957;23(1):15–22. doi: 10.1007/BF02545855. [DOI] [PubMed] [Google Scholar]
- 122.Boemare N, Thaler JO, Lanois A. Simple bacteriological tests for phenotypic characterization of Xenorhabdus and Photorhabdus phase variants. Symbiosis. 1997;22:167–175. [Google Scholar]
- 123.Rowe GE, Welch RA. Assays of hemolytic toxins. Methods Enzymol. 1994;235:657–67. doi: 10.1016/0076-6879(94)35179-1. [DOI] [PubMed] [Google Scholar]
- 124.Akhurst RJ. Antibiotic activity of Xenorhabdus spp., bacteria symbiotically associated with insect pathogenic nematodes of the families Heterorhabditidae and Steinernematidae. J Gen Microbiol. 1982;128(12):3061–5. doi: 10.1099/00221287-128-12-3061. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Genomes have been deposited in GenBank (http://www.ncbi.nlm.nih.gov/genbank/) (Table 1).